
Abstract
Nuclear clearance and cytoplasmic accumulations of the RNA-binding protein TDP-43 are pathological hallmarks in almost all patients with amyotrophic lateral sclerosis (ALS) and up to 50% of patients with frontotemporal dementia (FTD) and Alzheimer’s disease. In Alzheimer’s disease, TDP-43 pathology is predominantly observed in the limbic system and correlates with cognitive decline and reduced hippocampal volume. Disruption of nuclear TDP-43 function leads to abnormal RNA splicing and incorporation of erroneous cryptic exons in numerous transcripts including Stathmin-2 (STMN2, also known as SCG10) and UNC13A, recently reported in tissues from patients with ALS and FTD. Here, we identify both STMN2 and UNC13A cryptic exons in Alzheimer’s disease patients, that correlate with TDP-43 pathology burden, but not with amyloid-β or tau deposits. We also demonstrate that processing of the STMN2 pre-mRNA is more sensitive to TDP-43 loss of function than UNC13A. In addition, full-length RNAs encoding STMN2 and UNC13A are suppressed in large RNA-seq datasets generated from Alzheimer’s disease post-mortem brain tissue. Collectively, these results open exciting new avenues to use STMN2 and UNC13A as potential therapeutic targets in a broad range of neurodegenerative conditions with TDP-43 proteinopathy including Alzheimer’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease is a progressive neurodegenerative disorder, affecting up to 30% of the population over 65 [59]. Genetic and pathological studies have provided overwhelming evidence that Alzheimer’s disease has a heterogeneous etiology, thus a better characterization of patient subgroups is essential. Extracellular deposits of amyloid-β plaques, and intraneuronal accumulations of neurofibrillary tau tangles are canonical hallmarks of Alzheimer’s disease pathology [14]. Cytoplasmic inclusions of phosphorylated TAR DNA-binding protein-43 (TDP-43) are a pathological hallmark of several neurodegenerative disorders including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and limbic-predominant age-related TDP-43 encephalopathy (LATE) [6, 20, 48, 53, 66,67,68, 81]. Notably, TDP-43 proteinopathy has also been reported in approximately one-third of Alzheimer’s disease cases, with estimates varying from 19 to 57% [5, 7, 13, 21, 29, 34, 35, 38, 54, 85, 92, 100], also referred to as Alzheimer’s disease with LATE neuropathologic changes (LATE-NC) [65,66,67]. TDP-43 pathology burden in Alzheimer’s disease follows a stereotypic spread, starting in the amygdala and entorhinal cortex, with progression through stages that correlates with a decrease in cognition and hippocampal volume [32,33,34]. Moreover, TDP-43 pathology in Alzheimer’s disease is associated with loss of cognitive resilience, higher odds of clinical dementia diagnosis [17, 18, 32, 36, 37, 99], and faster brain atrophy rates [33]. While disruption of TDP-43 defines a distinct subgroup of Alzheimer’s disease patients, the impact of TDP-43 pathology and its associated loss of function remains largely unexplored in this condition.
TDP-43 is an RNA-binding protein encoded by the TARDBP gene that regulates fundamental aspects of RNA metabolism including splicing [20, 48, 49, 53, 71, 74, 90]. In normal conditions, TDP-43 is mainly nuclear and represses the inclusion of mostly non-conserved cryptic exons [52]. In disease, TDP-43 is cleared from the nucleus and accumulates in phosphorylated and poly-ubiquitinated cytoplasmic inclusions [6, 68]. Loss of TDP-43 nuclear function leads to the aberrant incorporation of cryptic exons into messenger RNAs, which are detectable in tissues from patients with ALS [16, 42, 52, 57, 62], FTD [16, 57, 76], and Alzheimer’s disease [22, 87]. We and others have identified that the human RNA most affected by TDP-43 disruption encodes a neuronal protein called stathmin-2 (STMN2, also known as SCG10) [42, 62]. Indeed, TDP-43 is required to prevent the inclusion of a cryptic exon within the first intron of STMN2 pre-mRNA. Abnormal inclusion of this cryptic exon and usage of a premature polyadenylation signal produces a non-functional truncated mRNA, leading to a striking loss of stathmin-2 protein [10, 42, 62]. Misprocessing of STMN2 is detected in post-mortem tissues of patients with sporadic and familial forms of ALS and FTD [42, 62, 76]. Remarkably, disruption of STMN2 mRNA is critical for neuronal dysfunction caused by TDP-43 nuclear loss-of-function. Restoring STMN2 protein levels either by lentivirus-mediated expression of STMN2 [62] or by antisense oligonucleotides (ASO)-mediated steric blockage of STMN2 misprocessing [10] rescues axonal regeneration and transport defects in iPSC derived human motor neurons depleted for TDP-43 [10, 42, 62]. In addition, germline deletion of Stmn2 in mice [26, 45, 50] or chronic reduction of Stmn2 in the nervous system of otherwise normal adult mice [56] leads to motor deficits with denervation of the neuromuscular junctions that recapitulate clinical manifestations observed in patients with ALS. Hence, restoration of stathmin-2, including with ASOs that sterically block the abnormal splicing [10], emerges as an attractive therapeutic strategy in TDP-43 proteinopathies. Recently the transcript encoding UNC13A, a synaptic protein essential for neurotransmission [8, 95], was also shown to be mis-spliced in ALS and FTD [16, 57]. Loss of TDP-43 leads to the abnormal inclusion of a cryptic exon between exon 20 and 21 of the UNC13A transcript that results in a premature stop codon and nonsense-mediated decay of UNC13A RNA. Interestingly, non-coding polymorphisms within the UNC13A gene previously associated with an increased risk of ALS [93] were shown to reduce the affinity of TDP-43 for UNC13A RNA and perturb splicing repression of this intronic region [16, 57].
Here, we titrated different levels of TDP-43 downregulation within human neuroblastoma cells to determine the sensitivity of STMN2 and UNC13A pre-mRNA processing to TDP-43 loss. We also show, that both STMN2 and UNC13A RNAs are mis-spliced in the amygdala and entorhinal cortex of a substantial fraction of patients with Alzheimer’s disease. We demonstrate, for the first time, that accumulation of STMN2 and UNC13A cryptic exons correlates with TDP-43 pathology in Alzheimer’s disease, independently of amyloid-β or tau pathological burden. Similarly, our analysis of large RNA-seq datasets from post-mortem tissues also unravels the disruption of STMN2 and UNC13A in Alzheimer’s disease patients. These results highlight the potential of TDP-43-dependent splicing alterations as molecular markers to subgroup Alzheimer’s disease patients. Importantly, ongoing efforts to develop therapeutic strategies preventing TDP-43-associated splicing defects [10] may benefit patients with a range of neurological conditions including ALS, FTD and Alzheimer’s disease.
Materials and methods
RNA extraction and complementary DNA synthesis
Total RNA was extracted from 20 to 40 mg of frozen post-mortem brain tissue, or neuroblastoma SH-SY5Y cells, and lysed using Trizol reagent (ThermoFisher, Cat# 10,296,010). Post-mortem frozen brain samples were obtained from the Massachusetts Alzheimer’s Disease Research Center Neuropathology Core operated under the Massachusetts General Hospital IRB guidelines. A mechanical tissue homogenizer was used for brain tissue. RNA was then purified using Phase Lock Gel Heavy tubes (QuantaBio, Cat# 10847-802) and Isolate RNA Mini Kit (Bioline, Cat#BIO-52073). RNA concentration and quality were measured using nanodrop and 750–1000 ng of total RNA used for cDNA synthesis, using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher, Cat# 4374966) according to the manufacturer’s instructions.
RT-PCR and quantitative PCR
Reverse transcription polymerase chain reactions (RT-PCRs) were performed using Q5 High-Fidelity DNA polymerase (New England BioLabs, Cat# M0492S) according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using iTaq Universal SYBR Green (Bio-rad, Cat#1725121), or Taq™ Universal Probes Supermix (Bio-rad, Cat#1725131). In order to increase specificity of the signal, an optimized combination of primers and probes were used for both STMN2 and UNC13A cryptic exons. Probes and primers used for these reactions are listed in Supplementary Table 1. Reactions were run in triplicates in CFX96 real-time PCR machine (Biorad). For analysis, target RNA levels were normalized to the RNA levels of GAPDH (reference transcript) and to Alzheimer’s disease patients. As STMN2 and UNC13A cryptic exons are normally not expressed, certain samples did not display any amplification of these aberrant transcripts. A sample was considered positive for cryptic exons when there was product amplification in all 3 triplicates evaluated.
