
Abstract
Tauopathies, including Alzheimer’s disease (AD) and frontotemporal lobar degeneration with Tau pathology (FTLD-tau), are a group of neurodegenerative disorders characterized by Tau hyperphosphorylation. Post-translational modifications of Tau such as phosphorylation and truncation have been demonstrated to be an essential step in the molecular pathogenesis of these tauopathies. In this work, we demonstrate the existence of a new, human-specific truncated form of Tau generated by intron 12 retention in human neuroblastoma cells and, to a higher extent, in human RNA brain samples, using qPCR and further confirming the results on a larger database of human RNA-seq samples. Diminished protein levels of this new Tau isoform are found by Westernblotting in Alzheimer’s patients’ brains (Braak I n = 3; Braak II n = 6, Braak III n = 3, Braak IV n = 1, and Braak V n = 10, Braak VI n = 8) with respect to non-demented control subjects (n = 9), suggesting that the lack of this truncated isoform may play an important role in the pathology. This new Tau isoform exhibits similar post-transcriptional modifications by phosphorylation and affinity for microtubule binding, but more interestingly, is less prone to aggregate than other Tau isoforms. Finally, we present evidence suggesting this new Tau isoform could be linked to the inhibition of GSK3β, which would mediate intron 12 retention by modulating the serine/arginine rich splicing factor 2 (SRSF2). Our results show the existence of an important new isoform of Tau and suggest that further research on this less aggregation-prone Tau may help to develop future therapies for Alzheimer’s disease and other tauopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tau is a microtubule-associated protein (MAP) that acts as a neurite microtubule stabilizer [9, 61]. Tau overexpression, especially in its modified forms (i.e. phosphorylated, aggregated or truncated) may result in a toxic gain of function [5]. Human Tau protein isoforms are expressed from a unique gene (MAPT) located at chromosome 17 [30, 42] and has at least 16 exons [2, 3]. Exons 1, 4, 5, 7, 9, 11, 12 and 13 are constitutive while the others are subjected to alternative splicing [2, 60]. Importantly, over 90% of human genes allegedly undergo alternative splicing [59], although recent evidence point out that this may not be the main mechanism to explain proteome complexity [55]. However, there is robust evidence for certain genes being subjected to alternative splicing, yielding well-studied spliced variants, being MAPT one of the most relevant [1, 55]. Noteworthy, aberrant splicing has been associated with numerous diseases, including Alzheimer’s disease and other age-related disorders [51]. In accordance with that, mutations altering alternative splicing of exon 10 of MAPT cause some tauopathies, including FTDP-17 [16, 25, 32], and may be associated with others such as Huntington’s disease, whose patients display a four-repeat tauopathy with nuclear rods [19].
Truncated Tau proteins lead to a toxic gain of function, promoting abnormal microtubule assembly and inducing aggregation, being a key feature of Alzheimer's disease, especially in the sporadic form [43, 65].
Tau truncation can take place at N-terminal or C-terminal regions [6, 22, 41, 44, 46, 48, 52, 63, 64]. Recently, a truncated toxic Tau fragment raised upon asparagine endopeptidase (AEP) cleavage, has been reported in mice model of AD and elderly and Alzheimer’s disease human brains [63]. This toxic fragment contains residues 1–368 of the human Tau molecule and the last residue (asparagine 368) coincides with the end of exon 12.
Here, we present evidence of a new, human-specific truncated form of Tau similar to Tau 1–368 generated by intron 12 retention in human neuroblastoma cell lines and brain. An analogous mechanism of truncation has been described for a toxic huntingtin fragment [52]. This intron-retaining RNA species would generate a truncated Tau protein similar to the one resulting from AEP cleavage, but followed by 18 extra amino acids, which dramatically reduces its ability to aggregate.
Strikingly, the resulting Tau truncated isoform is reduced in Alzheimer’s disease patients’ brain, especially in later stages of the disease, in contrast to increasing total Tau levels. Despite the similarity of its sequence to that of the AEP truncated isoform, this new Tau isoform is able to bind to microtubules and is less prone to aggregate, suggesting a beneficial role in the pathology.
Materials and methods
Human brain samples
Hippocampal and frontal lateral cortex brain samples from sporadic Alzheimer’s disease patients and control subjects were kindly provided by Dr. A. Rabano from Banco de Tejidos (Fundación CIEN, Instituto de Salud Carlos III, Madrid, Spain). Based on quantitative pathological features, the Alzheimer’s brain specimens were classified according to Braak stages I (n = 3), II (n = 6), III (n = 4), IV (n = 1), V (n = 10) and VI (n = 8), and non-demented control subjects (n = 10) (see Supplementary table 1, online resource for further subject information). Written informed consent premortem was obtained from all patients.
Nomenclature
Classical nomenclature [2] for MAPT gene exon/intron numbers has been used consistently throughout the paper, although some of the databases (such as ENSEMBL: https://www.ensembl.org/index.html) employ a different nomenclature assigning different numbers to exons and introns (according to ENSEMBL, intron retention would take place in intron 13).
As for the new Tau isoform described in this work, the transcript including the translation of intron 12 up to the first stop codon has been named TIR-MAPT (Truncated by Intron Retention MAPT), while the protein generated from such transcript has been termed W-Tau, due to the appearance of two characteristic tryptophan residues (W) in this isoform, an amino acid that does not appear elsewhere within the Tau molecule.
In addition, Tau truncated by asparagine endopeptidase, as previously described by Zhang et al. [63] is referred to as ET-Tau (Endopeptidase Truncated Tau) throughout the text.
Cell culture
HEK293T (CRL-11268, ATCC), SK-N-MC (HTB-10, ATCC), and SH-SY5Y (CRL-2266, ATCC) cells were cultured in DMEM or MEM supplemented with 10% fetal bovine serum, 2 mM glutamine, non-essential amino acids, 10 U/ml penicillin and 10 µg/ml streptomycin, at 37 °C and 5% CO2.
TIR-MAPT and ET-MAPT cloning
TIR-MAPT transcript was obtained from an SH-SH5Y RNA extract, using specific oligos (TauNt and TauD, see Supplementary table 2, online resource) and was cloned into pBlueScript-SK + (212,205, Agilent Technologies), thanks to a TA-cloning strategy [37]. Using specific oligos that include the appropriate restriction sites (A1 and TIR-T-BglII and ET-T-BglII, see Supplementary table 2, online resource) TIR-MAPT and ET-MAPT were subcloned into a eukaryotic expression vector pSG5 (216201, Agilent technologies). Similarly, Tau isoforms were cloned into a prokaryotic expression vector pRK172 using specific oligos (TAU-PRK172 fw, TIR-T-pRKpWPI rv and ET-T-pRKpWPI rv; see Supplementary table 2, online resource). After cloning, all vectors were sequenced using the described oligos (Supplementary table 2, online resource).
RNA-seq data analysis
RNA-seq raw data from 363 samples of 3 brain regions (frontal cortex, dorsolateral prefrontal cortex and hippocampus) of 180 human brain healthy donors were retrieved from the Genotype-Tissue Expression (GTEx) project [36] (Supplementary table 3, online resource). SRA files were converted to FASTQ files and reads were re-mapped to human genome GRCh38 using STAR version 2.5.2a [18]. Gene expression quantification was performed with RSEM version 1.3.1 [35].
The annotation file was retrieved from GENCODE (gencode.v23.annotation.gtf) and was modified to include a new MAPT-related gene (TIR-MAPT) whose genome coordinates are chr17:45894382–46018851, which includes part of the intron 12 (chr17:46018731–46018851) as the 3′ end of the gene in the region mapping the oligonucleotide sequence TauD (Supplementary Table 2, online resource and Fig. 1). Gene expression levels were obtained from RSEM as transcripts per kilobase million (TPM) values. Expression levels of MAPT and TIR-MAPT genes were analyzed per brain region. For those donors with more than one sample in the same brain region, analysis was done only in the sample with highest MAPT expression.

