
Abstract
At the time of their clinical manifestation, the heterogeneous group of adult and pediatric gliomas carries a wide range of diverse somatic genomic alterations, ranging from somatic single-nucleotide variants to structural chromosomal rearrangements. Somatic abnormalities may have functional consequences, such as a decrease, increase or change in mRNA transcripts, and cells pay a penalty for maintaining them. These abnormalities, therefore, must provide cells with a competitive advantage to become engrained into the glioma genome. Here, we propose a model of gliomagenesis consisting of the following five consecutive phases that glioma cells have traversed prior to clinical manifestation: (I) initial growth; (II) oncogene-induced senescence; (III) stressed growth; (IV) replicative senescence/crisis; (V) immortal growth. We have integrated the findings from a large number of studies in biology and (neuro)oncology and relate somatic alterations and other results discussed in these papers to each of these five phases. Understanding the story that each glioma tells at presentation may ultimately facilitate the design of novel, more effective therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gliomas encompass a very diverse group and account for the great majority of tumors originating in the parenchyma of the central nervous system (CNS) [166]. Two larger glioma groups are recognized: so-called diffuse gliomas, characterized by extensive infiltrative growth into the surrounding CNS parenchyma, and more circumscribed (non-diffuse) gliomas such as pilocytic astrocytoma and ependymomas. Diffuse gliomas, by far the most frequent gliomas in adult patients, are traditionally classified according to their microscopic similarities with (precursors of) glial cells and then designated as diffuse astrocytomas, oligodendrogliomas or mixed gliomas/oligoastrocytomas. Additionally, a malignancy grade is assigned to these tumors based on the presence/absence of especially marked mitotic activity, florid microvascular proliferation (MVP), and necrosis [175, 233]. For over a century, such microscopic evaluation has provided the gold standard for the diagnosis of gliomas, assessment of prognosis and formed the basis for therapeutic management. However, multiple studies showed that a purely histopathologic classification suffers from considerable inter- and intraobserver variability [3, 46, 217].
As with other human cancers, the pathogenesis and molecular evolution of gliomas are often characterized by somatic chromosomal aberrations, widespread or focal copy number changes and targeted gain and loss of function events in oncogenes and tumor suppressor genes [211, 224, 225]. Various permutations of somatic alterations were shown to be associated with distinct tumor entities and differential sensitivities to treatment, such as a chromosome 1p/19q-codeletion in oligodendrogliomas conferring increased sensitivity to chemotherapy [26, 27]. In the course of the last two decades, it became increasingly clear that such molecular characteristics may provide a more robust and objective basis for subtyping of diffuse gliomas and both scientists and clinicians turned towards molecular markers to aid diagnosis [30, 34, 59, 234, 244]. Indeed, the International Society for Neuropathology—Haarlem Consensus Guidelines and the most recent edition of the WHO classification of CNS tumors (published in 2016) embraced the notion of an integrated histo-molecular classification of diffuse gliomas [130, 131].
In adult patients three major subgroups of diffuse glioma are now defined based on the presence or absence of mutations in the isocitrate dehydrogenase 1 (IDH1) or IDH2 gene and of complete, combined loss of the short arm of chromosome 1 and of the long arm of chromosome 19 (complete 1p/19q-codeletion):
-
IDH-wildtype: most of these histologically represent astrocytic tumors, a large percentage belonging to the highest malignancy grade, i.e., glioblastomas.
-
IDH-mutant and 1p/19q-non-codeleted: these tumors also generally have an astrocytic phenotype, but a much larger percentage is at first diagnosis histologically lower grade/WHO grade II or III.
-
IDH-mutant and 1p/19-codeleted: most of these are characterized by a prominent oligodendroglial phenotype of the tumor cells.
Following this strategy, the diagnosis of mixed glioma/oligoastrocytoma can be expected to largely disappear, except when additional molecular tests cannot be performed or do not provide unequivocal results; in that situation, not-otherwise-specified (NOS) should be added to the diagnosis to indicate that ideally such samples require further workup [132]. Rare ‘dual genotype’ oligoastrocytomas have been reported that show polymorphic phenotypes with both a complete 1p/19q-codeletion component and non-codeleted component [93, 235]. Diffuse midline glioma, H3 K27M-mutant, was added in the WHO 2016 classification as a separate entity [116, 191, 242]. This type of diffuse glioma, which most often occurs in children, is by definition located in the ‘midline’ of the CNS (brainstem, thalamus, cerebellum and/or spinal cord) and considered as highly aggressive (WHO grade IV) irrespective of the malignancy grade assigned by histology [129]. By far, the most frequent astrocytic tumors in children though are pilocytic astrocytomas that generally are more circumscribed (therefore, grouped under non-diffuse gliomas) and show an indolent, WHO grade I behavior [44, 105, 190]. Meanwhile, the transition from a purely histological to a histo-molecular classification of especially diffuse gliomas represents a paradigm shift and necessitates re-evaluation of histologic criteria used for grading and guidance of therapeutic decisions [164, 181].
In this review, we summarize current knowledge and propose five phases in gliomagenesis that occur sequentially and ultimately lead to its clinical manifestation (Fig. 1). Each phase is characterized by distinct molecular alterations and phenotypic characteristics, such as differences in growth dynamics and evolutionary mechanisms. A critical assumption in our model is the existence of two growth barriers, which we refer to as oncogene-induced and replicative senescence. Similar barriers have been described in detail in the context of cultured epithelial cells and fibroblasts and much of this work has paved the road for our understanding of these mechanisms in gliomagenesis [77, 183, 239]. In the model we propose, the first phase of initial growth (phase I) follows the acquisition of a glioma initiation event and is characterized by aberrant proliferation of pre-tumor cells. Continued oncogenic exposure may impede tumor growth and trigger a durable form of cell cycle arrest termed oncogene-induced senescence (phase II) in a majority of tumor cells. Some phase I/II cells may acquire molecular changes to bypass oncogene-induced senescence and continue growth in spite of unfavorable and stressful conditions including DNA damage and dysfunctional telomeres. Continued growth despite incremental genomic instability marks the third phase of stressed growth (phase III). This second round of glioma cell growth under harsh conditions triggers a second round of durable growth arrest termed replicative senescence (phase IV) and in some cases, brings forth a state of cellular crisis characterized by widespread cell death. Rare cells may acquire stem-like characteristics and a means to continue growth indefinitely, giving rise to a final phase of immortal growth (phase V). Such phase V glioma stem-like cells uphold the tumor progenitor cell population via their capacity for self-renewal and may also give rise to more differentiated and growth-arrested stage IV cells, losing their stem-like properties. In this manuscript, we systematically review the evidence for this model and propose candidate mechanisms where definitive evidence is lacking. Acknowledging that this model is an abstract simplification of gliomagenesis, we also provide some examples of exceptions and conflicting evidence. While the focus of this review is on various molecular categories of diffuse glioma recognized by the most recent WHO classification, a few examples in the realm of non-diffuse gliomas are touched upon as well.

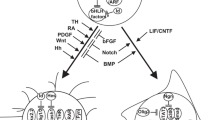
Model of the molecular life history of gliomas, prior to becoming clinically manifest. The temporal sequence of events can be subdivided into five phases (I–V) represented in different colors. a The number of dividing cells (or proliferation rate) across each phase. Proliferation peaks towards the end of growth phases and dips going into senescence phases. b The tumor mass across each phase. Tumor mass increases exponentially during growth phases and logarithmically during senescence phases. c Telomere length across each phase. Telomere length over time follows a pattern that is inverse to tumor mass. d Cell doubling diagram indicating the growth barriers (senescence phases) and resulting selection bottleneck. e Somatic alterations associated with different phases in gliomagenesis. The timing of each event is indicated on the x-axis of panel C. Genomic instability events are accumulated during phase III–IV. Of note, this model is a simplified representation of true gliomagenesis. The x-axis is not drawn to scale, in part because the duration of the phases likely varies from cell to cell and between various tumor types. Furthermore, the position of the curves is arbitrary as cells in a tumor may not be in sync. BFB breakage–fusion–bridge, DM double minute, ALT alternative lengthening of telomeres
A model for the temporal molecular pathogenesis of gliomas
Phase I: initial growth
The theory that cancer results from accumulation of mutations over time, in a subset of patients combined with contribution of inherited risk factors, has been around for over six decades and has been refined over the years [5, 79, 80, 111, 158, 160]. For the purpose of this review we will consider a glioma initiation event to be the first acquired (somatic) event towards developing glioma. This event should provide a competitive growth advantage, either by directly increasing proliferation or by creating the conditions in which increased proliferation may happen. Cells and their progeny characterized by such an event will be primed to outcompete neighboring cells giving rise to an initial submicroscopic tumor mass. While germline events that contribute to glioma risk precede such glioma initiation events, their incomplete penetrance suggests that they cannot be considered as causal for glioma formation and instead prime the environment for tumor formation [196]. Furthermore, even if a glioma initiation event marks the first somatic event in the formation of a tumor it may not be responsible for initiating growth directly. Instead, this event may promote tumorigenesis indirectly via stochastic activation of oncogenes or repression of tumor suppressors. Genetic or epigenetic selection pressures will prioritize daughter cells with growth advantages over those without and daughter cells with lethal genotypes will rapidly disappear [64, 150, 245].
IDH-mutant diffuse gliomas
Mutations in IDH1/IDH2 are commonly considered to be glioma initiating. Several studies have shown that they are amongst the few alterations highly shared amongst gliomas at first presentation and their recurrences [9, 100, 204] which is explained by their presence in the cell of origin and all cells derived thereof. Comparing multiple biopsies from the same tumor, IDH mutation can be confidently detected in each tumor segment and thus fit the proposed criteria of a glioma initiation event [107, 124, 204]. In vitro experiments have demonstrated that IDH mutations alone are sufficient to reprogram the transcriptome and epigenome of normal cells to prevent these cells from entering a terminally differentiated state [134, 147, 213].