SH-SY5Y cell culture and siRNA treatment
SH-SY5Y human neuroblastoma cells (ATCC Cat# CRL-2266) were cultured in DMEM F12 (Gibco, Cat#1132-033) containing 1% of Penicillin–Streptomycin (Gibco, Cat#15140-122) and 10% of Fetal Bovine Serum (Sigma, Cat# F0926) and kept at 37 °C and 5% CO2. TDP-43 knockdown was achieved by transfecting cells with pooled siRNAs against TARDBP (ON-TARGETplus siRNA, Dharmacon, Cat# L-012394) or control siRNA (ON-TARGETplus Non-targeting Control Pool, Dharmacon, Cat# D001810–10) using Lipofectamine RNAiMAX Transfection Reagent (Cat# 13778075) in Opti-MEM™ (Gibco, Cat#31985070). For the dose-dependency analyses we used decreasing amounts of siRNAs ranging from 25 to 0.125 pmol per well. Cells were treated with siRNA either for 3 or 5 days, as previous studies reported decrease in UNC13A transcripts after longer knockdown of TDP-43 [16, 57].
Generation of genetically modified SH-SY5Y cells
Cells containing the N352S mutation in both TDP-43 alleles were previously reported [62]. In short, a plasmid containing a single guide RNA (GCGGGTAATAACCAAAACCA) was electroporated in SH-SY5Y cells using a Amaxa Nucleofector (assay A-023), along with a 180-nucleotide long single-stranded donor oligonucleotides (IDT), containing the desired ALS-causing mutation and four synonymous single-nucleotide replacements to avoid DNA cleavage recurrence by Cas9. Cells were sorted and single-cell seeded into a 96-well plate to obtain individual clones. Replacement of AAT to AGT (c.1055A > G) was confirmed by Sanger sequencing.
To obtain cells overexpressing heterogeneous nuclear ribonucleoprotein L (hnRNP L), SH-SY5Y cells were treated with polybrene (Santa Cruz, Cat# sc-134220) and transduced with lentiviral particles encoding the HNRNP L transcript (Origene Cat# CW307794V) or a GFP control (Origene Cat# CW307797V). Transduced cells were then selected using 1 μg/mL puromycin (ThermoFisher, Cat#A1113803). Overexpression of hnRNP L was verified using qRT-PCR (Forward 5′-GTGGAGATGGCTGATGGCTAC-3′, Reverse 5′ GCTCATCGCAGATCTCAAAGAAG-3′) and Western blot (Antibody Sigma Cat#R4903) with GFP overexpressing cells used as control. SH-SY5Y cells were transfected using lipofectamine with 2.5 pMol of TDP-43 or control siRNAs and harvested 3 days later for qRT-PCR analysis.
Cell toxicity assay
Cell toxicity was assessed using the CellTox Green Toxicity assay (Promega, Cat#G8742) according to manufacturer’s instructions. Cells were seeded in a 96-well plate, and toxicity measured after 5 days of siRNA treatment. Cells were treated with CellTox Green Reagent for 15 min at room temperature and shielded from light. As positive control, the cells were lysed for 30 min using the lysis buffer provided by the Promega. Fluorescence was measured on a Synergy2 plate reader (BioTek Instruments) using 485 nm excitation and 528 nm emission.
Immunostaining of post-mortem tissues and image analysis
Formalin fixed paraffin-embedded brain sections from control or Alzheimer’s disease patients were obtained from the Massachusetts Alzheimer’s Disease Research Center Neuropathology Core operated under the Massachusetts General Hospital IRB guidelines. Brains donated to the Massachusetts Alzheimer’s Disease Research Center, are cut in half, one hemisphere is fresh frozen while the contralateral hemisphere is fixed in 10% neutral buffered formalin. The fixed hemisphere was sectioned and an average of 19–22 distinct regions of interest were mounted in blocks for histological examination. Alzheimer’s disease patients were classified using clinical and neuropathological criteria [30], and aged and sex matched control subjects were chosen that had no significant clinical or neuropathological abnormalities. Demographic characteristics of Alzheimer’s disease patients and control individuals selected are described in Supplementary Table 2. The cohort of patients in this study did not include any patient with pure LATE-NC (i.e. without Alzheimer’s disease neuropathological changes).
For immunohistochemistry and immunofluorescence of TDP-43, sections were first dehydrated in an oven for 1 h at 70 °C, followed by sequential steps of deparaffinization and rehydration using Citrosolv (Decon Laboratories, Cat# 04-355-121), 100% Ethanol (Fisher Scientific, Cat# BP28184), 90% Ethanol, 70% Ethanol and demineralized water. Antigen retrieval was performed using Sodium Citrate Buffer (Fisher Scientific, Cat#S279-500) pH 6.0 for 20 min at 120 °C. Endogenous peroxidases were quenched with a treatment of 3% H2O2 (Millipore Sigma, Cat# HX0635) for 30 min. Sections were blocked for one hour in a solution of 1:10 donkey serum (Jackson Immuno Research Labs, Cat#NC9624464) in TBS-0.1% Triton (Tris Buffered Saline, Boston BioProducts, Cat# BM-301; Triton Sigma, Cat#T8787). Sections were incubated overnight at 4 °C with phosphorylated TDP-43 primary antibody (CosmoBio Mouse Polyclonal, Cat#CAC-TIP-PTD-M01, 1:10,000), or TDP-43 primary antibody (Proteintech, Rabbit Polyclonal, Cat# 10782-2-AP, 1:500) diluted in blocking solution. The next day sections were washed 3× using TBS-0.1% Triton, and then incubated with corresponding secondary antibodies for 2 h at room temperature. For immunohistochemistry the EnVision + Single Reagent HRP Mouse (Agilent, Cat# K400111-2) system was used according to manufacturer’s instructions, and cell nuclei were stained for 30 s using Hematoxylin (Fisher Scientific, Cat#3530-32). For immunofluorescence, slides were incubated with Alexa Fluor anti-rabbit 488 and Alexa Fluor anti-mouse 568 secondary antibodies, and cell nuclei were stained using DAPI. Finally, slides were dehydrated by a sequential treatment with demineralized water, 70% Ethanol, 90% Ethanol, 100% Ethanol and Citrosolv, and then coverslipped with Epredia mounting media (Epredia, Cat#4112). At the end, immunofluorescent slides were treated with 0.1% Sudan Black, to quench the human tissue auto-fluorescence. Total tau (1:6000 dilution, Agilent/Dako, #A002401-2, 1), and amyloid-β 6F/3D (1:600 dilution, Agilent/Dako, #M087201-2) were stained using a Leica BOND-RX automated stainer.
For immunohistochemistry, whole-slide images were taken using brightfield 40× objective (NanoZoomer, Hamamatsu), and a confocal Nikon C2 Confocal was used to image slides stained by immunofluorescence at a 60× magnification. To quantify the percentage of area occupied by phosphorylated TDP-43, Tau or amyloid-β, five images of the same area were selected within the amygdala for each slide and extracted for quantification using FIJI [82]. The Color Threshold and Analyze particles plugins from FIJI were used to quantify the area stained for each protein. Investigators were blinded to the disease status during the experimental procedure and analysis.
TDP-43 staging was determined according to the simplified staging method proposed by Nelson et al. [67]. In short, patients were classified according to the presence of TDP-43 proteinopathy in the amygdala (stage I), hippocampus (stage II) and frontal cortex (stage III). The information obtained for each individual is included in Supplementary Table 3.
Bioinformatics analysis of RNA-seq data
The Mayo Clinic RNA-seq cohort has been made available to the research community and described in detail previously [2, 3, 98]. In brief, Alzheimer’s disease and control subjects were diagnosed neuropathologically at autopsy [61]. Alzheimer’s disease subjects from the Mayo Clinic Brain Bank had definite neuropathologic diagnosis according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA) criteria and had Braak neurofibrillary tangle (NFT) stage ≥ 4.0. Control subjects, either from the Mayo Clinic Brain Bank or the Banner Sun Health Institute, had Braak NFT stage of 3.0 or less, Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic and cortical plaque densities of 0 (none) or 1 (sparse) and lacked neuropathologic diagnoses for neurodegenerative diseases. Temporal cortex samples underwent RNA extractions via the Trizol/chloroform/ethanol method, followed by DNase and clean-up of RNA using Qiagen RNeasy Mini Kit and Qiagen RNase-Free DNase Set. The quantity and quality of RNA samples were determined by the Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Chip. Samples included in this study all have RIN ≥ 5.0. After quality control procedures, final samples included in this study consisted of 80 Alzheimer’s disease patients and 69 controls. In a subset of 38 Alzheimer’s disease samples (18 TDP-43 positive and 20 negative) the presence of TDP-43 inclusion bodies was determined by immunohistochemistry with antibodies directed against pathological TDP-43 [5].
