
Abstract
Base metal sulfides (Fe–Ni–Cu–S) are ubiquitous phases in mantle and subduction-related lithologies. Sulfides in the mantle often melt incongruently, which leads to the production of a Cu–Ni-rich sulfide melt and a solid residue called monosulfide solid solution (mss). Even though peridotite-hosted sulfides, which tend to be more Ni-rich, are likely completely molten at mantle potential temperatures, the same is not true for eclogite-hosted Ni-poor, Fe-rich sulfides. Because of this, solid crystalline mss may persist at higher pressures and equilibrate with co-existing sulfide melt along colder geotherms, like those associated with subduction zones. Because highly siderophile elements (HSE—Pt, Pd, Rh, Ru, Os, Ir, and Re) are known to fractionate as a result of mss/sulfide-melt equilibrium, the persistence of an mss/sulfide-melt assemblage to higher pressures may lead to the fractionation of these elements during the subduction process. In this contribution, we carried out an experimental investigation of the partitioning behavior of the HSE, as well as Cu and Ni, between mss and sulfide melt over a pressure and temperature range relevant to equilibration between Earth’s surface and transition zone depths (0.1 MPa to 14 GPa; 930–1530 \(^{\circ }\)C), and variable Ni contents in sulfide. Results show that at higher pressures, the HSE are considerably less fractionated as a result of mss and sulfide melt equilibrium compared to lower pressure conditions. This is exemplified by a lowering of the \(D_{i}^\mathrm{mss/melt}\) for the more compatible HSE (Ru, Os, Ir, Rh and Re) from around 10 at 0.1 MPa to just above or below unity at 14 GPa. Moreover, the higher the Ni content of the bulk sulfide assemblage, the larger the degree of change in the magnitude of HSE fractionation seen over the pressure range studied. The exchange coefficient (\(K_D^{\textrm{Ru}-\textrm{Pt}}\)) between highly compatible HSE (Ru) and less compatible Pt illustrates a notable contrast. In the Ni-poor composition (E1), \(K_D^{\textrm{Ru}-\textrm{Pt}}\) changes from 27 at 0.1 MPa to 6 at 14 GPa. In contrast, the Ni-rich composition exhibits a broader range, with \(K_D^{\textrm{Ru}-\textrm{Pt}}\) ranging from 150 to 17 across the same pressure interval. Our results highlight key differences between experimental data obtained at lower and higher pressure, and how composition, namely the Ni content of sulfide, affects HSE partitioning behavior.
Similar content being viewed by others


Introduction
Magmatic sulfides play a significant role in controlling the behavior of highly siderophile elements (HSE) like Pt, Pd, Rh, Ru, Os, Ir, and Re. They both concentrate and fractionate these elements, with Ru, Os, Ir, Re, and Rh generally preferring crystalline sulfide, whereas Pt and Pd tend to remain in sulfide liquids (cf. Fleet et al. 1993). Consequently, extensive research has focused on understanding HSE partitioning during high-temperature equilibria involving sulfides, especially the fractionation of HSE at the monosulfide solid solution (mss) and sulfide melt equilibrium (e.g., Brenan 2002; Barnes et al. 1997; Fleet et al. 1993, 1999; Li et al. 1996; Ballhaus et al. 2001, 2006; Ballhaus and Sylvester 2000; Mungall et al. 2005; Helmy et al. 2013b; Liu and Brenan 2015; Fonseca et al. 2017, to name a few). These studies have shown that Os, Ir, Ru, and Rh tend to favor mss, whereas Pt and Pd, and to a lesser extent Re, remain in the sulfide liquid. Most previous studies were conducted at room pressure (0.1 MPa) because incongruent melting or fractional crystallization of sulfides is primarily expected to occur in the Earth’s crust or uppermost mantle (Ryzhenko et al. 1973; Bockrath et al. 2004; Zhang and Hirschmann 2016). Despite some early controversy (see Bockrath et al. 2004), the broad consensus up to this point is that sulfides are, for the most part, completely molten at upper mantle pressures up to 8 GPa (Zhang and Hirschmann 2016), such that equilibria involving co-existing mss and sulfide melt are likely of minor relevance to the behavior of HSE in Earth’s mantle. Yet, according to Beyer et al. (2022), in situations involving higher pressures or colder geotherms like subduction zones, the persistence of monosulfide solid solution (mss) alongside sulfide melt is likely. This finding underscores the importance of closely examining HSE partitioning in scenarios where both phases coexist. To our knowledge, apart from two experiments conducted at 3 GPa by Ballhaus et al. (2006), no other partitioning studies involving monosulfide solid solution (mss) and sulfide melt have been conducted at higher pressures, especially those pertinent to subduction geotherms, Archean/early Proterozoic geotherms, and the deeper upper mantle near the top of the transition zone. Potential changes to the structure of both mss and sulfide melt in response to higher pressures will likely affect the partitioning behavior of the HSE, as elastic strain associated with cation substitution into either phase is likely different at transition zone pressures (\(> 13\) GPa) compared to lower pressure condition (0.1 MPa–8 GPa).
Here we report the results of a series of experiments carried out over a range of pressures (0.1 MPa–14 GPa), temperatures (970–1530 \(^{\circ }\)C), and variable Fe/Ni (ca. 0.4 to 9 molar) that aimed to investigate the partitioning of the HSE between mss and sulfide melt. Four sulfide compositions (E1, P1, P2 and P3—Table 1) were chosen to reflect those typically found as inclusions in diamonds hosted by peridotitic and eclogitic mantle xenoliths, for which their melting relationships have been thoroughly characterized (Beyer et al. 2022).
Methods
Experimental methods
Bulk compositions were prepared by initially adding Pt, Pd, Rh, Ru, Os, Ir, and Re from Alfa Aesar acid solutions (in 20% HCl, 1000 \(\upmu\)g/mL concentration) to elemental sulfur powder (Alfa Aesar, 99.9% purity) at a ratio of approximately 40–50 \(\upmu\)g/g. The mixture was dried at 60 \(^{\circ }\)C in an oven overnight. Subsequently, metallic powders of Fe, Ni, and Cu (Alfa Aesar, 99.9% purity) were incorporated into the HSE-doped sulfur following the proportions listed in Table 1, using a procedure similar to the one described in prior works, including (Beyer et al. 2022). The powder mixes were thoroughly ground in an agate mortar and then placed inside 6 mm outer and 4 mm inner diameter SiO\(_{2}\) glass tubes. These tubes were sealed at 1 Pa under vacuum using a hydrogen-oxygen torch. To form crystalline sulfide phases, sample powders were stepwise heated to 900 \(^{\circ }\)C. The resulting sulfides were crushed and this process was repeated once more to ensure material homogeneity. The pre-reacted sulfides were then ground and subsequently used in experiments conducted at 0.1 MPa, as well as in both the piston cylinder and multi-anvil apparatuses.
Low-pressure experiments were conducted at 0.1 MPa using each of the four bulk compositions to obtain HSE partitioning data at atmospheric pressure. Approximately 150 mg of sample powders were evacuated at 1 Pa inside SiO\(_{2}\)-sealed glass tubes and reacted in high-temperature furnaces heated using MoSi\(_2\) (Nabertherm LHT17). The temperature range chosen for these experiments allowed for the coexistence of mss and sulfide melt, following the methodology outlined in Beyer et al. (2022). To ensure precise temperature control, a Type-B thermocouple was positioned within 2 cm of the samples. Temperature fluctuations during the experiments were limited to within 3 \(^{\circ }\)C of the set temperature. The experiments were quenched by rapidly removing them from the oven and immersing them in a beaker filled with cold water.
High-pressure experiments were conducted using two different apparatuses: a 2 MN end-load piston-cylinder system (Voggenreiter Mavopress LPC 250/30) and a 630 t Walker-type multi-anvil apparatus (8 and 14 GPa), located at Ruhr-University Bochum. In both cases, the experiments began with subsolidus sintering at temperatures between 800 and 1000 \(^{\circ }\)C, lasting up to 48 h (see Table 2). This step aimed to reduce capsule porosity and prevent sulfide liquid loss along grain boundaries. For piston-cylinder experiments, 1/2" talc-Pyrex pressure assemblies were employed, wrapped in 0.025 mm lead foil or Teflon foil to minimize friction. Temperature was monitored via the use of a type-D thermocouple (W\(_{97}\)Re\(_3\)-W\(_{75}\)Re\(_{25}\)) introduced just above the sample capsule, and insulated with a quadbore alumina rod surrounded by a standard machining (not stainless) steel plug and a crushable MgO plug beneath it. The experiments followed the hot-piston in method. Initially, the sample was pressurized to 90% of the target pressure, followed by heating to 800 \(^{\circ }\)C for sintering. Subsequently, the temperature was increased to 100 \(^{\circ }\)C below the target temperature, and then both pressure and temperature were simultaneously raised to the final run conditions. The experiments were held at these conditions for a specified duration (refer to Table 2), and quenching was achieved by cutting electrical power to the heater.