IDH dysregulation likely contributes to gliomagenesis via the accumulation of the oncogenic metabolite R(−)-2-hydroxyglutarate (2HG) [114, 176]. Wildtype IDH enzymatically converts isocitrate into α-ketoglutarate (α-KG) as part of the citric acid cycle, whereas mutant IDH metabolizes α-KG into 2HG [54, 231]. Both mutant and wildtype IDH alleles are, therefore, essential for the oncogenic function of IDH. IDH mutations in glioma result in genome-wide hypermethylation [159, 213], most likely due to effects of 2HG on the ten–eleven translocation methylcytosine dioxygenase (Tet) family of proteins [61, 134, 243]. This hypermethylation may provide a growth advantage to cancer cells due to the epigenetic activation of oncogenes via stochastic activation of alternative gene regulatory programs, some conferring added fitness [64]. One such mechanism in glioma may be through methylation-induced disruption of a CCCTC-binding factor (CTCF) binding site, resulting in aberrant activation of platelet-derived growth factor receptor alpha (PDGRFA) [63].
Although most experimental models of IDH involve overexpression of mutant IDH in vitro, several transgenic mouse models have been described [28, 125]. Early transgenic models showed that conditional knock-in of mutant IDH in the murine brain led to perinatal lethality [188]. A more recent inducible model demonstrated that mutant IDH led to increased proliferation and infiltration in the CNS parenchyma of murine neural stem cells [10]. Though these mice eventually died due to hydrocephalus and did not develop malignant tumors, they showed symptoms of the initial phases of gliomagenesis. Thus, while experimental models of mutant IDH are generally insufficient to cause glioma, mutant IDH leads to changes that could be interpreted as early tumor development.
IDH-wildtype diffuse astrocytomas
Approximately 70% of IDH-wildtype diffuse astrocytomas are characterized at the molecular level by a single copy loss of chromosome 10 and gain of chromosome 7 (+ 7/− 10) [16, 34]. Loss or diploid loss of heterozygosity of chromosome 10 and of chromosome arm 10p in particular has been reported to occur more frequently and may precede gain of chromosome 7 [94]. Based on evolutionary modeling using primary-recurrent tumor pairs and multisector tumor sampling, several independent groups have found that + 7/− 10 is homogeneous and longitudinally preserved and thus likely the first and glioma initiation event in a large fraction of IDH-wildtype diffuse astrocytomas/glioblastomas [69, 107, 168, 198, 229]. A recent study suggested that gains of chromosome 7 likely occur early in tumorigenesis, amongst the first 10% of driver events [69]. In the past several years, there has been a lot of interest in the role of TERT promoter mutations in oncogenesis and an increasing body of evidence suggests that these mutations precede + 7/− 10 [106]. Nevertheless, a potential role of TERT promoter mutations to promote proliferation in the initial growth phase and as a glioma initiation event is speculative and will be discussed later in this review.
Chromosome 7 is home to several oncogenes that have been implicated in gliomagenesis such as cyclin-dependent kinase 6 (CDK6), MET proto-oncogene (MET) and epidermal growth factor receptor (EGFR), while chromosome 10 hosts several tumor suppressor genes, including Tet family member Tet methylcytosine dioxygenase 1 (TET1) and phosphatase and tensin homolog (PTEN). Though these genes comprise the prime suspects, it is unlikely that they alone are responsible for initiating glioma development [168]. Studies across different diseases and in various model organisms have shown that large chromosomal copy number changes led to gross gene dosage fluctuations impacting various specific and general cellular functions [206]. Such changes may, therefore, act in concert to promote tumor development.
Some IDH-wildtype diffuse gliomas show cytogenetically intact chromosomes 7 and 10, implying that other initiation events give rise to these tumors as well. Such events may include activating or inactivating alterations in the phosphoinositide 3-kinase (PI3K), receptor tyrosine kinase (RTK) and mitogen-activated protein kinase (MAPK) pathways [142]. PI3K pathway alterations include mutations in PI3-kinase subunit alpha (PIK3CA), PI3-kinase subunit P85-alpha (PIK3R1), or inactivation the aforementioned tumor suppressor PTEN [127, 173, 199]. Glioma initiation events may also include point mutations in RTK pathway genes such as EGFR and PDGFRA or in MAPK pathway genes such as neurofibromin 1 (NF1) [220]. Much is already known about the effect of these mutations on cancer growth but additional research is needed to secure their potential role as glioma initiation events.
A particular subgroup of diffuse IDH-wildtype gliomas is characterized by mutations in H3 histone family members and these gliomas occur most often in children [191, 202, 241]. The diffuse midline glioma, H3 K27M-mutant, shows a lysine to methionine substitution at position 27 of the H3 histone family member 3A (H3F3A) or histone cluster 1 H3 family member 3B (HIST1H3B) gene and is included in the WHO 2016 classification as a separate entity. Other H3-mutant diffuse gliomas in children and adolescents occurring predominantly in the cerebral hemispheres often show H3 G34R/V mutation (implying a glycine 34 to arginine or valine substitution) [115, 201, 202, 241]. In contrast to hypermethylated IDH-mutant gliomas, these H3-mutant gliomas have a general DNA hypomethylation phenotype [14]. A recent study showed that expression of mutant H3 K27M in neonatal mice brains led to ectopic proliferation, indicating a possible pre-cancerous change [126]. Although additional support is needed, combined with their apparent mutual exclusivity with mutations in IDH and changes characteristic for IDH-wildtype tumors, these findings indicate that H3 K27M and H3 G34R/V mutations may be glioma initiation events [32].
Non-diffuse gliomas
Recent studies have shown that pilocytic astrocytomas near universally harbor abnormalities in the MAPK pathway, and most commonly a tandem duplication targeting chromosome 7q, which gives rise to a KIAA1549–BRAF fusion gene consisting of the N terminus of KIAA1549 and the kinase domain of v-RAF murine sarcoma viral oncogene homolog B1 (BRAF) [44]. Alternative alterations include the oncogenic V600E missense mutation also targeting BRAF [190]. The BRAF V600E mutation results in an activating change due to a substitution of valine with glutamic acid at codon 600. In a non-cancer setting, BRAF activates kinases MEK and ERK, which in turn activate transcriptional machinery to promote differentiation, proliferation, growth and apoptosis [31]. Both BRAF V600E mutation and BRAF fusion genes contribute to tumorigenesis by constitutively activating the kinase domain of BRAF, resulting in overactive signaling activity and a selective growth advantage for affected cells [55, 67, 102]. In most pilocytic astrocytomas (even after thorough analysis), an activating change in BRAF or other MAPK pathway members is the only genomic change that can be confidently detected, implying that it is the glioma initiation event in this disease [187].
Phase II: oncogene-induced senescence
Continued oncogenic signaling in the initial growth phase prompts the activation of tumor suppressive signaling via activation of the p16INK4a/p14ARF–RB–p53 cell cycle and cell stress pathways (Fig. 2), slowing tumor growth and transitioning a majority of cells with intact pathways into a terminal state called oncogene-induced senescence [43]. First discovered over five decades ago in cultured fibroblasts, senescence is a stress-induced durable cell cycle arrest [82]. The role of senescence in cancer has been reviewed extensively [29, 42, 78, 83, 174, 193]. Briefly, senescence provides a major tumor suppressive barrier and dividing tumor cells are put under selection pressure to acquire molecular events to prevent or overcome its onset. Hallmarks of senescence include durable growth arrest; short, dysfunctional telomeres; and a marked increase in DNA damage and stress signaling [42]. Although senescent cells are growth arrested, they are metabolically active and release a plethora of signaling molecules to the microenvironment, also known as the senescence-associated secretory phenotype [174]. Distinction must be made between oncogene-induced senescence which is discussed here and is triggered by chronic oncogenic signaling, and replicative senescence discussed later, describing senescence triggered by telomere dysfunction following extensive replicative cycles [193]. Oncogene-induced senescence poses a significant growth barrier and most cells will not acquire molecular alterations that allow them to bypass this barrier and will, therefore, become senescent [43]. However, rare cells may acquire such alterations, eliciting a selective sweep by a subclone that will rapidly dominate the neoplastic cell population.

Process diagram indicating the p16INK4a/p14ARF–RB–p53 pathway in normal conditions. Disruption of one or multiple components through mutation or copy number change may prevent or suppress the onset of senescence. Various stimuli use different routes to activate the senescence response, leaving compensatory mechanisms in place in case components fail. For example, if oncogene-induced senescence is repressed via CDKN2A/B inactivation, DNA damage and telomere shortening could still trigger replicative senescence via ATM and ATR. CDKs cyclin-dependent kinases (e.g., CDK2), MDMs murine double minutes (e.g., MDM2)
In some cancer types, a senescent precursor stadium can be identified, such as intestinal polyps in colon cancer and dysplastic naevi in melanoma [192]. In contrast to the catastrophic karyotypes demonstrated by their later stage derivatives, these growth-arrested senescent precursor lesions entered a senescent state via the activation of single oncogene such as BRAF and are otherwise genetically unremarkable [72, 117, 151, 153]. This begs the question if such premalignant precursor changes exist for diffuse gliomas as well. If so, such precursor lesions may, however, well be (sub)microscopic in size and go unnoticed in imaging or autopsy studies.
The tumor suppressor proteins p16INK4a, p14ARF, RB and p53 can be considered as gatekeepers of senescence. The INK4a/ARF locus on chromosome 9 contains both cyclin dependent kinase inhibitor 2A (CDKN2A) and 2B (CDKN2B), combined encoding for both p16INK4a and p14ARF via alternative splicing. Tumor suppressors RB and p53 on the other hand are encoded for by the genes retinoblastoma 1 (RB1) and tumor protein 53 (TP53) on chromosomes 13 and 17, respectively. Inactivation of one or multiple of these genes via genomic deletion and/or inactivating mutations has been linked to repression of senescence signaling and is a common event in all cancers including gliomas [39, 83, 195, 240]. For the purpose of this review, we define the term ‘senescence bypass event’ as any molecular alteration that suppresses the onset of oncogene-induced senescence.
