
Abstract
Lewy body disorders are heterogeneous neurological conditions defined by intracellular inclusions composed of misshapen α-synuclein protein aggregates. Although α-synuclein aggregates are only one component of inclusions and not strictly coupled to neurodegeneration, evidence suggests they seed the propagation of Lewy pathology within and across cells. Genetic mutations, genomic multiplications, and sequence polymorphisms of the gene encoding α-synuclein are also causally linked to Lewy body disease. In nonfamilial cases of Lewy body disease, the disease trigger remains unidentified but may range from industrial/agricultural toxicants and natural sources of poisons to microbial pathogens. Perhaps due to these peripheral exposures, Lewy inclusions appear at early disease stages in brain regions connected with cranial nerves I and X, which interface with inhaled and ingested environmental elements in the nasal or gastrointestinal cavities. Irrespective of its identity, a stealthy disease trigger most likely shifts soluble α-synuclein (directly or indirectly) into insoluble, cross-β-sheet aggregates. Indeed, β-sheet-rich self-replicating α-synuclein multimers reside in patient plasma, cerebrospinal fluid, and other tissues, and can be subjected to α-synuclein seed amplification assays. Thus, clinicians should be able to capitalize on α-synuclein seed amplification assays to stratify patients into potential responders versus non-responders in future clinical trials of α-synuclein targeted therapies. Here, we briefly review the current understanding of α-synuclein in Lewy body disease and speculate on pathophysiological processes underlying the potential transmission of α-synucleinopathy across the neuraxis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lewy body disorders are multisystemic conditions with clinical symptoms spanning from mild to severe deficits in olfaction, peripheral autonomic and enteric function, mood/affect, sleep/wake cycles, movement, memory, and/or cognition. Lewy body disorders include, but are not limited to, Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), dementia with Lewy bodies (DLB), incidental Lewy body disease (ILBD), and primary autonomic failure (PAF). Lewy body disorders are histologically categorized by abnormal inclusions known as Lewy bodies in neuronal somata and Lewy neurites within neuronal processes. Lewy pathology is not uncommon at autopsy. Although estimates vary, ~ 8 to 17% of subjects sixty years or older display ILBD [25, 60, 81, 122], and Lewy pathology is present in ~ 40 to 57% of Alzheimer’s patients [51, 94, 136, 182, 249]. Lewy bodies may also be observed in some cases with Type I Gaucher disease and parkinsonism or in neurodegeneration with brain iron accumulation type 1 [6, 173, 273].
α-Synuclein is a key—Albeit partial—component of Lewy bodies
In this review, we will focus on a critical component of Lewy inclusions, the protein α-synuclein. Lewy bodies are partly composed of abnormal, filamentous aggregates of α-synuclein [110, 232, 233]. The objective of this review is to encourage speculation on mechanisms underlying the potential transmission of α-synuclein pathology across the central nervous system. This is an important topic because the molecular components of Lewy bodies, their morphology, and the spatiotemporal patterns of inclusion formation and neurodegeneration may dictate the type of Lewy body disorder that the patient develops. Although neurodegeneration is a key driver of neurological deficiencies, this does not rule out the possibility that living, α-synuclein aggregate-bearing neurons are functionally compromised in patients with Lewy body disease and contribute to maladaptive neurological traits. On the other hand, the region-specific pattern of α-synuclein aggregation does not strictly parallel regional patterns of cell death [87, 241]. Similarly, if all neurons housing Lewy inclusions were sufficiently dysfunctional, one would expect a robust positive correlation between Lewy-related pathologies and neurodegeneration or movement deficits, but this is not universally observed. Although Lewy-related pathology across the brain was shown to be correlated with neuronal loss in the substantia nigra and with motor deficits in cases without concomitant Alzheimer’s disease [14], other studies report a lack of correlation of nigral neurodegeneration with Lewy body distribution or density [183]. A third study demonstrated a negative association between nigral neuronal densities and α-synuclein+ profiles [61], but the latter report also showed that the demise of nigral neurons and loss of dopaminergic markers can precede the emergence of nigral α-synuclein aggregates, confirming earlier work [61, 164]. Some brain regions, such as the supraoptic and paraventricular hypothalamic nuclei, and selective parts of the hippocampal formation and presupplementary motor cortex may also display neuronal loss with minimal Lewy-related pathologies [4, 20, 150, 194]. Hence, despite being the namesake, Lewy bodies are not the sole source of pathology in Lewy body disease. Among other possibilities, the potential uncoupling of cell death and α-synuclein aggregation may reflect a protective nature of some of the diverse types of Lewy bodies, at least within neurons that are already diseased, as speculated below.
α-Synuclein is encoded by the phylogenetically conserved SNCA gene. SNCA expression is normally expressed in presynaptic terminals of the vertebrate nervous system, but it is also highly enriched in other tissues, such as blood (Fig. 1). As with many genes, SNCA is pleiotropic, and α-synuclein plays subtle roles in an array of synaptic functions, including neuroplasticity, neurotransmitter release, SNARE complex assembly, vesicle trafficking and clustering, fusion pore formation, endocytosis, tyrosine hydroxylase activation, and dopamine synthesis and transport [36, 37, 44, 79, 132, 190, 217, 220, 221, 239, 245, 266, 269]. Although Snca gene deletion in mice does not elicit dramatic changes in neurotransmission, neuronal viability, and/or behavioral outcomes (even when combined with deletions of other members of the synuclein family), laboratory mice are typically evaluated when young, whereas the impact of loss of α-synuclein is more evident in old animals [5, 89, 174]. Thus, when inclusions materialize in Lewy body disease, the manifestation of neurological symptoms may depend on age-dependent loss of normal α-synuclein functions in the synapse and other subcellular locations. Aside from this loss-of-function phenotype, there may also be a toxic gain of function of the abnormal α-synuclein aggregates, including interference with axonal transport [257, 259, 274] and clogging of protein degradation systems [53, 154, 235, 256]. The gain-of-function scenario does not exclude the possibility that the growth and maturation of Lewy bodies also benefits the diseased neuron [69]. For example, the formation of Lewy bodies may lower the concentration of soluble α-synuclein oligomers with more harmful properties than mature α-synuclein filaments. Lewy bodies may thus serve to sequester damaged cellular components that escape or saturate degradation systems (e.g., lysosomal and proteasomal digestion). In other words, there may be both advantages and disadvantages to formation of inclusions within diseased neurons, and the net outcome of these diverse responses may be the final arbiter of cell fate.

Bulk tissue expression of SNCA according the GTEx online data portal hosted by the Broad Institute of MIT and Harvard. The Y axis is shown on a linear scale and the X axis is sorted alphabetically according to tissue type or brain region. Note the high median expression of SNCA within human cerebellar tissues, discussed below alongside Fig. 2. These violin plots were retrieved from the GTEx portal on July 27, 2024 at this link: https://www.gtexportal.org/home/gene/SNCA
The gene SNCA lies along the long arm of chromosome 4q21-q23, and point mutations (e.g., V15A, A30P, E46K, H50Q, G51D, A53T, and A53V) or wildtype gene duplications and triplications have been identified in familial cases of Lewy body disease [23, 24, 45, 67, 74, 104, 106, 126, 191, 226, 277]. The number of SNCA alleles is dose-dependently linked to Lewy body disease severity, with cases of duplication causing milder forms of late-onset PD, whereas triplications cause severe clinical manifestations, with parkinsonism, autonomic dysfunction, dementia and, in some cases, vacuolation of cortical tissue [71, 107, 170, 203, 227]. Although more than 90% of PD cases do not adhere to Mendelian patterns of inheritance, all cases of Lewy body disease likely have a genetic component that intersects with disease triggers in the environment [227]. Numerous risk loci for PD exist, but genome-wide association studies consistently demonstrate that specific sequence variations in SNCA are risk factors for Lewy body disease (and an earlier age of disease onset), particularly when the variants raise SNCA expression levels [23, 24, 57, 206, 223]. In cases with SNCA triplication, a 1.8-fold increase in levels of soluble α-synuclein protein was observed in blood samples, along with a doubling of SNCA mRNA in brain tissues, with corresponding increases in insoluble α-synuclein species of high molecular mass [165], suggestive of aggregate formation.
Emerging experimental evidence suggests that the backbones of the inclusion filaments, the abnormal α-synuclein aggregates, are transmitted from cell to cell across interconnected brain regions and serve as nucleation sites for additional Lewy inclusion formation [84, 86, 189, 250]. Thus, aggregated forms of α-synuclein stoke the fibrillization of neighboring α-synuclein monomers into β-sheet-rich fibrils typical of amyloid material [85, 212, 218]. Although intense α-synuclein immunoreactivity is observed within Lewy bodies and Lewy neurites [75, 128, 232, 233], it is not clear if α-synuclein is truly the most abundant component by mass [219]. Indeed, Lewy inclusions contain numerous other proteins, including those related to PD risk, such as DJ1, parkin, and PINK1. The inclusions house proteins involved in proteasomal and autophagic degradation systems (e.g., ubiquitin, ubiquitin C-terminal hydrolase, proteasome activators 28 and 700, and autophagic adaptor p62/sequestosome-1), proteins associated with aggresomes (pericentrin, γ-tubulin, and HDAC6), as well as microtubule-associated proteins, heat shock proteins, and kinases [135, 140, 263]. Lewy bodies also harbor lipids and fragments of lysosomes, mitochondria, proteasomes, and the cytoskeleton [168, 219, 263]. Thus, the inclusions are remarkably heterogeneous, as may be reflected in their potential cytotoxic versus cytoprotective roles, and in their diverse histological staining properties. Lewy bodies can be eosinophilic, weakly eosinophilic (pale bodies), or hematoxylin and eosin-negative and they can react with thioflavin dyes and lipid-binding agents [86, 168]. Lewy bodies are also found at varying stages of compaction and maturity in different neurons; some Lewy bodies may develop a dense core and peripheral halo where α-synuclein immunoreactivity is concentrated, whereas others may harbor α-synuclein immunoreactivity without a core and shell [263].
Properties of α-synuclein in the context of Lewy body disease
α-Synuclein is widely recognized as a soluble protein, but it likely exists in equilibrium between soluble cytosolic forms and phospholipid-bound forms along membranous structures, where it affects their curvature [43, 162, 255]. An 11-residue sequence (XKTKEGVXXXX) repeated seven times in the 140 amino acid sequence of α-synuclein facilitates the formation of an amphipathic α-helical fold, which associates with phospholipid membranes and displays preference for smaller vesicles, such as ~ 40 nm synaptic vesicles [37]. The SNCA missense mutations associated with Lewy body disease tend to code for amino acids within these eleven-residue repeats. Residues 1 to 60 along the amino terminus of α-synuclein house the membrane-binding motifs important for formation of the α-helices. The central non-β-amyloid component (NAC) region along residues 61–95 is hydrophobic and crucial for β-sheet formation, and the unstructured, negatively charged acidic and glutamate-harboring carboxyl terminus is subject to numerous post-translational modifications that may affect α-synuclein hydrophobicity and its interactions with proteins and lipids [37, 234].
α-Synuclein was commonly reported as intrinsically disordered, based on biochemical assays, but in human brain tissue subjected to protein cross-linking methods, approximately 75% of α-synuclein normally adopts an α-helical multimeric form [58]. The latter multimeric form is not pathologic and plays a physiologic role in synaptic vesicle clustering [266] but it is destabilized and less abundant in the presence of Lewy body disease-causing missense mutations [58, 59]. Thus, low physiologic α-synuclein multimer/monomer ratios may contribute to disease progression, as suggested by loss of dopaminergic neurons and emergence of L-DOPA responsive motor symptoms in mice forced to express multiple E → K mutations in the KTKEGV motifs or similar multiplications of the G51D mutation in α-synuclein [176, 177].