TIR-MAPT RNA expression. a Schematic representation of the MAPT gene and MAPT and TIR-MAPT mRNAs generated from it. A, C, D and E represent the hybridization sites of the primers designed for semi-quantitative PCR employed for the PCRs in b (Supplementary Table 2, online resource). The fragment of intron 12 that would remain upon retention is represented with colored stripes. b Representative images of agarose gels showing of semi-quantitative PCR results using total or cytoplasmic-enriched RNA of SH-SY5Y cells. Results showed the existence of RNA species from exon 11 to intron 12 where intron 11 was spliced out (PCR 5 and 6). Controls of the addition (RT+) or no addition of retrotranscriptase (RT−) were included. PCR 9 shows MAPT in which both intron 11 and 12 are spliced out. Detailed information of all semi-quantitative PCR combinations and amplicon sizes is provided in Supplementary Table 5, online resource. c Schematic representation of the MAPT gene including the hybridization sites of the oligos used for quantitative PCR. d TIR-MAPT RNA levels by qPCR in cytoplasmic-enriched fraction or whole extracts (total) of SH-SY5Y cells. e Comparison of TIR-MAPT level in SH-SY5Y cells and hippocampus and frontal lateral cortex of human brain. Graphs show means and SE of technical triplicates. f Percentage of brain samples having expression of MAPT or TIR-MAPT genes. Data are shown for three different regions within brains (see Supplementary Table 3, online resource). g Scatter dot blot of expression values of MAPT and TIR-MAPT genes in brain regions having TPM (transcripts per kilobase million) > 0. Number of samples f, g: cortex = 122; frontal cortex = 113; hippocampus = 98. Graphs represent mean and SEM. p values were calculated using a T test, ****p value ≤ 0.0001
Bacteria culture and Tau purification
Upon cloning, pRK172 vectors encoding different Tau isoforms were transformed in BL21 E. coli competent cells by electroporation, from which Tau was purified, as described elsewhere [49]. These bacteria were cultured in LB medium with 100 ng/ml of ampicillin at 37 °C overnight. Bacterial suspensions were transferred to 1L of LB with 100 ng/ml of ampicillin and further incubated at 37 °C, up until optical density readings at 600 nm ranged between 0.6 and 0.8. At this point, 0.4 mM of IPTG was added to each sample so as to trigger transcription, and incubated once again at 37 °C for 2 h. The samples were centrifuged at 2950×g at 4 °C for 20 min and pellets were resuspended in Tau buffer, which consists of buffer A (0.1 M MES pH 6.4, 0.5 mM MgCl2 and 2 mM EGT) supplemented with 1 mM PMSF; 0.5 NaCl and 5 mM β-mercaptoethanol. Resuspended pellets were sonicated on ice at 24 microns peak to peak (mpp) five times for 1 min, waiting 10 s between repetitions. Upon sonication, samples were centrifuged at 13,850×g at 4° C for 10 min and the resultant supernatants were subjected to 5 min at 100° C and 5 min on ice before centrifuging one more time at 13,850×g at 4 °C for 30 min. Supernatants were kept and Tris 1 M was added until pH reaches a level of 11, to get rid of residual DNA. Ammonium sulfate 50% was added to the samples and they were incubated agitating at 4 °C for at least 1 h before centrifuging again at 13,850×g at 4 °C for 1 h. Pellets containing purified Tau were resuspended in buffer A and the purification process was checked by SDS-PAGE and Coomassie blue staining to check if any other protein bands appeared upon Coomassie blue staining.
Tau protein quantitation
Protein concentration for each Tau isoform was first determined by means of absorbance measurements corrected by individually calculated extinction coefficients (ε), based on the specific amino acid sequence of each isoform [24] (Supplementary Table 4, online resource). Namely, we measured Tau concentration as indicated by Kundel and collaborators [34]: absorbance at 280 nm (A280) was measured for each Tau isoform and concentration was determined as the ratio between A280 and ε. Extinction coefficients can be estimated from the amino acid sequence of the protein [24], according to the equation: ε280 = number of tryptophan residues (W) × 5500 + tyrosine residues × 1490 [17]. Using this equation and the amino acid composition of each Tau isoform, the extinction coefficients were calculated for each one (Supplementary Table 4, online resource). Calculated values for T42 and T30 coincide with those calculated by Kundel [34], which has been typically used for all CNS human Tau isoforms [21], but are not valid for W-Tau isoforms that contain 2 extra tryptophan residues nor for ET-Tau isoforms, which lack 1 tyrosine that appear on the rest of the isoforms. Coomassie blue staining of samples containing the same amount of protein was performed to confirm their concentrations.
RNA extraction and purification
Either total RNA or cytosolic enriched fraction RNA was purified from cells using RNAeasy Mini Kit (74104, Qiagen) following the protocols described in Qiagen handbook. For brain tissue, previous homogenization using a TissueLyser (Retsch MM300, Qiagen, Hilden, Germany) (30 Hz, 5 min) with 5-mm stainless steel beads (69989, Qiagen) in 700 µl QIAzol Lysis Reagent (79306, Qiagen) was performed. RNA integrity numbers (RIN) were calculated using the Agilent 2100 Bioanalyzer system (Agilent Technologies), and only RNAs with RIN > 5 were used for RT-qPCR.
RNA was purified using RNAeasy Mini Kit (74104, Qiagen) with the following modifications. Cells were pelleted and frozen at -80 °C. Pellets were carefully resuspended in 175 μl of precooled (4 °C) buffer: 50 mM TrisHCl pH 8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% (v/v) Nonidet P40 (1.06 g/ml) plus 1 mM DTT just before use and incubated on ice for 5 min. Lysates were centrifuged at 4 °C for 2 min at 300 × g. Supernatants were kept as cytoplasmic-enriched fraction. To each fraction, 600 μl of Buffer RLT were added. After vortexing, 430 μl of ethanol 100% were added to the homogenized lysate. Samples were transferred to RNeasy spin columns, centrifuged for 15 s at 9000×g and the flow-through was discarded. Samples were treated with 10 µl of DNase in 70 µl of buffer RDD (RNase-free DNase Set 79254, Qiagen) for 15 min at room temperature. The rest of the extraction was performed following the protocol and RNA was collected in 30 µl of RNAse free water twice.
Semi-quantitative PCR
Total RNA was purified using RNAeasy Mini Kit (74104, Qiagen) following provider’s guidelines. Cytoplasmic RNA fractions were purified as previously described. Retrotranscription was performed using 40 ng/µl of RNA with the Transcriptor First Strand cDNA Synthesis Kit (04379012001, Roche) using oligo(dT)18 primer. Semi-quantitative PCR was performed using 1 µl of cDNA (0.5 ng/µl) supplemented with 2.5 mM MgCl2, 0.2 mM each dNTP, 1.3 M betaine, 0.5 mM of each primer (described in supplementary table 2, online resource) and 0.025 U/ml of GoTaq® Flexi DNA Polymerase (M829, Promega), in the following conditions: 95 °C for 2 min, and 35 cycles (PCR1-8) or 30 cycles (PCR9-13) of 95 °C for 45 s, 58.6 °C for 45 s and 72 °C for 45 s, followed by a final extension of 10 min at 72 °C. PCR combinations are described in supplementary table 5, online resource.
Detection of Tau levels by quantitative RT-PCR
RNA was retrotranscribed with the Transcriptor First Strand cDNA Synthesis Kit (04379012001, Roche) using 20 ng/µl RNA with oligo(dT)18 primer. Quantitative PCR was performed in a LightCycler480 (Roche) in the following conditions: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Specific intron-spanning oligonucleotides against MAPT or TIR-MAPT transcript were designed (MAPT-E11-E13-fw/rv and TIR-T-fw/rv, respectively; see Supplementary table 2, online resource). Gene expression was normalized to GAPDH expression using TaqMan primer human GAPDH (Hs02758991_g1, Applied Biosystems). For every RT-qPCR experiment, only samples with RIN above 5 were used.