For experiments using the multi-anvil press, we utilized a 14/8 (14 mm octahedron edge length, 8 mm truncated edge length) octahedral pressure medium at 8 GPa and a 10/4 octahedral pressure medium at 14 GPa. This octahedron consisted of Cr\(_{2}\)O\(_{3}\)-doped magnesium oxide (from Ceramic Substrates, Isle of Wight), a zirconium oxide insulator (OZ8C, Mino Ltd., Japan), and, depending on pressure, either a stepped graphite heater (up to 8 GPa) or a 50 \(\upmu\)m thick rhenium-foil heater (at 14 GPa). Crushable MgO (RM98TE, Rauschert GmbH) and ZrO\(_{2}\) (10/4) plugs were used inside the pressure medium. To monitor temperature, we inserted a type-D thermocouple axially using a quadbore alumina tube, surrounded by a crushable MgO sleeve. Cavities around the thermocouple tip were filled with alumina powder to protect it during compression and prevent deformation of the pressure assembly. Both single- and multi-chamber sample containers made from crushable MgO and SiO\(_{2}\) glass were employed. Refer to Beyer et al. (2022) for detailed dimensions of the assemblies and capsules. Each experiment underwent a slow compression to the target pressure (4–6 h) followed by the aforementioned sintering step. After sintering, the temperature was ramped up to its target value at a rate of 100 \(^{\circ }\)C/min and maintained at the desired temperature for 20–360 min (see Table 2). Quenching was achieved by cutting off electrical power, causing the temperature to drop to below 80 \(^{\circ }\)C within 2–3 s (monitored by the Eurotherm temperature controller).
The multi-anvil press calibration at room temperature involved the use of the electrical resistivity changes of Bi I–II (2.55 GPa) (Bean et al. 1986), Bi III–V (7.7 GPa) (Bean et al. 1986), Zr \(\alpha\)–\(\omega\) (7.96 GPa) (Tange et al. 2011), Pb I–II (13.4 GPa) (Bean et al. 1986), and ZnS (15 GPa) (Piermarini and Block 1975), utilizing 4-wire sensing. For temperature calibration, phase transitions in CaGeO\(_{3}\) (garnet-perovskite, 1200 \(^{\circ }\)C, 5.61 GPa), SiO\(_{2}\) (coesite-stishovite, 1200 \(^{\circ }\)C, 9.27 GPa), and the olivine-wadsleyite (Stixrude and Lithgow-Bertelloni 2011) and olivine-ahrsenite (Chanyshev et al. 2021) binary loops at 1200 and 1400 \(^{\circ }\)C were employed. The pressure calibration was complemented by a temperature calibration using the melting curve of silver (Errandonea 2010) and gold (Akella and Kennedy 1971). Temperature offset corrections were applied based on the thermal model of the assemblies (Hernlund et al. 2006), resulting in a +40 \(^{\circ }\)C correction for the 10/4 experiments and +20 \(^{\circ }\)C for the 14/8 experiments. Pressure effects on thermocouple electromotive force were not considered.
In the piston-cylinder press, pressure calibration utilized the albite breakdown reaction at 900 \(^{\circ }\)C (Holland 1980) and the quartz-coesite phase transition at 1000 \(^{\circ }\)C (Bose and Ganguly 1995), with a 20% friction correction. Temperature monitoring employed a type-D thermocouple with a corundum sleeve, and a hollowed conic pyrophyllite plug was added to prevent thermocouple extrusion during compression and relaxation, ensuring reliable temperature measurements during experiments.
Sulfur and oxygen fugacity (fS\(_2\) and fO\(_2\)) were not explicitly controlled during the experiments, as compositions were chosen to reflect the natural range found in inclusions in diamonds. It is possible to calculate sulfur fugacity from the molar fraction of FeS found in mss, this is only possible for the 0.1 MPa experiments using the calibration of Mengason et al. (2010), values of which can be seen in Table 2. This approach to calculate fS\(_2\), which relies on the measured molar fractions of Fe, Ni, Cu and S, and their uncertainties, results on a large error associated with the calculated fS\(_2\) once uncertainties are propagated (see Table 2. Sulfur fugacities were not calculated for the higher pressure experiments, because activity composition relations for the Fe–FeS system were only determined at high pressures for the metal-rich side of the eutectic of that system (Buono and Walker 2011), which do not cover the compositional range of our experiments.
After the experiments, samples were embedded in epoxy and longitudinally ground to reveal a cross-section. High-quality polished surfaces were obtained through a series of epoxy-bound diamond sanding plates and hard polishing discs with diamond paste down to 1 \(\upmu\)m with a water-free lubricant/cooling liquid (Struers DP Blue). All samples were stored in a desiccator to prevent oxidation of the exposed sulfides.
Aliquots of the twice-sintered bulk compositions were recovered, polished, and analyzed for HSE concentrations and major elements using both electron microprobe (EMPA) and laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), as detailed in Table 1. Although the target values for both major and trace elements were achieved, Pd concentrations in all bulk compositions turned out to be significantly lower than expected, ranging from 0.3 to 5 \(\upmu\)g/g. This discrepancy likely resulted from an inadvertent overdilution of the Alfa Aesar Pd solution used during bulk composition synthesis. Nevertheless, since Pd concentrations were successfully measured in the majority of the experiments, the lower doping levels for this element did not pose an issue, and partitioning data for Pd were obtained. Further details can be found in the companion manuscript by Beyer et al. (2022) published in this journal.
Analytical methods
The textures of sulfide phases in each experiment were examined using optical and electron microscopy. Specifically, a JEOL JSM-7200F SEM at the Center for Interface-Dominated High-Performance Materials (ZGH) of Ruhr-University Bochum (Germany) was used. The major element composition of sulfide phases in the experiments was determined using a JEOL 8530F Field-Emission Electron Microprobe (EMPA) and a Cameca Field-Emission SXFiveFE EMPA, hosted at the Institute for Mineralogy of the University of Münster (Germany) and the Institute for Geology, Mineralogy, and Geophysics of Ruhr-University Bochum (IGMG), respectively. Measurements were conducted at 15 kV accelerating voltage and 15 nA beam current, using a 1–20 \(\upmu\)m-diameter beam to account for quenched sulfide melt heterogeneity. Samples and reference materials were coated with a 10 nm carbon coat to ensure reliable measurements of their oxygen content. Reference materials included pyrite, chalcopyrite, and pentlandite for S, Fe, and Ni, respectively, utilizing peak-to-background calibrations. Oxygen was measured using PC0 and LDEP1 light-element detector crystals in Bochum and Münster respectively. Oxygen content was referenced to periclase (MgO), while oxygen backgrounds were monitored on nominally oxygen-free phases (metals and sulfide reference materials). The detection limit for oxygen under these analytical conditions was 0.1 wt.%. Major element contents for monosulfide solid solution (mss) and sulfide melt in each experiment can be found in Table 3.