IDH-mutant diffuse astrocytomas
IDH-mutant astrocytomas are often characterized by loss of one allele of TP53, combined with a loss-of-function mutation in the remaining allele. The frequency of TP53 mutations in IDH-mutant (‘secondary’) glioblastomas is comparable to that in lower grade IDH-mutant astrocytomas from which these glioblastomas are derived via malignant progression, suggesting that TP53 aberrations are early lesions in these tumors [162]. Furthermore, and in contrast to IDH-wildtype gliomas, TP53 mutations were shared between all TP53-mutant cases of primary and recurrent tumors in a recent study [100]. Analysis of multiple biopsies from the same IDH-mutant tumors indicated that samples mutant for TP53 were always IDH mutant, while some IDH-mutant samples lacked TP53 mutations, suggesting that IDH mutations precede TP53 inactivation [232]. IDH mutation and TP53 inactivation, therefore, both comprise early events in gliomagenesis, with TP53 inactivation generally following mutation in IDH.
The p53 tumor suppressor protein is involved in many different functions, and especially its role in cell cycle arrest and senescence is very well understood [81]. While enzymatically active wildtype p53 triggers senescence in response to oncogenic stress, mutant p53 inadequately blocks proliferation thereby bypassing the onset of oncogene-induced senescence. In addition, p53 takes a prominent role in the senescence pathway (Fig. 2) and loss of its enzymatic activity impacts replicative senescence, triggering crisis [195]. This is discussed further in phase IV (“Phase IV: Replicative senescence/crisis” section).
IDH-mutant oligodendrogliomas, 1p/19q-codeleted
The majority of IDH-mutant tumors wildtype for TP53 demonstrate a combined single copy loss of the complete chromosome arms 1p and 19q (complete 1p/19q-codeletion) [20, 27]. These tumors are the canonical oligodendrogliomas according to the revised WHO criteria [130]. Though it has been suggested that 1p/19q-codeletions are the result of an unbalanced translocation, much about the contribution of this event to oncogenesis remains to be resolved [95]. 1p/19q-codeletion was found to be stable across longitudinal samples and multiple biopsies, suggesting that they are early events [1, 97, 218]. The finding that codeleted tumors are almost exclusively IDH-mutant, while the reverse is not true, suggests that mutations in IDH precede codeletion. This suggestion may implicate a common cell of origin for both astrocytoma (IDH-mutant, non-codeleted) and oligodendroglioma (IDH-mutant and 1p/19q-codeleted). Indeed, recent evidence was provided that substantiates this hypothesis, demonstrating that differences in inferred cell identity between oligodendrogliomas and astrocytomas may be entirely explained by different microenvironment makeup and the impact of the 1p/19q-codeletion on the expression of genes on these chromosome arms [219]. The authors found that although these histological tumor types differ in morphology, these histological subtypes share a comparable developmental hierarchy and glial lineage. Transcription factors involved in oligodendrocyte differentiation are also expressed in both histologies [6]. Several case reports have highlighted ‘dual genotype’ oligoastrocytomas, demonstrating molecular features of both bona fide astrocytic and bona fide oligodendroglial tumor cells [93, 235]. Such a dual genotype may be explained by assuming that very early in gliomagenesis a subset of IDH-mutant cells experiences a complete 1p/19q-codeletion and a TERT promoter mutation, while another IDH-mutant subset acquires a mutation in TP53 and/or ATRX. Detailed and sufficiently powered longitudinal studies of primary and recurrent diffuse gliomas may help to elucidate the actual frequency of such ‘dual genotypes’, how they are related to mixed histological appearances, and how they evolve over time [2].
Additional loss-of-function mutations in far-upstream element binding protein (FUBP1) on 1p31.1 and capicua transcriptional repressor (CIC) on 19q13.2 are observed in over 60% of 1p/19q-codeleted gliomas [15]. A paired analysis of primary and recurrent 1p/19q-codeleted oligodendrogliomas described distinct alterations in CIC and FUBP1 in the primary and the recurrent tumor [9]. Another report described that these events were frequently private to either primary or recurrence, but not both [1]. Both findings corroborate observations of CIC and FUBP1 mutation heterogeneity across nine distinct samples from the same oligodendroglioma, including finding five distinct CIC mutants across nine tumor samples [204]. Loss-of-function mutations in these genes led to a loss of protein expression and the FUBP1 mutation was associated with adverse survival compared to wildtype tumors [35, 90]. These findings suggest an important role for these genes and indicate that in cases in which these events were not found in one sample of the tumor, they still might be present elsewhere. Despite their apparent importance, the role of these events in gliomagenesis remains to be understood.
It is not clear whether 1p/19q-codeleted oligodendrogliomas undergo senescence or acquire mechanisms to bypass senescence. 1p/19q-codeleted gliomas generally lack alterations in genes associated with oncogene-induced senescence such as BRAF or senescence bypass such as TP53, RB1 or CDKN2A. Nevertheless, given their continued clinical growth it appears that these tumors have somehow evaded growth arrest and senescence barriers. Perhaps the 1p/19q-codeletion allows pre-cancerous cells to avoid senescence through the mono-allelic inactivation of tumor suppressor genes on these chromosome arms [197]. One mechanism that was previously proposed may be via a dosage-dependent repression of chromodomain helicase DNA-binding domain 5 (CHD5) on 1p36 [7, 8, 222]. A study that used genetic engineering to create mouse models with gains and losses of a region corresponding to human 1p36 found that duplication of this region led to decreased proliferation and senescence whereas a single-copy deletion led to immortalization [8].
IDH-wildtype diffuse astrocytomas
Amongst IDH-wildtype astrocytomas/glioblastomas, one of the most frequent alterations is a homozygous loss of CDKN2A and CDKN2B [128]. Mathematical modeling has suggested that homozygous CDKN2A/B loss occurs after +7/−10 but before other molecular events [168]. Homozygous CDKN2A/B loss alone is insufficient for tumor formation in mice, requiring the activation of an oncogene to generate tumors in vivo [215]. As such, homozygous CDKN2A/B loss is likely a second event in the tumorigenesis of IDH-wildtype astrocytoma or glioblastoma. The role of protein products p16INK4a and p14ARF in senescence is very well understood. Indeed, astrocytes with a homozygous deletion of CDKN2A/B can grow indefinitely in culture, and introduction of p16INK4a in immortal human glioma cell lines with this deletion leads to cell cycle arrest and senescence [87, 216]. Taken together, these results indicate that loss of CDKN2A/B may provide adult IDH-wildtype astrocytomas with a reliable means for senescence bypass.
Mutations in TP53 sometimes co-occur with homozygous CDKN2A/B loss in IDH-wildtype glioma [163]. While TP53 mutations are often shared across all tumor cells in IDH-mutant astrocytoma, TP53 mutations in IDH-wildtype astrocytomas are frequently unique to one or a few tumor subclones [107]. In this same study, it was found that amongst IDH-wildtype astrocytomas, whereas CDKN2A/B is consistently deleted, TP53 mutations are frequently lost or gained at tumor recurrence. These observations suggest that, in IDH-wildtype astrocytoma, CDKN2A/B may be primarily important for senescence regulation
Pediatric H3-mutant/IDH-wildtype diffuse gliomas are TP53 mutant in about 50% of cases, which may act as a senescence bypass event in these tumors [191]. Amongst remaining H3-mutant tumors, approximately 20–30% of H3 K27 mutant diffuse gliomas show mutations in Activin A Receptor Type 1 (ACVR1) [23, 65, 208, 242]. There is no evidence that ACVR1 has any role in sentence regulation; however, ACVR1 alterations were found to be mostly mutually exclusive with alterations in TP53 and PPM1D. PPM1D is a protein phosphatase downstream of p53 involved in apoptosis regulation following DNA damage and thus likely involved in senescence [62]. Expression of ACVR1 mutants in Tp53null murine astrocytes implanted in mouse brains failed to induce tumors, likely because H3 K27 mutations are required as a tumor initiation event and thus suggesting that ACVR1 does not meet the criteria for a tumor initiation event [242]. Thus, while about half of pediatric H3-mutant gliomas demonstrate TP53 mutations that may act to bypass senescence, it remains unclear if and how TP53-wildtype H3-mutant pediatric gliomas bypass senescence.
Non-diffuse gliomas
Several lines of evidence suggest that pilocytic astrocytomas (WHO grade I, IDH-wildtype) are arrested in a senescent phase II state and do not advance to later phases. First, these tumors were found to frequently demonstrate several biomarkers of senescence at tumor detection, including widespread β-galactosidase activity and p16INK4a staining [88, 112, 177]. Second, these tumors are very quiescent genetically, often demonstrating but a single activated oncogene, such as a BRAF fusion, BRAF V600E mutation or rarely an activating mutation in FGFR1 or PTPN11 [101, 212, 246]. Third, these tumors generally grow slowly, have excellent outcomes and sometimes regress, perhaps because the tumor cells do not immortalize [24, 25, 75, 185]. Fourth, expression of activated BRAF V600E alone does not lead to tumor development in in vitro and in in vivo mouse models, while the combined activation of BRAF and loss of CDKN2A/B is transforming, suggesting that additional mutations to bypass senescence are required to advance to phase III [92, 182, 189]. The clinical presentation of pilocytic astrocytoma may be the result of senescence-mitigating circumstances, such as a cell of origin with proliferative potential in the absence of senescence-bypass.
Phase III: stressed growth
Cells presenting with continued proliferative signaling beyond the oncogene-induced senescence barrier are generally characterized by defective DNA damage response signaling and continued growth in a stressed environment. During this phase the repetitive DNA at the telomeric terminal ends of chromosomes become increasingly important [161]. Telomeres progressively shorten as cells divide due to the linear conformation of chromosomes and directional replication machinery, a phenomenon that is critically important for diseases like cancer which are characterized by often rampant proliferation [165]. Telomeric DNA takes on a lasso conformation called the t-loop, and these loops are bound by the shelterin DNA-binding protein complex. Together, these characteristics protect chromosome ends from being recognized as DNA double-strand breaks and prevent inadvertent activation of DNA damage response pathways [56].