Detection of Tau levels by Western blot
Frozen brain tissue was homogenized using a TissueLyser (Retsch MM300, Qiagen, Hilden, Germany) (30 Hz, 5 min) with 5-mm stainless steel beads (69,989, Qiagen) in total extraction buffer: 50 mM Tris–HCl pH 7.5, 300 mM NaCl, 0.5% SDS (sodium dodecyl sulphate) and 1% Triton X-100 (Fig. 2d, e and Supplementary Fig. 6b, online resource). The same buffer was also used for cells (Figs. 2b, c, 3b, 4a, 5b and Supplementary Fig. 4b, online resource and Supplementary Fig. 5a, b, online resource). For human cortex brain extracts, a strong lysis buffer (50 mM Tris–HCl pH 7.6, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% SDS) was also used in Fig. 7b. The homogenates are incubated for 15 min at 95 °C, centrifuged (16,100×g, 10 min) and the supernatant was considered the brain extract. Protein concentration was measured using the DC protein assay kit (500-0111, Bio-Rad). From each sample, equal amount of total protein was resolved on a 10% Bis–Tris gel and transferred to nitrocellulose membranes. Blots are probed with the corresponding primary antibodies (see below), followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibody (Dako, Glostrup, Denmark). Protein expression was quantified by measuring the ECL signal with ImageJ software (Fig. 7 and Supplementary Fig. 6, online resource) (http://rsbweb.nih.gov/ij/) or with Quantity One® 1-D analysis software from Bio-Rad.

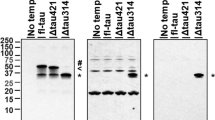
W-Tau antibody validation and protein expression in human brain. a Immunoprecipitation assay of HEK239T cells overexpressing W-T30 using W-Tau (Abyntek) or total Tau (NOVUSBIO, NB100-82247) antibodies and Western blot detection of immunoprecipitated Tau using 7.51 antibody. b Immunoprecipitation of the same cells overexpressing W-T30 and control untransfected cells using W-Tau antibody and detection of immunoprecipitated Tau using total Tau (Tau 7.51) antibody. c HEK293T cells were transfected with eukaryotic expression vectors empty or encoding different MAPT isoforms (T42, T30, W-T42, W-T30, ET-42 and ET-T30). Representative Western Blot for Tau 7.51 and W-Tau antibodies proving W-Tau specificity. d Western blot detection of W-Tau in frontal lateral cortex and hippocampus of one human subject that show bands at 50 kDa (W-T42), 38 KDa (W-T30), and 31 KDa (W-Tau truncated). e Representative Western blot of frontal lateral cortex and hippocampus samples of the same three human subjects (a, b, c) showing the presence of W-Tau and total Tau (Tau5)

W-Tau phosphorylation pattern. a Schematic representation of different Tau protein isoforms: full-length isoform with four repeats (R) and two insertions (N) (T42), full-length isoform with three repeats and no insertions (T30), the truncated by intron retention isoforms W-Tau with four repeats, two insertions and an extra peptide (W-T42) or with three repeats, no insertions and the extra peptide (W-T30); and the correspondent asparagine-endopeptidase-truncated isoforms (ET-T42 and ET-T30). Representation of the antibodies recognizing the Tau molecule at their corresponding epitopes: Antibodies recognize all isoforms of Tau (Tau12 on amino acids 6–18; Tau 5 on amino acids 210–241 and Tau 7.51 on amino acids 315–376) or specific against dephosphorylated Tau in residues Ser195, 198, 199 and 202 (Tau1), and phospho-Tau in residues Ser202/Thr205 (AT8), Thr231 (AT180), Ser404 (Tau404), Ser396 (Tau396) and Ser396/Ser404 (PHF1). W-Tau antibody recognizes the unique peptide present on W-Tau isoforms. b Samples from HEK293T cells transfected with the different isoforms were probed with different antibodies for phosphorylated and non-phosphorylated Tau. c Quantification of the data of different Tau epitopes with respect to total Tau measured with Tau 7.51 antibody, showing mean and SEM (n = 4). One-way ANOVA for multiple comparisons followed by a Kruskal–Wallis test was performed to compare each isoform and T42 full-length level of phosphorylation

W-Tau aggregation capacity. a Representative Western blot of the presence of Tau in 1% sarkosyl-soluble and insoluble cell fractions of HEK293T cell overexpressing different Tau isoforms (T42, T30, W-T42, W-T30, ET-42 and ET-T30) detected with Tau 5 antibody. b, c Quantification of the signal obtained from Tau 5 showing sarkosyl-soluble vs insoluble fractions (b) or soluble/insoluble ratio (c). Graphs show means and SEM (N = 3). One-way ANOVA for multiple comparisons followed by a Kruskal–Wallis test was performed to compare each isoform with the correspondent full-length isoform, *p ≤ 0.05. d Representative electron microscopy images of Tau aggregates obtained upon in vitro incubation from partially purified T42, W-T42, and ET-42 extracts from bacteria in the presence of heparin to prompt aggregates formation. Scale bars show 500 nm wide. e Western blot showing soluble (supernatant) and aggregated (pellet) protein from the incubation in d, upon centrifugation to separate both fractions. Quantification shows the ratio between soluble and aggregated protein

Modulation of TIR-MAPT and W-Tau by GSK3. a. Detection of mature cytoplasmic RNA levels of TIR-MAPT, total MAPT and the ratio TIR-MAPT/total MAPT in SK-N-MC human neuroblastoma cell line in the absence or presence of 5 µg/ml amyloid-β1–42 and the GSK3 inhibitor AR-014418 (10 µM) for 24 h. Graphs show mean and SEM. One-way ANOVA and Dunnett’s multiple comparisons test were performed and statistical significance between untreated and cells treated with either Aβ1–42 and AR-014418 was given. ***p ≤ 0.001; ****p ≤ 0.0001. b Western blot analysis of the protein levels of W-Tau (W-Tau antibody) in SK-N-MC cells treated with SB216763 (25 µM), Aβ1–42 (1.1 µM) or AR-14418 (10 µM). Right panel shows the quantification of the signal of complete lanes obtained with W-Tau antibody. Graphs show mean and SEM. One-way ANOVA for multiple comparisons followed by a Kruskal–Wallis test was performed to compare treated and untreated cells. *p ≤ 0.05. c Schematic representation of the modulation of TIR-MAPT by splicing factor SC35, regulated by GSK3-mediated phosphorylation. Blue arrows represent activation; red, truncated arrows represent inhibition
Antibodies
Different antibodies were used for the different essays, as indicated in each one. Altogether, Tau 12 (Millipore, MAB2241; amino acids 6-18, diluted 1/1000), Tau 5 (Calbiochem, D00139295, amino acids 210-241, diluted 1/1000), Tau antibody (NOVUSBIO, NB100-82247, epitope around amino acid 231, diluted 1/500) and Tau 7.51 ([44], amino acids 315-376, diluted 1/100) were used as antibodies recognizing all Tau isoforms; Tau 1 (Chemicon, MAB3420, diluted 1/1000) recognizing dephosphorylated residues Ser195, 198, 199 and 202 and AT8 (phospho-Ser202/Thr205; Innogenetics, MN1020, diluted 1/100), AT180 (phospho-Thr231; Innogenetics, MN1040, diluted 1/100), Tau 404 (phospho-Ser404; Life Technologies, 44758G, diluted 1/1000), Tau 396 (phospho-Ser396; Life Technologies, 44752G, diluted 1/1000) and PHF1 (phospho-Ser396/Ser404, kind gift of Peter Davies [26], diluted 1/100) as antibodies recognizing phosphorylated Tau isoforms. In addition, β-actin (SIGMA A5441, diluted 1/20000) or GAPDH (Cell signalling, 2118, diluted 1/1000 antibodies were used as loading control.
A peptide composed of the W-Tau unique sequence (KKVKGVGWVGCCPWVYGH) (W-Tau peptide) was synthesised and a polyclonal IgG antibody against that peptide was obtained from Abyntek (Bizkaia, Spain), using New Zealand rabbit as host strain. This antibody was named W-Tau antibody and its specificity was tested (diluted 1/1000) to ensure it reacts exclusively with TIR-Tau but not with other Tau isoforms (Fig. 2a–c).
