
Abstract
Subthreshold psychiatric disorders do not fully meet the diagnostic criteria of syndromal disorders but may be associated with comparable disability. To investigate the anxiolytic effect of Silexan, an active substance from lavender oil for oral administration, in patients with subthreshold anxiety, a meta-analysis that included all published trials with Silexan in this indication was performed. Three randomised, placebo-controlled trials in subthreshold anxiety disorders (anxiety disorder not otherwise specified, restlessness and agitation, mixed anxiety and depressive disorder) were included. Eligible participants with a baseline Hamilton Anxiety Rating Scale (HAMA) total score ≥ 18 points received 1 × 80 mg/day Silexan or placebo for 10 weeks. Outcomes included the HAMA, the Pittsburgh Sleep Quality Index, the Zung Self-rating Anxiety Scale, the Clinical Global Impressions questionnaire and the SF-36 health status inventory. Data were analysed using meta-analysis based on pooled raw data of individual patients (random effects models). A total of 697 patients were assessed for efficacy. Silexan was superior to placebo in reducing the HAMA total score during 10 weeks’ treatment [mean value difference, 95% confidence interval: 3.83 (1.28; 6.37) points]. Superiority was comparably pronounced for psychic and somatic anxiety as well as for observer- and self-rated anxiety. Silexan had a beneficial effect on sleep (secondary to the anxiolytic effect) without causing sedation and improved the patients’ health-related quality of life. Adverse event incidence in both treatment groups was comparable [risk ratio: 1.06 (0.85; 1.33)]. Silexan has a significant and clinically meaningful anxiolytic effect in subthreshold anxiety. The results cannot be generalised to other lavender oil products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Subthreshold anxiety disorder (SSAD, subsyndromal anxiety disorder) is a very common but nevertheless often underdiagnosed and undertreated condition that refers to individuals with clinically relevant symptoms of anxiety who meet several, but not all diagnostic criteria of ‘syndromal’ generalised anxiety disorder (GAD) according to the standards of the Diagnostic and Statistical Manual of Mental Disorders (DSM) of the American Psychiatric Association or of the WHO’s International Classification of Diseases (ICD) [1]. It is in fact so poorly recognised that the current versions of the applicable classification systems do not even offer a specific diagnostic category; instead, clinicians have to code SSAD as Other Specified Anxiety Disorder (300.09) or Unspecified Anxiety Disorder (300.00) in DSM-5 or as Anxiety Disorder, Unspecified (F41.9) or Mixed Anxiety and Depression (F 41.2; in case of additional subthreshold but clinically relevant depressive symptoms) in ICD-10.
Epidemiological data suggest that the population prevalence of SSAD may exceed that of GAD in Europe and North America by a factor of about three [1,2,3]. Moreover, a recent study showed that nearly three out of four adult individuals seeking treatment for clinically relevant worry failed to meet the criteria for ‘syndromal’ GAD only by a single criterion [4]. There is almost uniform agreement between researchers that SSAD causes relevant functional impairment and suffering, has a detrimental effect on quality of life, is associated with a high level of co-morbidity and bears a considerable risk of exacerbation to threshold anxiety, mood disorder or substance use disorders [1, 5, 6]. Moreover, SSAD, in contrast to GAD, was found to be associated with a significant decrease in perceived social support [7]. These findings underline the need for an early and adequate treatment of SSAD, which could also efficiently and cost-effectively prevent a progression to the syndromal level [8, 9].
Silexan, which has been licensed in 14 countries worldwide and is the active substance of a medicinal product marketed in Germany, is produced from lavender and causes a potent inhibition of voltage dependent calcium channels (VOCCs) in synaptosomes, primary hippocampal neurons and stably overexpressing cell lines [10], which have been shown to play an important role in both anxiety and depression. Inhibition of VOCCs can lead to an attenuation of the overreaching, situationally inadequate stress response of the central nervous system associated with anxiety and mood disorders [11]. Moreover, Silexan significantly reduces the 5-HT1A binding potential in the brain clusters encompassing the temporal gyrus, the fusiform gyrus, the hippocampus, the insula and the anterior cingulate cortex, which may lead to an increase of extracellular serotonin levels [12].
In randomised, double-blind, placebo-controlled clinical trials Silexan showed a pronounced anxiolytic effect in threshold GAD and also in indications having subthreshold anxiety as a principal feature (SSAD, mixed anxiety and depressive disorder (MADD), anxiety-related restlessness and agitation) [13,14,15,16]. In order to obtain a comprehensive overview of the anxiolytic efficacy of Silexan in subthreshold anxiety, we performed a meta-analysis of randomised, placebo-controlled clinical trials that investigated the effect of the active substance in these indications.