RNA sequencing Library preparation and sequencing of the samples were conducted at the Mayo Clinic Genome Analysis Core using TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA). The library was sequenced on Illumina HiSeq2000 instruments, generating 101 base-pair, paired-end raw reads. Raw reads were processed through MAPR-Seq pipeline v1.0 [39]. Reads of low base-calling Phred scores were removed, the remaining reads were aligned to the human reference genome build hg19 using Tophat v2.0.12 [91], and were counted for genes using Subread 1.4.4 [51]. Quality control measures were obtained from both pre-alignment and post-alignment reads using RSeQC toolkit and fastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Samples that had high RNA degradation, low reads mappability, or inconsistency between recorded sex and estimated sex using RNA-seq chromosome Y expression were removed from downstream analysis. Additional information is also available in our previous publications [2, 3, 98].
The Mount Sinai Brain Bank (MSBB) RNA-seq data was downloaded from the Accelerating Medicines Partnership–Alzheimer’s Disease (AMP–AD) Knowledge Portal (syn3157743) as previously described [97]. For the MSBB RNA-seq data, pre-mortem clinical diagnosis was used to categorize Alzheimer’s disease patients and controls. We used trimmed mean of M-values (TMM) provided by the MSBB study which were already adjusted for batch, sex, race, age at death, PMI, RNA integrity number, exonic rate and ribosomal RNA rate.
For both RNA-seq datasets, reads of full-length STMN2 (chr8: 80,523,049–80,578,410 in hg19), UNC13A (chr19: 17,712,145–17,799,163 in hg19), the cryptic exon of STMN2 (chr8: 80,529,057–80,529,284 in hg19) and the cryptic exon of UNC13A (chr19: 17,753,223–17,753,350 in hg19) were counted for each sample using the bedtools program (v2.27.1) [77] with at least 75% overlap in the region of interest. These counts were then normalized by the total number of reads of all genes (counts per million reads, CPM), followed by Log2 transformation to obtain the expression levels in Log2(CPM). Plots were created using the ggplot2 package in R and p-values were obtained using the Wilcoxon rank-sum test. Chi-Square tests were performed using the chisq.test function and the plots were created using the ggplots package (v3.36) in R. In the plot of stratified Braak for the Mayo Clinic cohort, non-integer scores (*0.5) were rounded to the previous integer, leading to 69, 6, 34 and 40 subjects with scores 0–III, IV, V and VI, respectively. The Braak stages 0 to III were considered controls and pooled together for the analysis. Information about the presence or absence of TDP-43 was available only for 38 cases, most of them with Braak stages of V (4 TDP-43 positive and 9 negative) or VI (13 TDP-43 positive and 10 negative).
Data visualization of iCLIP data
Individual-nucleotide resolution cross linking and immunoprecipitation (iCLIP) for TDP-43 in SH-SY5Y cells (ERR039854) previously generated by Tollervey et al. [90] was mapped to the human genome GRCh38/hg38 using star/2.7.9a. Sorted bam files were visualized and plotted using sashimi plot function in IGV (version 2.12.3) for the respective genes of interest (UNC13A and STMN2).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0 software. Normality was tested using D’Agostino & Pearson test. When all groups were normally distributed, differences between groups were tested using an unpaired t-tests (for comparison of two groups) or one-way ANOVA, followed by Holm-Šídák’s Multiple comparisons post-hoc test (for comparison of more than two groups). When at least one group was not normally distributed, differences between groups were tested using Mann–Whitney test (for comparison of two groups) or Kruskal–Wallis, followed by a Dunn’s Multiple comparisons post-hoc test (for comparison of more than two groups). Correlations were tested using the Spearman’s rank correlation coefficient. A p-value of ≤ 0.05 was considered statistically significant. The following code was used to indicate the level of significance: * ≤ 0.05; ** ≤ 0.01; *** ≤ 0.001.
Data availability
The data from this study are available from the corresponding authors, upon request. Mayo Clinic brain RNA-seq data are available from the AD Knowledge Portal (syn5550404). MSBB RNA-seq data are available from the AD Knowledge Portal (syn3157743).
Results
Different sensitivity of STMN2 and UNC13A RNAs to TDP-43 loss
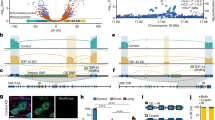
Both STMN2 and UNC13A RNAs are mis-spliced upon reduction of nuclear TDP-43 with incorporation of cryptic exons to their mRNAs, resulting in decreased levels of full-length transcripts (Fig. 1a,b) [16, 42, 57, 62]. Titration of TDP-43 suppression via a small-interfering RNA (siRNA) dose response achieved a nearly continuous range of TDP-43 expression from 100 to 18% of the level in cells treated with a control siRNA (Fig. 1c,d). This approach enabled direct comparison of relative rates of cryptic exon incorporation into UNC13A and STMN2 transcripts and revealed that STMN2 pre-mRNA is more sensitive to TDP-43 disruption than UNC13A (Fig. 1e–g). Indeed, misprocessing of STMN2 RNA and significant reduction of full-length STMN2 were detected following only 33% reduction of TDP-43 level (Fig. 1d–f; 0.125 pmol siRNA leading to 67% of normal TDP-43 levels). In contrast, UNC13A cryptic exon was only detected following 60% reduction of TDP-43 and was associated with significant loss of full-length UNC13A only after 80% of TDP-43 loss (Fig. 1d,f,g; 0.75 and 25 pmol of siRNA leading to 40% and 18% of normal TDP-43 levels, respectively). Five days after treating cells with the highest dose of siRNA (25 pmol), 70% reduction of TDP-43 led to 60% reduction in full length STMN2 but only 30% reduction in UNC13A (Sup. Fig. S1). Notably, treatment with 25 pmol of siTDP-43 was not associated with cellular toxicity (Sup. Fig S2a). Consistently, we detected misprocessing of STMN2 (Sup. Fig. S2b) but not UNC13A (Sup. Fig. S2c) in neuroblastoma SH-SY5Y cells homozygous for a N352S mutation in TARDBP which results in a decrease of TDP-43 mRNA level to around 60% of wild-type cells (Sup. Fig. S2d) [62]. Analysis of previously published RNA sequencing datasets [44, 55, 62] corroborated these results (Sup. Fig. S3). Indeed, SH-SY5Y cells with TDP-43 downregulation to 25% of control cells [62], yielded a 85% reduction of full-length STMN2; whereas, full-length UNC13A was only reduced by 20% (Sup. Fig. S3a). Notably, this relative resistance of UNC13A to TDP-43 loss occurs in SH-SY5Y cells while sequencing of this line revealed homozygosity for the ALS-associated risk allele (G) at the SNP rs12973192 that leads to an increased likelihood of UNC13A cryptic exon incorporation [16, 57]. In addition, laser-microdissected motor neurons of sporadic ALS patients [44] have significantly reduced full-length STMN2 transcripts [62] while UNC13A RNA levels were not significantly impacted (Sup. Fig. S3b). Finally, while both STMN2 and UNC13A cryptic exons were detected in FAC sorted cortical neurons lacking nuclear TDP-43 [16, 55, 57], only full-length STMN2 transcripts were significantly reduced (Sup. Fig. S3c, left panel) while UNC13A RNA levels were not significantly impacted (Sup. Fig. S3c, right panel). Collectively, these results indicate that while STMN2 and UNC13A pre-mRNAs are both misprocessed upon TDP-43 loss, STMN2 is more sensitive to mild TDP-43 disruption than UNC13A.