Dysfunctional telomeres are critically short and improperly protected telomeres lacking t-loops and shelterin complexes. They trigger the activation of DNA damage response pathways via ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinase, which are triggered by exposed and unprotected double-stranded and single-stranded DNA break ends, respectively. The exposed ends then fall victim to homology directed repair (HDR) and non-homologous end joining (NHEJ) repair processes, intended to repair accidental DNA breaks but lead to gross genomic instability when triggered by dysfunctional telomeres. When telomeres are unprotected, these repair processes prompt sister chromatids to fuse with one another, forming a dicentric chromosome. During the anaphase, the dicentric chromosome will form a bridge spanning the mitotic spindle and connecting the two daughter cells. Resolution of the chromatin bridge via cytoplasmic 3′ nuclease TREX1 results in breakage of the dicentric chromosome at a locus not necessarily at the site where the fusion had occurred, resulting in an unbalanced inheritance of genetic material between the two daughter cells. Because the resulting daughter cells also lack telomeres, this process of breakage–fusion–bridge (BFB) cycles (Fig. 3a) will repeat itself every subsequent cell division until telomeres are restored [4, 70, 144, 148, 149]. The detrimental genomic instability acquired via telomere dysfunction and BFB cycles endows these cells with powerful stochastic mutational mechanisms to acquire changes that provide a survival benefit under selective pressure.

Genomic instability related to telomere stress. a Schematic illustrating BFB cycles. Following a single BFB cycle, daughter cells are left with unequal DNA content, leading to a deletion in A and an amplification in B. BFB cycles may also involve fusion of non-sister telomeres (not shown). b Repetitive BFB cycles form palindromes demonstrating high intra-segmental homology. This can lead to intra-chromosomal fusions and formation of double minutes. c Following chromothripsis segments can be rearranged, lost or circularized. TSG tumor suppressor gene, OG oncogene, DDR DNA damage response, DM double minutes, BFB breakage–fusion–bridge
The intensity of genomic instability endured during the stressed growth phase may depend on the severity of senescence pathway dysregulation incurred overcoming oncogene-induced senescence (Fig. 2). In the case of H3-mutant, IDH-wildtype and IDH-mutant astrocytoma/glioblastoma this pathway is perturbed close to the source via the direct loss of RB, p53 or p16INK4a protein function. In IDH-mutant and 1p/19q-codeleted oligodendroglioma this pathway may be repressed indirectly, for example, via the modulation of p14ARF activity through a partial deletion of CHD5. This may explain why the latter group of tumors shows significantly less genomic instability compared to the former tumor types. Moreover, in pilocytic astrocytoma this pathway may not be affected at all and these tumors may not advance beyond oncogene-induced senescence. While genomic instability incurred during phase III demonstrates some patterns that are unique to a certain glioma subtype, these features are generally shared across all gliomas regardless of subtype. We, therefore, did not separate this section according to tumor type as we did for the other phases. More research is needed to carefully delineate the selective pressures at play to better understand differences and similarities between various glioma entities in this respect.
The advent of high-throughput sequencing has led to remarkable progress in understanding the complexity of genomic instability in cancer, including complex deletions, amplifications and translocations [245]. It is important to note that the genomic organization that can be reconstructed using sequencing at the time of analysis are those changes that resulted in viable cells and were selected for. Recent work has shown that telomere dysfunction directly leads to catastrophic genomic events, including genome shattering (chromothripsis), clustered regions of focal hypermutation (kataegis) and whole genome doubling (tetraploidization) [60, 137, 138]. Although the incidence of chromothripsis across the spectrum of gliomas is not known, a recent report suggests that chromothripsis is very common in glioblastoma [139]. Comparison of primary and recurrent tumors across various tumor types including gliomas demonstrated that recurrent tumors lack additional genomic instability, suggesting that these events occurred during a stressful period, the initial development of the tumor that was later stabilized [60].
Recently, there has been a renewed interest in circular extrachromosomal DNA elements called double minute (DM) chromosomes in cancer, and it was shown that such DMs are common in gliomas [57, 214]. Although DM chromosomes have been long recognized as a cytogenetic feature of cancer, relatively little was known about its biological relevance. DM chromosomes have a predisposition to involve cancer oncogenes such as MYC proto-oncogene protein (MYC), MDM2 proto-oncogene (MDM2) or cyclin-dependent kinase 4 (CDK4) [53, 113, 156, 186, 247]. A unique feature of DM is that they lack centromeres and telomeres to dictate the organization of the mitotic spindle during mitosis and are, therefore, randomly distributed across daughter cells [103]. Interestingly, this feature hypothetically provides DM chromosomes with an impressive fitness advantage over linear chromosomes as they do not need telomeres to protect them from inadvertent DNA damage response pathways and are not subjected to detrimental BFB cycles. It has been proposed that DM chromosomes are the result of the fusion and circular assembly of stretches of linear DNA consisting of highly homologous sequences of inverted duplications in tandem following sequential BFB cycles (Fig. 3b) [205]. Others have proposed that ineffective DNA repair following chromothripsis can lead to linear DNA fragments getting pieced together in a circular fashion (Fig. 3c) [66]. A study by our group found evidence of chromothripsis in several glioblastoma samples localized to chromosome 12 involving MDM2 and CDK4 and suggested that these segments may be arranged in extrachromosomal DMs [247]. Because these are inherently random processes, it is possible that DM chromosomes that promote survival are positively selected for during telomere dysfunction. Compared to IDH-mutant gliomas, DMs in IDH-wildtype tumors more often involve established glioma oncogenes, despite what appears to be a comparable frequency of DMs in both glioma categories [57]. More research is needed to precisely determine the frequency of DMs in glioma subtypes and to pinpoint the genetic origin of these structures.
A recent study of paired primary and recurrent IDH-mutant gliomas reported that in some cases allelic imbalances of the IDH-mutant allele occurred upon recurrence, which led to a change in mutant protein expression and consequently decreased 2HG production [145]. Furthermore, IDH-mutant tumors are very hard to culture, and when it succeeds, IDH mutations that were present initially have been reported missing, raising the possibility that losing an IDH mutation is advantageous for survival in culture [135]. Introduction of mutant IDH in cell cycle checkpoint-deficient cells rapidly transforms these cells into competent tumor cells [99]. However, IDH inhibition in these cells after as little as 4 days after its first introduction did little to slow tumor growth. These findings suggest that some IDH-mutant gliomas may rapidly evolve and acquire additional driver events to uphold the tumor cell population.
To summarize, telomere dysfunction and stressed growth may promote the context-dependent evolution of glioma cells, sometimes rendering glioma initiation events redundant and providing gliomas with new fuel that rapidly increase intratumoral heterogeneity and can deal with various toxic stresses and bottlenecks. While stochastic mutational mechanisms in the stressed growth phase provide ample selection pressure to acquire beneficial changes, the detrimental genomic instability under which cells must operate acts as a powerful tumor suppressive barrier. Unchecked growth will rapidly lead to another round of DNA damage-induced replicative senescence, or when those checkpoints fail completely, cell crisis.
Phase IV: replicative senescence/crisis
Sustained stressed growth is not durable and will eventually lead tumor cells down to one of two possible roads. Tumor cells with a partially intact senescence response (i.e., functional p53 and RB) may undergo a second round of senescence called replicative senescence in response to dysfunctional telomeres. Tumor cells with a completely dysfunctional senescence response (i.e., loss-of-function mutation in TP53 or RB1) instead continue proliferating in a state of cellular crisis leading to cell death in a vast majority of cells [51]. Replicative senescence and crisis both pose a second population bottleneck to further tumor formation. It is essential that tumor cells transition to a less stressful environment with proper telomere maintenance to prevent further BFB cycles and other catastrophic events. Cell culture experiments have demonstrated that direct immortalization of cells prior to a stressed growth phase enables them to bypass genomic instability and immortalize lacking the wild karyotypes typically associated with malignant transformation [68, 154]. These observations suggest that genomic instability in cancer development generally precedes immortal growth and is required to generate errors enabling telomere maintenance [68].
Acquisition of a telomere maintenance mechanism endows cancer cells with immortal growth, meaning that they are bestowed a limitless replicative potential [108]. Telomere maintenance is established once a tumor cell has reactivated telomerase or activated alternative lengthening of telomeres. Moreover, restoration of telomere function may prevent further BFB cycles and restore genome stability. The canonical pathway involves the reactivation of the ribonucleoprotein telomerase which is transcriptionally silent in differentiated adult cells [74]. The telomerase catalytic component telomerase reverse transcriptase (TERT) is expressed in over 80% of human cancers and is thought to be rate limiting for telomerase activity [194]. In the alternative pathway tumors become immortalized via a recombination-driven mechanism called alternative lengthening of telomeres (ALT) [21].
IDH-mutant diffuse astrocytomas
IDH-mutant diffuse astrocytomas almost universally demonstrate ALT [85]. ALT cells present with several defining characteristics, including a heterogeneous distribution of telomere length across chromosomes, extrachromosomal telomeric DNA fragments in a circular configuration (c-circles), increased expression of telomeric repeat-containing RNA (TERRA) from telomeres, the formation of ALT-associated promyelocytic leukemia bodies (APBs), frequent telomere sister chromatid exchanges (T-SCEs) and recombination between telomeres from different chromosomes [58]. Telomeres in ALT cells are heterogeneous in length and relatively long, demonstrating telomere lengths much longer than telomerase-positive cells on average [22]. ALT provides cancer cells with stabilizing telomere maintenance in a telomerase-negative setting. Although it remains unclear how ALT becomes activated, its presence has been tightly associated with loss-of-function events targeting the α-thalassemia/mental retardation syndrome X-linked (ATRX) or death-domain-associated protein (DAXX) genes and these events are also a hallmark feature of IDH-mutant astrocytomas [84, 98, 104]. ATRX functions as an ATP-dependent helicase within the SWI/SNF family and combined these two genes form the ATRX–DAXX complex, which functions as a histone chaperone to deposit the histone variant H3.3 at telomeres [71]. Telomeric DNA has a tendency to form secondary quadruplex structures that challenge the replication machinery and need to be resolved for proper replication [170]. How exactly these two genes protect telomeres from recombination and ALT is still unknown. It has been suggested that the combined helicase activity of ATRX and the histone chaperone capabilities of the ATRX–DAXX complex can resolve the secondary quadruplex structure at telomeres, thereby enabling proper progression of the replication fork during S-phase and preventing the inadvertent activation of recombination (ALT) mechanisms [121, 122].
The presence or absence of inactivating ATRX and DAXX mutations present a strong correlation with ALT in many tumor types including gliomas [84, 133, 191]. Recent in vitro studies have shown that knockout of ATRX alone is insufficient to cause ALT; however, ATRX knockout combined with inactivation of p53 and RB enzymatic activity led to an increased incidence of ALT after enduring several cycles of telomere induced crisis [40, 155, 180]. Furthermore, the reintroduction of ATRX expression in ATRX mutant ALT cells led to a repression of T-SCE, APBs and c-circle formation [40, 155].