Methods
Searches, study and participant characteristics
For our meta-analysis individual patient data of three double-blind, randomised, placebo-controlled, multicentre phase III clinical trials in patients with subthreshold anxiety [14,15,16] were obtained from the manufacturer of Silexan. To identify any additional studies performed with Silexan in patients with subthreshold anxiety, we performed free-text searches of all fields of the MEDLINE database as well as of the ClinicalTrials.gov registry, the EMA Clinical Trials Register, and the ISRCTN registry for any records that included the search term ‘Silexan’ in combination with ‘anxiety’ and had been entered before 31 March 2017 (no further restrictions applied). In addition to the original publications presenting the results of the three trials above [14,15,16], the MEDLINE search retrieved a total of 14 other publications. Four described randomised, controlled trials with Silexan in other psychiatric indications [13, 17,18,19], and three covered non-clinical studies with Silexan [10, 12, 20]. Retrieved publications also included a contraceptives interaction study [21] and a case report [22]. The remaining five publications were review articles from which no additional studies with Silexan in patients with subthreshold anxiety could be identified [14, 23,24,25,26]. Searches performed in clinical trials registries also did not reveal any additional studies with Silexan performed in the indication of interest. The search results thus indicate that the three randomised, controlled trials performed in patients with psychiatric diagnoses characterised by subthreshold anxiety appear to be the only randomised, controlled studies performed with Silexan in the population of interest by the time when our searches were completed. The remaining identified trials covered different indications and were thus excluded.
Among the eligible trials, Study A assessed the efficacy of Silexan patients with SSAD (classified as ‘anxiety disorder not otherwise specified’ according to the DSM-IV 300.00 and as ‘anxiety disorder, unspecified’ according to ICD-10 F41.9) [14]. The participants of Study B suffered from restlessness and agitation (ICD-10 R45.1) as well as from disturbed sleep [16]. Study C was performed in patients diagnosed to be suffering from MADD (ICD-10 F41.2) [15].
The main characteristics and criteria for participant selection of the included trials are summarised in Table 1. The participants of all studies were female or male, adult out-patients who consulted a general practitioner or a psychiatrist. Each trial started with a 3–7 days qualification phase after which eligible patients were randomised and received Silexan or placebo for a scheduled period of 10 weeks, with assessments performed at baseline (randomisation) and at the end of weeks two, four, six, eight, and ten (Studies A and B), or at the end of weeks one, two, four, seven, and ten (Study C). For eligibility, patients of all trials had to meet the diagnostic criteria for the condition under investigation and had to present with a total score ≥ 18 points for the Hamilton Anxiety Rating Scale (HAMA) as well as with minimum scores for two individual HAMA items as shown in Table 1 at both inclusion and baseline.
Ethical conduct
All trials included into our meta-analysis were performed following the principles of Good Clinical Practice and the Declaration of Helsinki after obtaining approval from an Independent ethics committee. The studies were registered in the EudraCT database (all studies) as well as in the ISRCTN registry (studies A and C). All participants provided written informed consent.
Interventions
Silexan is a special active substance with an essential oil produced from Lavandula angustifolia flowers by steam distillation that complies with the monograph Lavender oil of the European Pharmacopoeia and exceeds the quality requirements of the monograph. Batch to batch consistency is assured by a well-defined, standardised manufacturing process. Immediate release soft gelatine capsules containing 80 mg of Silexan or identically matched placebo capsules were used. The smell of the investigational treatments was matched by flavouring the capsules containing placebo with 1/1000 of the amount of lavender oil contained in the Silexan capsules. In all studies randomised patients received Silexan monotherapy and had to administer one capsule per day unchewed in the morning. The daily dose was chosen in accordance with recommended dose of the marketed product.
Meta-analysis outcomes
The anxiolytic effect of Silexan was assessed by analysing HAMA total score change between baseline and treatment end, which was the pre-defined main outcome in all eligible studies. Moreover, we analysed the change of the HAMA psychic and somatic anxiety subscores [27] as well as of the individual item scores for anxious mood, tension, and sleep. Anxiety self-ratings were included into the meta-analysis using the change of the total score of the Zung Self-rating Anxiety Scale (SAS; studies A and B) or of the anxiety rating of the Hospital Anxiety and Depression Scale (HADS; study C). Sleep quality was assessed by analysing the total score change of the Pittsburgh Sleep Quality Index (PSQI; studies A and B only). Meta-analyses were also performed for the changes of the mental health and the physical health subscores of the SF-36 Health Survey Questionnaire which assesses health-related quality of life (studies A and C only), and of Items 1 and 2 of the Clinical Global Impressions scale (CGI).
Our meta-analysis also included assessments of treatment response and remission. We defined response as a decrease of the HAMA total score by at least 50% of the baseline value or as a score equal to or less than two points for CGI item 2 (i.e., much or very much improved compared to project admission), both assessed at treatment end. Remission was defined as a HAMA total score of less than ten points or of less than or equal to seven points at treatment end.
In all trials tolerability and safety were primarily assessed by monitoring adverse events (AEs).
Bias assessment
Bias assessment on the study level was performed by an independent assessor who was not involved in the planning, conduct, analysis or interpretation of any of the eligible trials, using the Cochrane Collaboration’s tool for assessing risk of bias [28]. Assessments were based upon the applicable publications, the patient raw data, and on the original protocols and the full integrated study reports made available to the authors and to the assessor.
Statistical methods
The meta-analysis of treatment efficacy was based on the original (raw) data of the included trials and was performed for the primary efficacy analysis data sets (full analysis set, FAS) of the original protocols. For comparability with the published results of the trials, missing data for efficacy outcomes were imputed by carrying forward the last valid observation.