Experimental run products were measured for their HSE abundances using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). An initial batch of experiments were measured at the Institute for Mineralogy of the University of Münster using a Photon Machines Analyte G2 193 nm Excimer laser ablation system attached to a Thermo Scientific Element 2 sector field ICP-MS mass spectrometer. The bulk of the experiments were measured at the IGMG using a ESI NWRImageGEO 193 nm excimer laser ablation system coupled to an Agilent 8900 triple quadrupole ICP-MS run in single-quadrupole mode. Following (Wohlgemuth-Ueberwasser et al. 2007), laser fluence and repetition rate were kept low (4–5 J/cm\(^2\), and 5 Hz, respectively) to enhance ablation yield. Spot sizes varied between 40 and 70 \(\upmu\)m depending on the size of the phases measured, as well as the coarseness of sulfide melt quench textures in each experiment. Sample aerosols were carried to the mass spectrometers using a constant He flow of 500–1000 mL/min. For both instrument configurations, the reference materials used for external calibration (synthetic Ni-bearing pyrrhotite—PGE-Ni7b—described in Wohlgemuth-Ueberwasser et al. 2007), and internal standard used for normalization (\(^{57}\)Fe) were the same. The count rates for the nuclides of \(^{34}\)S, \(^{56}\)Fe, \(^{57}\)Fe, \(^{60}\)Ni, \(^{61}\)Ni, \(^{63}\)Cu, \(^{65}\)Cu, \(^{99}\)Ru, \(^{101}\)Ru, \(^{103}\)Rh, \(^{105}\)Pd, \(^{106}\)Pd, \(^{108}\)Pd, \(^{185}\)Re, \(^{189}\)Os, \(^{193}\)Ir and \(^{195}\)Pt were measured. Measurements were carried out via standard/sample bracketing, with no more than 15 spots measured between each standard measurement. Data reduction was carried out using the procedure described by Longerich et al. (1996) in similar fashion to what is described by Fonseca et al. (2017). Possible argide molecular interferences on \(^{101}\)Ru (\(^{61}\)Ni\(^{40}\)Ar\(^+\)), \(^{103}\)Rh (\(^{63}\)Cu\(^{40}\)Ar\(^+\)) and \(^{105}\)Pd (\(^{65}\)Cu\(^{40}\)Ar\(^+\)) were evaluated and, except for \(^{105}\)Pd, found to be negligible given the good agreement between concentrations obtained from multiple isotopes of the same element (i.e., Ru and \(^{106}\)Pd and \(^{108}\)Pd). For Pd, only the concentrations obtained from \(^{106}\)Pd and \(^{108}\)Pd were considered, as those obtained from \(^{105}\)Pd were systematically higher (around 500 ng/g on average), likely due to the aforementioned \(^{65}\)Cu\(^{40}\)Ar\(^+\) interference. Assuming that argide production affects \(^{63}\)Cu to the same extent as it does \(^{65}\)Cu, and considering each isotope’s natural relative abundances, \(^{63}\)Cu\(^{40}\)Ar\(^+\) production will add around 1 \(\upmu\)g/g to the abundances derived from \(^{103}\)Rh count rates, despite the overall low Cu abundances in each bulk composition (5700 to 8200 \(\upmu\)g/g). However, with the exception of a single experiment (P3_2022_2), where sulfide melt has 10 \(\upmu\)g/g of Rh and therefore an interference correction was applied to it, all other experimental run products, had higher than 22 \(\upmu\)g/g, and on average 65 \(\upmu\)g/g of Rh (see Table 4), meaning that the effect of the \(^{63}\)Cu\(^{40}\)Ar\(^+\) interference on \(^{103}\)Rh was mostly negligible and did not affect its derived concentrations beyond measurement uncertainty. The trace element contents for mss and sulfide melt for each experiment are given in Table 4.
Results
Experimental run products
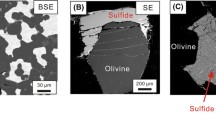
All experiments resulted in two distinct phases: mss and sulfide melt. In subsolidus regions, mss exhibited large subhedral grains with uniform composition (Fig. 1). In low Ni experiments (composition E1—Fig. 1A), microgranular quench textures were observed, where distinguishing grain boundaries between quench crystals and subsolidus mss were challenging through backscatter electron imaging (BSE). The primary differentiation often relied on their response to polishing; mss crystals were more susceptible to removal during polishing than quenched sulfide melt. In some instances, particularly for Ni-poor compositions like E1 (Fig. 1A), clear identification of mss and sulfide melt required X-ray elemental mapping. Here, the slightly more Ni-rich, and Fe-poor melt was distinguishable from the more Ni-poor and Fe-rich mss (see Fig. 5A in Beyer et al. (2022)). Conversely, experiments with higher Ni compositions (Fig. 1B) displayed quenched sulfide melt characterized by Ni-rich intergranular regions surrounding relatively Ni-poor quenched mss grains. Additional details on interpreting sulfide textures in such experiments can be found in Beyer et al. (2022). The boundary between mss and sulfide melt was typically sharp and slightly concave, as observed in Figs. 1A, B. However, in cases where one phase dominated over the other, a clear meniscus did not form, and the subordinate phase was typically intergrown with the dominant one, as shown in Fig. 1C, where the subordinate sulfide melt is integrated into crystalline mss.

Backscatter electron images of typical experimental run products. Coexisting sulfide melt and monosulfide solid solution (mss) in low- and high-Ni experiments (A and B, respectively) with a sharp concave meniscus forming at the contact between both phases. In some instances, no meniscus formed, and both phases appear intergrown (C—see text for more detail)
Partitioning of HSE, Ni, and Cu between mss and sulfide melt
We have summarized all mss/sulfide-melt partition coefficients for HSE, Ni, and Cu in Table 5, and these data are also depicted in Figs. 2 and 3. It is noteworthy that, with a few exceptions (e.g., 3 GPa data from Ballhaus et al. 2006), the majority of experimental data on HSE partitioning between mss and sulfide melt in the literature has been conducted at low pressure (Ballhaus et al. 2006; Mungall et al. 2005; Brenan 2002; Liu and Brenan 2015; Fleet et al. 1993; Fonseca et al. 2017). We conducted a series of experiments at 0.1 MPa using all bulk compositions for comparison with previous literature data. It is important to note that we excluded the experimental data from Helmy et al. (2013b) and Helmy et al. (2013a) since those studies focused on the complexing effect of sub-metals (e.g., As, Sb, Se, Te, etc.) on \(D_\textrm{HSE}^\mathrm{mss/melt}\), which falls outside the scope of our study. \(D_\textrm{HSE}^\mathrm{mss/melt}\) values for Ru, Os, and Ir, and to a lesser extent for Re and Rh, are up to two orders of magnitude higher than those for Pt and Pd, as well as \(D_\textrm{Ni}^\mathrm{mss/melt}\) and \(D_\textrm{Cu}^{mss/melt}\) (Fig. 2). Notably, only the most Ni-rich composition (P3) closely matched literature values for \(D_\textrm{HSE}^{mss/melt}\) (black circles in Fig. 2A). Compositions with intermediate Ni contents (e.g., P2) exhibited \(D_\textrm{HSE}^{mss/melt}\) values at the lower end of the literature range (green circles in Fig. 2A), while experiments with the Ni-poor composition (E1) yielded \(D_{HSE}^{mss/melt}\) values lower than the literature range, except for Pd and Pt (purple symbols in Fig. 2A).
At higher pressures (i.e., at and above 8 GPa), the effect of Ni becomes less pronounced, with no discernible difference in \(D_\textrm{HSE}^\mathrm{mss/melt}\) values at 8 and 14 GPa, regardless of the Ni content (Fig. 2B, C). At higher pressures, \(D_\textrm{HSE}^{mss/melt}\) for Ru, Os, Ir, Re, and Rh approaches unity (between 0.75 and 4.3). Notably, \(D_\textrm{Ir}^\mathrm{mss/melt}\) can be at or below unity for certain compositions (E1, P1, and P2) above 8 GPa, lower than neighboring HSE such as Os and Re, which exhibit similar compatibility in mss at ambient pressure.
Pressure appears to modify \(D_\textrm{HSE}^\mathrm{mss/melt}\) more significantly for the more Ni-rich composition (P3) compared to the most Ni-poor, Fe-rich composition (E1) (Fig. 3A, C). For composition P3, a decrease in \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Ru, Os, Ir, Re, and Rh is evident at 2 GPa, intensifying at 14 GPa. Conversely, composition E1 shows only a moderate decrease in \(D_\textrm{HSE}^\mathrm{mss/melt}\) with increasing pressure, with \(D_\textrm{Pd}^\mathrm{mss/melt}\) and \(D_\textrm{Pt}^\mathrm{mss/melt}\) appearing to increase with pressure (Fig. 3C). Experiments with intermediate Ni bulk contents, like P2, exhibit no significant differences in \(D_{HSE}^{mss/melt}\) above 8 GPa, except for \(D_\textrm{Pd}^\mathrm{mss/melt}\), which does not exhibit a systematic change with pressure. A single experiment at 2 GPa resulted in higher \(D_\textrm{HSE}^\mathrm{mss/melt}\) values (specifically, Ru, Os, Ir, Re, and Rh) compared to other P2 experiments at various pressures, including those conducted at 0.1 MPa. These experiments show a slight negative correlation between \(D_\textrm{HSE}^\mathrm{mss/melt}\) for the most compatible HSE and temperature, with the exception of Pt and Pd, which display significant scatter (Fig. S1). In general, experiments using composition P2 yielded more variable results, likely due to a more heterogeneous bulk starting composition (see Table 1), despite employing the same synthesis procedure as the other three compositions. This heterogeneity may have contributed to the greater variability in \(D_\textrm{HSE}^\mathrm{mss/melt}\) values obtained from P2 experiments. Nevertheless, the overall pattern of decreasing \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Ru, Os, Ir, and Rh with increasing pressure remains consistent with the other compositions.