IDH-mutant oligodendrogliomas, 1p/19q-codeleted
Oligodendrogliomas, IDH-mutant and 1p/19q-codeleted almost universally use telomerase to maintain telomeres and virtually all of these tumors carry an activating TERT promoter mutation [106]. These TERT promoter mutations are amongst the most common non-coding mutations in cancer [89, 91, 106, 223]. Recurrent hotspot point mutations substitute a cytosine at − 228 or − 250 relative to the promoter to a thymine (C228 > T or C250 > T) to create a de novo e-twenty-six (ETS) transcription factor binding site that recruits the ETS family member GA-binding protein alpha chain (GABPA) to activate transcription [13].
Although the timing of TERT promoter mutations is still under debate, several lines of evidence suggest that TERT promoter mutations arise early in gliomagenesis and perhaps even occur prior to the glioma initiation event. TERT promoter mutations preferentially occur in tissues with a lower rate of self-renewal and there are numerous reports on the extra-telomeric functions of TERT, including effects on the NF-κB and WNT/β-catenin pathway promoting tumor growth and invasiveness [106, 141, 169]. Combined, this raises the possibility that these mutations may contribute to tumorigenesis via other pathways than its effect on telomerase alone, providing a biological reason for these mutations to contribute to gliomagenesis early in phase I and before the onset of dysfunctional telomeres. In a glioma-specific analysis, it was found that nearly all tumors with the ‘phase I event’ + 7/− 10 or ‘phase II event’ 1p/19q-codeletion have TERT promoter mutations, whereas not all TERT promoter mutant gliomas have + 7/− 10 or 1p/19q-codeletions, which may indicate that TERT promoter mutations even precede + 7/− 10 and 1p/19q-codeletions [34]. Another group studied the mutation fraction using multisector sequencing in 1p/19q-codeleted oligodendrogliomas and found that TERT promoter mutations indeed are early events and may occur before IDH mutations [204]. The idea that TERT promoter mutation occurs early is further corroborated by the finding that genetically engineered TERT promoter mutations in telomerase-positive embryonic stem cells do not affect telomerase activity, while upon differentiation these engineered cells remain telomerase positive and acquire immortality [36]. More recently, it was found that TERT promoter mutations in melanoma initially do not support telomere maintenance and telomeres shorten to critically short length despite harboring promoter mutations [37]. The effect on telomere length was not observed until later, which the authors attributed to a two-step immortalization process. One study even reported canonical (− 228 and − 250) somatic TERT promoter mutations in the blood of multiple non-cancer patients, indicating that these events could even occur before the onset of cancer and act to prime the tumor bed [143]. Although roles for TERT outside of telomere maintenance remain to be understood, these observations provide a sound argument that TERT promoter mutations can occur early in or even before gliomagenesis while providing a means towards immortalization at a later stage.
IDH-wildtype diffuse astrocytomas
The majority of IDH-wildtype diffuse gliomas use telomerase for telomere maintenance [120]. Re-analysis of previously published samples reclassified according to WHO 2016 criteria demonstrated that approximately 75% of diffuse IDH-wildtype gliomas are TERT promoter mutant [172]. Thus, TERT promoter mutations are common in both the most and the least aggressive diffuse gliomas (IDH-wildtype diffuse astrocytomas and IDH-mutant and 1p19q-codeleted oligodendrogliomas, respectively), suggesting that TERT promoter mutations are not dictating their biological behavior. It was further found that ATRX mutations occur in approximately 5% of IDH-wildtype diffuse astrocytomas [172]. The prevalence of ALT in IDH-wildtype diffuse gliomas is higher than the frequency of ATRX mutations, suggesting that some of these tumors may use ATRX-independent mechanisms to activate ALT [85]. In a similar fashion, the prevalence of telomerase activity is higher than the prevalence of TERT promoter mutations in this tumor type, suggesting that these tumors may use TERT promoter-independent mechanisms for the reactivation of telomerase [120]. Several candidate mechanisms have been previously described in glioma, including TERT promoter methylation or TERT amplifications [11]. Contrary to adult IDH-wildtype diffuse glioma, H3-mutant malignant pediatric glioma frequently demonstrates ALT [140], and several studies reported frequent co-occurrence of ATRX mutations in both H3 K27 and G34 mutant gliomas. However, reports of co-occurrence vary between 30 and 60% for K27 and 75–100% for G34, suggesting that there is a role for telomerase in many of these tumors as well [123].
Phase V: immortalization and dedifferentiation
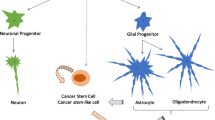
The glioma stem cell theory states that amongst all cancerous cells in a tumor, a subset of cells act as progenitor or stem cells with reproductive capabilities and sustaining the cancer, much like normal bone marrow stem cells are responsible for replenishing the population of circulating leukocytes [221]. It has often been contrasted to the theory of clonal evolution, which suggests that cancers evolve through an iterative process of clonal expansion from a single cell [73]. Recent advances in single-cell sequencing and lineage tracing have unveiled multiple populations of tumor cells in bulk tumor samples, providing fuel for the cancer stem cell hypothesis [119, 171, 210, 219]. One study used single-cell RNA sequencing on IDH-mutant and 1p/19q-codeleted oligodendroglioma patient samples and uncovered distinct cell populations of undifferentiated tumor stem cells and cells that are more differentiated along various glial lineages [210]. In a similar study of IDH-mutant astrocytoma the authors were able to detect the same cellular populations but with a higher ratio of stem-like to differentiated cells that increased with increasing WHO grades [219]. Another study used DNA barcoding of repeatedly in vivo-transplanted glioblastoma cells to trace the lineage during their engraftment and found a population of progenitor cells that sustained the tumor and gave rise to differentiated non-proliferative cells [119].
These studies all provide support for a cancer stem cell hypothesis and raise the question how these findings fit with our model, which leans towards a model of clonal evolution. In fact, current evidence may suggest that both mechanisms are acting together (Fig. 4a). Whereas clonal evolution is important to establish the initial cancer stem cell population, neutral evolution (in line with the cancer-stem cell hypothesis) may fit better once the initial cancer core has been established, especially so when further evolutionary stimuli (e.g., senescence barriers, hypoxia, treatment) are lacking. The concept of neutral evolution holds that most molecular changes are not caused by natural selection but rather by the stochastic allelic variation that are neutral and do not affect cellular fitness [109]. A recent study analyzed cancer genomes from TCGA and found neutral evolution in approximately one-third of a wide spectrum of over 900 tumors, including 35 gliomas of which 8 (23%) suggested evidence in support of a neutral evolution process [236]. The authors conclude that all clonal selection must have occurred before the onset of cancer growth and not in later arising subclones. Several groups have since challenged these findings and suggested that their analysis does not univocally prove neutral evolution starting from the first malignant cell [157, 207, 237]. While it may be possible that tumors evolve neutrally beyond the most recent selective sweep we do not agree with the conclusion that these findings suggest that all clonal selection must occur before the onset of tumor growth. The authors do not take into account that at the time the tumor presents itself and is surgically removed, all remnants of a selection process that have been outcompeted or died will have completely disappeared, in contrast to “neutral” variants which do not affect fitness and will remain. According to our simplified model, tumorigenesis sequentially follows phase I–V. Once the cancer stem cell population has been established, tumor cells will follow neutral evolution, as long as new evolutionary or other stimuli for opportunistic growth are lacking (Fig. 4a).

a Integration of a clonal evolution and cancer stem cell model for gliomagenesis. This model assumes that sequential mutations and selection pressure drive the evolution of cancer stem-like cells. At the same time, these stem-like cells may give rise to more differentiated (i.e., phase IV) offspring that may divide further but rapidly become growth arrested. b According to this model these cells may be senescent and contribute to the cancer phenotype by eliciting a microenvironment response via SASP. SASP senescence-associated secretory phenotype; ILs interleukins; CXCLs chemokines (C–X–C motif); CCLs chemokines (C–C motif)
Aforementioned studies supporting the cancer stem cell hypothesis suggest that multiple cellular populations exist within a tumor, including a self-renewing cancer stem cell population and a less-proliferative differentiated population. A recent paper demonstrated that cells derived from glioma stem cells may differentiate and subsequently undergo senescence [167]. We speculate that phase V tumor cells represent cancer stem cells that may give rise to more differentiated, phase IV cells (Fig. 4a). The transition from phase IV to V and vice versa is likely volatile in nature owing to transcriptional reprogramming including the activation of the stemness factors oligodendrocyte transcription factor 2 (OLIG2), sex-determining region Y-box 2 (SOX2) and the reactivation of telomerase [203]. On the other hand, the transition from phase I to phase IV is more rigid in nature, involving various changes on a genomic level, including somatic mutations and copy number changes as described. This important distinction in flexibility led us to believe that phase V cells may re-differentiate and assume a phase IV state.
Although they are growth arrested, senescent cells are metabolically active and release a plethora of signaling molecules to the microenvironment. The senescence-associated secretory phenotype (SASP) is a feature of senescent cells that curtails these cells with the release of proinflammatory cytokines [47]. SASP does not depend on p16INK4a or p21 activity and senescence with intact p16INKa function actually suppresses SASP [49]. Similarly, activated p53 signaling also suppresses SASP while TP53 loss induces SASP [48, 136, 178]. These findings suggest that the SASP response is stronger when senescence pathway genes are lost. Indeed, IDH-wildtype astrocytomas often harbor homozygous deletions in CDKN2A/B and are known to have a highly active microenvironment [230]. Genes associated with SASP were shown to be overexpressed in higher grades of glioma and older patients, the latter group more likely to be affected by high-grade IDH-wildtype astrocytoma [50]. Moreover, it was found that primary glioblastoma cells retain a functional senescence program despite mutations in the TERT promoter and CDKN2A/B locus [118]. These findings imply that senescent and differentiated phase IV cells may be crucial for shaping the immune microenvironment in gliomas (Fig. 4b).