Patient age, sex, and premature withdrawal rate were analysed using descriptive statistics. Within each trial continuous outcomes were analysed using analysis of covariance (ANCOVA) with treatment as a factor, the intraindividual difference between treatment end and baseline for the outcome of interest as the dependent variable, and the baseline value of the outcome as a covariate. Rating scales were analysed as continuous outcomes. For the analysis of CGI item 2 (‘Change from project admission’), which inherently includes change from baseline and which was thus assessed only at post-baseline visits, the baseline value of CGI item 1 (‘Severity of illness’) was used as a covariate.
Meta-analysis methods were pre-defined in a statistical analysis plan. We used a two-stage individual participant data (IPD) meta-analysis approach according to which the outcomes of interest were first analysed equally within each study and then combined using ‘traditional’ meta-analysis [29, 30]. For continuous outcomes, we used the marginal (adjusted) mean values and their estimated standard deviations as input for the meta-analysis and computed random effects models based on the treatment group mean value difference. Inverse variance weighting was used for combining the results of the single trials. We applied the DerSimonian–Laird method to calculate the variance between the trials. In the meta-analysis of self-rated anxiety, which combined ratings originating from the SAS and the HADS clinical questionnaires, the bias corrected Hedges’ G was calculated as an estimate for the combined, standardised mean value difference between the treatments in order to account for the different scales. For the HAMA total score difference and the clinical global impression of change from baseline (CGI item 2) the bias corrected Hedges’ G was calculated in addition in order to facilitate the comparison of results with other published work. Meta-analyses of binary outcomes (response, remission, and AE rates) were based on relative risk. For response and remission, random effects models were used, and trial results were combined according to the inverse variance method. Meta-analyses AE rates were performed using fixed effects models with Mantel–Haenszel weighting for combining the trial results. Additionally meta-analyses of binary outcomes based on risk differences were performed to calculate numbers needed to treat and numbers needed to harm.
For all analyses two-sided p values ≤ 0.05 were considered descriptively significant.
Heterogeneity between the trials was assessed using the I2 statistic in accordance with the criteria proposed in section 9.5.2 of the Cochrane Handbook for Systematic Reviews of Interventions [31].
Meta-analyses were computed with the R software package meta (version 4.3.2) using functions metacont and metabin for continuous and binary data, respectively. All other analyses were performed in SAS statistical software version 9.3.
Results
Study and participant characteristics
Risk of within-study bias assessments are provided in Table 2. All eligible studies [14,15,16] were conceived by the same working group and were performed according to very similar protocols except for the diagnosis for inclusion. Elevated risk of bias related to incomplete outcome data in study B [16] was attributable to a somewhat higher premature withdrawal rate for lack of efficacy in the placebo group (5/84 patients compared to 3/86 for Silexan) and a higher rate of withdrawals for adverse events in the Silexan group (4/86 versus none for placebo), in combination with last observation carried forward missing data imputation.
Across all trials included into our analyses 709 patients (Silexan 356; placebo 353) were randomised, 704 (353 and 351) were assessed for safety, and 697 (349 and 348) were analysed for efficacy. Pooled premature withdrawal rates were 12.6 and 10.5% for Silexan and placebo, respectively.
Demographic characteristics and baseline values of efficacy outcomes are shown in Table 3. More than two-thirds of the participants of all studies were female. Across all trials, the patients in both treatment groups were on average about 47 years old.
Within-trial baseline mean value differences for the HAMA total score did not exceed one point, and the mean value differences for HAMA ‘core’ items Anxious mood, Tension, and insomnia never exceeded 0.1 points (data not shown). The baseline values of the remaining efficacy outcomes presented in Table 3 also indicate that the treatment groups within the included trials were essentially comparable at baseline.
Anxiolytic effect
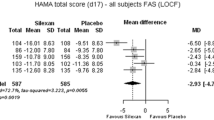
The main results for the studies’ pre-defined primary or co-primary outcome measure, HAMA total score change between baseline and treatment end, are presented in Fig. 1. In the meta-analysis Silexan was significantly superior to placebo. The figure also shows that Silexan was superior to placebo in each of the three included trials, with the largest treatment effect in trial A. The standardised mean value difference (Hedges’ G) was 0.45 (95% CI 0.15; 0.74, p = 0.003).

Hamilton Anxiety Rating Scale total score—change between baseline and treatment end (SD standard deviation, MD mean value difference, CI confidence interval, W weight)
Table 4 shows that the items conventionally assigned to the psychic (items 1–6, 14) and somatic anxiety subscores of the HAMA (items 7–13) contributed comparably to the over-all effect observed for the total score. Significant advantages for Silexan over placebo were also observed in the meta-analyses of items Anxious mood, Tension, and Insomnia (other items were not analysed separately). For all analyses of HAMA scores and items heterogeneity (ranging between I2 = 0% and I2 = 78.5%) was caused by between-trial differences regarding treatment effect size whereas the direction of the treatment group differences always favoured Silexan.