As noted above, α-synuclein is subject to post-translational modifications that include, but may not be limited to, ubiquitination at lysine residues, phosphorylation, oxidation, acetylation, O-GlcNAcylation (e.g., [12]), sumoylation, nitration (e.g., [35]), and N- or C-terminal truncation (e.g., [141]). Spillantini and colleagues originally observed that antibodies raised against ubiquitin tend to label fewer Lewy inclusions than antibodies against α-synuclein [232, 233], consistent with the notion that ubiquitination of α-synuclein occurs subsequent to deposition in Lewy inclusions [3, 83]. This may also be true of phosphorylation events, but the probability of a post-translational modification will naturally rise with the age of the protein and with impediments to protein turnover/clearance, as will the probability that the protein denatures and precipitates into an insoluble inclusion.
Candidate kinases playing a role in α-synuclein phosphorylation include casein kinase I and II, G-protein-coupled receptors 1, 2, 5, and 6, and polo-like kinases [37, 39, 118]. The phosphorylation sites along α-synuclein include serine 87 or 129, and tyrosine 125, 133, or 136 [37], but the hyperphosphorylation of serine 129 is perhaps the most useful modification in distinguishing Lewy body disease. In brain tissues from patients with Lewy body disease, nearly the entire fraction of aggregated α-synuclein is phosphorylated at serine 129, whereas a low percentage of the soluble α-synuclein fraction is phosphorylated at this residue in control and DLB brains [3, 75, 178, 181]. The latter, physiological phosphorylation of synaptic α-synuclein at serine 129 is linked with neuronal activity and facilitates the association of α-synuclein with other synaptic proteins, such as VAMP2 and synapsin [185, 197]. There are numerous conflicting reports of the causal impact of phosphorylation of α-synuclein on neurotoxicity and Lewy inclusion formation (e.g., [10, 46, 72, 88, 159, 270]), but recent work suggests that phosphatases are unable to effectively dephosphorylate α-synuclein molecules that are sheltered within inclusions [49] and that the hyperphosphorylation event may serve to prevent further aggregation of α-synuclein [80].
Although there is utility in employing pSer129 antibodies to identify Lewy-related pathologies, the use of pSer129 antibodies to identify Lewy bodies and Lewy neurites is associated with technical artifacts, as is common with phospho-specific antibodies (see [2]). Therefore, in situ or biochemical confirmation of Lewy or Lewy-like inclusions is recommended via colabelling with various antibodies that react with Lewy inclusions (e.g., multiple antibodies against α-synuclein, ubiquitin, and/or p62), use of electron microscopy and amyloid stains (e.g., Thioflavin derivatives), and leveraging α-synuclein insolubility in nonionic detergents, resistance to proteinase K, and proteolytic truncation of its carboxyl terminus (e.g., [141]).
SNCA is highly expressed in the central nervous system, but its mRNA is also present in peripheral tissues, such as blood, uterine, ovary, prostrate, colon, esophageal, and skin tissues, with highest bulk expression in cerebellar tissues and lowest expression within skeletal muscle (Fig. 1). Some peripheral tissues may release α-synuclein protein into the interstitial fluid and plasma, but the source of transmissible α-synuclein aggregates in Lewy body disease is likely to originate in neurons, given SNCA expression patterns at the tissue level in Fig. 1 and enrichment in synaptic terminals at the cellular level.
Aside from α-synuclein expression in diverse tissue types, the intracellular heterogeneity of Lewy inclusions was originally recorded by their discoverer, Fritz Lewy, who drew various shapes of inclusions along cellular processes, within neuronal somata, and inside cell nuclei (see Fig. 3 of [86]). Since then, α-synuclein has been reported in nonsynaptic compartments such as the Golgi apparatus and nucleus [19, 125, 209, 271]. Indeed, the term α-synuclein was coined as a portmanteau of the words synapse and nucleus, as it was reported to lie along the inner lining of the neuronal nuclear envelope, aside from being concentrated in synaptic terminals [156]. Controversies surrounding the histological work [240] could reflect lack of specificity of the older antibodies or higher levels of α-synuclein in the synaptic terminal compared to nucleus.
Given the broad tissue distribution of α-synuclein and breadth of neurological symptoms in Lewy body disorders, it is unsurprising that Lewy-related aggregates form not only in the brain and spinal cord, but also in the skin [82, 96], submandibular gland [16, 179], autonomic ganglia [15], heart [113, 179], adrenal medulla [76], and gastrointestinal plexuses of Meissner and Auerbach [193, 262]. Although the anatomical reach of Lewy pathologies may not be as spatially expansive as the normal expression of SNCA, the latter may also have diagnostic significance.
α-Synuclein aggregation is prompted by a stealthy disease trigger
Braak and colleagues originally speculated that Lewy pathology is initiated by exposure of cranial nerve endings in the gastrointestinal and nasal mucosal linings [31,32,33, 98, 99]. Thus, once cranial nerves I and X are exposed to the causative agent and aggregates of α-synuclein take shape, the aggregates were proposed to spread from cell to cell via neuroanatomical fiber tracts, perhaps explaining olfactory and autonomic symptoms at the earliest stages of PD [29,30,31,32,33, 56]. Prior to this unconventional notion, PD had often been described as a motor condition with confined, selective degeneration of the substantia nigra in the ventrolateral mesencephalon and perhaps the locus coeruleus of the pons (another catecholaminergic structure housing melanin pigment). Historical biases towards catecholamine-releasing neurons and melanin-rich brain regions understandably reflect the evolution of histochemical and microscopic techniques in the last century. In addition, animal studies also supported the view that selective loss of nigrostriatal fibers underlies the motor deficits of PD [279], while clinical evidence confirmed the utility of L-DOPA to help replace dopamine stores.
The idea that attractions between proteins culminate in their ordered assembly into filaments that are deposited in the intra- or extracellular space also arose decades ago (e.g., [114, 214, 247, 264, 265]. The molecular conversion event of α-synuclein monomers into filaments that precipitate and help form Lewy inclusions is akin to the prion diseases (e.g., Kuru and fatal familial insomnia) [38, 231, 268]. Although α-synucleinopathies are not (yet) viewed as infectious conditions with inter-organismal transmission in the wild (see [70, 130]), human fetal brain cells transplanted into the brains of patients with PD eventually develop Lewy bodies [124, 138, 139]. The latter findings strongly support human cell-to-cell transmission of Lewy-related pathologies but may also reflect a response to the diseased microenvironment of the host, leading to structural modifications and precipitation of otherwise normal α-synuclein molecules within grafted cells.
Although inclusions harboring α-synuclein filaments are the main hallmark of Lewy body disease, the aggregation of α-synuclein may be secondary to exposure to the disease trigger, even in inherited cases. Unless there is stochastic misfolding of α-synuclein, some other stimulus, such as environmental factors or SNCA overexpression/mutation, likely initiates the first aggregation event. During disease initiation, an environmental trigger may breach not only epithelial barriers within nasal, oral, esophageal, or gastrointestinal mucosa, but perhaps also the epidermis of the skin. One can further speculate that, although everyone is exposed to the disease trigger—including the spouses and coworkers of those with Lewy body disease—those with greater genetic predispositions (e.g., old age, male sex, defects in protein quality control pathways, inferior immune function) are more likely to develop a clinical syndrome. In addition, individuals could be exposed to the same disease trigger but at varying frequencies, durations, and concentrations (amplitude), as well as at different circadian phases. The temporal qualities and intensity of these environmental factors, when combined with compromised genetic defenses, conceivably influence the probability of α-synuclein aggregation. An alternative scenario is that only few individuals are exposed to the disease trigger, and only they will develop Lewy body disease.
The disease trigger(s) for idiopathic Lewy body disease has not been confirmed, but the inhalation and swallowing of (or perhaps skin contact with) toxicants such as trichloroethylene, paraquat, or natural toxins such as rotenone are possible culprits [54, 55, 172, 242], as are head trauma and dairy product ingestion [8, 196]. By suppressing energetic resources, eliciting oxidative stress, and disabling protein quality control mechanisms in parts of the neuraxis that contact peripheral boundaries, exposures to environmental vectors conceivably shift the shapes of α-synuclein molecules towards the pathologic forms. Based on emerging evidence, one can also speculate that environmental disease triggers encourage the first α-synuclein nucleation event by promoting liquid–liquid phase separation prior to droplet formation, followed by potential conversion into a perinuclear aggresome, and then maturation into a relatively insoluble inclusion [168, 171, 198, 199]. Host factors, such as polymorphisms in the amino acid sequence of α-synuclein could influence cellular thermodynamics in this scenario [152], particularly when genetically determined defenses are naturally low, cells are exposed to chronic environmental stressors, and SNCA transcription rates are higher in select neuron populations [91], leading to possible molecular crowding and phase changes of α-synuclein.
Aside from toxicant and toxin exposures, head trauma, and dairy product ingestion as potential disease triggers, a fourth possibility is that infections with microbial pathogens set the stage for immune cell dysregulation and α-synucleinopathy in peripheral and central neurons [97, 131, 137, 142, 188]. Recent experimental evidence suggests that, (1) α-synuclein plays a role in immunity and inflammation, (2) microbial gut amyloids stimulate α-synuclein aggregation, and (3) α-synuclein harbors antimicrobial properties under conditions of viral or bacterial infection after nasal or systemic inoculation [1, 17, 117, 204, 236, 243, 244, 246]. The evidence for inflammation and immune cell dysregulation in human Lewy body disease is partly based on imaging of translocator protein 18kDa (TSPO) (reviewed in [143, 213, 243, 244, 276]), but it has been difficult to ascertain if the net impact of immune cell function in Lewy body disease is helpful or harmful (it may also vary across genetically diverse human subjects).
Innate immune cells are the first line of defense within injured and diseased tissues, and in the immunospecialized CNS, this essential role is primarily executed by microglia/macrophages [77]. Neurodegenerative disorders are believed to be accompanied by microgliosis (i.e., recruitment, hyperplasia, and stimulation of microglia) and the hypersecretion of proinflammatory cytokines, chemokines, eicosanoids, free radicals, and proteases. However, microglia/macrophages are essential for CNS repair and recovery, particularly under conditions of injury and/or infection. Postmortem studies reveal numerous microglia-like cells immunopositive for the antigen-presenting cell marker, major histocompatibility complex class II antigen (HLA-DR) in nigral tissues of subjects with PD [160] and in the transentorhinal cortex of subjects with DLB [151], compared to control subjects. In addition, the area occupied by the microglia/macrophage lysosome marker CD68 (i.e., macrosialin) is higher in DLB brain tissues [237]. However, these effects were not observed for the pan-microglia/macrophage marker Iba1, which may indicate glial cell senescence, with no enhanced proliferation or reactivity [237]. Indeed, when microglia/macrophages are stimulated and turn reactive, they tend to hypertrophy, but this stress response is also not observed in brains with DLB [237]. On the other hand, higher levels of dystrophic (beaded/fragmented) microglia/macrophage processes are evident in DLB [11] and mRNA levels for the cytokine gene Il-6 are higher in the hippocampi of subjects with PD and DLB [109]. The evidence for immune cell dysregulation in Lewy body disease reveals, at the least, a rich pleomorphism of microglia/macrophages [11].
α-Synucleinopathy may be transmitted across the neuraxis
Following the seeding of α-synuclein aggregates in the rostral forebrain and caudal brainstem, Lewy pathology may be transmitted deeper into the brain, eventually affecting the locus coeruleus of the pons, the raphe complex, the pedunculopontine tegmentum, the substantia nigra and ventral tegmental area of the mesencephalon, the magnocellular basal forebrain nuclei, the amygdala and other limbic regions of the telencephalon, such as the entorhinal cortex and hippocampal subfield CA2 [29,30,31,32,33, 56]. Lewy pathology was originally hypothesized by Braak et al. to emerge within neocortical regions at final stages of PD, perhaps spreading to these destinations from meso- and allocortical sites, with the heavily myelinated primary motor and sensory cortices as the last to be affected [31, 32, 34].