Immunoprecipitation was performed with W-Tau and total Tau (NOVUSBIO, NB100-82247) antibodies using SantaCruz Immunoprecipitation Kit (SC-2003) using 500 µg of whole cell extracts and following provider’s guidelines.
Tau phosphorylation determination
HEK293T cells were transfected with pSG5 plasmids (2 µg/p60) encoding different Tau isoforms using Lipofectamine and Plus reagents following instructions of the supplier (#18324 and #11514, respectively, Life Technologies). 48 h after transfection, cells were collected, pelleted and homogenized in 200 µl of total extraction buffer. Protein concentrations were measured using the DC protein assay kit (500-0111, Bio-Rad) and samples containing equivalent amounts of protein were analyzed by Western blot, as previously described, using nitrocellulose membranes. Immunodetection was performed with antibodies Tau 7.51 and Tau 5 for total Tau, Tau 1 for dephosphorylated Tau and AT8, AT180, Tau 404, Tau 396 and PHF1 for phosphorylated Tau.
Tau solubility determination
Once again, HEK293T cells were transfected with pSG5 plasmids (2 µg/p60) encoding different Tau isoforms using Lipofectamine and Plus reagents (Life Technologies). After 48 h, cells were collected, pelleted and homogenized in 150 µl of lysis buffer (50 mM Tris–HCl (pH 7,4), 150 mM NaCl, 20 mM NaF, 1 mM Na3VO4, 0.5 mM MgSO4; supplemented with protease inhibitors cocktail, 04693159001 Roche) at 4 °C. Homogenates were centrifuged at 27,000×g for 20 min at 4 °C. Pellets were discarded and 1% sarkosyl was added to the supernatants, being incubated for 1.5 h agitating at room temperature. Upon incubation, samples were centrifuged at 150,000×g during 45 min at 4 °C. Supernatants were kept as sarkosyl-soluble fraction and pellets were resuspended in 100 µl of a mix of total extraction buffer and loading buffer (1:1) and considered sarkosyl-insoluble fraction.
Results were further confirmed by carrying out a similar protocol to measure Triton X-100 solubility. Namely, pelleted cells were homogenized in 500 µl of lysis buffer (1% Triton X-100, 50 mM Tris–HCl pH 7, 100 mM NaCl and 1 mM EDTA) and incubated 20 min at 4 °C. The samples were centrifuged at 18,000×g for 5 min and the supernatants were kept as soluble fraction. Pellets were resuspended in total extraction buffer (see point 11 of methods) and considered Triton-insoluble fraction.
In both cases, equivalent volumes of each sample were then analyzed by Westernblotting.
Tau in vitro aggregation determination
Tau aggregates were grown for the different Tau isoforms by vapor diffusion in hanging drops in the standard way used for protein crystallizations [15]. The different Tau isoforms were purified from bacteria as described above and their concentration was estimated by measuring absorbance at 280 nm and taking into account individually calculated extinction coefficients for each isoform (Supplementary table 4, online resource) and confirmed by means of Coomassie Blue staining. Equivalent quantities of each isoform were added to the corresponding volume of buffer A (0.1 M MES pH 6.4, 0.5 mM MgCl2 and 2 mM EGT) plus 50 mM NaCl in the presence of heparin, reaching a concentration of 1 mg/ml. The reservoir contained 0.2 M NaCl in buffer A. Aggregates were obtained after incubation for 10 days at room temperature (see also [49]). An aliquot was kept for electron microscopy analysis, and the rest of the samples were centrifuged in Airfuga at 28 lb per square inch (psi) for 30 min at room temperature to separate soluble and aggregated protein. The fractionated proteins were characterized by electrophoresis and Western blot.
Tau microtubule-binding capacity assay
For microtubule preparation, the brains of 12 2-month-old C57BL/6J mice were homogenized with a potter in isotonic buffer (0.32 M sucrose, 1 mM EGTA, 1 mM MgCl2, 10 mM phosphate buffer pH 7 and 1 mM PMSF) at 4 °C. Supernatant was collected after 40 min ultracentrifugation at 100,000×g at 4 °C in an Optima L-100 XP Ultracentrifuge (Beckman Coulter). 100 μl of this supernatant were incubated with equal quantity of protein for the different Tau isoforms, purified from bacteria as mentioned above during 30 min at 37 °C, in the presence of 30% glycerol, 1 mM PMSF and 1 mM GTP to promote tubulin polymerization. After that, samples were ultracentrifuged for 60 min at 100,000×g at 25 °C.
Equivalent volumes of pellets containing microtubules and microtubule-bound Tau were analyzed by Western Blot, using Tau 5 and tubulin antibodies and the ratio between Tau 5 and tubulin signal was calculated for each isoform as their microtubule-binding ratio, normalized to the corresponding full-length isoform.
GSK3 modulation
SK-N-MC human neuroblastoma cells (ATCC® HTB-10™) were treated with GSK3 inhibitors SB216763 (25 µM, GlaxoSmithKline compounds [13]) and AR-14418 (10 µM, AstraZeneca compound [7]) or Aβ1–42 (1.1 µM; Neosystem Laboratoire, Strasbourg, France) for 24 h and whole cell extracts were used for Western blot analysis, probing the blots with W-Tau antibody. From those cells treated with AR-14418 and Aβ1-42, RNA-enriched cytoplasmic fraction was also retrieved and TIR-MAPT RNA levels were assessed by quantitative RT-PCR as previously described.
Splicing factor binding site prediction analysis
Exonic and intronic human MAPT sequences were retrieved from ENSEMBL (ENSG00000186868). Exon 12–exon 13 and exon 12–intron 12 sequences (Exon 13–exon 14 and exon 13–intron 13, according to ENSEMBL nomenclature of Tau exons/introns) were analyzed using ESEFinder 3.0 (http://exon.cshl.edu/ESE/) [53]. ESEFinder settings were adjusted to show binding sites with score 4.0 or higher. The analysis was completed by a detailed bibliographic search comparing SRSF2 binding sites described in Masaki et al., 2019 and Cavaloc et al., 1999 [11, 38] with junctions MAPT Exon 12–exon 13 and exon 12–intron 12 sequences.
Statistical analysis
Quantitative data, represented as mean ± SD or SEM, were compared between groups using the two-tailed Student’s t test. For multiple comparisons, one-way ANOVA and Dunnett's test were performed to compare each isoform and the correspondent full-length isoform. When the distribution of the data was not Gaussian a nonparametric Kruskal–Wallis test was used. The differences are given with their corresponding statistical significance or p value, which is the probability that the difference occurred merely by chance under the null hypothesis (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001; N.S. not significant). The method used for each experiment is specified in the corresponding figure legend.
Results
New alternative splicing variant of MAPT present in mature human transcriptome
Recently, the presence of new protein isoforms generated by intron retention in neurodegenerative diseases such as Alzheimer’s disease [45] and Huntington’s disease [52] has been described. This, together with evidence of an asparagine-endopeptidase-truncated Tau isoform [63], whose cleavage point coincides with the end of exon 12 led us to seek possible intron retention events within the intron between exons 12 and 13 (intron 12). Curiously, human intron 12 includes a canonical stop codon sequence followed by a polyadenylation canonical site, so intron 12-containing species would be translated as a truncated protein similar to the truncated isoform mediated by AEP (ET-Tau), with 18 more amino acids, which we have named W-Tau (Fig. 1a). Importantly, this isoform would be human-specific, since this polyadenylation site cannot be found in other species, such as mice.