The results for the observer-rated HAMA were supported by the study participants’ anxiety self-ratings obtained with the Zung SAS (trials A and B) and the HADS (trial C; Fig. 2), the values of which cannot be compared directly due to the different ranges of the 2 self-rating scales. The meta-analysis model again showed a significant treatment group difference (p = 0.004) favouring Silexan while only minor heterogeneity was observed.

Anxiety self-rating (studies A and B: Zung Anxiety Self-rating Scale total score; study C: Hospital Anxiety and Depression Scale, anxiety subscore)—change between baseline and treatment end (SD standard deviation, SMD standardised mean value difference, CI confidence interval, W weight)
Over-all clinical condition
In the included trials the patients’ over-all severity of impairment rated with CGI item 1 decreased by mean values between 1.14 and 2.06 points for Silexan and by between 0.66 and 1.19 points for placebo. The meta-analysis showed a significant over-all effect favouring Silexan (Table 5). Heterogeneity was again caused by differences regarding the magnitude, not the direction of the treatment effect.
Figure 3 shows the meta-analysis results for CGI item 2 (global change between project admission and treatment end) which were similar to those for item 1. In addition to the over-all significant mean value difference (p = 0.004), two out of the three trials also showed a significant treatment group difference favouring Silexan. The standardised mean value difference (Hedges’ G) was 0.49 (95% CI 0.17; 0.81, p = 0.003).

Clinical Global Impressions, item 2, change from project admission—assessment at treatment end (SD standard deviation, MD mean value difference, CI confidence interval, W weight)
Response and remission
The percentage of patients whose HAMA total score decreased by at least 50% of the baseline value during randomised treatment was significantly larger in participants treated with Silexan as compared to those who received placebo (Fig. 4; p = 0.002). The corresponding number needed to treat (NNT) derived from the meta-analysis was 6. When response was defined by a score of 1 or 2 for CGI item 2 (i.e., much or very much improved), a risk ratio of 1.69 (95% CI 1.44; 2.00, p < 0.001) favouring Silexan was determined by meta-analysis, with individual study risk ratios ranging between 1.53 and 1.84 (meta-analysis NNT 5).

Analysis of response—number of patients with a Hamilton Anxiety Rating Scale total score reduction by at least 50% between baseline and treatment end (RR risk ratio, CI confidence interval, W weight)
Remission, defined by a HAMA total score of less than ten points at treatment end, was also significantly more frequent in the Silexan group as compared to placebo (p = 0.008; Fig. 5; meta-analysis NNT 8). When the threshold for remission was tightened from less than ten to less than or equal to seven points, two out of the three trials still showed advantages for Silexan, and the meta-analysis model resulted in a pooled risk ratio of 1.37 (95% CI 1.05; 1.79, p = 0.022) favouring Silexan.

Analysis of remission—number of patients with a Hamilton Anxiety Rating Scale total score < 10 points at treatment end (RR risk ratio, CI confidence interval, W weight)
Effect on disturbed sleep related to SSAD
According to HAMA item ‘Insomnia’ (defined by difficulty in falling asleep, broken sleep, unsatisfying sleep, fatigue on waking, dreams, nightmares, and night terrors) 10 weeks’ administration of Silexan led to a significantly more pronounced symptom reduction than placebo treatment (Table 4). More detailed insight into the effect of the investigational treatments on sleep was obtained by means of the PSQI which was administered in studies A and B. The meta-analysis of total score of the scale showed significantly more pronounced symptom alleviation in the Silexan group as compared to placebo (Table 5). Advantages for Silexan were also observed in the meta-analyses for PSQI components Sleep quality (p = 0.051), Sleep latency (p < 0.001), Sleep disturbances (p = 0.014), and Daytime dysfunction (p < 0.001), whereas the remaining components showed no significant differences between Silexan and placebo.
Health-related quality of life
Health-related quality of life was assessed in studies A and C using the SF-36 health status questionnaire. The pooled meta-analysis mean value differences for the mental and physical health subscores of the SF-36 are presented in Table 5. Both scales showed significant treatment group differences favouring Silexan. Moreover, for both domains, superiority of Silexan over placebo was also observed in each study evaluated independently.
Safety
For Silexan and placebo similar percentages of patients with any AEs during randomised treatment were observed for all included trials, with two trials slightly favouring placebo and one trial slightly favouring Silexan (p = 0.589; Fig. 6) and a meta-analysis number needed to harm (NNH) of 55.

Number of patients with any adverse events (RR risk ratio, CI confidence interval, W weight)
Serious AEs were reported in two patients in each treatment group of trial A and in one patient in the Silexan group as well as in two patients in the placebo group of trial C, whereas there were no serious events in trial B (meta-analysis risk ratio: 0.75 favouring Silexan, 95% CI 0.17; 3.33, p = 0.710).
Across all trials, a total of six out of 356 patients randomised to Silexan and four out of 353 randomised to placebo were withdrawn due to an AE (meta-analysis risk ratio: 1.43 favouring placebo, 95% CI 0.43; 4.76, p = 0.557; NNH 182). AEs leading to premature withdrawal in Silexan-treated patients were female genital pain (not related), two cases of nausea (one not related, one possibly related), gastritis (unlikely related), eructation (probably related), and oral discomfort (possibly related). In the placebo group patients were withdrawn for subileus, anorexia, nausea and dizziness, as well as for diverticular perforation.