The clinical staging of Lewy body disorders continues to be refined since Braak criteria were originally proposed. In 2009, Beach et al. reported a Unified Staging System, in which DLB and Alzheimer’s disease with Lewy bodies (ADLB) were found to be associated with limbic-predominant Lewy pathology in early stages of disease, whereas early PD was linked to brainstem-predominant pathology, with both types of disease breaching the neocortex at end stages [14]. Importantly, Beach and colleagues also suggested that the first brain region likely to be affected is the olfactory bulb rather than medulla oblongata [13, 14]. The staging of Lewy pathology by other comprehensive histological systems, such as that of McKeith or Leverenz et al., and more recently, the Lewy pathology consensus criteria by Attems et al., supports parcellation into olfactory-only, amygdala-predominant, limbic, brainstem, and neocortical stages [9, 136, 161]. Borghammer’s Brain-First vs. Body-First staging scheme also distinguishes cases with Lewy pathology in the autonomic nervous system or lower brainstem—which rarely display Lewy pathology in olfactory nuclei—from cases with amygdala-predominant Lewy pathology, which nearly always have Lewy pathology in olfactory nuclei [26,27,28]. Chahine and colleagues recently offered a biologic staging system for PD that also accounts for the presence of prodromal disease and proposes α-synuclein aggregation as a major risk factor for dopaminergic dysfunction [41, 225].
Although retrograde and anterograde transport of α-synuclein both contribute to intracellular expansion of aggregates, retrograde transport to the somata appears to predominate [101, 210]. Thus, neuritic forms of Lewy inclusions tend to appear before somal Lewy bodies, including the Lewy-like inclusions in experimental models (e.g., see [62]). Indeed, α-synuclein aggregation has been reported to commence in axon collaterals, supporting retrograde transfer to the cell body prior to formation of somal inclusions [116]. Retrograde degeneration of neurons also seems more likely, given that synaptic and axonal deficits in Lewy body disease also precede loss of neuronal somata. Thus, there is greater fractional loss of dopaminergic terminals in the striatum compared to degeneration of nigral neurons in PD [47, 78, 274].
In both rodents and nonhuman primates, infusions of preformed α-synuclein fibrils into dorsal striata precipitate the (retrograde) onset of Lewy-like inclusions in nigral efferent neurons and other (anterogradely and retrogradely) anatomically connected structures [50, 144,145,146,147,148, 187, 201, 210, 251]. Nigral cell loss, deficiencies of dopaminergic tone in the striata, and L-DOPA-responsive behavior deficits are thereby induced, suggesting that striatal infusions of α-synuclein fibrils model mid-to-late disease, according to Braak staging criteria. Infusions of oligomeric forms tend to be less successful in recapitulating disease [73].
As Lewy pathologies may emerge earlier in the olfactory bulb than in nigral efferents, the olfactory bulb has also been infused with preformed fibrils in mice, rats, and monkeys to model Lewy body disease [21, 22, 49, 50, 157, 158, 166, 167, 201, 202, 208]. As expected, dense Lewy-like pathologies emerge in limbic regions of the telencephalon connected to the olfactory site of infusion, such as the cortical amygdala, piriform (primary olfactory) and entorhinal cortices, and the ventral hippocampal formation (first sector of Ammon’s horn). The inclusion patterns appear like a moonlike crescent along lateral, ventral, and dorsolateral boundaries of the telencephalon, as is readily appreciated along sagittal or horizontal planes of section. A limitation of this type of work is that naturally formed, human wildtype fibrils differ structurally from fibrils synthesized in vitro from rodent Snca, in ways that may alter templating and propagation properties, as well as the kinetics of cell-to-cell transfer [251, 278]. It should be acknowledged that animals do not develop the Lewy bodies that are found in human brains, and that it is difficult to faithfully recapitulate Lewy body disease in the lab. The attributes of α-synucleinopathy that are described in short-lived rodents may therefore not translate to age-related human conditions. Another limitation of the animal models is that rodents are macrosmatic creatures with possibly stronger, more active connections in brain regions involved in olfaction, compared to microsmatic (visual) primates. As discussed below, the density of synaptic terminals and synaptic activity levels (including pinocytic rate during vesicular recycling) may influence the probability of developing Lewy-related pathologies.
In cynomolgus monkeys, preformed fibril infusions in the striata elicit mild loss of nigral neurons, with a parallel rise in the dopaminergic transporter (DAT) [50]. Increases in DAT and tyrosine hydroxylase are also observed after fibril infusions in the bulbar anterior olfactory nucleus (Fig. 11 in [158]). These observations in mice and nonhuman primates may be analogous to the Parkinson's Progression Marker Initiative (PPMI) showing higher DAT in as yet-unaffected mutant GBA carriers—prior to the onset of clinical parkinsonism [224]. However, in most studies with long survival periods after infusions of fibrils into the striatum, dopaminergic markers are lowered, and mild to moderate loss of nigral neurons is observed [63, 147, 186, 187, 252]. It is still unclear whether patients who are exposed to a disease trigger but have not developed ILBD display compensatory increases in enzymes that lie along the biosynthetic pathway for dopamine and/or show compensatory sprouting of dopaminergic terminals in the striatum. These types of adaptive changes could be manifested as increases in tyrosine hydroxylase and DAT markers, at least in the earliest stages of disease.
Current assumptions on Lewy body disease
With the intention of accelerating research on Lewy body disease, we will list some of the additional factors known or speculated to influence α-synucleinopathy:
-
1.
Intracellular expression of α-synuclein must be available to feed the aggregate pool but may not always scale linearly with Lewy-related pathologies. The availability of α-synuclein monomers as substrates for inclusion formation will depend on a balance between α-synuclein transcription/translation rates, clearance, and multimer formation and aggregation in both patients with Lewy body disease and experimental models thereof. SNCA expression in humans varies considerably across tissue types and brain subregions (Fig. 1). In the C57 mouse that is commonly used to model α-synucleinopathies, the Snca gene is also not uniformly expressed across brain subregions (Fig. 2). For example, subcortical structures display less Snca mRNA, with exception of the dorsomotor nucleus of the vagus and the substantia nigra, pars compacta—both of which are notably vulnerable to Lewy pathology in humans (arrows in Fig. 2).
Fig. 2 
Allen Brain Atlas in situ mRNA hybridization for Snca, the mouse gene encoding α-synuclein [134]. A–D Sagittal brain sections from a 56-day-old C57BL/6 J male mouse are shown from medial to lateral, with high expression of α-synuclein mRNA in the dorsomotor nucleus of the vagus (DMV), the substantia nigra pars compacta (SNpc), the pyramidal and granule cells of the hippocampus (HP), and the mitral cell layer of the olfactory bulb (OBml). Moderate expression is seen in the ventral tegmental area (VTA), anterior olfactory nucleus (AON), the locus coeruleus (LC), the subiculum (Sub), the cortical amygdala (CoA), the piriform cortex (Piri CTX). There is low expression of Snca in the olfactory tubercle (OT), cerebellum (CB), and white matter (unlabeled). Antisense data were retrieved from the Allen Institute at this link: https://mouse.brain-map.org/experiment/siv?id=79904550&imageId=79764405&initImage=expression&contrast=0.5,0.5,0,255,4
The cerebellum does not routinely develop Lewy pathologies [68] and has overall low Snca expression in mice (Figure 2) but unexpectedly high SNCA expression in humans (Figure 1), suggestive of species differences. On the other hand, mRNA levels may not scale in proportion to protein expression levels. Thus, the human cerebellum displays low expression of pan-α-synuclein protein [68] despite relatively high SNCA mRNA (Figure 1).
It is notable that Snca is expressed at low levels in the mouse cerebellum of Fig. 2 while α-synuclein protein levels are relatively high in rat cerebellar tissues (see Fig. 2a in [112]). However, Fig. 3 shows single-cell RNA sequencing data from the DropViz database, and lists mouse cerebellar interneurons as harboring high Snca expression [207]. In this respect, it is worth noting that the cerebellum also contains a high proportion of white matter, and myelinated neurons do not typically develop Lewy pathology [31, 32, 34]. Thus, some cellular subtypes within the cerebellum will have more α-synuclein expression than others, potentially leading to variance across studies that rely on different methods and have varying degrees of cellular resolution.
Fig. 3 
DropViz database ranking of top ten mouse brain cell clusters with the highest expression of Snca [207]. Note the high expression of Snca in cerebellar neurons and in hippocampal neurons, consistent with mouse data in Fig. 2d. Retrieved from http://dropviz.org
Other heterogeneous brain regions, such as the hippocampus, are also not major flashpoints for somal Lewy pathology (i.e., Lewy bodies) in humans, and yet express abundant α-synuclein mRNA in both humans (Fig. 1) and mice (Fig. 2 and Fig. 3; also see [112]). Thus, intracellular α-synuclein concentration is only one of the drivers of hippocampal Lewy pathology, alongside the existence of neuronal connections as roadways for spatial transmission [52, 149, 163, 175, 195]. On the other hand, characteristic Lewy-like structures and cell loss fail to develop in experimental models after knockout of Snca [120, 200, 258, 260], which has two important implications. First, the presence of α-synuclein is a prerequisite for formation of pathologic inclusions in mice. Second, preformed fibrils per se do not disrupt membranes and other cell components sufficiently to cause frank cell loss in the absence of α-synuclein. It would be helpful to evaluate Lewy-related pathologies in Snca knockout mice with tools not inherently dependent on α-synuclein expression. If the gene is absent, so will be the protein, and it will therefore be useful to test, at both microscopic and ultrastructural (cryo-electron microscopic) levels, if filamentous, eosinophilic, and lipid-filled inclusions can form in the absence of the gene encoding α-synuclein.
-
2.
α-Synuclein is present in both cytosolic and interstitial compartments. The majority of α-synuclein substrate in the central nervous system is likely to be synthesized inside neurons, but α-synuclein is also released into the interstitial matrix and is therefore present in the cerebrospinal fluid compartment, including in the oligomeric form [64, 65, 153, 184]. Both intracellular and extracellular α-synuclein molecules, including those found inside nanosized extracellular vesicles, may serve as templates for further aggregate formation [92, 253]. Thus, emerging evidence suggests that α-synuclein, including aggregated species of high molecular mass, can be packaged into extracellular vesicles, secreted in a calcium-dependent manner, and transmit pathology to recipient cells [66, 90, 93, 95, 127, 133, 238]. Conditions that encourage extracellular vesicle release may influence the spread of α-synucleinopathy across the neuraxis and open a path for potential entry of α-synuclein aggregates across the blood–brain, gut-to-plasma, and nose-to-brain barriers.
-
3.
The extracellular aggregate pool requires access to plasma membranes for passage into the cytosol and cell-to-cell propagation. The probability of uptake of α-synuclein aggregates by a particular neuron may be influenced by how often the neuron samples the extracellular environment by micro- or macropinocytosis, receptors and transporters housed in its plasma membrane, and the binding of cell surface heparan sulfate glycans [103, 108]. One might expect neuronal uptake and release of aggregates to be highest at the synaptic membrane, especially of neurons with extensive synaptic arborization, although the synaptic space is somewhat separated from the remaining interstitial fluid by adhesion molecules and membrane proteins. Given that vesicular membrane recycling is accompanied by pinocytosis of surrounding fluids, the electrical activity of synapses may play a crucial role in uptake and release of aggregates [275]. Human dopamine neurons have been estimated to arborize into more than a million synapses each, possibly extending more than four meters in length, which also places a heavy energetic burden on the mitochondrion [192]. If aggregate uptake occurs along the length of the axon, the degree of myelination may also influence entry of aggregates into neurons [31, 32, 34]. The myelin sheath is not only associated with lower energy expenditures and therefore, less leakage of reactive oxygen species from the electron transport chain, but it also endows the axon with a simple physical barrier. Hence, intersubject differences in active neuronal circuity, density of synaptic terminals, plasma membrane molecules that bind/transport α-synuclein, pinocytic rates, membrane surface area, axon length, and the degree of myelin coverage could underlie variations in α-synuclein aggregate engulfment and partly explain the diverse clinical outcomes of Lewy body disease.