We could detect intron 12 retaining RNA species in which intron 11 was spliced out in SH-SY5Y mature (polyA positive) total RNA and cytoplasmic-enriched fraction (Fig. 1b, Supplementary Fig. 1 and Supplementary Tables 2 and 5, online resources). This was observed by means of semi-quantitative PCR using an exon 11-matching forward oligo and intron 12 reverse oligos (Fig. 1a, b, Supplementary Table 5, online resource, PCR 5 and 6). As expected, we could also detect the equivalent amplicon using an exon 12-matching reverse oligo in which intron 12 was spliced out (Fig. 1a, b, Supplementary Table 5, online resource, PCR 9). Additionally, we could observe similar results using an exon 12 forward oligo and intron 12 reverse oligos (Supplementary Fig. 1a, b, Supplementary Table 5 PCR 1 and 2, online resources). Genomic DNA contamination was ruled out using intron-spanning forward oligos (Supplementary Fig. 1a, b and Supplementary Table 5, PCR 3, 4, 7 and 8; online resources). Curiously, intron-spanning primers matching exon 10 to detect RNA species containing intron 12 never detected these as mature cytoplasmic polyA positive RNA (Supplementary Fig. 1a, b and Supplementary Table 5, PCR 7 and 8; online resources). Exon 10 is spliced out in Tau isoforms with three repeats (Tau 3R) and it has been previously described that proliferative SH-SY5Y cells mainly express these Tau isoforms [57], so detection of RNA species with partial inclusion of exon 10 was not expected a priori. As a control, mature polyA positive RNA species including only exons were found in our samples (Supplementary Fig. 1a, b and Supplementary Table 5, PCR 10–12; online resources), even detecting transcripts that include exon 10 (Tau 4R), although with decreased efficiency (Supplementary Fig. 1a, b and Supplementary table 5, PCR 12; online resources). These results were also validated by qPCR using the same oligos (Supplementary Fig. 1c, online resource) and different, qPCR-optimized ones (Fig. 1c, d, Supplementary Table 2, online resource). Moreover, we were able to amplify whole-length TIR-MAPT cDNA from starting codon to intron 12 using specific primers (TauNt and TauD, Supplementary Table 2, online resource) in SH-SY5Y cells. This cDNA would correspond to TIR-MAPT with three repeats and no inserts (TIR-T30).
More importantly, TIR-MAPT polyA positive mRNA was found in both hippocampus and frontal cortex in human samples (Fig. 1e). Remarkably, TIR-MAPT expression levels in human brain were more than 100-fold the expression found in SH-SY5Y, suggesting a higher relevance of this RNA species in human brains than in cultured cells.
Additionally, RNA-seq data from 363 samples from 3 different brain regions of 180 healthy donors were used to confirm the existence of TIR-MAPT transcripts (Supplementary Table 3, online resource). TIR-MAPT gene mature mRNA containing the first part of intron 12 up until the canonical stop site was expressed in the majority of samples (Fig. 1f) supporting our previous findings in a big cohort of human samples. Gene expression of MAPT mature mRNA was compared with TIR-MAPT (Fig. 1g). Even though TIR-MAPT is expressed at lower levels, the great number of samples expressing this intron-retaining species suggests a relevant function in human brain. Among the RNA species containing intron 12, some of the reads would correspond to a putative Tau protein without inserts, and with 3 tandem repeats (T30), but substituting the 3′-terminal of the RNA for the sequence of intron 12 until the first stop codon; what we have named TIR-T30. The putative translation of this RNA species into a protein has been named W-T30, due to the presence of two tryptophan residues (W) in the sequence of intron 12 translation (Supplementary Fig. 2, online resource). This RNA species would be equivalent to that found in SH-SY5Y cells.
W-Tau expression in human brain
With the aim of determining whether this new Tau isoform is present in human brain as a protein, we obtained a specific antibody against the unique 18 amino acid sequence of W-Tau peptide (KKVKGVGWVGCCPWVYGH; W-Tau antibody), corresponding to the translation of intron 12 until the canonical stop codon. To confirm the specificity of this antibody, we cloned TIR-MAPT cDNA obtained from SH-SY5Y (TIR-T30, which would correspond to W-T30 as a protein) into the expression vector pSG5 (216201, Agilent technologies) and used it to overexpress this isoform in HEK293T cells. We demonstrated that W-T30 could be specifically immunoprecipitated with both W-Tau (Abyntek) or Total Tau (NOVUSBIO, NB100-82247) antibodies (Fig. 2a) and then that W-Tau antibody only precipitates W-Tau without any background on control untreated cells (Fig. 2b). In both cases, the immunoprecipitated protein was confirmed to be Tau by detecting it by Western blot with Tau 7.51 antibody.
To confirm this specificity and contrast it with other Tau isoforms, we further modified the eukaryotic vector pSG5 to include exon 10 corresponding to the fourth tandem repeat of the microtubule-binding domain and the two Tau inserts (thus obtaining W-T42). Additionally, asparagine-endopeptidase-truncated Tau corresponding isoforms were also cloned into pSG5 expression vector (ET: Endopeptidase truncated; ET-T42 and ET-T30); mimicking the cut of endopeptidase by including a stop codon after the one that encodes asparagine 368. We used this vector encoding W-Tau, ET-Tau and the full-length equivalents (T42 and T30) to overexpress these proteins on HEK293T cells, together with control cells transfected with the empty pSG5 vector. We observed that, while total Tau antibodies (such as Tau 7.51) recognize all these isoforms, W-Tau antibody only shows signal for W-Tau isoforms, without any background on other species (Fig. 2c).
Once the specificity of the antibody was confirmed, we probed human hippocampal and frontal lateral cortex samples with it, confirming the existence of this novel Tau isoform as a protein in human brain (Fig. 2d, e). In the first image, we show an example of a human subject that exhibits a predominant band close to 52 KDa that may correspond to a 4R form of W-Tau (Fig. 2d, 50 kDa band). Also, a smaller band at 38 KDa suggest the presence of W-T30 isoform in the hippocampal sample. Moreover, there is another band around 31 KDa that is recognized by W-Tau antibody, that might be explained as a proteolytic cleavage of the above bands, although further studies will be necessary to unravel the identity of this band. Then, we studied W-Tau expression in another three human subjects by comparing hippocampal and frontal lateral cortex samples (Fig. 2e). We could observe a predominant expression of 52 KDa 4R W-Tau bands, especially in frontal lateral cortex whose levels vary among individuals, and the 31 KDa band that may correspond to the truncated W-Tau form. All these results together indicate that intron 12 retaining Tau isoforms are expressed in human brain, of which two of them show similar molecular weights to those of W-T42 and W-T30 in cells upon overexpression.
W-Tau phosphorylation
The pSG5 vectors encoding the different isoforms were employed to induce overexpression of these isoforms, to test W-Tau properties and compare them to those of full-length isoforms (T42 and T30) and those of asparagine-endopeptidase-truncated isoforms (ET-42 and ET30) (Fig. 3a). First, we studied the phosphorylation of W-Tau isoform, comparing it with the phosphorylation found in other Tau isoforms, expressed in HEK293T cells. We expected differential detections according to the epitopes that remain despite truncation and those that are lost because of it (Fig. 3a). Indeed, phosphorylation of AT8 sites Ser202/Thr205 and AT180 (phospho-Thr231) still occurred in truncated isoforms (Fig. 3b, c). However, as expected, PHF1 phosphorylation sites Ser396/Ser404 were not observed in truncated isoforms because these sequences are not present in those species. Also, some of the modifiable residues remained in dephosphorylated state, as shown by Tau 1 antibody label (dephospho- Ser195, 198, 199 and 202) (Fig. 3b, c) and negative staining for phospho-Thr212 and phospho-Ser214 (AT100) (data not shown).
These results point out a similar post-translational processing of W-Tau when compared to other Tau isoforms, at least regarding its phosphorylation on the mentioned epitopes.
W-Tau microtubule stabilization and binding capacity
Then, we analyzed the effect of W-Tau on the polymerization of mouse brain microtubules in vitro by means of electron microscopy in the presence or absence of W-Tau, showing a higher proportion of microtubules in the presence of W-Tau (W-T42) (Supplementary Fig. 3a, online resource).