Discussion
Subthreshold anxiety is not only a diagnostic entity in its own right, but also an important feature or co-morbidity symptom of several psychiatric (most notably depression) and somatic diseases. Our meta-analysis demonstrates that Silexan is superior to placebo in reducing the anxiety-associated symptoms in patients suffering from SSAD, including MADD or restlessness and agitation disorder. Significant superiority of Silexan was observed for psychic as well as for somatic manifestations of anxiety. While no empirically derived minimal clinically important difference (MCID) has yet been published for HAMA total score change or for the difference between drug and placebo, the protocols of the included trials have assumed a treatment group mean value difference in HAMA total score reduction of 2.5 or three points to be clinically important. The 3.8-point difference observed in our meta-analysis actually exceeds this margin. The clinical relevance of the observed effect is also supported by significant superiority of Silexan over placebo regarding the percentage of patients who showed an at least 50% reduction of their baseline HAMA total score, or who had a rating of one or two points for CGI item 2 (i.e., very much or much improved) at treatment end, criteria that are commonly used in pharmacotherapy studies as indicators of an important treatment effect [32].
The effect size point estimates of 0.45 for HAMA total score change and of 0.49 for CGI global improvement observed in this trial are not easily compared to the literature because published data on treatment effect sizes in SSAD are sparse. For GAD, Hidalgo and colleagues have published a review of 21 double-blind, placebo-controlled trials which shows effect sizes of 0.50 for pregabalin, 0.45 for hydroxyzine, 0.42 for venlafaxine, 0.38 for all benzodiazepines (alprazolam, diazepam, lorazepam), 0.36 for all SSRIs (paroxetine, sertraline, fluvoxamine, escitalopram), and 0.17 for buspirone for HAMA total score change [33]. For Silexan, effect sizes of 0.37 and of 0.50 have been reported in GAD for dosages 80 and 160 mg/day, respectively [13]. Moreover, the NNT of six patients exposed to observe one additional treatment responder defined by a ≥ 50% decrease of the HAMA total score was comparable to that determined for second-generation antipsychotics (NNT: 6) [34] and lower than that observed in a recent study on vilazodone (NNT: 10) [35] in patients with GAD (no NNTs could be found in the literature for patients with SSAD). NNTs published for remission [35,36,37] were mainly based on criteria substantially different from our prospectively defined criteria and were thus not comparable. Depping and colleagues [34] determined an NNT of ten for quetiapine when remission was defined by a HAMA total score < 17 points, compared to an NNT of eight in our meta-analysis for Silexan although we required a HAMA total score of less than 10 points for remission. These results suggest that the therapeutic effect of Silexan 80 mg/day in SSAD may likely be within the range of that of synthetic anxiolytic drugs that are recommended as first-line treatments in anxiety disorders [38].
It is important to note that the anxiolytic effect of Silexan was evident both in the observer ratings as well as in the patients’ anxiety self-ratings. Patient relevance is supported by significant superiority of the product over placebo regarding health-related quality of life, where a beneficial drug effect was again observed for both mental and physical aspects of disease-associated impairment.
Disturbed sleep has been recognised as an important co-morbidity of anxiety [39]. Our results indicated that Silexan has a beneficial effect on anxiety-related sleep impairments. Detailed results of trials A and B published elsewhere indicate a clear association between the alleviation of anxiety symptoms and the improvement of sleep, with a slight delay in PSQI total score decrease as compared to that of the HAMA [14, 16]. These results indicate that the improvement of sleep is secondary to the anxiolytic effect of Silexan. Importantly, while the product improves the quality and shortens the latency of sleep, it also has a favourable effect on sleep-related daytime dysfunction. The observation is consistent with previous research according to which Silexan has a calming but not a sedating effect [24, 40].
Our literature search showed that information regarding the anxiolytic efficacy of Silexan in patients with subsyndromal anxiety is sparse as the three trials included into our meta-analysis are the only ones performed in this patient population to date. Moreover, only one of the trials investigated patients with ‘pure’ SSAD, i.e., without relevant psychiatric co-morbidity. While this imposes certain limitations regarding generalisability, our results are nevertheless based on the whole body of evidence that pertains to the efficacy of Silexan in subthreshold anxiety.
Sedation has been described for many of the drugs currently recommended as first-line treatments of anxiety disorders, including selective serotonin reuptake inhibitors (SSRIs) and the antihistamine hydroxyzine [38]. Unwanted sedation may cause significant limitations in a patient’s ability to pursue essential activities of daily living, e.g., to operate machinery or to drive a vehicle. Other common side effects of SSRIs and/or hydroxyzine include anticholinergic reactions, gastrointestinal disturbances, weight gain, sexual dysfunction, or agitation and irritability [41, 42]. For hydroxyzine the European Medicines Agency’s Pharmacovigilance Risk Assessment Committee has recently issued a warning according to which the drug may cause serious cardiac side effects including QT interval prolongation and torsades de pointes [43]. Anxiolytic drugs may thus cause disturbing, partly serious side effects, which may be one of the reasons for the undertreatment of subthreshold anxiety [44]. Our meta-analysis shows that the rates of serious and non-serious adverse events observed during 10 weeks’ treatment with Silexan differed hardly from those reported for patients exposed to placebo. Indeed, reports published for Silexan to date suggest that eructation and dyspeptic symptoms as well as allergic skin reactions may be the only specific adverse effects associated with the product [24]. Patients may perceive the absence of side effects that are likely to interfere with essential aspects of daily living as an important contribution to their quality of life. This interpretation is supported by the marked improvements observed in the SF-36 quality of life questionnaire in patients treated with Silexan.