The effect of Ni content on mss/sulfide-melt partitioning at low (0.1 MPa—A), intermediate (7–8 GPa—B), and higher pressures (14 GPa—C). Except for Cu and Pt, increasing the bulk Ni content appears to lead to an increase in \(D_\textrm{HSE}^\mathrm{mss/melt}\) at low pressure (0.1 MPa), promoting HSE fractionation. However, at higher pressures (above 7 GPa), the bulk Ni content of each experiment appears to make no difference to the value of \(D_\textrm{HSE}^\mathrm{mss/melt}\). Shaded area represents literature data (Ballhaus et al. 2006; Mungall et al. 2005; Brenan 2002, 2008; Liu and Brenan 2015; Fleet et al. 1993; Fonseca et al. 2017) collected from experiments at 0.1 MPa

Effect of pressure on the mss/sulfide-melt partitioning behavior of the HSE for three different compositions, P2 (A), P3 (B), and E1 (C). For all compositions, higher pressures result in a decrease in the extent of HSE fractionation, although this pressure effect is more pronounced for Ni-rich compositions compared to Ni-poor compositions (see text for discussion)
Discussion
Comparison to previous studies
Past experimental studies selected bulk compositions to match the compositional range of magmatic sulfides found in the Earth’s crust. These crustal sulfides typically exhibit a narrower range of Ni and higher Cu contents compared to our experimental compositions (Fig. 4). This choice was driven by the recognition that the primary mechanism for HSE fractionation in the Earth’s crust, particularly in the context of magmatic sulfide deposits, involves the fractional crystallization of mss from a residual sulfide melt (e.g., Naldrett 1969; Naldrett et al. 1967; Craig and Kullerud 1969; Mungall et al. 2005). Consequently, extensive data on HSE mss/sulfide-melt partitioning have been accumulated from experiments conducted at 0.1 MPa using compositions richer in Cu and featuring a narrower Ni range. In contrast, our chosen bulk compositions span the compositional spectrum observed in sulfide inclusions within peridotitic (P1, P2, and P3) and eclogitic (E1) diamonds (Bulanova et al. 1996; Westerlund et al. 2006). Therefore, our compositions better represent deeper mantle domains compared to prior studies.
Comparing our \(D_\textrm{HSE}^\mathrm{mss/melt}\) values obtained at 0.1 MPa with literature data reveals significant disparities arising from differences in our bulk composition range compared to previous studies. Notably, \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Ru, Os, Ir, Re, and Rh largely align with the literature range for moderate- to high-Ni compositions (P2 and P3). Conversely, \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Fe-rich compositions, such as E1, consistently exhibit lower values. Prior research by Brenan (2008) and Fonseca et al. (2011) observed that the solubility of Os and Ir in sulfide melt increased with Ni content, resulting in lower \(D_\textrm{Os}^\mathrm{alloy/melt}\) and \(D_\textrm{Ir}^\mathrm{alloy/melt}\) in more Ni-rich experiments. A similar trend was identified for Ru by Andrews and Brenan (2002), who demonstrated that \(D_\textrm{Ru}^\mathrm{alloy/melt}\) could increase by up to a factor of three when sulfide melt contained higher Ni concentrations. This phenomenon is likely coupled to a decrease in the activity coefficients of the HSE species present in the sulfide liquid, as suggested for example by Andrews and Brenan (2002) and later by Fonseca et al. (2012). Based on these findings, it is reasonable to infer that the solubilities of Ru, Os, and Ir increase with rising Ni content in sulfide melt, and therefore the \(D_\textrm{HSE}^\mathrm{mss/melt}\) is expected to decrease with melt Ni content, whereas the data indicate that the opposite is true. Results indicate that a concomitant increase in the Ni content of mss both leads to an increase in the \(D_\textrm{HSE}^\mathrm{mss/melt}\) values for these elements, and seems to outweigh the positive effect that Ni has on their solubility sulfide melt. In their study, Fleet et al. (1993) pointed out that Pt and Pd tend to exhibit an affinity for Ni, behaving somewhat like Ni during mss/sulfide-melt equilibria. When it comes to partition coefficients, \(D_\textrm{Pt}^\mathrm{mss/melt}\) does not exhibit a consistent correlation with Ni content, whereas \(D_\textrm{Pd}^\mathrm{mss/melt}\) tends to increase with higher bulk Ni content in the experiment at 0.1 MPa. This suggests that the high-Ni content either in mss or the melt leads Pd to preferentially substitute into the former rather than having its solubility enhanced in the latter. An overview of literature data (see compilation in Supplementary Table S1) does not reveal any systematic changes in \(D_\textrm{HSE}^\mathrm{mss/melt}\) with Ni content (Figure S2 in the Supplement). This lack of clarity is compounded by the relatively narrow range of Ni contents in most past studies (Fig. 4). For instance, when comparing experiments from Ballhaus et al. (2006) and Fleet et al. (1993), which have varying Ni contents (i.e., 16 wt.% for Ballhaus et al. 2006 and approximately 7 wt.% for Fleet et al. 1993) but similar Fe and Cu contents, no clear difference in their \(D_\textrm{HSE}^\mathrm{mss/melt}\) is evident.
The influence of Cu on \(D_\textrm{HSE}^\mathrm{mss/melt}\) appears more evident in the literature, particularly in the experiments conducted by Liu and Brenan (2015), which had a relatively high bulk Cu content of 16.5 wt.%. These experiments displayed the highest \(D_\textrm{HSE}^\mathrm{mss/melt}\) values for Ru, Os, Ir, Re, and Rh, while exhibiting the lowest values for Pt and Pd in our compilation of literature data. However, it is worth noting that these elevated \(D_\textrm{HSE}^\mathrm{mss/melt}\) values reported by Liu and Brenan (2015) are less relevant for mantle conditions, as mantle sulfides, at least those found as inclusions in eclogitic and peridotitic diamonds, are Cu-poor (e.g., 0.5–2 wt.% Westerlund et al. 2006; Bulanova et al. 1996; Aulbach et al. 2009) when compared to crustal sulfides, which can have Cu contents in the tens of wt.%.
Interestingly, our experiments reveal that both \(D_\textrm{Ni}^\mathrm{mss/melt}\) and \(D_\textrm{Cu}^\mathrm{mss/melt}\) values are lower than those found in the literature, regardless of the Ni content. This behavior may be linked to the fact that all our bulk compositions have very low Cu concentrations (< 1 wt.% Cu) compared to previous studies, causing Cu to behave more like a trace element. Consequently, Cu partitioning follows Henry’s law, resulting in lower \(D_\textrm{Cu}^\mathrm{mss/melt}\) values compared to those reported in the literature where experiments tend to have higher bulk Cu contents. However, this explanation does not seem to apply to Ni. As stated previously, neither fS\(_2\) nor fO\(_2\) have been explicitly controlled in our experiments as we opted to use bulk compositions that reflect that seen in natural sulfide diamond inclusions. As such, variations in these two parameters could potentially explain our \(D_\textrm{Ni}^\mathrm{mss/melt}\) values. However, \(D_\textrm{Ni}^\mathrm{mss/melt}\) shows no covariation with calculated fS\(_2\) in the experiments carried out at 0.1 MPa. It should be noted, however, that the propagated uncertainties for the calculated fS\(_2\) are very high, such that they could obfuscate any correlation between that parameter and \(D_\textrm{Ni}^\mathrm{mss/melt}\). Moreover, the vast majority of the experimental run products were nominally oxygen-free, and only one experiment (E314\(\_\)P3) had more than 1 wt.% O. At is stands, neither redox variable can account for the observed discrepancy between our \(D_\textrm{Ni}^\mathrm{mss/melt}\) and literature values.