-
4.
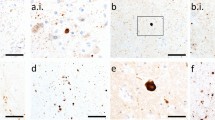
Aggregates may be released from damaged or dying cells and attract immune cells by chemotactic means. If dying cells are not quickly and fully cleared by microglia, astrocytes, or phagocytic peripheral immune cells that infiltrate the brain, aggregated forms of α-synuclein could be released through damaged membranes into the extracellular compartment for further seeding and expansion of the aggregate pool. In this scenario, a robust inflammatory response may not be as toxic as often assumed but may help isolate and destroy α-synuclein aggregates. Therefore, the hunt for and destruction of damaged or dead neurons harboring α-synuclein aggregates and other toxic cargo by immune cells may slow the spread of Lewy-related pathologies, a scenario consistent with the chemoattractant properties of α-synuclein [100, 236, 267]. These speculations are supported by in vivo observations of microglia/macrophage efferocytosis of neuronal somata housing exogenous α-synuclein fibrils (Fig. 4), and raise the possibility that neurodegeneration is not always harmful in the diseased brain. On the other hand, microglia also discharge α-synuclein within extracellular vesicles that encourage the cell-to-cell transmission of pathology, hyperphosphorylation of α-synuclein, and dopaminergic neuron loss [93]. These collective data likely reflect the wide spectrum of immune cell functions.
Fig. 4 
Microglia/macrophages may engulf neurons that house exogenous, preformed fibrils. Phosphate-buffered saline (PBS) or ATTO647 preformed fibrils (PFFs) were sonicated and infused in the bulbar anterior olfactory nucleus in genetically outbred CD-1 mice of both sexes, as described [21]. Mice were sacrificed 3 h or 3–6 days later for immunostaining with antibodies against the pan-microglia/macrophage marker Iba1 (Wako 01919741) and the neuronal nuclear marker NeuN (Millipore ABN90). The Hoechst reagent was applied to label all nuclei. A Infusion site in the dorsal anterior olfactory nucleus and the caudal olfactory bulb. B The labeled markers are shown in isolation (for clarity) or in merged form at three days post-infusion. Note the Iba1+ cell partially enfolding a NeuN+ cell. Most PFFs at the site of infusion were in NeuN+ neurons rather than NeuN− profiles. Thus, we speculate that neurons may be unable to fully degrade aggregates—or else the PFF model would not work. C Shown is a confocal Z-slice at the intersection of an Iba1+ object engulfing a NeuN+ object that houses fluorescent PFFs. D–E Three-dimensional Imaris rendering of the cell from panel C. A second representative example is included on the left of panel D
-
5.
Aggregates are incompletely removed from extracellular and intracellular compartments by the available clearance mechanisms. The clearance of protein aggregates by autophagic or proteasomal degradation, the sequestration of α-synuclein in mature Lewy bodies, and the extrusion of toxic aggregates into the extracellular matrix for uptake by neighboring glia (or removal by glymphatic clearance) all conceivably impact the course of Lewy body disease but are not adequate to fully contain the spread of protein aggregates. Undigested aggregates can rupture lysosomal membranes, leading to autophagic failure, unless lysophagic clearance systems can be fully engaged [115]. Each of these defensive systems will closely depend on the metabolic and energetic resources available to the cell for repair and recovery. Other defenses against protein aggregates include tunneling nanotubes across glial cells, perhaps as a means of diluting the toxicity and sharing the burden of degrading α-synuclein aggregates with other cells by autophagy [48, 211]. However, high concentrations of α-synuclein may saturate microglial autophagic systems [248]. It seems worth cautioning that the presence of α-synuclein mRNA and protein within glial and other non-neuronal cells does not necessarily imply that these cells transcribe Snca mRNA from nuclear DNA. Rather, single-cell transcriptomic sequencing and flow cytometric analyses of single cells may also detect mRNAs and proteins engulfed from non-autonomous sources that are not yet degraded, including via uptake of extracellular vesicles or glial efferocytosis of neurons containing α-synuclein (Fig. 4).
-
6.
The properties of extracellular matrices influence aggregate diffusion. There are few studies of the extracellular compartment in Lewy body disease, but α-synucleinopathy has been demonstrated to alter the structure and function of extracellular matrix components, including forcing the degradation of hyaluronan [229]. The flow of protein aggregates alongside neuronal membranes may be influenced by adhesion to components of the extracellular matrix (including lipophilic components), binding to plasma membrane proteins and carbohydrates that protrude deep into the interstitial fluid, repulsion from extracellular water, and the flow and shear dynamics of the extracellular compartment [111, 169, 229].
-
7.
Aggregates accumulate with age and/or the passage of time. In several models of Lewy body disease, older animals tend to develop more pathology than younger animals [42, 105, 121, 254]. Advanced age is the chief risk factor for Lewy body disease, perhaps due to loss of protein quality control with aging and accumulation of undigested protein aggregates, following cumulative lifetime exposures to environmental and intrinsic insults (e.g., reactive oxygen species from oxidative phosphorylation). In the absence of a degenerative disorder, aging is not accompanied by robust, nonspecific neuronal loss [261]. However, aging may be accompanied by a decay of immune cell function [272], which could permit the escape of α-synuclein aggregates from one cell into another (see above). Another major impact of aging and the passage of time is the amassment of DNA damage [215], particularly in postmitotic neurons that cannot divide and that escape repair mechanisms, such as base-excision repair. This accruing burden might lead to diversion of energetic resources and structural changes in proteins translated from damaged sequences in nuclear or mitochondrial genomes. Over time, age-related proteomic shifts may surpass the threshold for a functional neuronal or glial cell, leading to age-related neurological deficiencies and engagement of cellular senescence programs.



Conclusions
Research on α-synuclein has progressed to the point that patient-tailored anti-α-synuclein therapies may be on the horizon. Clinicians can measure α-synuclein aggregation propensities through α-synuclein seed amplification assays conducted on patients’ cerebrospinal fluid, blood, or biopsies of solid tissues where Lewy-related pathologies tend to form [40, 123, 155, 180, 222]. The mechanism of the α-synuclein seed amplification assay is easy to envisage, because filamentous structures tend to naturally get nested together with vigorous shaking in a confined box, and the rate of alignment is accelerated the more filaments are aligned at the beginning of the reaction. As the structural aspects of the filaments (e.g., smooth or bumpy) will influence the kinetics of such reactions, one can also envision that diverse spatial qualities of α-synuclein strains influence clinical phenotypes and rates of disease progression [129, 162]. The contribution of polymorphic strains of α-synuclein filaments to disease progression is particularly evident when comparing Lewy body disorders to multiple system atrophy, an α-synucleinopathic condition that is not classified as a Lewy body disorder [102, 119, 216, 228].
Although spatial patterns of Lewy inclusions are not uniformly correlated with neurodegeneration (e.g., see discussions by [241] versus [14]), the α-synuclein seeding capacities of antemortem cerebrospinal fluid samples appear to correlate with postmortem evaluations of the presence and anatomical reach of Lewy inclusions in the brain, suggesting that the number (and shapes) of pathogenic seeds are associated at least to some degree with the transmission of Lewy-related pathology across the neuraxis [7, 18, 205]. Because these tests can be carried out on live subjects, they may be useful to stratify patients prior to clinical trials as α-synuclein-targeted therapies become available [230]. For example, patients with parkinsonism but without positive α-synuclein seed amplification may be inappropriate to include in clinical trials of monoclonal antibodies against α-synuclein.
In conclusion, aside from processes listed above, α-synuclein aggregates may form, spread across cells, exert toxicity, and induce cell loss depending upon the following factors:
-
The number of α-synuclein molecules that crowd the cell and are available as nucleation points for additional seeding and phase separation
-
The intracellular location of α-synuclein aggregates (e.g., possibly more toxic or disruptive if in a thin axon or small synapse, rather than tucked within Lewy bodies in a sequestered corner of the roomier somata)
-
The compactness/maturity/insolubility of the inclusion and its interactions at inclusion boundaries with cytoskeletal proteins and mitochondria, lysosomes, and proteasomes
-
The tendencies of noncompact inclusions to break apart and leach soluble oligomers
There are many other factors we have not considered here. Additional research on the biophysical and biochemical underpinnings of Lewy body disease, the expansion of α-synucleinopathic lesions over space and time, and the molecular means of initiation of cell death programs will likely be needed to conduct effective clinical trials and slow or prevent Lewy body disease.
Data availability
Data will be made available upon request to leakr@duq.edu.