Lavender oil is a complex, multi-ingredient mixture in which more than 160 different substances have been identified [24]. The anxiolytic properties of the drug have been ascribed to different ingredients, among them linalool and linalyl acetate [45]. Marketed oils from lavender differ greatly with regard to quality linalool or linalyl acetate content [46]. It is, therefore, important to note that the results presented in this paper apply to Silexan but not to other lavender oil containing products.
In conclusion, our meta-analysis shows that Silexan has a statistically significant and clinically meaningful anxiolytic effect in subthreshold anxiety. The product had a beneficial effect on disturbed sleep secondary to anxiety and was associated with improvements in health-related quality of life. Silexan was well tolerated, with adverse event rates similar to those observed for placebo.
References
Haller H, Cramer H, Lauche R, Gass F, Dobos GJ (2014) The prevalence and burden of subthreshold generalized anxiety disorder: a systematic review. BMC Psychiatry 14:128. https://doi.org/10.1186/1471-244X-14-128
Cohen CI, Magai C, Yaffee R, Walcott-Brown L (2006) The prevalence of anxiety and associated factors in a multiracial sample of older adults. Psychiatr Serv 57(12):1719–1725. https://doi.org/10.1176/appi.ps.57.12.1719
Wittchen HU (2002) Generalized anxiety disorder: prevalence, burden, and cost to society. Depress Anxiety 16(4):162–171. https://doi.org/10.1002/da.10065
Lawrence AE, Brown TA (2009) Differentiating generalized anxiety disorder from anxiety disorder not otherwise specified. J Nerv Ment Dis 197(12):879–886. https://doi.org/10.1097/NMD.0b013e3181c29992
Karsten J, Penninx BW, Verboom CE, Nolen WA, Hartman CA (2013) Course and risk factors of functional impairment in subthreshold depression and anxiety. Depress Anxiety 30(4):386–394. https://doi.org/10.1002/da.22021
Diefenbach GJ, Hopko DR, Feigon S, Stanley MA, Novy DM, Beck JG, Averill PM (2003) ‘Minor GAD’: characteristics of subsyndromal GAD in older adults. Behav Res Ther 41(4):481–487
Kertz SJ, Woodruff-Borden J (2011) Human and economic burden of GAD, subthreshold GAD, and worry in a primary care sample. J Clin Psychol Med Settings 18(3):281–290. https://doi.org/10.1007/s10880-011-9248-1
Kanuri N, Taylor CB, Cohen JM, Newman MG (2015) Classification models for subthreshold generalized anxiety disorder in a college population: implications for prevention. J Anxiety Disord 34:43–52. https://doi.org/10.1016/j.janxdis.2015.05.011
Karsten J, Hartman CA, Smit JH, Zitman FG, Beekman AT, Cuijpers P, van der Does AJ, Ormel J, Nolen WA, Penninx BW (2011) Psychiatric history and subthreshold symptoms as predictors of the occurrence of depressive or anxiety disorder within 2 years. Br J Psychiatry 198(3):206–212. https://doi.org/10.1192/bjp.bp.110.080572
Schuwald AM, Nöldner M, Wilmes T, Klugbauer N, Leuner K, Müller WE (2013) Lavender oil-potent anxiolytic properties via modulating voltage dependent calcium channels. PLoS One 8(4):e59998. https://doi.org/10.1371/journal.pone.0059998
Satpute AB, Mumford JA, Naliboff BD, Poldrack RA (2012) Human anterior and posterior hippocampus respond distinctly to state and trait anxiety. Emotion 12(1):58–68. https://doi.org/10.1037/a0026517
Baldinger P, Hoflich AS, Mitterhauser M, Hahn A, Rami-Mark C, Spies M, Wadsak W, Lanzenberger R, Kasper S (2014) Effects of Silexan on the serotonin-1A receptor and microstructure of the human brain: a randomized, placebo-controlled, double-blind, cross-over study with molecular and structural neuroimaging. Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyu063
Kasper S, Gastpar M, Müller WE, Volz H-P, Möller H-J, Schläfke S, Dienel A (2014) Lavender oil preparation Silexan is effective in generalized anxiety disorder—a randomized, double-blind comparison to placebo and paroxetine. Int J Neuropsychopharmacol 17(6):859–869. https://doi.org/10.1017/S1461145714000017
Kasper S, Gastpar M, Müller WE, Volz HP, Möller HJ, Dienel A, Schläfke S (2010) Silexan, an orally administered Lavandula oil preparation, is effective in the treatment of ‘subsyndromal’ anxiety disorder: a randomized, double-blind, placebo controlled trial. Int Clin Psychopharmacol 25(6):277–287. https://doi.org/10.1097/YIC.0b013e32833b3242