Impact of reduced TDP-43 levels on the processing of STMN2 and UNC13A mRNAs. a, b Schemes representing STMN2 (a) and UNC13A (b) constitutive exons (black) and cryptic exons (CE, red and purple, respectively) that are included upon TDP-43 loss of function. Forward (F) and reverse (R) primers’ location used to quantify the different transcripts by qRT-PCR in e–g, j, k are depicted. c Scheme of experimental design to test the sensitivity of UNC13A and STMN2 cryptic exons to TDP-43 knockdown in neuroblastoma (SH-SY5Y) cells treated with increasing amounts of siRNA against TDP-43 (siTDP43) for 3 days. d–g qRT-PCR quantification of the RNA levels of TDP-43 (d, blue), STMN2 transcripts with cryptic exon (e, red), STMN2 full-length (e, light gray), UNC13A transcripts with cryptic exon (g, purple) and UNC13A full-length (g, dark gray). As control, cells were treated with a scramble siRNA. f Levels of TDP-43 (blue), STMN2 cryptic exon (red) and UNC13A cryptic exon (purple) in response to TDP-43 knockdown. Data were fitted using a non-linear curve-fit in Prism, and the x axis is plotted with a log10 scale. h–k SH-SY5Y cells genetically modified to overexpress hnRNP L or a GFP control and treated with siRNA against TDP-43 for 3 days (2.5 pmol). qRT-PCR was used to determine the expression levels of TDP-43 (blue, h), hnRNP L (i, gray), UNC13A transcripts with (j, purple) and without cryptic exon (j, dark gray) and STMN2 transcripts with (k, red) and without cryptic exon (k, light gray). d–k RNA levels were normalized to GAPDH and to the control group. Levels of full-length RNAs were normalized to cells treated with the control siRNA and transcripts with cryptic exons were normalized to cells treated with highest dose of TDP-43 siRNA. Results from 4 to 5 independent experiments were plotted with each dot representing a technical replicate and bars representing mean ± SEM. Normal distribution of data was tested using D’Agostino & Pearson test and one-way ANOVA, followed by Holm-Šídák’s Multiple comparisons post-hoc test (parametric) or Kruskal–Wallis, followed by a Dunn’s Multiple comparisons post-hoc test (non-parametric) were performed accordingly
Several factors may underly the different sensitivities of STMN2 and UNC13A to TDP-43 loss-of-function including the binding patterns of TDP-43 on the pre-mRNAs and the level of expression of both transcripts. Analysis of a previously published individual-nucleotide resolution cross linking and immunoprecipitation (iCLIP) dataset in SH-SY5Y cells [90] identified a specific binding of TDP-43 on STMN2 cryptic exon while no read mapped in the region surrounding the UNC13A cryptic exon (Sup. Fig. S4a,b). This result may be due to the low level of UNC13A expression compared to STMN2 in SH-SY5Y cells (200 STMN2 transcripts per million compared to 20 UNC13A transcripts per million in control SH-SY5Y cells, Sup. Fig. S3a). Notably, iCLIP from SH-SY5Y cells overexpressing UNC13A minigenes demonstrated TDP-43 binding on a large region of intron 20 spanning approximately 800 bp [16] (Fig. S5a). Consistently, the region bound by TDP-43 is highly UG-rich with not only UG repeats but also motifs interrupted by adenines that were previously identified to be bound by TDP-43 [16, 74, 90]. This binding pattern strikingly contrasts with the discreet binding site of TDP-43 on STMN2 that spans only 24 nucleotides [10, 62] (Fig. S5b), likely contributing to the high sensitivity of STMN2 transcripts to TDP-43 loss.
It is also possible that other RNA binding proteins influence TDP-43-dependent cryptic splicing events. Indeed, overexpression of the heterogeneous nuclear ribonucleoprotein L (hnRNP L) was recently shown to mitigate the incorporation of cryptic exon in UNC13A upon TDP-43 loss [43]. We used genetically modified SH-SY5Y cells over-expressing either hnRNP L or a GFP control to determine whether hnRNP L also represses the incorporation of STMN2 cryptic exon upon siRNA-mediated reduction of TDP-43 (Fig. 1h). hnRNP L expression was not altered by TDP-43 loss (Fig. 1i), but as previously reported [43], significantly repressed the aberrant incorporation of UNC13A cryptic exon and increased the level of full length UNC13A (Fig. 1j). In contrast, both full length and cryptic exon-containing STMN2 transcripts were increased by overexpression of hnRNP L (Fig. 1k).
STMN2 and UNC13A RNAs are mis-processed in Alzheimer’s disease patients with TDP-43 proteinopathy
Aberrant accumulations of TDP-43 are found in approximately half of patients with Alzheimer’s disease, with the amygdala and entorhinal cortex, two anatomically adjacent regions, being the most frequently affected areas [5, 34]. To test whether TDP-43 pathology in Alzheimer’s disease patients leads to RNA mis-processing of STMN2 and UNC13A, we performed qRT-PCR using RNA from the amygdala and entorhinal cortex of 38 Alzheimer’s disease patients and 14 age-matched control individuals without significant confounding neuropathology (Sup. Table 2). Abnormal STMN2 transcripts were detected in the amygdala of 20 out of 38 patients and in the entorhinal cortex of 13 out of 27 patients with Alzheimer’s disease, and none of the control individuals (Sup. Fig. S6a–c and Sup. Table 3). We also tested misprocessing of UNC13A RNA in these areas and detected UNC13A cryptic exon in 19/38 amygdala samples and 13/27 entorhinal cortex samples from Alzheimer’s disease patients and at very low levels (qRT-PCR cycle thresholds Cts above 35 cycles) in the amygdala of 3 control individuals (Sup. Fig. S6d–f and Sup. Table 3). We then performed immunostaining to test the presence of hyperphosphorylated TDP-43 aggregates in the amygdala of these patients, and determined that the detection of STMN2 and UNC13A cryptic exons correlated with the presence of TDP-43 pathology (Fig. 2a–e and Sup. Table 3). Indeed, STMN2 cryptic exon was detected in the amygdala of 18 out of 21 Alzheimer’s disease patients positive for phosphorylated TDP-43 and was detected at low levels (Cts above 37; Sup. Fig. S6f) in only 2 out of 17 patients without detectable phosphorylated TDP-43 aggregates (Fig. 2a,b and Sup. Table 3). UNC13A cryptic exon was identified in the amygdala of 15 out of 21 Alzheimer’s disease patients with phosphorylated TDP-43 aggregates and only 4 patients without detected aggregates (Fig. 2a,d and Sup. Table 3). Similar results were obtained in the entorhinal cortex (Fig. 2c,e and Sup. Table 3). Notably, we observed a correlation between the levels of STMN2 and UNC13A cryptic exons detected in the amygdala of Alzheimer’s disease patients (Fig. 2f, Spearman r = 0.78, p < 0.0001), strengthening the link between detection of cryptic exons and loss of TDP-43 function. Finally, our observations were confirmed by RT-PCR showing the amplification of cryptic exons of both STMN2 and UNC13A transcripts in the amygdala of Alzheimer’s disease patients with TDP-43 pathology, but not in the controls or Alzheimer’s disease patients without TDP-43 pathology (Fig. 2g).

STMN2 and UNC13A RNAs are mis-processed in post-mortem brain of approximately half of patients with Alzheimer’s disease. a Table depicting the detection of STMN2 (red) or UNC13A (purple) cryptic exons in the amygdala of controls and Alzheimer’s disease patients with (+) and without (−) phosphorylated TDP-43 pathology. b–e Levels of truncated STMN2 RNA (red) or UNC13A cryptic exon (purple) measured by qRT-PCR in post-mortem amygdala (b, d) or entorhinal cortex (c, e) from patients with Alzheimer’s disease (AD) associated ( +) or not (−) with accumulation of phosphorylated TDP-43. Normal distribution of data was tested using D’Agostino & Pearson test and a one-way ANOVA followed by a Sidak’s multiple comparisons post-hoc test (parametric), or a Kruskal–Wallis, followed by a Dunn’s Multiple comparisons post-hoc test (non-parametric) were performed accordingly. f Log–log plot of STMN2 and UNC13A cryptic exon levels detected in the amygdala of Alzheimer’s disease patients. Correlation was determined using spearman rank coefficient and curve fitting with non-linear regression. g RT-PCR confirming the detection of STMN2 and UNC13A cryptic exons in a subset of the patients with Alzheimer’s disease associated with phosphorylated TDP-43 (pTDP-43) pathology
TDP-43 pathology follows a stereotypic spread from amygdala to other brain areas as Alzheimer’s disease progress [34, 67]. We classified postmortem tissues according to the presence of TDP-43 proteinopathy in the amygdala (stage I), hippocampus (stage II) and frontal cortex (stage III) following recent recommendations [67]. Staging was possible in 14 TDP-43 positive patients for which tissues were available from all three brain regions (Sup. Table 3). Only 4 out of 14 patients displayed phosphorylated TDP-43 aggregates in the frontal cortex (stage 3). In these cases, only a scarce number of inclusions were observed in the frontal cortex compared to amygdala and entorhinal cortex (Sup. Fig. S7). Consistently, STMN2 cryptic exon was only rarely detected in frontal cortex (1/19), occipital cortex (1/19) and cerebellum (4/19) in Alzheimer’s disease patients (Sup. Fig. S6a and Sup. Table 3). Notably, all the patients with cryptic exons detected in frontal cortex, occipital cortex and cerebellum also had cryptic exons in the amygdala (Sup. Table 3).