comparison between the bulk starting compositions depicted in this study, compared to those from the experimental literature. Note how our bulk compositions, which cover the natural range of sulfide inclusions in eclogitic and peridotitic diamonds, display a much wider range in Ni contents, and are Cu-poor in comparison to previous work, which is more realistic for mantle-derived sulfides. Data sources from the experimental literature from Ballhaus et al. (2006); Mungall et al. (2005); Brenan (2002, 2008); Liu and Brenan (2015); Fleet et al. (1993) and Fonseca et al. (2017). Natural sulfide diamond inclusion data from Bulanova et al. (1996) and Westerlund et al. (2006)
The effect of pressure on HSE mss/sulfide-melt partitioning
The partitioning behavior of the HSE between mss and sulfide melt, as well as their spatial coordination within these phases, is well-understood at low pressures but less so at higher pressures. Elements like Ru, Os, Ir, Re, and Rh show a distinct preference for mss and likely occur in sixfold coordination in the NiAs-type structure (Raybaud et al. 1997; Liu and Brenan 2015). Conversely, Pt and Pd, which exist in fourfold coordination in their own sulfide phases (e.g., PtS and PdS), are probably more stable in square planar coordination in sulfide liquid (Liu and Brenan 2015); hence, their lower \(D_\textrm{HSE}^\mathrm{mss/melt}\). Moreover, at low pressure, HSE incorporation into mss is universally promoted by increased metal deficiency (i.e., lower molar metal/S) regardless of whether the HSE is incompatible into mss (Ballhaus and Ulmer 1995). Our results clearly show that the substitution of the HSE in either mss or sulfide melt, as characterized for lower pressures, cannot be extrapolated to higher pressures. It is clear that the overall structure of mss, sulfide melt, and probably the coordination polyhedra of the individual species in either phase must change with increasing pressure and Ni content, but the exact nature of this change is unclear.
It is known that near stoichiometric mss (i.e. FeS) transforms from an NiAs-type phase to an hexagonal superstructure at pressures above 5 GPa (Fei et al. 1995; Urakawa et al. 2002), and that it adopts near ideal close packing (c/a axial ratio of 1.63) (Fei et al. 1995). Fei et al. (1995) argued that this transformation results in shorter bonding distances between Fe–Fe pairs, and the overall metallization of mss. The change in the crystal packing and metallization of the bonding environment of mss above 5 GPa will undoubtedly have an effect on the partitioning behavior of the HSE as it may even out any steric effects that contributed to the fractionation of Pt and Pd from Ru, Os, Ir, Rh and Re (see discussion in Liu and Brenan 2015, for more details). This expectation is at least in part borne by our data, given that already at 8 GPa, there is a substantial pressure effect on \(D_\textrm{HSE}^\mathrm{mss/melt}\) and attenuation of the extent of HSE fractionation in our more Fe-rich compositions (E1 and P2—Fig. 2). However, the FeS phase transition at 5 GPa reported by Fei et al. (1995) cannot fully account for the attenuation of differences in \(D_\textrm{HSE}^\mathrm{mss/melt}\) seen at 2 GPa, and especially 14 GPa, for the most Ni-rich composition (P3—Fig. 3B).
Compared to Fe sulfides, there are fewer studies on the response of the Ni-S compositional end-members at high pressures and temperatures. Campbell and Heinz (1993) reported the equation of state of NiS and its phase transitions at higher pressures in the NiAs structure, and reported a moderate change in its c/a axial ratio between 2.8 and 13 GPa (1.539–1.523—or 1% change) and no phase transition within that pressure range. This contrasts with the response of FeS to higher pressures, where from the phase transition at 5 GPa, its c/a axial ratio changes from 1.69 down to the aforementioned 1.63 at 13 GPa (3.5% change). Urakawa et al. (2011) reported on the effect of pressure on the Ni\(_3\)S component, which purportedly stabilizes above 5.1 GPa and, like with NiS, no phase transition was observed over the range studied (up to 10 GPa). The c/a axial ratio of Ni\(_3\)S is shown to decrease by only 0.3% (between 0.5040 and 0.5024 at 0.02 and 8.82 GPa, respectively—Urakawa et al. 2011) implying no significant shortening of Ni–Ni interatomic distances as in the case of the FeS end-member, and thus a lower extent of metallization of its lattice. The observation that our more Ni-rich compositions display larger magnitudes of change in their \(D_\textrm{HSE}^\mathrm{mss/melt}\) compared to those rich in Fe is, therefore, hard to reconcile with the limited dataset for Ni sulfides, as these data do not suggest the existence of significant structural changes in crystalline Ni-rich mss to account for the variations in \(D_\textrm{HSE}^\mathrm{mss/melt}\) seen here. At face value, one would expect that at higher pressure, the Fe-rich compositions would then exhibit the more pronounced pressure effects on \(D_\textrm{HSE}^\mathrm{mss/melt}\), which does not appear to be the case, so an alternate explanation is necessary.
Sulfide liquids, which are highly compressible and poorly ordered liquids compared to pure molten Fe alloys (Sanloup et al. 2000, 2002), are also known to undergo structural changes with increasing pressure. For example, Urakawa et al. (1998) found that the near-neighbor structure of FeS melts does not change significantly for pressures up to 5.6 GPa. Namely, the overall coordination number (CN) in FeS melt and and its Fe–Fe, Fe–S, and S–S interatomic distances only see a slight increase (CN between 5.6 and 6.1), and decrease (2.8–2.48 Å for Fe–Fe, 2.39–2.34 Å for Fe–S, and 2.08 Å for S–S), respectively. However, Morard et al. (2007) studied the structure of eutectic melts in the Fe–S system up to 25 GPa and noted a non-linear stepwise increase in compacity below 5 GPa and again above 13 GPa, with no change in that parameter in between, which correlates with observed changes in eutectic S content (Fei et al. 1997; Usselman 1975). Morard et al. (2007) noted that the change in compacity with increasing pressure will undoubtedly result in changes in trace element partitioning behavior, as the sulfide melt’s bulk modulus, as well as the nature of the Fe–S bond change with pressure. Unfortunately, to date, no study has systematically looked at how sulfide melt structure affects HSE partitioning behavior, as most studies focused on Fe-rich, Ni-poor or Ni-free compositions in the context of planetary core formation (Morard et al. 2007). Nevertheless, it is of note that the first change in compacity with pressure takes place at 5 GPa, which is very close to the first phase transition observed for mss (Fei et al. 1995; Urakawa et al. 2002; Morard et al. 2007). In light of our results, the combined structural changes in mss and sulfide melt seemingly resulted in a lowering of the activity coefficients of Ru, Os, Ir, Re, and Rh species sulfide melt, which results in lower \(D_\textrm{HSE}^\mathrm{mss/melt}\). Interestingly, among the more compatible HSE, the effect of pressure in lowering \(D_\textrm{HSE}^\mathrm{mss/melt}\) seems to be more pronounced for Ir and, to a lesser extent Rh. Iridium, in particular, displays \(D_\textrm{Ir}^\mathrm{mss/melt}\) that are lower than neighboring Os and Re, and shows values closer to that of Rh at higher pressures, whereas no such fractionation occurred at lower pressures (see Fig. 3). One possible clue to resolve this disparate behavior may be related to the type of crystal packing adopted by the Ir and Rh end-members in the their ternary systems with Fe–S. Both Ir and Rh exhibit face-centered-cubic (fcc) packing in their respective ternaries, the same as the Fe end-member, which enables these elements to form complete solution with Fe at higher temperatures (Swartzendruber 1984; Schwerdtfeger and Zwell 1968). In contrast, the Ru, Os, and Re end-members display hexagonal-close-packed (hcp) packing, and have only limited solution with the Fe end-member (Swartzendruber and Sundman 1983b, a; Karup-Møller and Makovicky 2002, 1999). It is, therefore, possible that the phase transition at 5 GPa associated with the FeS end-member in mss is affecting the behavior of Ir, and to some degree also of Rh, to a greater extent than the hcp, Ru, Os and Re end-members. It is clear from the somewhat speculative nature of this discussion that more work is needed to understand the speciation of HSE in mss and sulfide melt over the pressure range studied. Additional in situ spectroscopic data are clearly needed to fully understand how changes in the structure of these phases affect HSE partitioning behavior.