Kasper S, Volz HP, Dienel A, Schlafke S (2016) Efficacy of Silexan in mixed anxiety-depression—a randomized, placebo-controlled trial. Eur Neuropsychopharmacol 26(2):331–340. https://doi.org/10.1016/j.euroneuro.2015.12.002
Kasper S, Anghelescu I, Dienel A (2015) Efficacy of orally administered Silexan in patients with anxiety-related restlessness and disturbed sleep—a randomized, placebo-controlled trial. Eur Neuropsychopharmacol 25(11):1960–1967. https://doi.org/10.1016/j.euroneuro.2015.07.024
Uehleke B, Schaper S, Dienel A, Schläfke S, Stange R (2012) Phase II trial on the effects of Silexan in patients with neurasthenia, post-traumatic stress disorder or somatization disorder. Phytomedicine 19(8–9):665–671. https://doi.org/10.1016/j.phymed.2012.02.020
Woelk H, Schläfke S (2010) A multi-center, double-blind, randomised study of the Lavender oil preparation Silexan in comparison to Lorazepam for generalized anxiety disorder. Phytomedicine 17(2):94–99
Gastpar M, Müller WE, Volz HP, Möller HJ, Schläfke S, Dienel A, Kasper S (2017) Silexan does not cause withdrawal symptoms even when abruptly discontinued. Int J Psychiatry Clin Pract. https://doi.org/10.1080/13651501.2017.1301488
Kumar V (2013) Characterization of anxiolytic and neuropharmacological activities of Silexan. Wien Med Wochenschr 163(3–4):89–94. https://doi.org/10.1007/s10354-012-0164-2
Heger-Mahn D, Pabst G, Dienel A, Schlafke S, Klipping C (2014) No interacting influence of lavender oil preparation silexan on oral contraception using an ethinyl estradiol/levonorgestrel combination. Drugs R&D 14(4):265–272. https://doi.org/10.1007/s40268-014-0065-5
Decker SO, Polarz H (2011) Silexan und Narkose: fallbericht und Möglichkeiten des prä- sowie perioperativen Managements. Anaesthesist 60(10):946–949. https://doi.org/10.1007/s00101-011-1916-x
Generoso MB, Soares A, Taiar IT, Cordeiro Q, Shiozawa P (2017) Lavender oil preparation (Silexan) for treating anxiety: an updated meta-analysis. J Clin Psychopharmacol 37(1):115–117. https://doi.org/10.1097/JCP.0000000000000615
Kasper S (2013) An orally administered lavandula oil preparation (Silexan) for anxiety disorder and related conditions: an evidence based review. Int J Psychiatry Clin Pract 17(Suppl 1):15–22. https://doi.org/10.3109/13651501.2013.813555
Kasper S, Gastpar M, Müller WE, Volz HP, Möller HJ, Dienel A, Schläfke S (2010) Efficacy and safety of silexan, a new, orally administered lavender oil preparation, in subthreshold anxiety disorder—evidence from clinical trials. Wien Med Wochenschr 160(21–22):547–556. https://doi.org/10.1007/s10354-010-0845-7
Quante A (2015) Akute pharmakologische Therapieoptionen von ängstlicher Symptomatik bei Patienten mit Depression. Fortschr Neurol Psychiatr 83(3):142–148. https://doi.org/10.1055/s-0034-1399243
Maier W, Buller R, Philipp M, Heuser I (1988) The Hamilton Anxiety Scale: reliability, validity and sensitivity to change in anxiety and depressive disorders. J Affect Disord 14(1):61–68
Higgins JP, Altman DG, Gotzsche PC, Juni P, Moher D, Oxman AD, Savovic J, Schulz KF, Weeks L, Sterne JA, Cochrane Bias Methods G, Cochrane Statistical Methods G (2011) The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 343:d5928. https://doi.org/10.1136/bmj.d5928
Riley RD, Lambert PC, Abo-Zaid G (2010) Meta-analysis of individual participant data: rationale, conduct, and reporting. BMJ 340:c221. https://doi.org/10.1136/bmj.c221
Burke DL, Ensor J, Riley RD (2017) Meta-analysis using individual participant data: one-stage and two-stage approaches, and why they may differ. Stat Med 36(5):855–875. https://doi.org/10.1002/sim.7141
Higgins JPT, Green S (eds) (2011) Cochrane handbook for systematic reviews of interventions, version 5.1.0 [updated March 2011]. The Cochrane Collaboration. www.handbook.cochrane.org. Accessed 13 Apr 2016
Bandelow B (2006) Defining response and remission in anxiety disorders: toward an integrated approach. CNS Spectr 11(10 Suppl 12):21–28
Hidalgo RB, Tupler LA, Davidson JR (2007) An effect-size analysis of pharmacologic treatments for generalized anxiety disorder. J Psychopharmacol 21(8):864–872. https://doi.org/10.1177/0269881107076996
Depping AM, Komossa K, Kissling W, Leucht S (2010) Second-generation antipsychotics for anxiety disorders. Cochrane Database Syst Rev 12:CD008120. https://doi.org/10.1002/14651858.CD008120.pub2
Zareifopoulos N, Dylja I (2017) Efficacy and tolerability of vilazodone for the acute treatment of generalized anxiety disorder: a meta-analysis. Asian J Psychiatr 26:115–122. https://doi.org/10.1016/j.ajp.2017.01.016