Detection of STMN2 and UNC13A cryptic exons in the amygdala correlates with the burden of phosphorylated TDP-43 but not tau or amyloid-β
Since Alzheimer’s disease neuropathology is characterized by the accumulation of the two canonical proteins, amyloid-β and tau [14], we used semi-automated quantification to determine the area occupied by pathological inclusions of phosphorylated TDP-43, tau and amyloid-β in post-mortem amygdala of Alzheimer’s disease patients with (CE +) or without (CE−) STMN2 or UNC13A cryptic exons (Fig. 3). TDP-43 pathology is characterized by both cytoplasmic inclusions and loss of nuclear TDP-43 that appears to be an early pathogenic mechanism in TDP-43 proteinopathies [64, 68, 96]. Immunofluorescence staining confirmed that cells containing phosphorylated TDP-43 cytoplasmic inclusions did have nuclear clearance of TDP-43 consistent with TDP43 loss of function in regulating splicing, while neighbouring cells without inclusions had normal nuclear TDP-43 (Fig. 3a). Quantification of TDP-43 nuclear loss is challenging compared to quantification of phosphorylated TDP-43 aggregates [5, 7, 13, 21, 29, 34, 35, 54, 92], hence we have correlated the presence of cryptic exons with the burden of phopho-TDP-43 pathology (Fig. 3b,c). As expected, there were higher levels of phosphorylated TDP-43, tau and amyloid-β accumulations in Alzheimer’s disease patients compared to control individuals (Fig. 3b–e). However, while the pathological burden in tau (Fig. 3d) or amyloid-β (Fig. 3e) were not significantly different in tissues with or without cryptic exons, we observed higher pathological burden of TDP-43 in patients with accumulation of STMN2 or UNC13A cryptic exons (Fig. 3c). Notably, the burden of TDP-43 pathology (percentage of area positive for TDP-43) positively correlated with the levels of both STMN2 (Spearman r = 0.52; p-value = 0.006) and UNC13A (Spearman r = 0.56; p-value = 0.002) cryptic exons in the amygdala. Collectively these results demonstrate that mis-processing of STMN2 and UNC13A RNAs is a pathological hallmark of TDP-43-associated Alzheimer’s disease, with amygdala and entorhinal cortex being the most affected areas.

Detection of cryptic exons in the amygdala from Alzheimer’s disease patients correlates with the burden of phosphorylated TDP-43 but not Tau or Amyloid-β. a Representative immunofluorescence images showing full-length TDP-43 (red), phosphorylated TDP-43 (green) and DAPI (blue) of two controls and six Alzheimer’s disease patients. Top panels show merged images and lower insets show grayscale images of TDP-43 or phosphorylated TDP-43. Scale bar is 10 µm. b Example micrographs of post-mortem brain tissue from one control and two Alzheimer’s disease patients after immunohistochemical detection of phosphorylated TDP-43 (pTDP43), total Tau and Amyloid-β. Scale bar is 100 µm in larger panels, and 10 µm in insets. c–e Percentage of area occupied by phosphorylated TDP-43 (c), tau (d) or amyloid-β (e) staining in the amygdala of controls or Alzheimer’s disease patients with or without detection of cryptic exons. For each case five regions of interest of equal size were selected and the positive signal for the different markers determined and averaged. Data represent mean ± SEM, each dot represents an individual patient. Normal distribution of data was tested using D’Agostino & Pearson test and one-way ANOVA, followed by Holm-Šídák’s Multiple comparisons post-hoc test (parametric) or Kruskal–Wallis, followed by a Dunn’s Multiple comparisons post-hoc test (non-parametric) were performed accordingly
RNA-seq identifies STMN2 and UNC13A disruption in a subset of Alzheimer’s disease patients
We analyzed STMN2 and UNC13A transcripts in bulk RNA-seq from post-mortem brain regions of control and Alzheimer’s disease patients from data available through the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB-AD) [97] and an independent dataset generated at Mayo Clinic [2, 3, 98]. In the Mount Sinai dataset, RNA-seq of the parahippocampal gyrus, inferior frontal gyrus, frontal pole, and superior temporal gyrus from more than 40 control individuals and 95 Alzheimer’s disease patients were analyzed (Fig. 4a–c, Sup. Figs. S8, S9). Of note, no information was available concerning the presence or absence of TDP-43 pathology in this cohort. However, we observed a significant increase of truncated STMN2 in the inferior frontal gyrus (Fig. 4a) of Alzheimer’s disease patients; whereas, full-length STMN2 RNA levels were significantly decreased in Alzheimer’s disease patients compared to controls, in all the regions tested (Fig. 4b and Sup. Fig. S8b). While the number of reads mapping to UNC13A cryptic exon was extremely low in all samples, a significant decrease in UNC13A full-length was detected in the inferior frontal gyrus, parahippocampal gyrus and superior temporal gyrus from Alzheimer’s disease patients compared to control individuals (Fig. 4c and Sup. Fig. S8c). The Mayo Clinic dataset [2, 3, 98] consisted of bulk RNA-seq of the temporal cortex (superior temporal gyrus) from 69 control individuals and 80 Alzheimer’s disease patients. Here, TDP-43 pathology was assayed in 38 of the Alzheimer’s disease patients with 18 showing accumulation of phosphorylated TDP-43, consistent with previous reports of approximately 50% of Alzheimer’s disease patients having TDP-43 pathology [5, 7, 13, 21, 29, 34, 35, 54, 92]. Analysis of the Mayo Clinic dataset showed a significant enrichment of STMN2 cryptic exon in the temporal cortex of Alzheimer’s disease patients compared to controls (Fig. 4d) and a significant decrease in full-length STMN2 only in Alzheimer’s disease patients with TDP-43 pathology compared to controls (Fig. 4e). UNC13A full-length was significantly decreased in patients with TDP-43 pathology (Fig. 4f). We further explored this dataset by subdividing Alzheimer’s disease cases (including cases with and without TDP-43 pathology) according to their Braak stages, a method to classify the evolution of the disease by evaluating the spread of tau pathology in Alzheimer’s disease brain [14, 15]. This analysis revealed a significant increase of STMN2 cryptic exon in the more advanced Braak stages (Braak stages V and VI), a trend for decrease in full-length STMN2 in the Mayo Clinic dataset, and a significant decrease in the full-length transcripts of STMN2 and UNC13A in the Mount Sinai dataset (Fig. 4g and Sup. Fig. S9). In the cases from Mayo Clinic with Braak stages V and VI for which the TDP-43 status was available, we observed a significant increase of STMN2 cryptic exon in the temporal cortex of patients with Braak stage VI and TDP-43 pathology (Sup. Fig. 10a). Full-length STMN2 and UNC13A transcripts were also significantly reduced in both Braak stages V and VI when comparing brains with and without phosphorylated TDP-43 (Sup. Fig. 10).

Analysis of STMN2 and UNC13A mRNAs in RNA-seq datasets from Alzheimer’s disease post-mortem tissues. Expression of STMN2 and UNC13A was analyzed using published RNA-seq datasets from the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB-AD) [97] (a–c) and from the Mayo Clinic Brain Bank [2, 3, 98] (d–g). Levels of STMN2 cryptic exon (red, a, d), STMN2 full-length transcript (red, b, e) and UNC13A full-length transcript expression (purple, c, f) were determined in the parahippocampal and inferior frontal gyri (a–c) or temporal cortex (d–g) of Alzheimer’s disease patients compared to controls. g Levels of STMN2 cryptic exon (left, red), STMN2 full-length (middle, red) and UNC13A full-length (right, purple) in post-mortem brain tissue from controls (Braak stage 0–III) and Alzheimer’s disease patients segregated according to their Braak stage (Braak stages IV–VI)
Collectively this analysis establishes, in a large sample population, that STMN2 and UNC13A transcripts are altered in Alzheimer’s disease patients with advanced Braak stages when the burden of TDP-43 pathology is high.
Discussion
Alzheimer’s disease is a heterogenous disease, and precision medicine approaches to fine-tune diagnosis and treatment offer real promise for patients. TDP-43 pathology co-occurs with Alzheimer’s disease in a subset of patients, which is also termed Alzheimer’s disease with LATE-NC, likely contributing to neural system dysfunction in those patients. Indeed, TDP-43 pathology in Alzheimer’s disease is associated with more severe cognitive decline and neurodegeneration [17, 18, 32,33,34, 36, 37, 99], underscoring the need to stratify Alzheimer’s disease patients according to their TDP-43 pathology status. In addition, the higher likelihood of TDP-43 lesions in patients with typical Alzheimer’s disease pathology compared to the general population raises the question of whether there are synergistic or underlying genetic or environmental factors that contribute to both Alzheimer’s disease and TDP-43 pathology, but this is currently unknown. Notably, TDP-43 pathology has also been described in limbic regions of older patients with and without dementia (LATE-NC) who lack tau and amyloid-β pathologies [66, 67, 81]. Our results support the use of TDP-43-dependent splicing alterations to sub-categorize patients with respect to TDP-43 dysfunction. RNA-seq has been used before to demonstrate disease-specific splicing alterations in Alzheimer’s disease [9, 27, 78, 94]. Recent reports have detected cryptic exons by using qRT-PCR or RT-PCR in post-mortem tissues from patients with Alzheimer’s disease [22, 87]. Our study extends on those findings by demonstrating that STMN2 and UNC13A disruption from TDP-43 loss-of-function is detected in large RNA-seq datasets and can distinguish between Alzheimer’s disease patients with and without TDP-43 pathology. Notably, previous studies observed a non-significant tendency for decreased levels of full-length STMN2 RNA in the temporal cortex and hippocampus of a subset of patients with Alzheimer’s disease [73], as well as a decrease in STMN2 RNA in inhibitory neurons and oligodendrocytes of Alzheimer’s disease patients [60]. Although these studies did not correlate the transcriptional changes to TDP-43 pathology, they independently confirm STMN2 as a relevant mechanistic disease target. Importantly, we now demonstrate that cryptic exon detection correlates with TDP-43 burden, but not with amyloid-β and tau in the amygdala of Alzheimer’s disease patients. TDP-43 pathology follows a stereotypic spread as Alzheimer’s disease progress [34], and we observe that STMN2 and UNC13A are indeed significantly disrupted in temporal cortex of patients with high Braak stages. The STMN2 cryptic exon was detected in most of the cases with phosphorylated TD-43 aggregates except for 3 out of 21 tissues tested. Although we cannot exclude that additional protective factors (genetic or environmental) may prevent mis-processing of RNA targets in a subset of patients, it is noteworthy that, in a given brain region, TDP-43 pathology is spatially heterogeneous and expression of cryptic exons may be below the detection limit in the assessed tissue sample. Inversely, STMN2 cryptic exon was detected in 2 out of 17 brains without apparent phosphorylated TDP-43 aggregates which is consistent with reports of TDP-43 nuclear loss without cytoplasmic aggregates in a subset of patients with FTD or Alzheimer’s disease [64, 87].