Implications
The results of our experiments have some important implications to our understanding of how HSE may or may not fractionate in Earth’s deeper mantle, namely at greater depths like those associated with cratonic roots (120–280 km depth), the subcontinental lithospheric mantle (SCLM) (120–410 km depth), and the uppermost transition zone (>410 km depth). The \(D_\textrm{HSE}^\mathrm{mss/melt}\) data of our high-pressure experiments suggest that \(D_\textrm{HSE}^\mathrm{mss/melt}\) obtained at 0.1 MPa are not applicable to sulfide melting processes that occurred in the deeper Earth’s mantle. First and foremost, equilibria between mss and sulfide melt result in only modest HSE fractionation above 7 GPa (90 km depth) (Fig. 3). When combined with recent data on the melting behavior of mantle sulfides (Beyer et al. 2022; Zhang and Hirschmann 2016), two scenarios are of particular interest. First, in subduction-related lithologies (i.e. eclogites) where sulfides tend to be more Fe-rich (as per the diamond sulfide inclusion record—see Beyer et al. 2022) moderate HSE fractionation is expected only at depth below \(\sim {}\)180 km, where these more refractory sulfides begin to melt. Thus, HSE fractionation is not expected at shallower depths sampled by lithospheric diamonds. Indeed HSE data on sulfide inclusions in eclogitic diamonds do not show HSE fractionation relative to the MORB-precursor (Aulbach et al. 2012). However, because of the extent of the mss–sulfide liquid field to the top of the transition zone, Fe-rich sulfide may fractionate over a wide depth interval in the SCLM, where the record of sulfide inclusions in diamonds is limited, likely not representative of the ambient mantle (see Stachel and Harris 2008 for a detailed discussion).
Second, although \(D_\textrm{HSE}^\mathrm{mss/melt}\) of peridotitic Ni-rich sulfides look similar to their Fe-rich counterparts, the pressure–temperature space where Ni-rich sulfides are partially molten broadly overlaps with the depth of lithospheric diamond formation (cf. Fig. 10 in Zhang and Hirschmann 2016 and Fig. 11b in Beyer et al. 2022). However, Ni-rich sulfides are likely entirely molten in the SCLM and transition zone (Beyer et al. 2022; Zhang and Hirschmann 2016), suggesting that if Ni-rich sulfides fractionate in the deeper Earth’s mantle, it probably occurs predominately along typical cratonic geotherms (30–50 mW/m\(^2\)) at depths not greater than 180 km. Thus, HSE fractionation is expected to be associated with thick lithospheric roots below Proterozoic and Archean cratons, where Ni-rich sulfides are below their solidus. Interestingly, this region is the source of the vast majority of mantle diamonds (Stachel et al. 2022, for a review) that sample co-existing sulfides. These sulfides probably reflect their primary composition because of the inert nature of the diamond host. It has to be mentioned that the melting depth of sulfides also depends on minor elements, namely C and O, which are known to depress the solidus (Zhang et al. 2015; Urakawa et al. 1987). However, given that the reported fO\(_2\) values for the deeper mantle are considerably more reducing than those for the upper mantle (Frost and McCammon 2008), it is unlikely that O will be an important factor. The same cannot be said for C, which has been shown to significantly depress the solidus of sulfide (Urakawa et al. 1987; Zhang et al. 2015). However, the solubility of C in sulfide liquids within the diamond stability field is not well constrained, as all experimental work has been carried out in graphite-saturated systems (e.g. Zhang et al. 2015, 2018, 2019). It is, thus, unclear how much C can dissolve in sulfide melt beyond the graphite-diamond transition, and to what extent C saturation within the diamond stability field depresses the sulfide solidus.
Consideration should be given to the potential implications of our experimental data on the Pt–Re–Os isotope system. Sulfides play a crucial role as carriers of radioactive isotopes such as \(^{190}\)Pt and \(^{187}\)Re, along with their radiogenic descendants \(^{186}\)Os and \(^{187}\)Os (cf. Luguet et al. 2003, 2008; Lorand and Luguet 2016). Processes that may induce fractionation between Pt, Re, and Os, such as mss-melt equilibria, could lead to isotope anomalies in these systems. In the context of the Re–Os system, our data suggest that the persistence of mss and sulfide melt at higher pressures actually reduces the degree of fractionation for both elements. This reduction occurs because \(D_i^\mathrm{mss/melt}\) values for both Re and Os converge at higher pressures, regardless of Ni content (Table 5). A slight change in K\(_D^\mathrm{Re-Os}\) is observed between mss and sulfide melt, from approximately 0.5 to 0.7 at pressures up to 2 GPa, and from 0.8 to 1 at pressures above 7 GPa. Consequently, our results suggest that minimal Re–Os fractionation is anticipated from mss-melt equilibria at higher pressures compared to lower pressure conditions, where sulfide melt would exhibit a higher Re/Os ratio compared to mss. Therefore, no significant isotope anomalies are expected from mss-sulfide melt equilibration at pressures exceeding 7 GPa, which are experienced in regions like the lithospheric mantle and cratonic roots and the sub-lithospheric mantle.
The Pt–Os system exhibits a similar trend, with K\(_D^\mathrm{Pt-Os}\) varying significantly between low- and high-pressure experiments. Notably, while Ni content has no impact on Re–Os fractionation, it does influence Pt–Os fractionation. For instance, K\(_D^\mathrm{Pt-Os}\) ranges from approximately 0.004–0.26 for Ni-poor compositions (e.g. E1), but only between 0.002 and 0.063 for Ni-rich compositions (e.g. P3). The transition from low to high K\(_D^{Pt-Os}\) values occurs above 7 GPa. In practical terms, similar to the Re–Os system, when both mantle sulfide solid (mss) and sulfide melt coexist, Pt–Os fractionation is reduced, though not entirely eliminated, at pressures above 7 GPa. In other words, at higher pressures, mss/melt equilibria are unlikely to yield significant Pt/Os fractionation in either mss or sulfide melt, preventing measurable \(^{186}\)Os production from such a long-lived decay system (450–469 Ga—Carlson 2005; Walker et al. 1997; Begemann et al. 2001).
Concluding remarks
Our results unequivocally show that HSE partitioning data between mss and sulfide liquid, which up until now have been obtained at lower pressure conditions, cannot be extrapolated to higher pressures. At higher pressures, above \(\sim\) 8 GPa, \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Ru, Os, Re, and especially Ir and Rh become significantly lower compared to lower pressure experiments. The extent of this decrease from lower pressure is a function of the bulk Ni content of the sulfide assemblage, with the most Ni-rich compositions experiencing the largest decrease in \(D_\textrm{HSE}^\mathrm{mss/melt}\) for these elements, and Fe-rich compositions the lowest. In contrast, the \(D_\textrm{HSE}^\mathrm{mss/melt}\) for Pt and Pd change little with pressure, as well as with bulk Ni content. The observed changes in \(D_\textrm{HSE}^\mathrm{mss/melt}\) with pressure are probably associated with a sudden change in the crystal structure of mss above 5 GPa, as well as stepwise changes in the bulk modulus of the sulfide melt between 5 and 13 GPa. However, the exact substitution mechanisms of the HSE, their spatial coordination, and speciation in mss and sulfide melt at higher pressures remain elusive and further investigation is required to obtain this information. Our results imply that at greater mantle pressures, the expected degree of fractionation between the compatible and incompatible HSE decreases for all type of sulfides by one to two orders of magnitude (here expressed as K\(_D^{(\mathrm Ru-Pt)}\), see Table 5). Furthermore, our data indicate that at pressures exceeding 7 GPa, equilibria between mantle mss and sulfide melt are unlikely to cause substantial fractionation in the Re–Os and Pt–Os systems. Consequently, such conditions are expected to yield minimal to no radiogenic Os isotope anomalies. Thus, the degree of elemental fractionation can be used as a gauge to discriminate between sulfides that originated in the uppermost mantle or in the lithospheric to sub-lithospheric mantle, but radiogenic Os isotope data are likely not useful for that purpose. This implies that Os isotope data obtained from sulfide inclusions in diamonds, at pressures characteristic of cratonic roots and the sub-lithospheric mantle (>7 GPa), remain largely unaltered at the time of entrapment. Consequently, any Os model age information derived from these sulfide inclusions accurately reflects processes that occurred prior to entrapment, or at least not the ages are not modified by mss-melt equilibration.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper or its supplementary information files.