In addition, we analyzed the capacity of T42, W-T42, and ET-T42 purified from Tau-expressing bacteria (Supplementary Fig. 3b, online resource) to bind to microtubules purified from mouse brain, separating microtubule-bound and free Tau by centrifugation and characterizing Tau enrichment with respect to tubulin in microtubule-bound fraction by Western blot with Tau 5 antibody. As expected, all the tested isoforms were able to bind to microtubules (Supplementary Fig. 3b, c, online resource), since all of them contain microtubule-binding repeats. Of note, W-T42 showed a high affinity to bind microtubules compared to other isoforms harboring four repeats and two inserts (Supplementary Fig. 3c, online resource). In accordance with previously published results [63], ET-T42 showed lower affinity to bind microtubules than full-length Tau.
W-Tau aggregation capacity in cultured cells
We compared the aggregation tendency of different Tau isoforms when overexpressed in HEK293T cells, by quantification of Tau in Sarkosyl soluble versus insoluble protein fractions, with the insoluble fraction including Tau aggregates (Fig. 4a). With respect to full-length T42 aggregation capacity, we could observe that a similar soluble/aggregated protein ratio was found in full-length Tau isoforms. However, in truncated Tau isoforms, the ratio was different in W-Tau isoforms, with a higher proportion of soluble protein when compared to both full-length and ET-Tau isoforms, being significatively higher for W-T30 with respect to its correspondent full-length T30 (Fig. 3b, c). These results were further confirmed by means of a similar assay that tested solubility with Triton X-100 (Supplementary Fig. 4a, online resource). A similar analysis could be performed with the signal obtained from W-Tau antibody (Supplementary Fig. 4b, c, online resource).
In vitro aggregation of W-Tau
In addition, we carried out the analysis of the formation of protein aggregates by in vitro incubation of purified T42, W-T42 and ET-T42 with heparin, observing a reduced proportion of Tau aggregates when W-T42 was tested, compared to the rest of the isoforms (Fig. 4d), as determined by densitometry of electrophoretically fractionated protein bands present upon centrifugation in supernatant (non-aggregated protein) versus those present in pellet (aggregated protein) (Fig. 4e). These results support that W-Tau displays diminished aggregation tendency.
Potential mechanism explaining intron 12 inclusion
We performed an in silico study, along with a bibliographic search, of the potential splicing factors involved in the process. Results obtained with ESEFinder 3.0 indicate the presence of a high-scored SRSF6 splicing binding site in the canonical MAPT sequence, close to the beginning of Exon 13 (Supplementary Fig. 5, online resource). This site is not present in the TIR-MAPT sequence. However, in its place, ESEFinder 3.0 indicates the appearance of a high-scored SRSF2 (also known as SC35) splicing binding site in intron 12, in contrast to MAPT sequence splicing out intron 12 (Supplementary Fig. 5, online resource). We re-analyzed MAPT exon 12–exon 13 and exon 12–intron 12 junction sequences using SRSF2 previously described binding sites [11, 38]. Surprisingly, we found that exon 12–intron 12 junction sequence, AGGTAAAG, is extremely similar to a specific SRSF2 binding site AGGTRAG (R for any Purine, A or G) previously described by Masaki et al. [38]. Both analyses suggested that SRSF2 may bind to TIR-MAPT sequence at the exon 12–intron 12 junction and/or at the beginning of intron 12 pointing to SRSF2 as a potential responsible for the regulation of this process. This evidence may be further supported by previous well-established evidence that SRSF2 is also involved in Tau exon 10 splicing [29], pointing out a regulatory function of this splicing factor on Tau alternative splicing that may be linked to the intron retention described in this work.
Additionally, previous work of our group suggests that the inhibition of GSK3-mediated SRSF2 phosphorylation increases the inclusion of exon 10 [29]. Thus, we tested the effect of the inhibition of SRSF2 phosphorylation carried out by the constitutively active GSK3 kinase [29] on the retention of MAPT intron 12. We detected increased TIR-MAPT cytoplasmic mature RNA levels in SK-N-MC cells in the presence of the GSK3β inhibitor AR-014418 [7], compared to both untreated and amyloid β treated cells (Fig. 5a). When we analyzed W-Tau protein levels, we could observe that SB216763, other GSK3β inhibitor also results in increased W-Tau levels, although this increase is more relevant for AR-014418 (Fig. 5b). These preliminary data indicate that SRSF2 may be involved in this process via GSK3β regulation (Fig. 5c).
Finally, it is worth noting that the presence of beta amyloid peptide, that could stimulate GSK3 [28], did not result in a change in the level of neither TIR-MAPT nor W-Tau (Fig. 5a, b), probably because GSK3 is constitutively active and, therefore, amyloid-treated cells behave as untreated control cells containing already SC35 in its phosphorylated form. However, as previously described, amyloid β did result in a significant increase of total MAPT RNA (Fig. 5a).
Expression of TIR-MAPT RNA in the brain of AD patients.
We studied TIR-MAPT mRNA levels in a cohort of hippocampal brain samples from non-demented individuals and AD patients classified according to their Braak stage (see Supplementary Table 1, online resource). The study of absolute levels of TIR-MAPT mRNA revealed that it slightly decreases in the first stages of the disease (Braak II and III) but increases in the advanced stages (Braak V and VI), although no significant differences were found (Fig. 6a). Total MAPT mRNA levels were similarly studied, finding in this case decreased expression in AD patients with respect to healthy individuals, more significantly in Braak stage VI (Fig. 6b). These data correlate with what we previously described in a human RNA-seq analysis [23]. Finally, when we studied TIR-MAPT/total MAPT ratio we could observe a significant increase in Braak VI samples as well as a tendency towards an increased ratio in AD patients with respect to non-demented individuals (Fig. 6c).

TIR-MAPT RNA expression in the brains of AD patients. a Measurement of TIR-MAPT, b total MAPT and c TIR-MAPT/total MAPT ratio of RNA levels by RT-qPCR of non-demented (n = 8) and AD hippocampal samples (n = 16) classified according to their Braak stage (Braak II n = 3, Braak III n = 2, Braak V n = 7 and Braak VI n = 4), and healthy vs. AD patients. Graphs show means and SE. One-way ANOVA and Dunnett’s multiple comparisons test were performed and statistical significance of each group with respect to non-demented control individuals was given. For comparisons between non-demented individuals and AD patients as groups, p values were calculated using a T test (*p ≤ 0.05; ***p ≤ 0.001, †p = 0,078)
W-Tau presence in patients’ brains
Finally, in accordance to what was found for control human brains (Fig. 2d, e), we confirmed that W-Tau antibody was able to specifically immunoprecipitate W-Tau protein from human brain in extracts from AD patients (Braak stages V and VI) that could be identified as Tau again with a total Tau antibody (Tau 7.51) (Fig. 7a).

Tau protein determination in AD patients’ brain. a Immunodetection of the presence of W-Tau isoforms using W-Tau antibody in two different frontal lateral cortex brain extracts derived from AD patients’ brains (Braak V and VI, respectively). The immunoprecipitate performed with W-Tau antibody was characterized by Westernblotting. Left panel shows the blot developed using W-Tau antibody; right panel using Tau 7.51 antibody. b Western blot analysis of the levels of W-Tau and total Tau in frontal lateral cortical samples of non-demented (n = 9) and AD patients classified according to their Braak stage (Braak I = 3; Braak II n = 6, Braak III n = 3, Braak IV n = 1, and Braak V n = 10, Braak VI n = 8). c Quantification of W-Tau and total Tau protein levels as well as W-Tau/total Tau ratio of each group. One-way ANOVA and Dunnett’s multiple comparisons test were performed and statistical significance of each group with respect to non-demented control individuals was given (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). A.U. arbitrary units
Once this was confirmed, we analyzed the levels of W-Tau protein in the cortex of healthy individuals and AD patients classified according to their Braak stage (Fig. 7b). We could observe that W-Tau is expressed in both healthy and AD individuals. The analysis of both W-Tau bands showed a strong diminished expression in all Braak stages with respect to non-demented individuals (Fig. 7c). Accordingly, the quantification of each W-Tau band separately also yielded significant differences in all Braak stages with respect to non-demented individuals (Supplementary Fig. 6a, online resource). On the other hand, the expression of total Tau seems to increase at later stages of the disease, but we could not detect significant differences among groups when individuals are separated by Braak stages in our samples (Fig. 7c). However, the ratio of W-Tau with respect to total Tau expression reveals an enrichment of W-Tau in advanced stages of the disease (Braak V and VI) (Fig. 7c).