Our systematic analysis indicates that STMN2 is more sensitive than UNC13A to decrease in TDP-43 levels; whereas, a more robust TDP-43 suppression is necessary to observe the dose response of UNC13A cryptic exon generation. This is consistent with previous observation that depletion of at least 50% of TDP-43 encoding RNA was required to induce UNC13A RNA mis-splicing [16]; whereas, STMN2 mis-processing is triggered by less than 30% reduction of TDP-43 [62]. In addition, cryptic exons are more commonly identified in STMN2 than UNC13A within post-mortem tissues from patients with either ALS and/or FTD [16, 25]. Our RNA-seq analysis also shows that the STMN2 cryptic exon is enriched in tissues of Alzheimer’s disease; whereas, no significant enrichment of UNC13A cryptic exon was detected. STMN2 expression levels are tenfold higher than UNC13A both in SH-SY5Y cells and human tissues, with STMN2 being among the 25 most expressed genes in laser-microdissected human motor neurons [44, 62], which may contribute to the differential detection of STMN2 and UNC13A cryptic exons. In addition, UNC13A cryptic exon-containing transcripts might escape detection due to a shorter half-life since they are subjected to nonsense-mediated decay while STMN2 truncated transcripts are not [16]. However, we propose that the higher detection of STMN2 cryptic exons is also due to an intrinsic different vulnerability of STMN2 and UNC13A transcripts to TDP-43 loss of function. Indeed, the binding pattern of TDP-43 on STMN2 and UNC13A pre-mRNAs is distinct, with a narrow binding site consisting of 3 ugugug motifs over 22 bp in the cryptic exon (exon 2a) of STMN2 [10, 62]; whereas, TDP-43 binds an 800 bp-long ug-rich region in intron 20 of UNC13A [16]. In addition, hnRNP L was recently found to mitigate the impact of TDP-43 loss on UNC13A splicing [43]. In contrast, we show that hnRNP L does not repress the inclusion of STMN2 cryptic exon.
Identification of STMN2 and UNC13A disruption in Alzheimer’s disease has major implications: (1) they are both enriched in the brain, (2) they have crucial roles in maintaining neuronal function [8, 26, 42, 45, 56, 62, 63, 80, 84, 95] and (3) they are two prominent targets of TDP-43 for which therapeutic approaches are in development [10, 16, 42, 57, 62]. STMN2 is a tubulin-binding protein [19] important for axonal maintenance and regeneration after injury [42, 62, 63, 80, 84]. Loss of STMN2 in vivo leads to motor and sensory behavioural deficits, as well as denervation of neuromuscular junctions [26, 45, 50, 56]. Finally, even though many RNAs are affected by TDP-43 loss-of-function [42, 49, 52, 62, 72, 74, 90], restoring STMN2 levels alone is sufficient to rescue axonal transport defects and the ability of iPSC-derived motor neurons to regrow after axotomy [10, 42, 62]. UNC13A has been extensively linked to synaptic function, and it is essential for neurotransmission [8, 95]. As such, both STMN2 and UNC13A are important components of neuronal function, and given the lack of cognitive resilience and increased brain atrophies associated with TDP-43 pathology in Alzheimer’s disease [17, 18, 32, 34, 36, 37, 99], future studies should focus on exploring how these targets may contribute to neurodegeneration in Alzheimer’s disease. Importantly, strategies to either target TDP-43 cellular mislocalization [1, 4, 11, 23, 47, 58, 70, 75, 88, 89] or to prevent cryptic splicing of specific TDP-43 targets [10] are emerging for clinical development in neurodegenerative diseases with TDP-43 proteinopathy. Indeed, the usage of ASOs chemically modified to alter pre-mRNA splicing without recruiting RNase H is an approved standard of care for spinal muscular atrophy (SMA) and has become an attractive therapeutic approach for neurodegenerative diseases [12, 24]. We recently demonstrated the ability of steric binding ASOs to prevent mis-splicing and restore stathmin-2 protein level in TDP-43 deficient neurons and mouse brain [10], providing support for restoration of STMN2 as a potential therapeutic approach in TDP-43 proteinopathies. Collectively, this further supports the use of cryptic exons to stratify Alzheimer’s disease patients to enable the development of targeted therapies. It is noteworthy that approaches are also emerging to use TDP-43-dependent splicing alterations as potential biomarkers in patients with TDP-43 proteinopathy. Indeed, while PET imaging for TDP-43 pathology is not available and detection of pathological TDP-43 in biofluids has yielded conflicting results [28, 40, 41, 46, 69, 79, 86], recent studies show the feasibility of detecting aberrant peptides generated from TDP-43-dependent cryptic splicing in CSF of patients with ALS/FTD [31, 83]. Even though there are not yet reliable biomarkers based on the detection of STMN2 or UNC13A cryptic exons, development of these approaches may have ground-breaking implications for subcategorization of patients and the development of personalized therapeutics in TDP-43 proteinopathies, including Alzheimer’s disease.
Abbreviations
- siRNA:
-
Small-interfering RNA
- STMN2:
-
Stathmin-2
- TDP-43:
-
TAR DNA-binding protein-43
- ALS:
-
Amyotrophic lateral sclerosis
- FTD:
-
Frontotemporal dementia
- LATE:
-
Limbic-predominant age-related TDP-43 encephalopathy
- LATE-NC:
-
LATE neuropathologic changes
- ASO:
-
Antisense oligonucleotide
- hnRNP L:
-
Heterogeneous nuclear ribonucleoprotein L
- iCLIP:
-
Individual-nucleotide resolution cross linking and immunoprecipitation
References
Afroz T, Chevalier E, Audrain M, Dumayne C, Ziehm T, Moser R et al (2023) Immunotherapy targeting the C-terminal domain of TDP-43 decreases neuropathology and confers neuroprotection in mouse models of ALS/FTD. Neurobiol Dis 179:106050. https://doi.org/10.1016/j.nbd.2023.106050
Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS et al (2016) Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data 3:160089. https://doi.org/10.1038/sdata.2016.89
Allen M, Wang X, Burgess JD, Watzlawik J, Serie DJ, Younkin CS et al (2018) Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement 14:352–366. https://doi.org/10.1016/j.jalz.2017.09.012
Alquezar C, Salado IG, de la Encarnacion A, Perez DI, Moreno F, Gil C et al (2016) Targeting TDP-43 phosphorylation by casein kinase-1delta inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol Neurodegener 11:36. https://doi.org/10.1186/s13024-016-0102-7
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. https://doi.org/10.1002/ana.21154
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K et al (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. https://doi.org/10.1007/s00401-008-0480-1
Augustin I, Rosenmund C, Sudhof TC, Brose N (1999) Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400:457–461. https://doi.org/10.1038/22768
Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ et al (2013) U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A 110:16562–16567. https://doi.org/10.1073/pnas.1310249110
Baughn MW, Melamed Z, Lopez-Erauskin J, Beccari MS, Ling K, Zuberi A et al (2023) Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science 379:1140–1149. https://doi.org/10.1126/science.abq5622
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P et al (2017) Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544:367–371. https://doi.org/10.1038/nature22038
Bennett CF, Kordasiewicz HB, Cleveland DW (2021) Antisense drugs make sense for neurological diseases. Annu Rev Pharmacol Toxicol 61:831–852. https://doi.org/10.1146/annurev-pharmtox-010919-023738
Bigio EH, Mishra M, Hatanpaa KJ, White CL 3rd, Johnson N, Rademaker A et al (2010) TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 120:43–54. https://doi.org/10.1007/s00401-010-0681-2
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. https://doi.org/10.1007/BF00308809
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16:271–278. https://doi.org/10.1016/0197-4580(95)00021-6
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC et al (2022) TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603:131–137. https://doi.org/10.1038/s41586-022-04436-3
Buciuc M, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Boeve BF et al (2020) Association between transactive response DNA-binding protein of 43 kDa type and cognitive resilience to Alzheimer’s disease: a case-control study. Neurobiol Aging 92:92–97. https://doi.org/10.1016/j.neurobiolaging.2020.04.001
Carlos AF, Tosakulwong N, Weigand SD, Boeve BF, Knopman DS, Petersen RC et al (2022) Frequency and distribution of TAR DNA-binding protein 43 (TDP-43) pathology increase linearly with age in a large cohort of older adults with and without dementia. Acta Neuropathol 144:159–160. https://doi.org/10.1007/s00401-022-02434-3
Charbaut E, Curmi PA, Ozon S, Lachkar S, Redeker V, Sobel A (2001) Stathmin family proteins display specific molecular and tubulin binding properties. J Biol Chem 276:16146–16154. https://doi.org/10.1074/jbc.M010637200