References
Alam MM, Yang D, Li XQ, Liu J, Back TC, Trivett A et al (2022) Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep 38:110090. https://doi.org/10.1016/j.celrep.2021.110090
Altay MF, Kumar ST, Burtscher J, Jagannath S, Strand C, Miki Y et al (2023) Development and validation of an expanded antibody toolset that captures alpha-synuclein pathological diversity in Lewy body diseases. NPJ Parkinsons Dis 9:161. https://doi.org/10.1038/s41531-023-00604-y
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ et al (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752. https://doi.org/10.1074/jbc.M600933200
Ansorge O, Daniel SE, Pearce RK (1997) Neuronal loss and plasticity in the supraoptic nucleus in Parkinson’s disease. Neurology 49:610–613. https://doi.org/10.1212/wnl.49.2.610
Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N et al (2011) Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci 31:7264–7274. https://doi.org/10.1523/JNEUROSCI.6194-10.2011
Arawaka S, Saito Y, Murayama S, Mori H (1998) Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 51:887–889. https://doi.org/10.1212/wnl.51.3.887
Arnold MR, Coughlin DG, Brumbach BH, Smirnov DS, Concha-Marambio L, Farris CM et al (2022) Alpha-synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological alpha-synuclein in the context of co-pathology and non-LBD diagnoses. Ann Neurol 92:650–662. https://doi.org/10.1002/ana.26453
Ascherio A, Schwarzschild MA (2016) The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15:1257–1272. https://doi.org/10.1016/S1474-4422(16)30230-7
Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G et al (2021) Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol 141:159–172. https://doi.org/10.1007/s00401-020-02255-2
Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T et al (2009) Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet 18:872–887. https://doi.org/10.1093/hmg/ddn417
Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ et al (2015) Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3:32. https://doi.org/10.1186/s40478-015-0209-z
Balana AT, Mahul-Mellier AL, Nguyen BA, Horvath M, Javed A, Hard ER et al (2024) O-GlcNAc forces an alpha-synuclein amyloid strain with notably diminished seeding and pathology. Nat Chem Biol 20:646–655. https://doi.org/10.1038/s41589-024-01551-2
Beach TG, White CL 3rd, Hladik CL, Sabbagh MN, Connor DJ, Shill HA et al (2009) Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174. https://doi.org/10.1007/s00401-008-0450-7
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. https://doi.org/10.1007/s00401-009-0538-8
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL et al (2010) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119:689–702. https://doi.org/10.1007/s00401-010-0664-3
Beach TG, Adler CH, Dugger BN, Serrano G, Hidalgo J, Henry-Watson J et al (2013) Submandibular gland biopsy for the diagnosis of Parkinson disease. J Neuropathol Exp Neurol 72:130–136. https://doi.org/10.1097/NEN.0b013e3182805c72
Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE et al (2015) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 90:2767–2782. https://doi.org/10.1128/JVI.02949-15
Bentivenga GM, Mammana A, Baiardi S, Rossi M, Ticca A, Magliocchetti F et al (2024) Performance of a seed amplification assay for misfolded alpha-synuclein in cerebrospinal fluid and brain tissue in relation to Lewy body disease stage and pathology burden. Acta Neuropathol 147:18. https://doi.org/10.1007/s00401-023-02663-0
Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernandez MA, Balbuena-Olvera AJ, Morales-Moreno ID, Arguero-Sanchez R et al (2019) Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci 13:1399. https://doi.org/10.3389/fnins.2019.01399
Bernstein HG, Johnson M, Perry RH, LeBeau FE, Dobrowolny H, Bogerts B et al (2011) Partial loss of parvalbumin-containing hippocampal interneurons in dementia with Lewy bodies. Neuropathology 31:1–10. https://doi.org/10.1111/j.1440-1789.2010.01117.x
Bhatia TN, Clark RN, Needham PG, Miner KM, Jamenis AS, Eckhoff EA et al (2021) Heat shock protein 70 as a sex-skewed regulator of alpha-synucleinopathy. Neurotherapeutics 18:2541–2564. https://doi.org/10.1007/s13311-021-01114-6
Bhatia TN, Jamenis AS, Abbas M, Clark RN, Miner KM, Chandwani MN et al (2023) A 14-day pulse of PLX5622 modifies alpha-synucleinopathy in preformed fibril-infused aged mice of both sexes. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2023.106196
Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrom L et al (2019) Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 34:866–875. https://doi.org/10.1002/mds.27659
Blauwendraat C, Nalls MA, Singleton AB (2020) The genetic architecture of Parkinson’s disease. Lancet Neurol 19:170–178. https://doi.org/10.1016/S1474-4422(19)30287-X
Bloch A, Probst A, Bissig H, Adams H, Tolnay M (2006) Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol 32:284–295. https://doi.org/10.1111/j.1365-2990.2006.00727.x
Borghammer P (2021) The alpha-synuclein origin and connectome model (SOC Model) of Parkinson’s disease: explaining motor asymmetry, non-motor phenotypes, and cognitive decline. J Parkinsons Dis 11:455–474. https://doi.org/10.3233/JPD-202481
Borghammer P, Horsager J, Andersen K, Van Den Berge N, Raunio A, Murayama S et al (2021) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161:105557. https://doi.org/10.1016/j.nbd.2021.105557
Borghammer P, Just MK, Horsager J, Skjaerbaek C, Raunio A, Kok EH et al (2022) A postmortem study suggests a revision of the dual-hit hypothesis of Parkinson’s disease. NPJ Parkinsons Dis 8:166. https://doi.org/10.1038/s41531-022-00436-2
Braak H, Muller CM, Rub U, Ackermann H, Bratzke H, de Vos RA, Del Tredici K (2006) Pathology associated with sporadic Parkinson's disease--where does it end? J Neural Transm Suppl 89–97
Braak H, Rub U, Sandmann-Keil D, Gai WP, de Vos RA, Jansen Steur EN et al (2000) Parkinson’s disease: affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol 99:489–495. https://doi.org/10.1007/s004010051150
Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 3(249 Suppl):III/1-5
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Braak H, Rub U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134. https://doi.org/10.1007/s00441-004-0956-9
Burai R, Ait-Bouziad N, Chiki A, Lashuel HA (2015) Elucidating the role of site-specific nitration of alpha-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J Am Chem Soc 137:5041–5052. https://doi.org/10.1021/ja5131726
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. https://doi.org/10.1126/science.1195227
Burre J, Sharma M, Sudhof TC (2018) Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a024091
Carlson GA, Prusiner SB (2021) How an infection of sheep revealed prion mechanisms in Alzheimer’s disease and other neurodegenerative disorders. Int J Mol Sci. https://doi.org/10.3390/ijms22094861
Cavallarin N, Vicario M, Negro A (2010) The role of phosphorylation in synucleinopathies: focus on Parkinson’s disease. CNS Neurol Disord Drug Targets 9:471–481. https://doi.org/10.2174/187152710791556140
Chahine LM, Beach TG, Adler CH, Hepker M, Kanthasamy A, Appel S et al (2023) Central and peripheral alpha-synuclein in Parkinson disease detected by seed amplification assay. Ann Clin Transl Neurol 10:696–705. https://doi.org/10.1002/acn3.51753
Chahine LM, Merchant K, Siderowf A, Sherer T, Tanner C, Marek K et al (2023) Proposal for a biologic staging system of Parkinson’s disease. J Parkinsons Dis 13:297–309. https://doi.org/10.3233/JPD-225111
Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM et al (2020) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23:327–336. https://doi.org/10.1038/s41593-020-0589-7
Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC (2003) A broken alpha -helix in folded alpha-synuclein. J Biol Chem 278:15313–15318. https://doi.org/10.1074/jbc.M213128200
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123:383–396. https://doi.org/10.1016/j.cell.2005.09.028
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S et al (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169. https://doi.org/10.1016/S0140-6736(04)17103-1
Chau KY, Ching HL, Schapira AH, Cooper JM (2009) Relationship between alpha synuclein phosphorylation, proteasomal inhibition and cell death: relevance to Parkinson’s disease pathogenesis. J Neurochem 110:1005–1013. https://doi.org/10.1111/j.1471-4159.2009.06191.x
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725. https://doi.org/10.1002/ana.21995
Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K et al (2020) Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun 11:1386. https://doi.org/10.1038/s41467-020-15119-w
Choi S, Tittle T, Garcia-Prada D, Kordower J, Melki R, Killinger B (2024) Alpha-synuclein aggregates are phosphatase resistant. bioRxiv. https://doi.org/10.1101/2023.11.20.567854
Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J et al (2019) Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142:3565–3579. https://doi.org/10.1093/brain/awz296
Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC (2015) Clinical features of Alzheimer disease with and without lewy bodies. JAMA Neurol 72:789–796. https://doi.org/10.1001/jamaneurol.2015.0606
Courte J, Bousset L, Boxberg YV, Villard C, Melki R, Peyrin JM (2020) The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci Rep 10:4895. https://doi.org/10.1038/s41598-020-61757-x
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295. https://doi.org/10.1126/science.1101738305/5688/1292[pii]
De Miranda BR, Greenamyre JT (2020) Trichloroethylene, a ubiquitous environmental contaminant in the risk for Parkinson’s disease. Environ Sci Process Impacts 22:543–554. https://doi.org/10.1039/c9em00578a
De Miranda BR, Goldman SM, Miller GW, Greenamyre JT, Dorsey ER (2022) Preventing Parkinson’s disease: an environmental agenda. J Parkinsons Dis 12:45–68. https://doi.org/10.3233/JPD-212922
Del Tredici K, Rub U, De Vos RA, Bohl JR, Braak H (2002) Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 61:413–426
Deng H, Wang P, Jankovic J (2018) The genetics of Parkinson disease. Ageing Res Rev 42:72–85. https://doi.org/10.1016/j.arr.2017.12.007
Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D (2015) Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6:7314. https://doi.org/10.1038/ncomms8314
Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D (2015) KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci U S A 112:9596–9601. https://doi.org/10.1073/pnas.1505953112
Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ et al (2008) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol 115:437–444. https://doi.org/10.1007/s00401-008-0345-7
Dijkstra AA, Voorn P, Berendse HW, Groenewegen HJ, Netherlands Brain B, Rozemuller AJ et al (2014) Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov Disord 29:1244–1251. https://doi.org/10.1002/mds.25952
Dues DJ, Nguyen APT, Becker K, Ma J, Moore DJ (2023) Hippocampal subfield vulnerability to alpha-synuclein pathology precedes neurodegeneration and cognitive dysfunction. NPJ Parkinsons Dis 9:125. https://doi.org/10.1038/s41531-023-00574-1
Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG et al (2018) Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation 15:129. https://doi.org/10.1186/s12974-018-1171-z
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ et al (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17:1945–1947. https://doi.org/10.1096/fj.03-0098fje
El-Agnaf OM, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA et al (2006) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20:419–425. https://doi.org/10.1096/fj.03-1449com
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30:6838–6851. https://doi.org/10.1523/JNEUROSCI.5699-09.2010
Eriksen JL, Przedborski S, Petrucelli L (2005) Gene dosage and pathogenesis of Parkinson’s disease. Trends Mol Med 11:91–96. https://doi.org/10.1016/j.molmed.2005.01.001
Erskine D, Patterson L, Alexandris A, Hanson PS, McKeith IG, Attems J et al (2018) Regional levels of physiological alpha-synuclein are directly associated with Lewy body pathology. Acta Neuropathol 135:153–154. https://doi.org/10.1007/s00401-017-1787-6
Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A et al (2019) Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 92:329–337. https://doi.org/10.1212/WNL.0000000000006926
Ezzat K, Espay AJ (2023) The allure and pitfalls of the prion-like aggregation in neurodegeneration. Handb Clin Neurol 193:17–22. https://doi.org/10.1016/B978-0-323-85555-6.00004-7
Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M et al (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. https://doi.org/10.1002/ana.10846
Febbraro F, Sahin G, Farran A, Soares S, Jensen PH, Kirik D et al (2013) Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson’s disease. Neurobiol Dis 56:47–58. https://doi.org/10.1016/j.nbd.2013.03.014