Baldwin DS, Asakura S, Koyama T, Hayano T, Hagino A, Reines E, Larsen K (2016) Efficacy of escitalopram in the treatment of social anxiety disorder: a meta-analysis versus placebo. Eur Neuropsychopharmacol 26(6):1062–1069. https://doi.org/10.1016/j.euroneuro.2016.02.013
Kapczinski F, Lima MS, Souza JS, Schmitt R (2003) Antidepressants for generalized anxiety disorder. Cochrane Database Syst Rev 2:003592. https://doi.org/10.1002/14651858.CD003592
Bandelow B, Sher L, Bunevicius R, Hollander E, Kasper S, Zohar J, Moller HJ, Care WTFoMDiP, WFSBP Task Force on Anxiety Disorders, O. C. D., Ptsd (2012) Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract 16(2):77–84. https://doi.org/10.3109/13651501.2012.667114
Ohayon MM, Shapiro CM, Kennedy SH (2000) Differentiating DSM-IV anxiety and depressive disorders in the general population: comorbidity and treatment consequences. Can J Psychiatry 45(2):166–172
Dimpfel W, Wedekind W, Dienel A (2015) Cerebral bioavailability of silexan—a quantitative EEG study in healthy volunteers. Open J Psychiatr 5:285–297. https://doi.org/10.4236/ojpsych.2015.53032
Ferguson JM (2001) SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry 3(1):22–27
Simons FE (1994) H1-receptor antagonists. Comparative tolerability and safety. Drug Saf 10(5):350–380
European Medicines Agency (2015) New restrictions to minimise the risks of effects on heart rhythm with hydroxyzine-containing medicines. EMA/149624/2015, 27 March 2015
Collins KA, Westra HA, Dozois DJ, Burns DD (2004) Gaps in accessing treatment for anxiety and depression: challenges for the delivery of care. Clin Psychol Rev 24(5):583–616. https://doi.org/10.1016/j.cpr.2004.06.001
Setzer WN (2009) Essential oils and anxiolytic aromatherapy. Nat Prod Commun 4(9):1305–1316
Binder G, König WA (2000) Ätherische Öle - Untersuchung zur Deklaration und Qualität ausgewählter Präparate. Dtsch Apoth Ztg 140(37):55–62
Acknowledgements
The research presented in this paper was funded by Dr. Willmar Schwabe GmbH & Co. KG, manufacturer of Silexan, who was also the sponsor of the trials included in the meta-analysis. The statistical analysis plan for the meta-analysis was conceived and the analyses were performed by the biostatistical department of Dr. Willmar Schwabe GmbH & Co. KG under the responsibility of co-author SS. The company also provided the raw data of the trials included into the analyses. A first draft of the manuscript was prepared by Andreas Völp, Ph.D., an independent medical writer who was reimbursed by Dr. Willmar Schwabe GmbH & Co. KG, who also independently performed the risk of bias assessments. Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Contributions
HJM, HPV and SK advised on the design of the studies. AD and SS planned and performed the studies. HJM, HPV and SK supervised the studies. SS performed the data analysis. All authors reviewed the results, commented on and approved the report.
Corresponding author
Ethics declarations
Conflict of interest
Hans-Jürgen Möller has received grant/research support, consulting fees and honoraria within the last years from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen Cilag, Lundbeck, MSD, Novartis, Organon, Otsuka, Pfizer, Schwabe, Sepracor, Servier, and Wyeth. Hans-Peter Volz has served as a consultant or on advisory boards for Astra/Zeneca, Eli Lilly, Lundbeck, Pfizer, Schwabe, Janssen, Otsuka, Merz, Wyeth, neuraxpharm and has served on speakers’ bureaus for Astra/Zeneca, Eli Lilly, Lundbeck, Schwabe, Janssen, Merz, Wyeth, Lichtwer, Steigerwald, Hormosan, neuraxpharm and Bristol-Myers Squibb. Angelika Dienel and Sandra Schläfke are employees of Dr. Willmar Schwabe GmbH & Co. KG, manufacturer of Silexan. Siegfried Kasper received grants/research support, consulting fees and/or honoraria within the last 3 years from Angelini, AOP Orphan Pharmaceuticals AG, AstraZeneca, Eli Lilly, Janssen, KRKA-Pharma, Lundbeck, Neuraxpharm, Pfizer, Pierre Fabre, Schwabe and Servier.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Möller, HJ., Volz, HP., Dienel, A. et al. Efficacy of Silexan in subthreshold anxiety: meta-analysis of randomised, placebo-controlled trials. Eur Arch Psychiatry Clin Neurosci 269, 183–193 (2019). https://doi.org/10.1007/s00406-017-0852-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-017-0852-4