Cook C, Petrucelli L (2019) Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101:1057–1069. https://doi.org/10.1016/j.neuron.2019.02.032
Davidson YS, Raby S, Foulds PG, Robinson A, Thompson JC, Sikkink S et al (2011) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122:703–713. https://doi.org/10.1007/s00401-011-0879-y
Estades Ayuso V, Pickles S, Todd T, Yue M, Jansen-West K, Song Y et al (2023) TDP-43-regulated cryptic RNAs accumulate in Alzheimer’s disease brains. Mol Neurodegener 18:57. https://doi.org/10.1186/s13024-023-00646-z
Fang MY, Markmiller S, Vu AQ, Javaherian A, Dowdle WE, Jolivet P et al (2019) Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103:802–819.e811. https://doi.org/10.1016/j.neuron.2019.05.048
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC et al (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388:3017–3026. https://doi.org/10.1016/S0140-6736(16)31408-8
Gittings LM, Alsop EB, Antone J, Singer M, Whitsett TG, Sattler R et al (2023) Cryptic exon detection and transcriptomic changes revealed in single-nuclei RNA sequencing of C9ORF72 patients spanning the ALS-FTD spectrum. Acta Neuropathol 146:433–450. https://doi.org/10.1007/s00401-023-02599-5
Guerra San Juan I, Nash LA, Smith KS, Leyton-Jaimes MF, Qian M, Klim JR et al (2022) Loss of mouse Stmn2 function causes motor neuropathy. Neuron 110:1671–1688.e1676. https://doi.org/10.1016/j.neuron.2022.02.011
Hardwick SA, Bassett SD, Kaczorowski D, Blackburn J, Barton K, Bartonicek N et al (2019) Targeted, high-resolution RNA sequencing of non-coding genomic regions associated with neuropsychiatric functions. Front Genet 10:309. https://doi.org/10.3389/fgene.2019.00309
Hosokawa M, Arai T, Yamashita M, Tsuji H, Nonaka T, Masuda-Suzukake M et al (2014) Differential diagnosis of amyotrophic lateral sclerosis from Guillain-Barre syndrome by quantitative determination of TDP-43 in cerebrospinal fluid. Int J Neurosci 124:344–349. https://doi.org/10.3109/00207454.2013.848440
Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC et al (2008) Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 116:215–220. https://doi.org/10.1007/s00401-008-0400-4
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8:1–13. https://doi.org/10.1016/j.jalz.2011.10.007
Irwin KE, Jasin P, Braunstein KE, Sinha I, Bowden KD, Moghekar A, Oh ES, Raitcheva D, Bartlett D, Berry JD et al (2023) A fluid biomarker reveals loss of TDP-43 splicing repression in pre-symptomatic ALS. bioRxiv. https://doi.org/10.1101/2023.01.23.525202
James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA (2016) TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 139:2983–2993. https://doi.org/10.1093/brain/aww224
Josephs KA, Martin PR, Weigand SD, Tosakulwong N, Buciuc M, Murray ME et al (2020) Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 143:3463–3476. https://doi.org/10.1093/brain/awaa299
Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR et al (2014) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127:441–450. https://doi.org/10.1007/s00401-013-1211-9
Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M et al (2008) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857. https://doi.org/10.1212/01.wnl.0000304041.09418.b1
Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM et al (2015) TAR DNA-binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Ann Neurol 78:697–709. https://doi.org/10.1002/ana.24493
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM et al (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127:811–824. https://doi.org/10.1007/s00401-014-1269-z
Kadokura A, Yamazaki T, Lemere CA, Takatama M, Okamoto K (2009) Regional distribution of TDP-43 inclusions in Alzheimer disease (AD) brains: their relation to AD common pathology. Neuropathology 29:566–573. https://doi.org/10.1111/j.1440-1789.2009.01017.x
Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA et al (2014) MAP-RSeq: Mayo analysis pipeline for RNA sequencing. BMC Bioinformatics 15:224. https://doi.org/10.1186/1471-2105-15-224
Kapaki E, Boufidou F, Bourbouli M, Pyrgelis ES, Constantinides VC, Anastassopoulou C et al (2022) Cerebrospinal fluid biomarker profile in TDP-43-related genetic frontotemporal dementia. J Pers Med 12(10):1747. https://doi.org/10.3390/jpm12101747
Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ et al (2009) Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol 117:55–62. https://doi.org/10.1007/s00401-008-0456-1
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA et al (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22:167–179. https://doi.org/10.1038/s41593-018-0300-4
Koike Y, Pickles S, Estades Ayuso V, Jansen-West K, Qi YA, Li Z et al (2023) TDP-43 and other hnRNPs regulate cryptic exon inclusion of a key ALS/FTD risk gene, UNC13A. PLoS Biol 21:e3002028. https://doi.org/10.1371/journal.pbio.3002028
Krach F, Batra R, Wheeler EC, Vu AQ, Wang R, Hutt K et al (2018) Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol 136:405–423. https://doi.org/10.1007/s00401-018-1870-7
Krus KL, Strickland A, Yamada Y, Devault L, Schmidt RE, Bloom AJ et al (2022) Loss of stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep 39:111001. https://doi.org/10.1016/j.celrep.2022.111001
Kuiperij HB, Versleijen AA, Beenes M, Verwey NA, Benussi L, Paterlini A et al (2017) Tau rather than TDP-43 proteins are potential cerebrospinal fluid biomarkers for frontotemporal lobar degeneration subtypes: a pilot study. J Alzheimers Dis 55:585–595. https://doi.org/10.3233/JAD-160386
Kumar S, Phaneuf D, Cordeau P Jr, Boutej H, Kriz J, Julien JP (2021) Induction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD. Mol Neurodegener 16:1. https://doi.org/10.1186/s13024-020-00420-5
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46–R64. https://doi.org/10.1093/hmg/ddq137
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497. https://doi.org/10.1038/nn.3230
Li Y, Tian Y, Pei X, Zheng P, Miao L, Li L et al (2023) SCG10 is required for peripheral axon maintenance and regeneration in mice. J Cell Sci 136(12):jcs260490. https://doi.org/10.1242/jcs.260490
Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108. https://doi.org/10.1093/nar/gkt214
Ling JP, Pletnikova O, Troncoso JC, Wong PC (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349:650–655. https://doi.org/10.1126/science.aab0983
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Lippa CF, Rosso AL, Stutzbach LD, Neumann M, Lee VM, Trojanowski JQ (2009) Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol 66:1483–1488. https://doi.org/10.1001/archneurol.2009.277
Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB (2019) Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep 27:1409–1421.e1406. https://doi.org/10.1016/j.celrep.2019.04.003
Lopez-Erauskin J, Bravo-Hernandez M, Presa M, Baughn MW, Melamed Z, Beccari MS, de Almeida Agra, Quadros AR, Arnold-Garcia O, Zuberi A, Ling K et al (2023) Stathmin-2 loss leads to neurofilament-dependent axonal collapse driving motor and sensory denervation. Nat Neurosci. https://doi.org/10.1038/s41593-023-01496-0
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F et al (2022) TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603:124–130. https://doi.org/10.1038/s41586-022-04424-7
Martinez-Gonzalez L, Rodriguez-Cueto C, Cabezudo D, Bartolome F, Andres-Benito P, Ferrer I et al (2020) Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1delta kinase inhibitor treatment. Sci Rep 10:4449. https://doi.org/10.1038/s41598-020-61265-y
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL (2015) Alzheimer’s disease. Nat Rev Dis Primers 1:15056. https://doi.org/10.1038/nrdp.2015.56
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ et al (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570:332–337. https://doi.org/10.1038/s41586-019-1195-2
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944. https://doi.org/10.1212/wnl.34.7.939
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y et al (2019) Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 22:180–190. https://doi.org/10.1038/s41593-018-0293-z
Morii H, Shiraishi-Yamaguchi Y, Mori N (2006) SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol 66:1101–1114. https://doi.org/10.1002/neu.20295
Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y et al (2019) Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol 137:27–46. https://doi.org/10.1007/s00401-018-1942-8
Nelson PT, Brayne C, Flanagan ME, Abner EL, Agrawal S, Attems J et al (2022) Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol 144:27–44. https://doi.org/10.1007/s00401-022-02444-1
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503–1527. https://doi.org/10.1093/brain/awz099
Nelson PT, Lee EB, Cykowski MD, Alafuzoff I, Arfanakis K, Attems J et al (2023) LATE-NC staging in routine neuropathologic diagnosis: an update. Acta Neuropathol 145:159–173. https://doi.org/10.1007/s00401-022-02524-2