Froula JM, Castellana-Cruz M, Anabtawi NM, Camino JD, Chen SW, Thrasher DR et al (2019) Defining alpha-synuclein species responsible for Parkinson’s disease phenotypes in mice. J Biol Chem 294:10392–10406. https://doi.org/10.1074/jbc.RA119.007743
Fujioka S, Ogaki K, Tacik PM, Uitti RJ, Ross OA, Wszolek ZK (2014) Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord 20(Suppl 1):S29-34. https://doi.org/10.1016/S1353-8020(13)70010-5
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS et al (2002) Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164. https://doi.org/10.1038/ncb748
Fumimura Y, Ikemura M, Saito Y, Sengoku R, Kanemaru K, Sawabe M et al (2007) Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in Lewy body disease. J Neuropathol Exp Neurol 66:354–362. https://doi.org/10.1097/nen.0b013e3180517454
Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29:357–365. https://doi.org/10.1016/j.it.2008.05.002
Gcwensa NZ, Russell DL, Cowell RM, Volpicelli-Daley LA (2021) Molecular mechanisms underlying synaptic and axon degeneration in Parkinson’s disease. Front Cell Neurosci 15:626128. https://doi.org/10.3389/fncel.2021.626128
George JM, Jin H, Woods WS, Clayton DF (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15:361–372. https://doi.org/10.1016/0896-6273(95)90040-3
Ghanem SS, Majbour NK, Vaikath NN, Ardah MT, Erskine D, Jensen NM et al (2022) Alpha-synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc Natl Acad Sci USA 119:e2109617119. https://doi.org/10.1073/pnas.2109617119
Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51:745–752. https://doi.org/10.1136/jnnp.51.6.745
Gibbons CH, Levine T, Adler C, Bellaire B, Wang N, Stohl J et al (2024) Skin biopsy detection of phosphorylated alpha-synuclein in patients with synucleinopathies. JAMA. https://doi.org/10.1001/jama.2024.0792
Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501. https://doi.org/10.1038/35081564
Goedert M (2015) NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349:1255555. https://doi.org/10.1126/science.1255555
Goedert M, Spillantini MG, Davies SW (1998) Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol 8:619–632
Goedert M, Spillantini MG, Del Tredici K, Braak H (2013) 100 years of Lewy pathology. Nat Rev Neurol 9:13–24. https://doi.org/10.1038/nrneurol.2012.242
Gonzalez-Rodriguez P, Zampese E, Surmeier DJ (2020) Selective neuronal vulnerability in Parkinson’s disease. Prog Brain Res 252:61–89. https://doi.org/10.1016/bs.pbr.2020.02.005
Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP et al (2008) The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci USA 105:763–768. https://doi.org/10.1073/pnas.0711053105
Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH et al (2010) alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci USA 107:19573–19578. https://doi.org/10.1073/pnas.1005005107
Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E et al (2015) Acceleration of alpha-synuclein aggregation by exosomes. J Biol Chem 290:2969–2982. https://doi.org/10.1074/jbc.M114.585703
Grundemann J, Schlaudraff F, Haeckel O, Liss B (2008) Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res 36:e38. https://doi.org/10.1093/nar/gkn084
Gualerzi A, Picciolini S, Bedoni M, Guerini FR, Clerici M, Agliardi C (2024) Extracellular vesicles as biomarkers for Parkinson’s disease: how far from clinical translation? Int J Mol Sci. https://doi.org/10.3390/ijms25021136
Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q et al (2020) Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 143:1476–1497. https://doi.org/10.1093/brain/awaa090
Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10:378–384
Han C, Xiong N, Guo X, Huang J, Ma K, Liu L et al (2019) Exosomes from patients with Parkinson’s disease are pathological in mice. J Mol Med (Berl) 97:1329–1344. https://doi.org/10.1007/s00109-019-01810-z
Han Y, Wu D, Wang Y, Xie J, Zhang Z (2022) Skin alpha-synuclein deposit patterns: a predictor of Parkinson’s disease subtypes. EBioMedicine 80:104076. https://doi.org/10.1016/j.ebiom.2022.104076
Harris MA, Tsui JK, Marion SA, Shen H, Teschke K (2012) Association of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov Disord 27:1111–1117. https://doi.org/10.1002/mds.25077
Hawkes CH, Del Tredici K, Braak H (2007) Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. https://doi.org/10.1111/j.1365-2990.2007.00874.x
Hawkes CH, Del Tredici K, Braak H (2009) Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci 1170:615–622. https://doi.org/10.1111/j.1749-6632.2009.04365.x
Heiden DL, Monogue B, Ali MDH, Beckham JD (2023) A functional role for alpha-synuclein in neuroimmune responses. J Neuroimmunol 376:578047. https://doi.org/10.1016/j.jneuroim.2023.578047
Henderson MX, Cornblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ et al (2019) Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22:1248–1257. https://doi.org/10.1038/s41593-019-0457-5
Holec SAM, Woerman AL (2021) Evidence of distinct alpha-synuclein strains underlying disease heterogeneity. Acta Neuropathol 142:73–86. https://doi.org/10.1007/s00401-020-02163-5
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K et al (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA 110:E3138-3147. https://doi.org/10.1073/pnas.1301440110
Hope AD, Myhre R, Kachergus J, Lincoln S, Bisceglio G, Hulihan M et al (2004) Alpha-synuclein missense and multiplication mutations in autosomal dominant Parkinson’s disease. Neurosci Lett 367:97–100. https://doi.org/10.1016/j.neulet.2004.05.100
Iba M, McDevitt RA, Kim C, Roy R, Sarantopoulou D, Tommer E et al (2022) Aging exacerbates the brain inflammatory micro-environment contributing to alpha-synuclein pathology and functional deficits in a mouse model of DLB/PD. Mol Neurodegener 17:60. https://doi.org/10.1186/s13024-022-00564-6
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P et al (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171. https://doi.org/10.1016/S0140-6736(04)17104-3
Ibanez P, Lesage S, Janin S, Lohmann E, Durif F, Destee A et al (2009) Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol 66:102–108. https://doi.org/10.1001/archneurol.2008.555
Ihse E, Yamakado H, van Wijk XM, Lawrence R, Esko JD, Masliah E (2017) Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7:9008. https://doi.org/10.1038/s41598-017-08720-5
Imamura K, Hishikawa N, Ono K, Suzuki H, Sawada M, Nagatsu T et al (2005) Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol 109:141–150. https://doi.org/10.1007/s00401-004-0919-y
Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF et al (1998) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J Neuropathol Exp Neurol 57:334–337. https://doi.org/10.1097/00005072-199804000-00005
Ito N, Tsuji M, Adachi N, Nakamura S, Sarkar AK, Ikenaka K et al (2023) Extracellular high molecular weight alpha-synuclein oligomers induce cell death by disrupting the plasma membrane. NPJ Parkinsons Dis 9:139. https://doi.org/10.1038/s41531-023-00583-0
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA et al (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14:467–475. https://doi.org/10.1016/0896-6273(95)90302-x
Iwanaga K, Wakabayashi K, Yoshimoto M, Tomita I, Satoh H, Takashima H et al (1999) Lewy body-type degeneration in cardiac plexus in Parkinson’s and incidental Lewy body diseases. Neurology 52:1269–1271. https://doi.org/10.1212/wnl.52.6.1269
Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21:1341–1349. https://doi.org/10.1038/s41593-018-0238-6
Kakuda K, Ikenaka K, Kuma A, Doi J, Aguirre C, Wang N et al (2024) Lysophagy protects against propagation of alpha-synuclein aggregation through ruptured lysosomal vesicles. Proc Natl Acad Sci USA 121:e2312306120. https://doi.org/10.1073/pnas.2312306120
Kanazawa T, Adachi E, Orimo S, Nakamura A, Mizusawa H, Uchihara T (2012) Pale neurites, premature alpha-synuclein aggregates with centripetal extension from axon collaterals. Brain Pathol 22:67–78. https://doi.org/10.1111/j.1750-3639.2011.00509.x
Kasen A, Houck C, Burmeister AR, Sha Q, Brundin L, Brundin P (2022) Upregulation of alpha-synuclein following immune activation: possible trigger of Parkinson’s disease. Neurobiol Dis 166:105654. https://doi.org/10.1016/j.nbd.2022.105654
Kawahata I, Finkelstein DI, Fukunaga K (2022) Pathogenic impact of alpha-synuclein phosphorylation and its kinases in alpha-synucleinopathies. Int J Mol Sci. https://doi.org/10.3390/ijms23116216
Khedmatgozar CR, Holec SAM, Woerman AL (2024) The role of alpha-synuclein prion strains in Parkinson’s disease and multiple system atrophy. PLoS Pathog 20:e1011920. https://doi.org/10.1371/journal.ppat.1011920
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S et al (2019) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103(627–641):e627. https://doi.org/10.1016/j.neuron.2019.05.035
Klaestrup IH, Just MK, Holm KL, Alstrup AKO, Romero-Ramos M, Borghammer P et al (2022) Impact of aging on animal models of Parkinson’s disease. Front Aging Neurosci 14:909273. https://doi.org/10.3389/fnagi.2022.909273
Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF et al (2006) Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology 66:1100–1102. https://doi.org/10.1212/01.wnl.0000204179.88955.fa
Kluge A, Schaeffer E, Bunk J, Sommerauer M, Rottgen S, Schulte C et al (2024) Detecting misfolded alpha-synuclein in blood years before the diagnosis of Parkinson’s disease. Mov Disord. https://doi.org/10.1002/mds.29766
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506. https://doi.org/10.1038/nm1747
Koss DJ, Erskine D, Porter A, Palmoski P, Menon H, Todd OGJ et al (2022) Nuclear alpha-synuclein is present in the human brain and is modified in dementia with Lewy bodies. Acta Neuropathol Commun 10:98. https://doi.org/10.1186/s40478-022-01403-x
Kruger R, Vieira-Saecker AM, Kuhn W, Berg D, Muller T, Kuhnl N et al (1999) Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol 45:611–617. https://doi.org/10.1002/1531-8249(199905)45:5%3c611::aid-ana9%3e3.0.co;2-x
Kunadt M, Eckermann K, Stuendl A, Gong J, Russo B, Strauss K et al (2015) Extracellular vesicle sorting of alpha-synuclein is regulated by sumoylation. Acta Neuropathol 129:695–713. https://doi.org/10.1007/s00401-015-1408-1
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48. https://doi.org/10.1038/nrn3406
Lazaro DF, Lee VM (2024) Navigating through the complexities of synucleinopathies: insights into pathogenesis, heterogeneity, and future perspectives. Neuron. https://doi.org/10.1016/j.neuron.2024.05.017
Leak RK, Frosch MP, Beach TG, Halliday GM (2019) Alpha-synuclein: prion or prion-like? Acta Neuropathol 138:509–514. https://doi.org/10.1007/s00401-019-02057-1
Leblanc P, Vorberg IM (2022) Viruses in neurodegenerative diseases: more than just suspects in crimes. PLoS Pathog 18:e1010670. https://doi.org/10.1371/journal.ppat.1010670
Lee FJ, Liu F, Pristupa ZB, Niznik HB (2001) Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J 15:916–926. https://doi.org/10.1096/fj.00-0334com
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. https://doi.org/10.1523/JNEUROSCI.0692-05.2005
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A et al (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. https://doi.org/10.1038/nature05453
Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW et al (2007) Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathol 17:139–145. https://doi.org/10.1111/j.1750-3639.2007.00048.x
Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB et al (2008) Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 18:220–224. https://doi.org/10.1111/j.1750-3639.2007.00117.x
Levine KS, Leonard HL, Blauwendraat C, Iwaki H, Johnson N, Bandres-Ciga S et al (2023) Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 111(1086–1093):e1082. https://doi.org/10.1016/j.neuron.2022.12.029
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503. https://doi.org/10.1038/nm1746
Li JY, Englund E, Widner H, Rehncrona S, Bjorklund A, Lindvall O et al (2010) Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord 25:1091–1096. https://doi.org/10.1002/mds.23012
Licker V, Turck N, Kovari E, Burkhardt K, Cote M, Surini-Demiri M et al (2014) Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 14:784–794. https://doi.org/10.1002/pmic.201300342
Lloyd GM, Quintin S, Sorrentino ZA, Gorion KM, Bell BM, Long B et al (2024) A multiverse of alpha-synuclein: investigation of prion strain properties with carboxyl-terminal truncation specific antibodies in animal models. Acta Neuropathol Commun 12:91. https://doi.org/10.1186/s40478-024-01805-z
Lotz SK, Blackhurst BM, Reagin KL, Funk KE (2021) Microbial infections are a risk factor for neurodegenerative diseases. Front Cell Neurosci 15:691136. https://doi.org/10.3389/fncel.2021.691136
Loveland PM, Yu JJ, Churilov L, Yassi N, Watson R (2023) Investigation of inflammation in Lewy body dementia: a systematic scoping review. Int J Mol Sci. https://doi.org/10.3390/ijms241512116