References
Akella J, Kennedy GC (1971) Melting of gold, silver, and copper-proposal for a new high-pressure calibration scale. J Geophys Res 76(20):4969–4977
Andrews DRA, Brenan JM (2002) The solubility of ruthenium in sulphide liquid: implications for platinum group mineral stability and sulfide melt-silicate melt partitioning. Chem Geol 192:163–181
Aulbach S, Creaser RA, Pearson NJ, Simonetti SS, Heaman LM, Griffin WL, Stachel T (2009) Sulfide and whole rock re-os systematics of eclogite and pyroxenite xenoliths from the slave craton, Canada. Earth Planet Sci Lett 283(1–4):48–58
Aulbach S, Stachel T, Seitz HM, Brey GP (2012) Chalcophile and siderophile elements in sulphide inclusions in eclogitic diamonds and metal cycling in a Paleoproterozoic subduction zone. Geochim Cosmochim Acta 93:278–299. https://doi.org/10.1016/j.gca.2012.04.027http://www.sciencedirect.com/science/article/pii/S0016703712002323
Ballhaus C, Sylvester P (2000) Noble metal enrichment processes in the Merensky Reef, Bushveld complex. J Pet 41(4):545–561
Ballhaus C, Ulmer P (1995) Platinum-group elements in the Merensky reef: II. Experimental solubilities of platinum and palladium in Fe\(_{1-x}\)S from 950 to 450\(^{\circ }\)C under controlled fS\(_{2}\) and fH\(_{2}\). Geochim Cosmochim Acta 59(23):4881–4888
Ballhaus C, Tredoux M, Späth A (2001) Phase relations in the Fe-Ni-Cu-PGE-S system at magmatic temperature and application to massive sulphide ores of the Sudbury igneous complex. J Pet 42(10):1911–1926
Ballhaus C, Bockrath C, Wohlgemuth-Ueberwasser C, Laurenz V, Berndt J (2006) Fractionation of the noble metals by physical processes. Contrib Miner Petrol 152(6):667–684. https://doi.org/10.1007/s00410-006-0126-z
Barnes SJ, Makovicky E, Karup-Møller S, Makovicky M, Rose-Hansen J (1997) Partition coefficients for Ni, Cu, Pd, Pt, Rh and Ir between monosulphide solid solution and sulphide liquid and the implications for the formation of compositionally zoned Ni-Cu sulphide bodies by fractional crystallization of sulphide liquid. Can J Earth Sci 34:366–374
Bean VE, Akimoto S, Bell PM, Block S, Holzapfel WB, Manghnani MH, Nicol MF, Stishov SM (1986) Another step toward an international practical pressure scale: 2nd AIRAPT IPPS task group report. Physica B+C 139-140:52–54, https://doi.org/10.1016/0378-4363(86)90521-8, https://www.sciencedirect.com/science/article/pii/0378436386905218
Begemann F, Ludwig KR, Lugmair GW, Min K, Nyquist LE, Patchett PJ, Renne PR, Shih CY, Villa IM, Walker RJ (2001) Call for an improved set of decay constants for geochronological use. Geochim Cosmochim Acta 65(1):111–121
Beyer C, Bissbort T, Hartmann R, Berndt J, Klemme S, Fonseca ROC (2022) High-pressure phase relations in the system Fe-Ni-Cu-S up to 14 GPa: implications for the stability of sulfides in the Earth’s upper mantle. Contrib Miner Petrol 177(10):99
Bockrath C, Ballhaus C, Holzheid A (2004) Fractionation of the platinum group elements during mantle melting. Science 305:1951–1953
Bose K, Ganguly J (1995) Experimental and theoretical studies of the stabilities of talc, antigorite and phase A at high pressures with applications to subduction processes. Earth Planet Sci Lett 136(3–4):109–121. https://doi.org/10.1016/0012-821X(95)00188-I , http://www.sciencedirect.com/science/article/pii/0012821X9500188I
Brenan JM (2002) Re-Os fractionation in magmatic sulfide melt by monosulfide solid solution. Earth Planet Sci Lett 199(3–4):257–268
Brenan MJ (2008) Re-Os fractionation by sulfide melt-silicate melt partitioning: a new spin. Chem Geol 248:140–165
Bulanova GP, Griffin WL, Ryan CG, Shestakova OY, Barnes SJ (1996) Trace elements in sulfide inclusions from Vakutian diamonds. Contrib Miner Petrol 124(2):111–125
Buono AS, Walker D (2011) The Fe-rich liquidus in the Fe-FeS system from 1 bar to 10 GPa. Geochim Cosmochim Acta 75(8):2072–2087
Campbell AJ, Heinz DL (1993) Equation of state and high pressure phase transition of NiS in the NiAs structure. J Phys Chem Solids 54(1):5–7
Carlson RW (2005) Application of the Pt-Re-Os isotopic systems to mantle geochemistry and geochronology. Lithos 82:249–272
Chanyshev A, Bondar D, Fei H, Purevjav N, Ishii T, Nishida K, Bhat S, Farla R, Katsura T (2021) Determination of phase relations of the olivine-ahrensite transition in the Mg2SiO4-Fe2SiO4 system at 1740 K using modern multi-anvil techniques. Contrib Miner Petrol 176(10):77. https://doi.org/10.1007/s00410-021-01829-x
Craig JR, Kullerud G (1969) Phase relations in the Cu-Fe-Ni-S system and their application to magmatic ore deposits. Econ Geol Monogr 4:344–358
Errandonea D (2010) The melting curve of ten metals up to 12 gpa and 1600 k. J Appl Phys 108(3):033517
Fei Y, Prewitt CT, Hk Mao, Bertka CM (1995) Structure and density of fes at high pressure and high temperature and the internal structure of mars. Science 268(5219):1892–1894
Fei Y, Bertka CM, Finger LW (1997) High-pressure iron-sulfur compound, Fe\(_3\)S\(_2\), and melting relations in the Fe-FeS system. Science 275(5306):1621–1623
Fleet ME, Chryssoulis SL, Stone WE, Weisener CG (1993) Partitioning of platinum-group elements and Au in the Fe-Ni-Cu-S system: Experiments on the fractional crystallization of sulfide melt. Contrib Mineral Petrol 115:36–44
Fleet ME, Liu M, Crocket JH (1999) Partitioning of trace amounts of highly siderophile elements in the Fe-Ni-S. Geochim Cosmochim Acta 63(17):2611–2622
Fonseca ROC, Mallmann G, O’Neill HSC, Campbell IH, Laurenz V (2011) Solubility of Os and Ir in sulphide melt: implications for Re/Os fractionation in the upper mantle. Earth Planet Sci Lett 311:339–350
Fonseca ROC, Laurenz V, Mallmann G, Luguet A, Hoehne N, Jochum KP (2012) New constraints on the genesis and long-term stability of Os-rich alloys in the Earth’s mantle. Geochim Cosmochim Acta 87:227–242
Fonseca ROC, Brückel K, Bragagni A, Leitzke FP, Speelmanns IM, Wainwright AN (2017) Fractionation of rhenium from osmium during noble metal alloy formation in association with sulfides: Implications for the interpretation of model ages in alloy-bearing magmatic rocks. Geochim Cosmochim Acta 216:184–200
Frost DJ, McCammon CA (2008) The Redox State of Earth’s Mantle. Annu Rev Earth Planet Sci 36(1):389–420. https://doi.org/10.1146/annurev.earth.36.031207.124322
Helmy HM, Ballhaus C, Fonseca ROC, Nagel TJ (2013a) Fractionation of platinum, palladium, nickel, and copper in sulfide-arsenide systems at magmatic temperature. Contrib Mineral Petrol 166(6):1725–1737
Helmy HM, Ballhaus C, Fonseca ROC, Wirth R, Nagel T, Tredoux M (2013b) Noble metal nanoclusters and nanoparticles precede mineral formation in magmatic sulphide melts. Nat Commun 4
Hernlund J, Leinenweber K, Locke D, Tyburczy JA (2006) A numerical model for steady-state temperature distributions in solid-medium high-pressure cell assemblies. Am Miner 91(2–3):295–305
Holland T (1980) The reaction albite= jadeite+ quartz determined experimentally in the range \(600-1200^{\circ }\)C. Am Miner 65(1–2):129–134
Karup-Møller S, Makovicky E (1999) The phase system Fe-Re-S at \(1200\,^{\circ }\), \(1100\,^{\circ }\), \(1000\,^{\circ }\) and \(900\,^{\circ }\)C. N Jb Miner Mh 6:265–280
Karup-Møller S, Makovicky E (2002) The system Fe-Os-S at \(1180\,^{\circ }\), \(1100\,^{\circ }\) and \(500\,^{\circ }\)c. The Can Mineral 40:499–507
Li C, Barnes SJ, Makovicky E, Rose-Hansen J, Makovicky M (1996) Partitioning of nickel, copper, iridium, rhenium, platinum, and palladium between monosulfide solid solution and sulfide liquid: Effects of composition and temperature. Geochim Cosmochim Acta 60(7):1231–1238