Additionally, we could also obtain hippocampal samples of some of these individuals, and we studied W-Tau expression (Supplementary Fig. 6b, online resource). In such conditions, similar to what we previously observed in Fig. 2d, e, we mainly detect 31 KDa (W-Tau truncated) and 50 KDa (W-T42) bands and, to a lesser extent, 38 KDa (W-T30) band. We could observe a trend towards diminished W-Tau expression in AD patients in the tree bands being significant in advanced stages of the disease for the 38 KDa (W-T30) band as well as in W-Tau/total Tau ratio (Supplementary Fig. 6c, d, online resource).
These data together indicate an inverse correlation between W-Tau levels and disease progression that opposes total Tau levels behavior, strongly suggesting a relevant role of this new intron-retaining isoform in healthy individuals, whose loss would be related to the progression of the disease. Taken RNA and protein data together (Figs. 6, 7) and considering the more marked presence of Tau aggregates in Total Tau but not in W-Tau, the changes on the amount of W-Tau in AD with respect to total Tau may be more related to differences on the protein turnover, rather than to their synthesis.
Discussion
Although recent evidence point out that a large proportion of predicted alternative transcripts might not be translated into proteins [55], MAPT gene has been proven to be genuinely subjected to alternative splicing [1, 55]. Also, MAPT contains a cryptic exonic sequence codifying for the protein saitohin nested in the Tau gene within intron 9 [14]. Thus, the removal of RNA sequences in Tau RNA is very usual, but the translation of intronic sequences could also take place. Curiously, it has been suggested that the region of “big” Tau corresponding to exon 4a arose, evolutionary, from an intron from another protein [20].
Tau alternative splicing is also regulated by several features such as development, aging, disease or brain region [8, 54, 56, 58]. Tau isoforms lacking exons 2, 3 and 10, can be mainly found at early developmental stages [2, 56] and alternative splicing of exon 10 has been related to some tauopathies [10, 50].
Recently, a toxic fragment containing residues 1–368 has been reported [63]. Since residue 368 coincides with the end of exon 12, in this work we have studied whether a failure in the removal of intron 12 could give rise to a novel Tau isoform. We found that intron 12 retention can take place, in low proportions in non-demented human control as well as AD patients’ brains. The failure of intron 12 removal results in the appearance of a Tau isoform containing 18 additional residues after exon 12. Strikingly, this new isoform is significantly reduced in AD, especially in the advanced stages, while total Tau is accumulated. It is worth emphasizing that this isoform is human-specific, since other species’ Tau orthologues do not contain the polyadenylation canonical site that allows intron retention to give rise to a truncated protein. This is especially noteworthy considering that Alzheimer’s disease is a human pathology that is not well reproduced in animal models.
A possible mechanism explaining intron 12 retention is that trans-acting splicing factors could be modified, for example by phosphorylation, and that the modified factor could change its function. Moreover, modification of intron splicing has been previously reported for SR-proteins [40] and, specifically, for SRSF2 (SC35) which, when phosphorylated by GSK3 kinase, favors the expression of Tau 3R (lacking exon 10) [29]. Additionally, it has been described that the absence of SRSF2 favors exon 10 inclusion increasing Tau 4R species [12]. Notably, intron splicing events have been previously reported for SRSF2 and other member of the SR-proteins family [40]. In this work, we found that GSK3 inhibition also induces the retention of intron 12, presumably by the inhibition of SRSF2 function, inducing the expression of TIR-MAPT mRNA and protein, while total Tau levels remain unmodified. SRSF2 could play a role in this process, since there are two open recognition sites for this factor in TIR-MAPT sequence, one of them located at the exon 12–intron 12 junction [11, 38]. Of note, W-Tau levels did not change in amyloid-treated cultured cells, even though amyloid beta peptide has been reported to stimulate GSK3 [12, 28]. This may be due to GSK3 being constitutively active and, consequently, amyloid-mediated activation does not imply further SRSF2 phosphorylation when comparison to untreated cells.
The novel Tau fragment described in this work is very similar to the previously reported fragment generated by cleavage of AEP [63]. However, contrary to what was expected, the isoforms truncated by intron retention exhibit a potentially protective behavior, with decreased aggregation capacity but conservation of microtubule assembly capacity. This novel Tau isoforms contain a unique 18-aminoacid extra peptide, including the sequence GVGWVG, which could be similar in nature to that of LYIWVQ, a recently described inhibitor of Tau and Aβ aggregation [27] (Supplementary Fig. 7, online resource). Thus, we suggest that the decreased aggregation capacity of W-Tau could be related to the presence of this extra peptide. Moreover, W-Tau lacks the 12 amino acids after the fourth repeat, present in exon 13, which are found in the core of Tau filaments isolated from the brain of patients suffering some tauopathies, including Alzheimer’s disease [62]. It is also worth noting that the specific W-Tau 18 amino acid sequence contains two adjacent cysteines (KKVKGVGWVGCCPWVYGH), whose oxidation to cystine can mediate the formation of a strained eight-member ring [47], a conformation that would substantially differ of that of other Tau isoforms and that has been proposed to be able to affect protein function and conformation stability [33, 39].
As for the relevance of this novel truncated isoform in AD’s development and progression, similar TIR-MAPT mRNA levels were found in non-demented control subjects and AD patients’ samples. However, on a protein level, we have detected higher levels of W-Tau protein in healthy individuals compared to AD patients. Discordant RNA and protein levels for Tau in AD have been previously described [23]. We hypothesize that, in this case, it may be related to a relatively faster turnover for W-Tau isoforms, maybe linked to a different conformation rising from its different sequence. These data also showed a clearly less aggregative pattern on W-Tau isoforms when compared to total Tau (Fig. 7), suggesting that this is a less aggregation-prone Tau isoform indeed.
Finally, therapeutic correction of aberrant splicing that could take place in Alzheimer’s disease has been suggested for some nuclear RNAs [4, 31]. Given the potential role of this new Tau truncated isoform on a possible AD therapy, we will look for future therapeutic strategies based on an increase of this less aggregation-prone Tau isoform or the prevention of its decrease.