Luk KC, Lee VM (2014) Modeling Lewy pathology propagation in Parkinson’s disease. Parkinsonism Relat Disord 20(Suppl 1):S85-87. https://doi.org/10.1016/S1353-8020(13)70022-1
Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR et al (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA 106:20051–20056. https://doi.org/10.1073/pnas.0908005106
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209:975–986. https://doi.org/10.1084/jem.20112457
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ et al (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. https://doi.org/10.1126/science.1227157
Luna E, Luk KC (2015) Bent out of shape: alpha-synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease. FEBS Lett 589:3749–3759. https://doi.org/10.1016/j.febslet.2015.10.023
Luna E, Decker SC, Riddle DM, Caputo A, Zhang B, Cole T et al (2018) Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol 135:855–875. https://doi.org/10.1007/s00401-018-1829-8
MacDonald V, Halliday GM (2002) Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson’s disease. Mov Disord 17:1166–1173. https://doi.org/10.1002/mds.10258
Mackenzie IR (2000) Activated microglia in dementia with Lewy bodies. Neurology 55:132–134. https://doi.org/10.1212/wnl.55.1.132
Mahapatra A, Newberry RW (2024) Liquid-liquid phase separation of alpha-synuclein is highly sensitive to sequence complexity. Protein Sci 33:e4951. https://doi.org/10.1002/pro.4951
Majbour NK, Vaikath NN, van Dijk KD, Ardah MT, Varghese S, Vesterager LB et al (2016) Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener 11:7. https://doi.org/10.1186/s13024-016-0072-9
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA (2010) Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 285:13621–13629. https://doi.org/10.1074/jbc.M109.074617
Manne S, Kondru N, Jin H, Anantharam V, Huang X, Kanthasamy A et al (2020) alpha-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Mov Disord 35:268–278. https://doi.org/10.1002/mds.27907
Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8:2804–2815. https://doi.org/10.1523/JNEUROSCI.08-08-02804.1988
Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC et al (2016) Transmission of alpha-synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener 11:49. https://doi.org/10.1186/s13024-016-0113-4
Mason DM, Wang Y, Bhatia TN, Miner KM, Trbojevic SA, Stolz JF et al (2019) The center of olfactory bulb-seeded alpha-synucleinopathy is the limbic system and the ensuing pathology is higher in male than in female mice. Brain Pathol 29:741–770. https://doi.org/10.1111/bpa.12718
McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT et al (2009) Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol 68:515–524. https://doi.org/10.1097/NEN.0b013e3181a24b53
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291. https://doi.org/10.1212/wnl.38.8.1285
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. https://doi.org/10.1212/01.wnl.0000187889.17253.b1
Meade RM, Fairlie DP, Mason JM (2019) Alpha-synuclein structure and Parkinson’s disease—lessons and emerging principles. Mol Neurodegener 14:29. https://doi.org/10.1186/s13024-019-0329-1
Mezias C, Rey N, Brundin P, Raj A (2020) Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: involvement of retrograde and anterograde axonal propagation. Neurobiol Dis 134:104623. https://doi.org/10.1016/j.nbd.2019.104623
Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW et al (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79:2307–2314. https://doi.org/10.1212/WNL.0b013e318278fe32
Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR et al (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838. https://doi.org/10.1212/01.wnl.0000127517.33208.f4
Miner KM, Jamenis AS, Bhatia TN, Clark RN, Rajasundaram D, Sauvaigo S et al (2022) alpha-synucleinopathy exerts sex-dimorphic effects on the multipurpose DNA repair/redox protein APE1 in mice and humans. Prog Neurobiol 216:102307. https://doi.org/10.1016/j.pneurobio.2022.102307
Miner KM, Jamenis AS, Bhatia TN, Clark RN, Abbas M, Luk KC et al (2023) The variance in phosphorylated, insoluble ⍺-synuclein in humans, rats, and mice is not mainly driven by biological sex. Acta Neuropathol 146:651–654. https://doi.org/10.1007/s00401-023-02621-w
Moors TE, Milovanovic D (2024) Defining a Lewy body: running up the hill of shifting definitions and evolving concepts. J Parkinsons Dis 14:17–33. https://doi.org/10.3233/JPD-230183
Moretto E, Stuart S, Surana S, Vargas JNS, Schiavo G (2022) The role of extracellular matrix components in the spreading of pathological protein aggregates. Front Cell Neurosci 16:844211. https://doi.org/10.3389/fncel.2022.844211
Muenter MD, Forno LS, Hornykiewicz O, Kish SJ, Maraganore DM, Caselli RJ et al (1998) Hereditary form of parkinsonism–dementia. Ann Neurol 43:768–781. https://doi.org/10.1002/ana.410430612
Mukherjee S, Sakunthala A, Gadhe L, Poudyal M, Sawner AS, Kadu P et al (2023) Liquid-liquid phase separation of alpha-synuclein: a new mechanistic insight for alpha-synuclein aggregation associated with Parkinson’s disease pathogenesis. J Mol Biol 435:167713. https://doi.org/10.1016/j.jmb.2022.167713
Nandipati S, Litvan I (2016) Environmental exposures and Parkinson’s disease. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph13090881
Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA (2000) Alpha-synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. Acta Neuropathol 100:568–574. https://doi.org/10.1007/s004010000224
Ninkina N, Tarasova TV, Chaprov KD, Roman AY, Kukharsky MS, Kolik LG et al (2020) Alterations in the nigrostriatal system following conditional inactivation of alpha-synuclein in neurons of adult and aging mice. Neurobiol Aging 91:76–87. https://doi.org/10.1016/j.neurobiolaging.2020.02.026
Nouraei N, Mason DM, Miner KM, Carcella MA, Bhatia TN, Dumm BK et al (2018) Critical appraisal of pathology transmission in the alpha-synuclein fibril model of Lewy body disorders. Exp Neurol 299:172–196. https://doi.org/10.1016/j.expneurol.2017.10.017
Nuber S, Rajsombath M, Minakaki G, Winkler J, Muller CP, Ericsson M et al (2018) Abrogating native alpha-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron 100(75–90):e75. https://doi.org/10.1016/j.neuron.2018.09.014
Nuber S, Zhang X, McCaffery TD, Moors TE, Adom MA, Hahn WN et al (2024) Generation of G51D and 3D mice reveals decreased alpha-synuclein tetramer-monomer ratios promote Parkinson’s disease phenotypes. NPJ Parkinsons Dis 10:47. https://doi.org/10.1038/s41531-024-00662-w
Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T et al (2000) Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem 275:390–397. https://doi.org/10.1074/jbc.275.1.390
Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T et al (2005) Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol 109:583–588. https://doi.org/10.1007/s00401-005-0995-7
Orru CD, Ma TC, Hughson AG, Groveman BR, Srivastava A, Galasko D et al (2021) A rapid alpha-synuclein seed assay of Parkinson’s disease CSF panel shows high diagnostic accuracy. Ann Clin Transl Neurol 8:374–384. https://doi.org/10.1002/acn3.51280
Oueslati A (2016) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinsons Dis 6:39–51. https://doi.org/10.3233/JPD-160779
Parkkinen L, Soininen H, Alafuzoff I (2003) Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol 62:363–367
Parkkinen L, O’Sullivan SS, Collins C, Petrie A, Holton JL, Revesz T et al (2011) Disentangling the relationship between lewy bodies and nigral neuronal loss in Parkinson’s disease. J Parkinsons Dis 1:277–286. https://doi.org/10.3233/JPD-2011-11046
Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O et al (2019) CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol 18:573–586. https://doi.org/10.1016/S1474-4422(19)30024-9
Parra-Rivas LA, Madhivanan K, Aulston BD, Wang L, Prakashchand DD, Boyer NP et al (2023) Serine-129 phosphorylation of alpha-synuclein is an activity-dependent trigger for physiologic protein-protein interactions and synaptic function. Neuron 111(4006–4023):e4010. https://doi.org/10.1016/j.neuron.2023.11.020
Patterson JR, Duffy MF, Kemp CJ, Howe JW, Collier TJ, Stoll AC et al (2019) Time course and magnitude of alpha-synuclein inclusion formation and nigrostriatal degeneration in the rat model of synucleinopathy triggered by intrastriatal alpha-synuclein preformed fibrils. Neurobiol Dis 130:104525. https://doi.org/10.1016/j.nbd.2019.104525
Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ et al (2015) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. https://doi.org/10.1016/j.nbd.2015.06.003
Peelaerts W, Mercado G, George S, Villumsen M, Kasen A, Aguileta M et al (2023) Urinary tract infections trigger synucleinopathy via the innate immune response. Acta Neuropathol 145:541–559. https://doi.org/10.1007/s00401-023-02562-4
Peng C, Trojanowski JQ, Lee VM (2020) Protein transmission in neurodegenerative disease. Nat Rev Neurol 16:199–212. https://doi.org/10.1038/s41582-020-0333-7
Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ (2002) A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci 22:3090–3099
Petrucci S, Ginevrino M, Valente EM (2016) Phenotypic spectrum of alpha-synuclein mutations: new insights from patients and cellular models. Parkinsonism Relat Disord 22(Suppl 1):S16-20. https://doi.org/10.1016/j.parkreldis.2015.08.015
Pissadaki EK, Bolam JP (2013) The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 7:13. https://doi.org/10.3389/fncom.2013.00013
Pouclet H, Lebouvier T, Coron E, Neunlist M, Derkinderen P (2012) Lewy pathology in gastric and duodenal biopsies in Parkinson’s disease. Mov Disord 27:708. https://doi.org/10.1002/mds.24993
Purba JS, Hofman MA, Swaab DF (1994) Decreased number of oxytocin-immunoreactive neurons in the paraventricular nucleus of the hypothalamus in Parkinson’s disease. Neurology 44:84–89. https://doi.org/10.1212/wnl.44.1.84
Rahayel S, Misic B, Zheng YQ, Liu ZQ, Abdelgawad A, Abbasi N et al (2022) Differentially targeted seeding reveals unique pathological alpha-synuclein propagation patterns. Brain 145:1743–1756. https://doi.org/10.1093/brain/awab440
Rajan S, Kaas B (2022) Parkinson’s disease: risk factor modification and prevention. Semin Neurol 42:626–638. https://doi.org/10.1055/s-0042-1758780
Ramalingam N, Jin SX, Moors TE, Fonseca-Ornelas L, Shimanaka K, Lei S et al (2023) Dynamic physiological alpha-synuclein S129 phosphorylation is driven by neuronal activity. NPJ Parkinsons Dis 9:4. https://doi.org/10.1038/s41531-023-00444-w
Ray S, Singh N, Kumar R, Patel K, Pandey S, Datta D et al (2020) Alpha-synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem 12:705–716. https://doi.org/10.1038/s41557-020-0465-9
Ray S, Mason TO, Boyens-Thiele L, Farzadfard A, Larsen JA, Norrild RK et al (2023) Mass photometric detection and quantification of nanoscale alpha-synuclein phase separation. Nat Chem 15:1306–1316. https://doi.org/10.1038/s41557-023-01244-8
Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A et al (2014) Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. https://doi.org/10.1002/ana.24066
Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ et al (2016) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213:1759–1778. https://doi.org/10.1084/jem.20160368
Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ et al (2018) Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135:65–83. https://doi.org/10.1007/s00401-017-1792-9
Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA et al (2008) Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol 63:743–750. https://doi.org/10.1002/ana.21380
Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG et al (2020) A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. Elife. https://doi.org/10.7554/eLife.53111
Samudra N, Fischer DL, Lenio S, Lario Lago A, Ljubenkov PA, Rojas JC et al (2024) Clinicopathological correlation of cerebrospinal fluid alpha-synuclein seed amplification assay in a behavioral neurology autopsy cohort. Alzheimers Dement. https://doi.org/10.1002/alz.13799
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M et al (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307. https://doi.org/10.1038/ng.485
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H et al (2018) Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174(1015–1030):e1016. https://doi.org/10.1016/j.cell.2018.07.028
Sawamura M, Onoe H, Tsukada H, Isa K, Yamakado H, Okuda S et al (2022) Lewy body disease primate model with alpha-synuclein propagation from the olfactory bulb. Mov Disord 37:2033–2044. https://doi.org/10.1002/mds.29161
Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW et al (2019) Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep 9:10919. https://doi.org/10.1038/s41598-019-47227-z