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Noto Y, Shibuya K, Sato Y, Kanai K, Misawa S, Sawai S et al (2011) Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: specificity, sensitivity, and a possible prognostic value. Amyotroph Lateral Scler 12:140–143. https://doi.org/10.3109/17482968.2010.541263
Nozal V, Martinez-Gonzalez L, Gomez-Almeria M, Gonzalo-Consuegra C, Santana P, Chaikuad A et al (2022) TDP-43 modulation by tau-tubulin kinase 1 inhibitors: a new avenue for future amyotrophic lateral sclerosis therapy. J Med Chem 65:1585–1607. https://doi.org/10.1021/acs.jmedchem.1c01942
Nussbacher JK, Batra R, Lagier-Tourenne C, Yeo GW (2015) RNA-binding proteins in neurodegeneration: Seq and you shall receive. Trends Neurosci 38:226–236. https://doi.org/10.1016/j.tins.2015.02.003
Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C (2019) Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 102:294–320. https://doi.org/10.1016/j.neuron.2019.03.014
Okazaki T, Wang H, Masliah E, Cao M, Johnson SA, Sundsmo M et al (1995) SCG 10, a neuron-specific growth-associated protein in Alzheimer’s disease. Neurobiol Aging 16:883–894. https://doi.org/10.1016/0197-4580(95)02001-2
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468. https://doi.org/10.1038/nn.2779
Pozzi S, Thammisetty SS, Codron P, Rahimian R, Plourde KV, Soucy G et al (2019) Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest 129:1581–1595. https://doi.org/10.1172/JCI123931
Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM et al (2020) Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest 130:6080–6092. https://doi.org/10.1172/JCI139741
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/bioinformatics/btq033
Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S et al (2018) Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet 50:1584–1592. https://doi.org/10.1038/s41588-018-0238-1
Ren Y, Li S, Chen S, Sun X, Yang F, Wang H et al (2021) TDP-43 and phosphorylated TDP-43 levels in paired plasma and CSF samples in amyotrophic lateral sclerosis. Front Neurol 12:663637. https://doi.org/10.3389/fneur.2021.663637
Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lutjens R et al (1997) Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A 94:741–745. https://doi.org/10.1073/pnas.94.2.741
Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E et al (2020) Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 143:2844–2857. https://doi.org/10.1093/brain/awaa219
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, Zhang Y, Prudencio M, Keuss MJ, Khandeshi A et al (2023) Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. bioRxiv. https://doi.org/10.1101/2023.01.23.525149
Shin JE, Geisler S, DiAntonio A (2014) Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252:1–11. https://doi.org/10.1016/j.expneurol.2013.11.007
Smith VD, Bachstetter AD, Ighodaro E, Roberts K, Abner EL, Fardo DW et al (2018) Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 28:264–273. https://doi.org/10.1111/bpa.12505
Suarez-Calvet M, Dols-Icardo O, Llado A, Sanchez-Valle R, Hernandez I, Amer G et al (2014) Plasma phosphorylated TDP-43 levels are elevated in patients with frontotemporal dementia carrying a C9orf72 repeat expansion or a GRN mutation. J Neurol Neurosurg Psychiatry 85:684–691. https://doi.org/10.1136/jnnp-2013-305972
Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y et al (2017) Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol 133:923–931. https://doi.org/10.1007/s00401-017-1701-2
Tamaki Y, Shodai A, Morimura T, Hikiami R, Minamiyama S, Ayaki T et al (2018) Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci Rep 8:6030. https://doi.org/10.1038/s41598-018-24463-3
Tanji K, Mori F, Shirai F, Fukami T, Seimiya H, Utsumi J et al (2021) Novel tankyrase inhibitors suppress TDP-43 aggregate formation. Biochem Biophys Res Commun 537:85–92. https://doi.org/10.1016/j.bbrc.2020.12.037
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14:452–458. https://doi.org/10.1038/nn.2778
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564. https://doi.org/10.1097/NEN.0b013e31817713b5
van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A et al (2009) Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 41:1083–1087. https://doi.org/10.1038/ng.442
Vaquero-Garcia J, Barrera A, Gazzara MR, Gonzalez-Vallinas J, Lahens NF, Hogenesch JB et al (2016) A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5:e11752. https://doi.org/10.7554/eLife.11752
Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K et al (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A 99:9037–9042. https://doi.org/10.1073/pnas.122623799
Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A et al (2016) Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 139:3202–3216. https://doi.org/10.1093/brain/aww250
Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q et al (2018) The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data 5:180185. https://doi.org/10.1038/sdata.2018.185
Wang X, Allen M, Is O, Reddy JS, Tutor-New FQ, Castanedes Casey M et al (2022) Alzheimer’s disease and progressive supranuclear palsy share similar transcriptomic changes in distinct brain regions. J Clin Invest 132(2):e149904. https://doi.org/10.1172/JCI149904
Wilson RS, Yu L, Trojanowski JQ, Chen EY, Boyle PA, Bennett DA et al (2013) TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 70:1418–1424. https://doi.org/10.1001/jamaneurol.2013.3961
Zhang X, Sun B, Wang X, Lu H, Shao F, Rozemuller AJM et al (2019) Phosphorylated TDP-43 staging of primary age-related tauopathy. Neurosci Bull 35:183–192. https://doi.org/10.1007/s12264-018-0300-0
Acknowledgements
We thank all members of the Lagier-Tourenne, Wainger, Albers, Cleveland and Lutz laboratories for helpful discussions and support, and Angelica Gaona, Alexandra Melloni and Patrick M. Dooley for technical assistance regarding human post-mortem tissue. Acknowledgement is made to the donors of the Alzheimer’s Disease Research Program, a program of the BrightFocus Foundation, for support of this research. We would like to thank the patients and their families for their participation, without whom this work would not have been possible. We thank the Massachusetts Alzheimer Disease Research Center (P30AG062421) for brain tissues and neuropathological assessments. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). These data were generated from post-mortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine.
Funding
ARAAQ is the recipient of a Postdoctoral Fellowship from the Alzheimer’s Disease Research Program of the BrightFocus Foundation (BrightFocus Grant Number A2022002F). ISN was the recipient of a fellowship from the Friedrich-Naumann Foundation. SA and MN were supported by the Cullen Education and Research Foundation Young Investigator Award. MN is supported by the Holloway Postdoctoral Fellowship from the Association for Frontotemporal Degeneration. MWB and MSB were both funded by Ruth Kirschstein Institutional National Research Service Awards (T32 GM008666 MWB and T32 AG 66596-2 MSB). MWB received support from the ALS association and ZM and JLE were awarded Muscular Dystrophy Association development grants. The Mayo RNA-seq study was led by Dr. Nilüfer Ertekin-Taner, Mayo Clinic, Jacksonville, FL as part of the multi-PI U01 AG046139 (MPIs Golde, Ertekin-Taner, Younkin, Price) using samples from the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, CurePSP Foundation, and support from Mayo Foundation. Study data included samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, USA. The Brain and Body Donation Program is supported by the NINDS (U24 NS072026, National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders); the NIA (P30 AG19610, Arizona Alzheimer’s Disease Core Center); the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center); the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901, and 1001, to the Arizona Parkinson’s Disease Consortium); and the Michael J. Fox Foundation for Parkinson’s Research. AUTTX LLC is supported by funds from the Center for Neurologic Study (Dr. Richard Smith, Director). This work was supported by grants from the NIH/NINDS (R01NS112503 to DWC and CLT; R01NS080820 to NET) and NIH/NIA (R01AG061796, U19AG074879, U01AG046139 to NET), the Packard Center for ALS Research (DWC), the ALS association (DWC), ALS finding a Cure (to CLT), the Sean M. Healey and AMG Center for ALS and the Massachusetts Center for Alzheimer Therapeutic Science (MassCATS to CLT). CLT is the recipient of the Araminta Broch-Healey Endowed Chair in ALS. NET is the recipient of Alzheimer’s Association Zenith Award.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors report no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Agra Almeida Quadros, A.R., Li, Z., Wang, X. et al. Cryptic splicing of stathmin-2 and UNC13A mRNAs is a pathological hallmark of TDP-43-associated Alzheimer’s disease. Acta Neuropathol 147, 9 (2024). https://doi.org/10.1007/s00401-023-02655-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-023-02655-0