References
Abascal F, Ezkurdia I, Rodriguez-Rivas J, Rodriguez JM, del Pozo A, Vázquez J et al (2015) Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput Biol 11:e1004325
Andreadis A (2005) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739:91–103. https://doi.org/10.1016/j.bbadis.2004.08.010
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human tau gene. Biochemistry 31:10626–10633
Avale ME, Rodriguez-Martin T, Gallo JM (2013) Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum Mol Genet 22:2603–2611. https://doi.org/10.1093/hmg/ddt108
Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84:361–384. https://doi.org/10.1152/physrev.00024.2003
Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F (2008) Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol 67:470–483. https://doi.org/10.1097/NEN.0b013e31817275c7
Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y et al (2003) Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem 278:45937–45945. https://doi.org/10.1074/jbc.M306268200
Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33:95–130
Caceres A, Kosik KS (1990) Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343:461–463. https://doi.org/10.1038/343461a0
Caffrey TM, Joachim C, Paracchini S, Esiri MM, Wade-Martins R (2006) Haplotype-specific expression of exon 10 at the human MAPT locus. Hum Mol Genet 15:3529–3537
Cavaloc Y, Bourgeois CF, Kister L, Stévenin J (1999) The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA 5:468–483
Chen K-L, Yuan R-Y, Hu C-J, Hsu CY (2010) Amyloid-β peptide alteration of tau exon-10 splicing via the GSK3β-SC35 pathway. Neurobiol Dis 40:378–385
Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ et al (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7:793–803
Conrad C, Vianna C, Freeman M, Davies P (2002) A polymorphic gene nested within an intron of the tau gene: implications for Alzheimer’s disease. Proc Natl Acad Sci USA 99:7751–7756. https://doi.org/10.1073/pnas.112194599
Crowther R, Olesen O, Smith M, Jakes R, Goedert M (1994) Assembly of Alzheimer-like filaments from full-length tau protein. FEBS Lett 337:135–138
D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD et al (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 96:5598–5603
Daniel BM, Edelstein S (1991) Protein methods. Wiley, New York
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Fernandez-Nogales M, Cabrera JR, Santos-Galindo M, Hoozemans JJ, Ferrer I, Rozemuller AJ et al (2014) Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat Med 20:881–885. https://doi.org/10.1038/nm.3617
Fischer I, Baas PW (2020) Resurrecting the Mysteries of Big Tau. Trends Neurosci 43:493–504
Furukawa Y, Kaneko K, Nukina N (2011) Tau protein assembles into isoform-and disulfide-dependent polymorphic fibrils with distinct structural properties. J Biol Chem 286:27236–27246
Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL et al (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA 100:10032–10037. https://doi.org/10.1073/pnas.1630428100
García-Escudero V, Gargini R, Martín-Maestro P, García E, García-Escudero R, Avila J (2017) Tau mRNA 3′ UTR-to-CDS ratio is increased in Alzheimer disease. Neurosci Lett 655:101–108
Gill SC, Von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182:319–326
Goedert M, Crowther RA, Spillantini MG (1998) Tau mutations cause frontotemporal dementias. Neuron 21:955–958
Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci 87:5827–5831
Griner SL, Seidler P, Bowler J, Murray KA, Yang TP, Sahay S et al (2019) Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife 8:e46924
Hernández F, de Barreda EG, Fuster-Matanzo A, Lucas JJ, Avila J (2010) GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol 223:322–325
Hernandez F, Perez M, Lucas JJ, Mata AM, Bhat R, Avila J (2004) Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J Biol Chem 279:3801–3806. https://doi.org/10.1074/jbc.M311512200
Himmler A (1989) Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol 9:1389–1396
Hinrich AJ, Jodelka FM, Chang JL, Brutman D, Bruno AM, Briggs CA et al (2016) Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol Med. https://doi.org/10.15252/emmm.201505846
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al (1998) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. https://doi.org/10.1038/31508
Kim BM, Schultz LW, Raines RT (1999) Variants of ribonuclease inhibitor that resist oxidation. Protein Sci 8:430–434
Kundel F, Hong L, Falcon B, McEwan WA, Michaels TC, Meisl G et al (2018) Measurement of tau filament fragmentation provides insights into prion-like spreading. ACS Chem Neurosci 9:1276–1282
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S et al (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45:580–585
Marchuk D, Drumm M, Saulino A, Collins FS (1991) Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res 19:1154. https://doi.org/10.1093/nar/19.5.1154
Masaki S, Ikeda S, Hata A, Shiozawa Y, Kon A, Ogawa S et al (2019) Myelodysplastic syndrome-associated SRSF2 mutations cause splicing changes by altering binding motif sequences. Front Genet 10:338
Miller SM, Moore MJ, Massey V, Williams CH Jr, Distefano MD, Ballou DP, Walsh CT (1989) Evidence for the participation of Cys558 and Cys559 at the active site of mercuric reductase. Biochemistry 28:1194–1205
Moon H, Cho S, Loh TJ, Jang HN, Liu Y, Choi N et al (2017) SRSF2 directly inhibits intron splicing to suppresses cassette exon inclusion. BMB Rep 50:423
Motoi Y, Sahara N, Kambe T, Hattori N (2010) Tau and neurodegenerative disorders. Biomol Concepts 1:131–145. https://doi.org/10.1515/bmc.2010.017
Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res 387:271–280
Novak M (1994) Truncated tau protein as a new marker for Alzheimer’s disease. Acta Virol 38:173–189
Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM (1991) Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA 88:5837–5841
Ong C-T, Adusumalli S (2020) Increased intron retention is linked to Alzheimer’s disease. Neural Regen Res 15:259–260. https://doi.org/10.4103/1673-5374.265549
Ozcelik S, Sprenger F, Skachokova Z, Fraser G, Abramowski D, Clavaguera F et al (2016) Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol Psychiatry. https://doi.org/10.1038/mp.2015.228
Park C, Raines RT (2001) Adjacent cysteine residues as a redox switch. Protein Eng 14:939–942
Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 25:5365–5375. https://doi.org/10.1523/JNEUROSCI.1125-05.2005
Perez M, Arrasate M, Montejo De Garcini E, Munoz V, Avila J (2001) In vitro assembly of tau protein: mapping the regions involved in filament formation. Biochemistry 40:5983–5991
Polanco JC, Li C, Bodea L-G, Martinez-Marmol R, Meunier FA, Götz J (2018) Amyloid-β and tau complexity—towards improved biomarkers and targeted therapies. Nat Rev Neurol 14:22
Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S et al (2018) Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet 50:1584
Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA 110:2366–2370. https://doi.org/10.1073/pnas.1221891110
Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR (2006) An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 15:2490–2508. https://doi.org/10.1093/hmg/ddl171
Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C et al (2012) MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet 21:4094–4103
Tress ML, Abascal F, Valencia A (2017) Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci 42:98–110
Tuerde D, Kimura T, Miyasaka T, Furusawa K, Shimozawa A, Hasegawa M et al (2018) Isoform-independent and-dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J Biol Chem 293:1781–1793
Uberti D, Rizzini C, Spano PF, Memo M (1997) Characterization of tau proteins in human neuroblastoma SH-SY5Y cell line. Neurosci Lett 235:149–153
Valenca GT, Srivastava GP, Oliveira-Filho J, White CC, Yu L, Schneider JA, Buchman AS et al (2016) The role of MAPT haplotype H2 and isoform 1N/4R in parkinsonism of older adults. PLoS ONE 11:e0157452
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C et al (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456:470
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:22–35. https://doi.org/10.1038/nrn.2015.1
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72:1858–1862
Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B et al (2020) Novel tau filament fold in corticobasal degeneration. Nature 580:1–5
Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM et al (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20:1254–1262. https://doi.org/10.1038/nm.3700
Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A et al (2015) Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 87:963–975. https://doi.org/10.1016/j.neuron.2015.08.020
Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M et al (2006) Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 580:3582–3588
Acknowledgements
This work was supported by the Ministerio de Ciencia, Innovación y Universidades from Spain (PGC2018-096177-B-00). Institutional grants from the Fundación Ramón Areces and Banco de Santander to CBMSO are also acknowledged. The Asociación Española Contra el Cáncer Scientific Foundation has financed Ricardo Gargini. We would like to acknowledge Daniela Rosiles for her technical support in cloning.
Author information
Authors and Affiliations
Contributions
JÁ, FH, and JJL conceived the original idea of the study. VG-E, DR-G, RG, MP, EG, RC, and IHH performed the experiments. VG-E, DR-G, RG, MP, and IHH designed the experiments. RG-E and JRC performed the computational analyses. VG-E, DR-G, RG, IHH, JRC, RG-E, JJL, FH, and JÁ analyzed the data. VG-E, DR-G, and JÁ wrote the paper with contribution from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests to disclose.
Ethical approval
All applicable international, national, and institutional animal welfare guidelines were followed for the experiments involving experimentation animals. All procedures were in accordance with ethical standards and were approved by the pertinent Ethics Committee. Mice were bred in the animal facility of Centro de Biología Molecular Severo Ochoa. They were housed in a specific pathogen-free colony facility under standard laboratory conditions, following European Community Guidelines (directive 86/609/EEC), and handled in accordance with European and local animal care protocols (PROEX 62/14 and 291/15). 4–5 mice were housed per cage with food and water available ad libitum, and maintained in a temperature-controlled environment on a 12 h/12 h light/dark cycle with light onset at 8 a.m.
Informed consent
Human brain samples were obtained from Banco de Tejidos (Fundación CIEN, Instituto de Salud Carlos III, Madrid, Spain). Written informed consent for tissue donation for research purposes was obtained for all individuals following their approval procedures.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Escudero, V., Ruiz-Gabarre, D., Gargini, R. et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol 142, 159–177 (2021). https://doi.org/10.1007/s00401-021-02317-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02317-z