Schaser AJ, Stackhouse TL, Weston LJ, Kerstein PC, Osterberg VR, Lopez CS et al (2020) Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an in vivo A53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathol Commun 8:150. https://doi.org/10.1186/s40478-020-01026-0
Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L et al (2021) Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184(5089–5106):e5021. https://doi.org/10.1016/j.cell.2021.09.007
Scheres SHW, Ryskeldi-Falcon B, Goedert M (2023) Molecular pathology of neurodegenerative diseases by cryo-EM of amyloids. Nature 621:701–710. https://doi.org/10.1038/s41586-023-06437-2
Schonhoff AM, Williams GP, Wallen ZD, Standaert DG, Harms AS (2020) Innate and adaptive immune responses in Parkinson’s disease. Prog Brain Res 252:169-216. https://doi.org/10.1016/bs.pbr.2019.10.006
Schulz JB, Dichgans J (1999) Molecular pathogenesis of movement disorders: are protein aggregates a common link in neuronal degeneration? Curr Opin Neurol 12:433–439. https://doi.org/10.1097/00019052-199908000-00010
Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ (2021) The central role of DNA damage in the ageing process. Nature 592:695–703. https://doi.org/10.1038/s41586-021-03307-7
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B et al (2020) Structures of alpha-synuclein filaments from multiple system atrophy. Nature 585:464–469. https://doi.org/10.1038/s41586-020-2317-6
Scott D, Roy S (2012) alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32:10129–10135. https://doi.org/10.1523/JNEUROSCI.0535-12.2012
Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci USA 97:4897–4902. https://doi.org/10.1073/pnas.97.9.4897
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP et al (2019) Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 22:1099–1109. https://doi.org/10.1038/s41593-019-0423-2
Sharma M, Burre J (2023) alpha-Synuclein in synaptic function and dysfunction. Trends Neurosci 46:153–166. https://doi.org/10.1016/j.tins.2022.11.007
Sharma M, Burre J, Sudhof TC (2011) CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol 13:30–39. https://doi.org/10.1038/ncb2131
Siderowf A, Concha-Marambio L, Lafontant DE, Farris CM, Ma Y, Urenia PA et al (2023) Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using alpha-synuclein seed amplification: a cross-sectional study. Lancet Neurol 22:407–417. https://doi.org/10.1016/S1474-4422(23)00109-6
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312. https://doi.org/10.1038/ng.487
Simuni T, Uribe L, Cho HR, Caspell-Garcia C, Coffey CS, Siderowf A et al (2020) Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol 19:71–80. https://doi.org/10.1016/S1474-4422(19)30319-9
Simuni T, Chahine LM, Poston K, Brumm M, Buracchio T, Campbell M et al (2024) A biological definition of neuronal alpha-synuclein disease: towards an integrated staging system for research. Lancet Neurol 23:178–190. https://doi.org/10.1016/S1474-4422(23)00405-2
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. https://doi.org/10.1126/science.1090278
Singleton AB, Farrer MJ, Bonifati V (2013) The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 28:14–23. https://doi.org/10.1002/mds.25249
So RWL, Watts JC (2023) Alpha-synuclein conformational strains as drivers of phenotypic heterogeneity in neurodegenerative diseases. J Mol Biol 435:168011. https://doi.org/10.1016/j.jmb.2023.168011
Soria FN, Paviolo C, Doudnikoff E, Arotcarena ML, Lee A, Danne N et al (2020) Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat Commun 11:3440. https://doi.org/10.1038/s41467-020-17328-9
Soto C (2024) alpha-Synuclein seed amplification technology for Parkinson’s disease and related synucleinopathies. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2024.01.007
Soto C, Estrada L, Castilla J (2006) Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci 31:150–155. https://doi.org/10.1016/j.tibs.2006.01.002
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Stefanis L (2012) Alpha-synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009399. https://doi.org/10.1101/cshperspect.a009399
Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK (2019) How is alpha-synuclein cleared from the cell? J Neurochem 150:577–590. https://doi.org/10.1111/jnc.14704
Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S et al (2017) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9:456–463. https://doi.org/10.1159/000477990
Streit WJ, Xue QS (2016) Microglia in dementia with Lewy bodies. Brain Behav Immun 55:191–201. https://doi.org/10.1016/j.bbi.2015.10.012
Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM et al (2016) Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139:481–494. https://doi.org/10.1093/brain/awv346
Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER et al (2019) Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci USA 116:11113–11115. https://doi.org/10.1073/pnas.1903049116
Surguchov A (2015) Intracellular dynamics of synucleins: “Here, There and Everywhere.” Int Rev Cell Mol Biol 320:103–169. https://doi.org/10.1016/bs.ircmb.2015.07.007
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113. https://doi.org/10.1038/nrn.2016.178
Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M et al (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119:866–872. https://doi.org/10.1289/ehp.1002839
Tansey MG, Romero-Ramos M (2019) Immune system responses in Parkinson’s disease: early and dynamic. Eur J Neurosci 49:364–383. https://doi.org/10.1111/ejn.14290
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V (2022) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22:657–673. https://doi.org/10.1038/s41577-022-00684-6
Tehranian R, Montoya SE, Van Laar AD, Hastings TG, Perez RG (2006) Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J Neurochem 99:1188–1196. https://doi.org/10.1111/j.1471-4159.2006.04146.x
Tomlinson JJ, Shutinoski B, Dong L, Meng F, Elleithy D, Lengacher NA et al (2017) Holocranohistochemistry enables the visualization of alpha-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. J Neural Transm (Vienna) 124:721–738. https://doi.org/10.1007/s00702-017-1726-7
Trojanowski JQ, Lee VM (2000) “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann N Y Acad Sci 924:62–67. https://doi.org/10.1111/j.1749-6632.2000.tb05561.x
Tu HY, Yuan BS, Hou XO, Zhang XJ, Pei CS, Ma YT et al (2021) alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 20:e13522. https://doi.org/10.1111/acel.13522
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697. https://doi.org/10.1097/01.jnen.0000225908.90052.07
Uemura N, Uemura MT, Luk KC, Lee VM, Trojanowski JQ (2020) Cell-to-cell transmission of tau and alpha-synuclein. Trends Mol Med 26:936–952. https://doi.org/10.1016/j.molmed.2020.03.012
Uemura N, Marotta NP, Ara J, Meymand ES, Zhang B, Kameda H et al (2023) Alpha-synuclein aggregates amplified from patient-derived Lewy bodies recapitulate Lewy body diseases in mice. Nat Commun 14:6892. https://doi.org/10.1038/s41467-023-42705-5
Uemura N, Marotta N, Ara J, Meymand E, Zhang B, Kameda H et al (2023) Distinct biological activity of Lewy body alpha-synuclein strain in mice. Res Sq. https://doi.org/10.21203/rs.3.rs-2579805/v1
Upadhya R, Shetty AK (2021) Extracellular vesicles for the diagnosis and treatment of Parkinson’s disease. Aging Dis 12:1438–1450. https://doi.org/10.14336/AD.2021.0516
Van Den Berge N, Ferreira N, Mikkelsen TW, Alstrup AKO, Tamguney G, Karlsson P et al (2021) Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 144:1853–1868. https://doi.org/10.1093/brain/awab061
Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC et al (2010) Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem 285:32486–32493. https://doi.org/10.1074/jbc.M110.139576
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283:23542–23556. https://doi.org/10.1074/jbc.M801992200
Volpicelli-Daley LA (2017) Effects of alpha-synuclein on axonal transport. Neurobiol Dis 105:321–327. https://doi.org/10.1016/j.nbd.2016.12.008
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A et al (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. https://doi.org/10.1016/j.neuron.2011.08.033
Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM (2014) Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 25:4010–4023. https://doi.org/10.1091/mbc.E14-02-0741
Volpicelli-Daley LA, Kirik D, Stoyka LE, Standaert DG, Harms AS (2016) How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem 139(Suppl 1):131–155. https://doi.org/10.1111/jnc.13627
von Bartheld CS (2018) Myths and truths about the cellular composition of the human brain: a review of influential concepts. J Chem Neuroanat 93:2–15. https://doi.org/10.1016/j.jchemneu.2017.08.004
Wakabayashi K, Takahashi H (1997) Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol 38(Suppl 2):2–7
Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H (2013) The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol 47:495–508. https://doi.org/10.1007/s12035-012-8280-y
Walker LC, Jucker M (2015) Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38:87–103. https://doi.org/10.1146/annurev-neuro-071714-033828
Walker LC, Levine H 3rd, Mattson MP, Jucker M (2006) Inducible proteopathies. Trends Neurosci 29:438–443. https://doi.org/10.1016/j.tins.2006.06.010
Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S (2014) alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24:2319–2326. https://doi.org/10.1016/j.cub.2014.08.027
Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H et al (2015) alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci USA 112:E1926-1935. https://doi.org/10.1073/pnas.1417883112
Watts JC (2019) Calling alpha-synuclein a prion is scientifically justifiable. Acta Neuropathol 138:505–508. https://doi.org/10.1007/s00401-019-02058-0
Wersinger C, Prou D, Vernier P, Sidhu A (2003) Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J 17:2151–2153. https://doi.org/10.1096/fj.03-0152fje
Weston LJ, Cook ZT, Stackhouse TL, Sal MK, Schultz BI, Tobias ZJC et al (2021) In vivo aggregation of presynaptic alpha-synuclein is not influenced by its phosphorylation at serine-129. Neurobiol Dis 152:105291. https://doi.org/10.1016/j.nbd.2021.105291
Weston LJ, Bowman AM, Osterberg VR, Meshul CK, Woltjer RL, Unni VK (2022) Aggregated alpha-synuclein inclusions within the nucleus predict impending neuronal cell death in a mouse model of Parkinsonism. Int J Mol Sci. https://doi.org/10.3390/ijms232315294
Weyand CM, Goronzy JJ (2016) Aging of the immune system. Mechanisms and therapeutic targets. Ann Am Thorac Soc 5(13 Suppl):422–428. https://doi.org/10.1513/AnnalsATS.201602-095AW
Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK et al (2004) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82:192–207. https://doi.org/10.1016/j.ymgme.2004.04.011
Wong YC, Luk K, Purtell K, Burke Nanni S, Stoessl AJ, Trudeau LE et al (2019) Neuronal vulnerability in Parkinson disease: should the focus be on axons and synaptic terminals? Mov Disord 34:1406–1422. https://doi.org/10.1002/mds.27823
Wu Q, Shaikh MA, Meymand ES, Zhang B, Luk KC, Trojanowski JQ et al (2020) Neuronal activity modulates alpha-synuclein aggregation and spreading in organotypic brain slice cultures and in vivo. Acta Neuropathol 140:831–849. https://doi.org/10.1007/s00401-020-02227-6
Yacoubian TA, Fang YD, Gerstenecker A, Amara A, Stover N, Ruffrage L, Collette C, Kennedy R, Zhang Y, Hong Het al (2023) Brain and Systemic Inflammation in De Novo Parkinson’s Disease. Mov Disord 38:743–754. https://doi.org/10.1002/mds.29363
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I et al (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. https://doi.org/10.1002/ana.10795
Zhang S, Dauer K, Strohaker T, Tatenhorst L, Caldi Gomes L, Mayer S et al (2023) Alpha-synuclein fibrils amplified from multiple system atrophy and Parkinson’s disease patient brain spread after intracerebral injection into mouse brain. Brain Pathol 33:e13196. https://doi.org/10.1111/bpa.13196
Zigmond MJ, Hastings TG, Abercrombie ED (1992) Neurochemical responses to 6-hydroxydopamine and L-dopa therapy: implications for Parkinson’s disease. Ann N Y Acad Sci 648:71–86. https://doi.org/10.1111/j.1749-6632.1992.tb24525.x
Acknowledgements
We thank the anonymous reviewers for constructive comments that enriched this review. RKL is grateful for support by R15 NS130532-01 (NINDS) and the School of Pharmacy at Duquesne University. JLB is supported by GM 131732 (NIGMS). JC is supported by the VA Senior Research Career Scientist Award. Conceptualization and writing: RKL. Revisions and/or feedback: KCL, JLB, XH, RNC, MA, FX, and JC. Figures: RKL, RNC, MA, and FX.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leak, R.K., Clark, R.N., Abbas, M. et al. Current insights and assumptions on α-synuclein in Lewy body disease. Acta Neuropathol 148, 18 (2024). https://doi.org/10.1007/s00401-024-02781-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02781-3