Liu Y, Brenan J (2015) Partitioning of platinum-group elements (PGE) and chalcogens (Se, Te, As, Sb, Bi) between monosulfide-solid solution (MSS), intermediate solid solution (ISS) and sulfide liquid at controlled \(f\)O\(_2\)-\(f\)S\(_2\) conditions. Geochim Cosmochim Acta 159:139–161
Longerich HP, Jackson SE, Günther D (1996) Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation. J Anal At Spectrom 11:899–904
Lorand JP, Luguet A (2016) Chalcophile and siderophile elements in mantle rocks: Trace elements controlled by trace minerals. Rev Mineral Geochem 81(1):441–488
Luguet A, Lorand JP, Seyler M (2003) Sulfide petrology and highly siderophile element geochemistry of abyssal peridotites: a coupled study of samples from the Kane Fracture Zone (45\(^{\circ }\) W 23\(^{\circ }\) 20N, MARK area, Atlantic Ocean). Geochim Cosmochim Acta 67(8):1553–1570
Luguet A, Pearson DG, Nowell GM, Dreher ST, Coggon JA, Spetsius ZV, Parman SW (2008) Enriched Pt-Re-Os isotope systematics in plume lavas explained by metasomatic sulfides. Science 319:453–456
Mengason MJ, Piccoli PM, Candela P (2010) An evaluation of the effect of copper on the estimation of sulfur fugacity (\(f\)S\(_2\)) from pyrrhotite composition. Econ Geol 105(6):1163–1169
Morard G, Sanloup C, Fiquet G, Mezouar M, Rey N, Poloni R, Beck P (2007) Structure of eutectic Fe-FeS melts to pressures up to 17 GPa: implications for planetary cores. Earth Planet Sci Lett 263(1–2):128–139
Mungall JE, Andrews DRA, Cabri LJ, Sylvester PJ, Ubrett M (2005) Partitioning of Cu, Ni, Au and platinum-group elements between monosulfide solid solution and sulfide melt under controlled oxygen and sulfur fugacities. Geochim Cosmochim Acta 69(17):4349–4360
Naldrett AJ (1969) A portion of the system Fe-S-O between 900 and 1080\(^{\circ }\)c. J Petrol 10:171–201
Naldrett AJ, Craig JR, Kullerud G (1967) The central portion of the Fe-Ni-S system and its bearing on pentlandite exsolution in iron-nickel sulfide ores. Econ Geol 62(6):826–847
Piermarini G, Block S (1975) Ultrahigh pressure diamond-anvil cell and several semiconductor phase transition pressures in relation to the fixed point pressure scale. Rev Sci Instrum 46(8):973–979
Raybaud P, Kresse G, Hafner J, Toulhoat H (1997) Ab initio density functional studies of transition-metal sulphides: i. Crystal structure and cohesive properties. J Phys Condens Matter 9(50):11085
Ryzhenko B, Kennedy GC et al (1973) The effect of pressure on the eutectic in the system Fe-FeS. Am J Sci 273(9):803–810
Sanloup C, Guyot F, Gillet P, Fiquet G, Mezouar M, Martinez I (2000) Density measurements of liquid Fe-S alloys at high-pressure. Geophys Res Lett 27(6):811–814
Sanloup C, Guyot F, Gillet P, Fei Y (2002) Physical properties of liquid fe alloys at high pressure and their bearings on the nature of metallic planetary cores. J Geophys Res Solid Earth 107(B11):ECV-4
Schwerdtfeger K, Zwell L (1968) Activities in solid iridium-iron and rhodium-iron alloys at 1200 \(^{\circ }\)C. Trans Meteorol Soc Aime 242(4)
Stachel T, Harris JW (2008) The origin of cratonic diamonds-constraints from mineral inclusions. Ore Geol Rev 34(1–2):5–32
Stachel T, Aulbach S, Harris JW (2022) Mineral inclusions in lithospheric diamonds. Rev Mineral Geochem 88(1):307–391. https://doi.org/10.2138/rmg.2022.88.06
Stixrude L, Lithgow-Bertelloni C (2011) Thermodynamics of mantle minerals—II. Phase equilibria. Geophys J Int 184(3):1180–1213. https://doi.org/10.1111/j.1365-246X.2010.04890.x. (://WOS:000287362500014)
Swartzendruber L, Sundman B (1983a) The Fe- Os (Iron-Osmium) system. J Phase Equilib 4(4):396–399
Swartzendruber L, Sundman B (1983b) The Fe-Ru (Iron-Ruthenium) system. J Phase Equilib 4(2):155–160
Swartzendruber LJ (1984) The Fe-Ir (iron-iridium) system. Bul Alloy Phase Diagr 5(1):48–52
Tange Y, Takahashi E, Ki Funakoshi (2011) In situ observation of pressure-induced electrical resistance changes in zirconium: pressure calibration points for the large volume press at 8 and 35 gpa. High Press Res 31(3):413–418
Urakawa S, Kato M, Kumazawa M (1987) Experimental study on the phase relations in the system Fe-Ni-O-S up to 15 GPa. High-Press Res Miner Phys Vol Honor of Syun-iti Akimoto 39:95–111
Urakawa S, Igawa N, Kusaba K, Ohno H, Shimomura O (1998) Structure of molten iron sulfide under pressure. Rev High Press Sci Technol 7:286–288
Urakawa S, Hasegawa M, Yamakawa J, Funakoshi KI, Utsumi W (2002) High-pressure phase relationships for FeS. High Press Res 22(2):491–494
Urakawa S, Matsubara R, Katsura T, Watanabe T, Kikegawa T (2011) Stability and bulk modulus of Ni\(_3\)S, a new nickel sulfur compound, and the melting relations of the system Ni-NiS up to 10 GPa. Am Miner 96(4):558–565
Usselman TM (1975) Experimental approach to the state of the core; Part I, The liquidus relations of the Fe-rich portion of the Fe-Ni-S system from 30 to 100 kb. Am J Sci 275(3):278–290
Walker RJ, Morgan JW, Beary ES, Smoliar MI, Czamanske GK, Horan MF (1997) Applications of the \(^{190}\)Pt-\(^{186}\)Os isotope system to geochemistry and cosmochemistry. Geochim Cosmochim Acta 61(22):4799–4807
Westerlund K, Shirey S, Richardson S, Carlson R, Gurney J, Harris J (2006) A subduction wedge origin for paleoarchean peridotitic diamonds and harzburgites from the panda kimberlite, slave craton: evidence from re-os isotope systematics. Contrib Miner Petrol 152(3):275–294
Wohlgemuth-Ueberwasser C, Ballhaus C, Berndt J, Stotter née Paliulionyte V, Meisel T (2007) Synthesis of PGE sulfide standards for laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). Contrib Mineral Petrol 154(5):607–617
Zhang Z, Hirschmann MM (2016) Experimental constraints on mantle sulfide melting up to 8 GPa. Am Miner 101(1):181–192
Zhang Z, Lentsch N, Hirschmann MM (2015) Carbon-saturated monosulfide melting in the shallow mantle: solubility and effect on solidus. Contrib Miner Petrol 170(5):1–13
Zhang Z, von der Handt A, Hirschmann MM (2018) An experimental study of Fe-Ni exchange between sulfide melt and olivine at upper mantle conditions: Implications for mantle sulfide compositions and phase equilibria. Contrib Miner Petrol 173(3):1–18
Zhang Z, Qin T, Pommier A, Hirschmann MM (2019) Carbon storage in Fe-Ni-S liquids in the deep upper mantle and its relation to diamond and Fe-Ni alloy precipitation. Earth Planet Sci Lett 520:164–174
Acknowledgements
ROCF and CB are grateful for financial support from the Deutsche Forschungsgemeinschaft (via grants number BE 6053/2–1 and FO 698/13–1). The authors also thank Nils Jöns and Sabine Weisel for their assistance during EMPA measurements at the Ruhr-University Bochum. We are likewise grateful to Stephan Klemme, Jasper Berndt-Gerdes and Beate Schmitte for their help during EMPA and LA-ICP-MS measurements carried out at the Institute of Mineralogy of the University of Münster. This manuscript is a contribution from the DFG-funded LA-ICP-MS laboratory of the Institute of Geology, Mineralogy and Geophysics of the Ruhr-University Bochum. We are very grateful to Editor Dante Canil, James Brenan, and an anonymous reviewer whose constructive comments greatly improved the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dante Canil.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fonseca, R.O.C., Beyer, C., Bissbort, T. et al. Partitioning of highly siderophile elements between monosulfide solid solution and sulfide melt at high pressures. Contrib Mineral Petrol 179, 17 (2024). https://doi.org/10.1007/s00410-023-02092-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-023-02092-y