A broader perspective
While there are not many known environmental risk factors that predispose to glioma, large cohort genome-wide association studies over the past two decades have identified multiple heritable polymorphisms conferring glioma risk [96, 110, 152, 179, 196, 238]. Notably, several of these risk loci are localized to genes involved in telomere maintenance, including the telomerase reverse transcriptase TERT, telomerase RNA component TERC, and other telomere maintenance-associated genes STN1, CST complex subunit (OBFC1), protection of telomeres 1 (POT1) and regulator of telomere elongation 1 (RTEL1) [152]. Moreover, there appears to be a significant increased glioma risk in people with increased leukocyte telomere lengths [45]. Unsurprisingly, glioma risk alleles at aforementioned genes are also associated with increased leukocyte telomere length [41, 226, 228]. Telomeres thus play an important role in not only the development of gliomas, but also in glioma risk [227]. In fact, the positive association between leukocyte telomere length and cancer risk is not specific to glioma and shared across many cancers. A recent Mendelian randomization study found that longer leukocyte telomere length was associated with an increased risk to cancer but a reduced risk to non-neoplastic disease such as cardiovascular disease [209]. It has been suggested that this difference is due to individuals with longer telomeres being more likely to acquire driver mutations due to an increased proliferative potential whereas the inverse relationship observed for non-neoplastic disease may be due to the impact of telomere shortening on tissue degeneration [19, 200].
Several hereditary disorders are associated with an increased risk of glioma development, including neurofibromatosis type 1 and type 2 (NF1, NF2) and the TP53 germline mutation/Li–Fraumeni syndrome. NF1 and NF2 are autosomal dominant hereditary disorders with germline mutations in NF1 and NF2 and clinically characterized by multiple benign nerve sheath tumors (especially neurofibromas in NF1, schwannomas in NF2), but also by a markedly increased risk on particular gliomas (especially pilocytic astrocytoma in NF1 and ependymomas in NF2) [33, 76, 184]. Both genes are well-known tumor suppressor genes and key components in the MAPK pathway [38]. It has been demonstrated that senescence commonly occurs in benign nerve sheath tumors and that prolonged NF1 disruption leads to oncogene-induced senescence in a model system, providing a rationale as to why these germline disorders present with tumors that are often relatively indolent [52]. A germline perturbation affecting NF1 or NF2 can be considered a tumor initiation event, explaining why this germline disorder guarantees the formation of multiple benign nerve sheath tumors over one’s lifetime.
Li–Fraumeni syndrome is a rare autosomal dominant hereditary disorder that is caused by the germline perturbation of TP53 or CHK2, which regulates p53 activity [12, 18, 146]. Whereas NF1 and NF2 guarantee the formation of especially multiple benign tumors (including non-diffuse gliomas) in a lifetime, Li–Fraumeni patients pose a greater risk to developing a malignant tumor, including a diffuse glioma [17]. This risk increases with age and is over 50% at age 30, with a lifetime cancer risk of over 70% in men and almost 100% in women [146]. Moreover, 15 and 4% of affected individuals were found to develop a second and third cancer [86]. Li–Fraumeni syndrome germline mutations affect phase II and prevent the onset of oncogene-induced senescence following the acquisition of a glioma initiation event, thus increasing the risk of developing cancer over a lifetime.
The germline mutations underlying NF1/NF2 and Li–Fraumeni syndrome represent pathways that both need to be disrupted for a malignant tumor to form. The fact that nearly all patients with NF1/NF2 develop one or more benign tumors can be understood by acknowledging that in these disorders a germline tumor initiation event is involved. Unless this pathway is supplemented by a senescence bypass event, these tumors do not readily proceed to malignancy. In contrast, Li–Fraumeni syndrome is characterized by an increased risk for malignant tumors in many (but not all) patients. In this syndrome, the germline senescence bypass event needs to be supplemented by a tumor initiation event for a tumor to develop, and such tumors may be more aggressive/malignant due to the defective senescence barrier, allowing the tumor (precursor) cell to instantly progress to phase III and instigate genomic instability.
Conclusion
Our knowledge on the molecular events driving cancer has grown exponentially over the years. This review has aimed to put this new knowledge into the perspective of the temporal molecular pathogenesis of glioma, starting from the first aberrant cell all the way to a symptom-causing glioma. To this end we have combined what is known on gene (mal)function, tumor evolution, genomic instability and telomere maintenance to develop a model of gliomagenesis. This model describes five sequential phases cancer cells undergo during their gliomagenesis. We speculate that transitions from one phase to the next can be characterized by acquisition of tumor-driving events that sequentially contribute to the hallmarks of cancer as previously proposed, including proliferation, evasion of apoptosis and limitless replicative potential [79, 80]. Our model is a simplified abstraction of what may be the truth and new insights will refine and improve our understanding. Meanwhile, we hope that our model will help foster hypotheses leading to new insights into the molecular life history of glioma that will help identify convincing therapeutic vulnerabilities.
References
Aihara K, Mukasa A, Nagae G, Nomura M, Yamamoto S, Ueda H, Tatsuno K, Shibahara J, Takahashi M, Momose T et al (2017) Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol Commun 5:18. https://doi.org/10.1186/s40478-017-0422-z
Aldape K, Amin SB, Ashley DM, Barnholtz-Sloan JS, Bates AJ, Beroukhim R, Bock C, Brat DJ, Claus EB, Costello JF et al (2018) Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the glioma longitudinal analysis consortium. Neurooncology. https://doi.org/10.1093/neuonc/noy020
Aldape K, Simmons ML, Davis RL, Miike R, Wiencke J, Barger G, Lee M, Chen P, Wrensch M (2000) Discrepancies in diagnoses of neuroepithelial neoplasms: the San Francisco Bay Area Adult Glioma Study. Cancer 88:2342–2349
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, De Pinho RA (2000) Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406:641–645. https://doi.org/10.1038/35020592
Ashley DJ (1969) The two “hit” and multiple “hit” theories of carcinogenesis. Br J Cancer 23:313–328
Azar S, Leventoux N, Ripoll C, Rigau V, Goze C, Lorcy F, Bauchet L, Duffau H, Guichet PO, Rothhut B et al (2018) Cellular and molecular characterization of IDH1-mutated diffuse low grade gliomas reveals tumor heterogeneity and absence of EGFR/PDGFRalpha activation. Glia 66:239–255. https://doi.org/10.1002/glia.23240
Bagchi A, Mills AA (2008) The quest for the 1p36 tumor suppressor. Can Res 68:2551–2556. https://doi.org/10.1158/0008-5472.CAN-07-2095
Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA (2007) CHD5 is a tumor suppressor at human 1p36. Cell 128:459–475. https://doi.org/10.1016/j.cell.2006.11.052
Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, Krischek B, Ozduman K, Omay SB, Sorensen EA et al (2016) Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet 48:59–66. https://doi.org/10.1038/ng.3457
Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L et al (2016) Expression of Idh 1(R132H) in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 30:578–594. https://doi.org/10.1016/j.ccell.2016.08.017
Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, Akdemir KC, Seth S, Song X, Wang Q et al (2017) Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 49:349–357. https://doi.org/10.1038/ng.3781
Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF et al (1999) Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 286:2528–2531
Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, Choi S, Hong C, He D, Pekmezci M et al (2015) Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348:1036–1039. https://doi.org/10.1126/science.aab0015
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W et al (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24:660–672. https://doi.org/10.1016/j.ccr.2013.10.006
Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G et al (2011) Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333:1453–1455. https://doi.org/10.1126/science.1210557
Bigner SH, Mark J, Burger PC, Mahaley MS Jr, Bullard DE, Muhlbaier LH, Bigner DD (1988) Specific chromosomal abnormalities in malignant human gliomas. Can Res 48:405–411
Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM (2001) Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 20:4621–4628. https://doi.org/10.1038/sj.onc.1204621
Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, Harris M, Jones PH, Binchy A, Crowther D et al (1994) Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Can Res 54:1298–1304
Blasco MA (2007) Telomere length, stem cells and aging. Nat Chem Biol 3:640–649. https://doi.org/10.1038/nchembio.2007.38
Bromberg JE, van den Bent MJ (2009) Oligodendrogliomas: molecular biology and treatment. Oncologist 14:155–163. https://doi.org/10.1634/theoncologist.2008-0248
Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3:1271–1274
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 14:4240–4248
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U et al (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46:451–456. https://doi.org/10.1038/ng.2936
Buder T, Deutsch A, Klink B, Voss-Bohme A (2015) Model-based evaluation of spontaneous tumor regression in pilocytic astrocytoma. PLoS Comput Biol 11:e1004662. https://doi.org/10.1371/journal.pcbi.1004662
Burkhard C, Di Patre PL, Schuler D, Schuler G, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H (2003) A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg 98:1170–1174. https://doi.org/10.3171/jns.2003.98.6.1170
Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W et al (2013) Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 31:337–343. https://doi.org/10.1200/JCO.2012.43.2674
Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Cairns RA, Mak TW (2013) Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3:730–741. https://doi.org/10.1158/2159-8290.cd-13-0083
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740. https://doi.org/10.1038/nrm2233
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci Met al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. New Engl J Med 372:2481–2498. https://doi.org/10.1056/nejmoa1402121
Cantwell-Dorris ER, O’Leary JJ, Sheils OM (2011) BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther 10:385–394. https://doi.org/10.1158/1535-7163.MCT-10-0799
Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pages M, Taylor KR, Saulnier P, Lacroix L et al (2015) Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827. https://doi.org/10.1007/s00401-015-1478-0
Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O’Connell P et al (1990) A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 62:193–201
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. https://doi.org/10.1016/j.cell.2015.12.028
Chan AK, Pang JC, Chung NY, Li KK, Poon WS, Chan DT, Shi Z, Chen L, Zhou L, Ng HK (2014) Loss of CIC and FUBP1 expressions are potential markers of shorter time to recurrence in oligodendroglial tumors. Mod Pathol 27:332–342. https://doi.org/10.1038/modpathol.2013.165
Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D (2015) Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. https://doi.org/10.7554/eLife.07918
Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, Ryu J, Darzacq X, Bastian BC, Hockemeyer D (2017) Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 357:1416–1420. https://doi.org/10.1126/science.aao0535
Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T (1999) Mouse models of tumor development in neurofibromatosis type 1. Science 286:2172–2176
Ciriello G, Cerami E, Sander C, Schultz N (2012) Mutual exclusivity analysis identifies oncogenic network modules. Genome Res 22:398–406. https://doi.org/10.1101/gr.125567.111
Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, Gibbons RJ (2015) Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6:7538. https://doi.org/10.1038/ncomms8538
Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I et al (2013) Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 45:422–427. https://doi.org/10.1038/ng.2528 (e421–422)
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130:223–233. https://doi.org/10.1016/j.cell.2007.07.003
Collado M, Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10:51–57. https://doi.org/10.1038/nrc2772
Collins VP, Jones DT, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:775–788. https://doi.org/10.1007/s00401-015-1410-7
Connelly JM, Malkin MG (2007) Environmental risk factors for brain tumors. Curr Neurol Neurosci Rep 7:208–214
Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK (1997) Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 79:1381–1393
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:2853–2868. https://doi.org/10.1371/journal.pbio.0060301
Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286:36396–36403. https://doi.org/10.1074/jbc.M111.257071
Coppola D, Balducci L, Chen DT, Loboda A, Nebozhyn M, Staller A, Fulp WJ, Dalton W, Yeatman T, Brem S (2014) Senescence-associated-gene signature identifies genes linked to age, prognosis, and progression of human gliomas. J Geriatr Oncol 5:389–399. https://doi.org/10.1016/j.jgo.2014.08.003
Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11:1921–1929
Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K (2006) A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 10:459–472. https://doi.org/10.1016/j.ccr.2006.10.003
Cox D, Yuncken C, Spriggs AI (1965) Minute chromatin bodies in malignant tumours of childhood. Lancet 1:55–58
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744. https://doi.org/10.1038/nature08617
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. https://doi.org/10.1038/nature00766
de Lange T (2009) How telomeres solve the end-protection problem. Science 326:948–952. https://doi.org/10.1126/science.1170633
de Carvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, Koeman J, Seth S, Protopopov A, Felicella M et al (2017) Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. bioRxiv: https://doi.org/10.1101/081158
Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000) Telomere maintenance by recombination in human cells. Nat Genet 26:447–450. https://doi.org/10.1038/82586
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. New Engl J Med 372:2499–2508. https://doi.org/10.1056/NEJMoa1407279
Ernst A, Jones DT, Maass KK, Rode A, Deeg KI, Jebaraj BM, Korshunov A, Hovestadt V, Tainsky MA, Pajtler KW et al (2016) Telomere dysfunction and chromothripsis. Int J Cancer J Int Cancer 138:2905–2914. https://doi.org/10.1002/ijc.30033
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567. https://doi.org/10.1016/j.ccr.2010.11.015
Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O’Connor PM, Appella E (1997) Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA 94:6048–6053
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529:110–114. https://doi.org/10.1038/nature16490
Flavahan WA, Gaskell E, Bernstein BE (2017) Epigenetic plasticity and the hallmarks of cancer. Science 357:l2380. https://doi.org/10.1126/science.aal2380
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA et al (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46:462–466. https://doi.org/10.1038/ng.2950
Forment JV, Kaidi A, Jackson SP (2012) Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer 12:663–670. https://doi.org/10.1038/nrc3352
Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, Jones TA, Aarum J, Dalton J, Bailey S et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. https://doi.org/10.1002/path.2558
Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, Brothman AR, Jackson M, Chin K, LaBarge MA, Watts G et al (2014) Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell cycle (Georgetown, Tex) 13:3423–3435. https://doi.org/10.4161/15384101.2014.954456
Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Mitchell TJ, Rubanova Y, Anur P, Rosebrock D, Yu K et al (2017) The evolutionary history of 2,658 cancers. bioRxiv: https://doi.org/10.1101/161562
Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N (2000) Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA 97:5357–5362. https://doi.org/10.1073/pnas.090013497
Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X et al (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140:678–691. https://doi.org/10.1016/j.cell.2010.01.003
Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D, Bennett DC (2006) Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer 95:496–505. https://doi.org/10.1038/sj.bjc.6603283
Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481:306–313. https://doi.org/10.1038/nature10762
Greider CW, Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43:405–413
Gunny RS, Hayward RD, Phipps KP, Harding BN, Saunders DE (2005) Spontaneous regression of residual low-grade cerebellar pilocytic astrocytomas in children. Pediatr Radiol 35:1086–1091. https://doi.org/10.1007/s00247-005-1546-z
Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D (1997) The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278:51–57
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400:464–468. https://doi.org/10.1038/22780
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319:1352–1355. https://doi.org/10.1126/science.1140735
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Harris SL, Levine AJ (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24:2899–2908. https://doi.org/10.1038/sj.onc.1208615
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
He S, Sharpless NE (2017) Senescence in health and disease. Cell 169:1000–1011. https://doi.org/10.1016/j.cell.2017.05.015
Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425. https://doi.org/10.1126/science.1207313
Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH et al (2011) Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179:1608–1615. https://doi.org/10.1016/j.ajpath.2011.06.018
Hisada M, Garber JE, Fung CY, Fraumeni JF Jr, Li FP (1998) Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 90:606–611
Holland EC, Hively WP, Gallo V, Varmus HE (1998) Modeling mutations in the G1 arrest pathway in human gliomas: overexpression of CDK4 but not loss of INK4a-ARF induces hyperploidy in cultured mouse astrocytes. Gene Dev 12:3644–3649
Horbinski C (2013) To BRAF or not to BRAF: is that even a question anymore? J Neuropathol Exp Neurol 72:2–7. https://doi.org/10.1097/NEN.0b013e318279f3db
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339:959–961. https://doi.org/10.1126/science.1230062
Hu X, Martinez-Ledesma E, Zheng S, Kim H, Barthel F, Jiang T, Hess KR, Verhaak RGW (2017) Multigene signature for predicting prognosis of patients with 1p19q co-deletion diffuse glioma. Neuro-oncology 19:786–795. https://doi.org/10.1093/neuonc/now285
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959. https://doi.org/10.1126/science.1229259
Huillard E, Hashizume R, Phillips JJ, Griveau A, Ihrie RA, Aoki Y, Nicolaides T, Perry A, Waldman T, McMahon M et al (2012) Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci USA 109:8710–8715. https://doi.org/10.1073/pnas.1117255109
Huse JT, Diamond EL, Wang L, Rosenblum MK (2015) Mixed glioma with molecular features of composite oligodendroglioma and astrocytoma: a true “oligoastrocytoma”? Acta Neuropathol 129:151–153. https://doi.org/10.1007/s00401-014-1359-y
Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP (1998) Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Gene Chromosomes Cancer 22:9–15
Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Can Res 66:9852–9861. https://doi.org/10.1158/0008-5472.can-06-1796
Jenkins RB, Xiao Y, Sicotte H, Decker PA, Kollmeyer TM, Hansen HM, Kosel ML, Zheng S, Walsh KM, Rice T et al (2012) A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet 44:1122–1125. https://doi.org/10.1038/ng.2388
Jeuken JW, Sijben A, Bleeker FE, Boots-Sprenger SH, Rijntjes J, Gijtenbeek JM, Mueller W, Wesseling P (2011) The nature and timing of specific copy number changes in the course of molecular progression in diffuse gliomas: further elucidation of their genetic “life story”. Brain Pathol 21:308–320. https://doi.org/10.1111/j.1750-3639.2010.00447.x
Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK et al (2012) Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3:709–722. https://doi.org/10.18632/oncotarget.588
Johannessen TA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R, Pieper RO (2016) Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol Cancer Res MCR 14:976–983. https://doi.org/10.1158/1541-7786.MCR-16-0141
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K et al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189–193. https://doi.org/10.1126/science.1239947
Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932. https://doi.org/10.1038/ng.2682
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Can Res 68:8673–8677. https://doi.org/10.1158/0008-5472.CAN-08-2097
Kanda T, Sullivan KF, Wahl GM (1998) Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol CB 8:377–385
Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT (2012) Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3:1194–1203. https://doi.org/10.18632/oncotarget.689
Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44:1839–1852
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026. https://doi.org/10.1073/pnas.1303607110
Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, Grimsby J, Sougnez C, Muller F, Hu J et al (2015) Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25:316–327. https://doi.org/10.1101/gr.180612.114
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266:2011–2015
Kimura M (1991) The neutral theory of molecular evolution: a review of recent evidence. Idengaku zasshi 66:367–386
Kinnersley B, Labussiere M, Holroyd A, Di Stefano AL, Broderick P, Vijayakrishnan J, Mokhtari K, Delattre JY, Gousias K, Schramm J et al (2015) Genome-wide association study identifies multiple susceptibility loci for glioma. Nat Commun 6:8559. https://doi.org/10.1038/ncomms9559
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, Lasitschka F, von Deimling A, Capper D (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891–900. https://doi.org/10.1007/s00401-013-1100-2
Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, Alt FW (1983) Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35:359–367
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S et al (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483:484–488. https://doi.org/10.1038/nature10898
Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, Sturm D, Meyer J, Jones C, Zheludkova O et al (2016) Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131:137–146. https://doi.org/10.1007/s00401-015-1493-1
Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA et al (2015) Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129:669–678. https://doi.org/10.1007/s00401-015-1405-4
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019–1031. https://doi.org/10.1016/j.cell.2008.03.039
Kumar R, Gont A, Perkins TJ, Hanson JEL, Lorimer IAJ (2017) Induction of senescence in primary glioblastoma cells by serum and TGFbeta. Sci Rep 7:2156. https://doi.org/10.1038/s41598-017-02380-1
Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, Guilhamon P, Lee L, Kushida MM, Pellacani D et al (2017) Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549:227–232. https://doi.org/10.1038/nature23666
Langford LA, Piatyszek MA, Xu R, Schold SC Jr, Shay JW (1995) Telomerase activity in human brain tumours. Lancet 346:1267–1268
Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A et al (2010) ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143:367–378. https://doi.org/10.1016/j.cell.2010.09.023
Lazzerini-Denchi E, Sfeir A (2016) Stop pulling my strings—what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol 17:364–378. https://doi.org/10.1038/nrm.2016.43
Lee J, Solomon DA, Tihan T (2017) The role of histone modifications and telomere alterations in the pathogenesis of diffuse gliomas in adults and children. J Neurooncol 132:1–11. https://doi.org/10.1007/s11060-016-2349-9
Lee JK, Wang J, Sa JK, Ladewig E, Lee HO, Lee IH, Kang HJ, Rosenbloom DS, Camara PG, Liu Z et al (2017) Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet 49:594–599. https://doi.org/10.1038/ng.3806
Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W (2017) Glioma: experimental models and reality. Acta Neuropathol 133:263–282. https://doi.org/10.1007/s00401-017-1671-4
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340:857–861. https://doi.org/10.1126/science.1232245
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R et al (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947
Liggett WH Jr, Sidransky D (1998) Role of the p16 tumor suppressor gene in cancer. J Clin Oncol 16:1197–1206. https://doi.org/10.1200/JCO.1998.16.3.1197
Louis DN, Giannini C, Capper D, Paulus W, Figarella-Branger D, Lopes MB, Batchelor TT, Cairncross JG, van den Bent M, Wick W et al (2018) cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. https://doi.org/10.1007/s00401-018-1826-y
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D, Perry A, Reifenberger G, Deimling AV (2016) WHO classification of tumours of the central nervous system. IARC Press, Lyon
Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A, Aldape K, Brat D, Collins VP, Eberhart C et al (2014) International Society Of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24:429–435. https://doi.org/10.1111/bpa.12171
Louis DN, Wesseling P, Paulus W, Giannini C, Batchelor TT, Cairncross JG, Capper D, Figarella-Branger D, Lopes MB, Wick W et al (2018) cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol 135:481–484. https://doi.org/10.1007/s00401-018-1808-0
Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M et al (2012) Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8:e1002772. https://doi.org/10.1371/journal.pgen.1002772
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478. https://doi.org/10.1038/nature10860
Luchman HA, Chesnelong C, Cairncross JG, Weiss S (2013) Spontaneous loss of heterozygosity leading to homozygous R132H in a patient-derived IDH1 mutant cell line. Neuro-oncology 15:979–980. https://doi.org/10.1093/neuonc/not064
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V et al (2013) Non-cell-autonomous tumor suppression by p53. Cell 153:449–460. https://doi.org/10.1016/j.cell.2013.03.020
Maciejowski J, de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18:175–186. https://doi.org/10.1038/nrm.2016.171
Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T (2015) Chromothripsis and Kataegis induced by telomere crisis. Cell 163:1641–1654. https://doi.org/10.1016/j.cell.2015.11.054
Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM (2013) Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 23:762–776. https://doi.org/10.1101/gr.143677.112
Mangerel J, Price A, Castelo-Branco P, Brzezinski J, Buczkowicz P, Rakopoulos P, Merino D, Baskin B, Wasserman J, Mistry M et al (2014) Alternative lengthening of telomeres is enriched in, and impacts survival of TP53 mutant pediatric malignant brain tumors. Acta Neuropathol 128:853–862. https://doi.org/10.1007/s00401-014-1348-1
Martinez P, Blasco MA (2011) Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer 11:161–176. https://doi.org/10.1038/nrc3025
Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E (2014) PI3K/AKT signaling pathway and cancer: an updated review. Ann Med 46:372–383. https://doi.org/10.3109/07853890.2014.912836
Maryoung L, Yue Y, Young A, Newton CA, Barba C, van Oers NS, Wang RC, Garcia CK (2017) Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. J Clin Investig 127:982–986. https://doi.org/10.1172/JCI91161
Maser RS, DePinho RA (2002) Connecting chromosomes, crisis, and cancer. Science 297:565–569. https://doi.org/10.1126/science.297.5581.565
Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, Smirnov IV, Marshall R, Souza CF, Shen Y et al (2017) Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci USA 114:10743–10748. https://doi.org/10.1073/pnas.1708914114
McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, Thomas DM, Mitchell G (2014) Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol 11:260–271. https://doi.org/10.1038/nrclinonc.2014.41
McCarthy N (2012) Metabolism: unmasking an oncometabolite. Nat Rev Cancer 12:229. https://doi.org/10.1038/nrc3248
McClintock B (1938) The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics 23:315–376
McClintock B (1941) The stability of broken ends of chromosomes in Zea Mays. Genetics 26:234–282
McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168:613–628. https://doi.org/10.1016/j.cell.2017.01.018
Meeker AK, Hicks JL, Iacobuzio-Donahue CA, Montgomery EA, Westra WH, Chan TY, Ronnett BM, De Marzo AM (2004) Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res 10:3317–3326. https://doi.org/10.1158/1078-0432.CCR-0984-03
Melin BS, Barnholtz-Sloan JS, Wrensch MR, Johansen C, Il’yasova D, Kinnersley B, Ostrom QT, Labreche K, Chen Y, Armstrong G et al (2017) Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat Genet 49:789–794. https://doi.org/10.1038/ng.3823
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436:720–724. https://doi.org/10.1038/nature03890
Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW (1999) Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet 21:115–118. https://doi.org/10.1038/5063
Napier CE, Huschtscha LI, Harvey A, Bower K, Noble JR, Hendrickson EA, Reddel RR (2015) ATRX represses alternative lengthening of telomeres. Oncotarget 6:16543–16558. https://doi.org/10.18632/oncotarget.3846
Nikolaev S, Santoni F, Garieri M, Makrythanasis P, Falconnet E, Guipponi M, Vannier A, Radovanovic I, Bena F, Forestier F et al (2014) Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 5:5690. https://doi.org/10.1038/ncomms6690
Noorbakhsh J, Chuang JH (2017) Uncertainties in tumor allele frequencies limit power to infer evolutionary pressures. Nat Genet 49:1288–1289. https://doi.org/10.1038/ng.3876
Nordling CO (1953) A new theory on cancer-inducing mechanism. Br J Cancer 7:68–72
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–28
O’Sullivan RJ, Karlseder J (2010) Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11:171–181. https://doi.org/10.1038/nrm2848
Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC et al (2004) Genetic pathways to glioblastoma: a population-based study. Can Res 64:6892–6899. https://doi.org/10.1158/0008-5472.CAN-04-1337
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453. https://doi.org/10.2353/ajpath.2007.070011
Olar A, Aldape KD (2014) Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol 232:165–177. https://doi.org/10.1002/path.4282
Olovnikov AM (1973) A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41:181–190
Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS (2015) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neurooncology 17(Suppl 4):iv1–iv62 https://doi.org/10.1093/neuonc/nov189
Ouchi R, Okabe S, Migita T, Nakano I, Seimiya H (2016) Senescence from glioma stem cell differentiation promotes tumor growth. Biochem Biophys Res Commun 470:275–281. https://doi.org/10.1016/j.bbrc.2016.01.071
Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC (2014) Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 26:288–300. https://doi.org/10.1016/j.ccr.2014.06.005
Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H et al (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460:66–72. https://doi.org/10.1038/nature08137
Parkinson GN, Lee MP, Neidle S (2002) Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 417:876–880. https://doi.org/10.1038/nature755
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL et al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344:1396–1401. https://doi.org/10.1126/science.1254257
Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, Sicotte H, Kollmeyer TM, McCoy LS, Sarkar G et al (2017) Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol 133:1001–1016. https://doi.org/10.1007/s00401-017-1690-1
Pennisi E (1997) New tumor suppressor found–twice. Science 275:1876–1878
Perez-Mancera PA, Young AR, Narita M (2014) Inside and out: the activities of senescence in cancer. Nat Rev Cancer 14:547–558. https://doi.org/10.1038/nrc3773
Perry A, Wesseling P (2016) Histologic classification of gliomas. Handb Clin Neurol 134:71–95. https://doi.org/10.1016/B978-0-12-802997-8.00005-0
Prensner JR, Chinnaiyan AM (2011) Metabolism unhinged: IDH mutations in cancer. Nat Med 17:291–293. https://doi.org/10.1038/nm0311-291
Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E et al (2011) BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res 17:3590–3599. https://doi.org/10.1158/1078-0432.CCR-10-3349
Raj N, Attardi LD (2013) Tumor suppression: p53 alters immune surveillance to restrain liver cancer. Curr Biol CB 23:R527–R530. https://doi.org/10.1016/j.cub.2013.04.076
Rajaraman P, Melin BS, Wang Z, McKean-Cowdin R, Michaud DS, Wang SS, Bondy M, Houlston R, Jenkins RB, Wrensch M et al (2012) Genome-wide association study of glioma and meta-analysis. Hum Genet 131:1877–1888. https://doi.org/10.1007/s00439-012-1212-0
Ramamoorthy M, Smith S (2015) Loss of ATRX suppresses resolution of telomere cohesion to control recombination in ALT cancer cells. Cancer Cell 28:357–369. https://doi.org/10.1016/j.ccell.2015.08.003
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V et al (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. https://doi.org/10.1007/s00401-014-1370-3
Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL (2010) Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene 29:335–344. https://doi.org/10.1038/onc.2009.333
Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD (2001) Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 409:633–637. https://doi.org/10.1038/35054579
Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B et al (1993) Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363:515–521. https://doi.org/10.1038/363515a0
Rozen WM, Joseph S, Lo PA (2008) Spontaneous regression of low-grade gliomas in pediatric patients without neurofibromatosis. Pediatr Neurosurg 44:324–328. https://doi.org/10.1159/000134925
Sanborn JZ, Salama SR, Grifford M, Brennan CW, Mikkelsen T, Jhanwar S, Katzman S, Chin L, Haussler D (2013) Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Can Res 73:6036–6045. https://doi.org/10.1158/0008-5472.CAN-13-0186
Sanoudou D, Tingby O, Ferguson-Smith MA, Collins VP, Coleman N (2000) Analysis of pilocytic astrocytoma by comparative genomic hybridization. Br J Cancer 82:1218–1222. https://doi.org/10.1054/bjoc.1999.1066
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J et al (2012) D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Gene Dev 26:2038–2049. https://doi.org/10.1101/gad.198200.112
Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS et al (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Can Res 70:512–519. https://doi.org/10.1158/0008-5472.CAN-09-1851
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M et al (2011) Analysis of BRAF V600E mutation in 1320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. https://doi.org/10.1007/s00401-011-0802-6
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. https://doi.org/10.1038/nature10833
Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B et al (2015) The genetic evolution of melanoma from precursor lesions. New Engl J Med 373:1926–1936. https://doi.org/10.1056/NEJMoa1502583
Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15:397–408. https://doi.org/10.1038/nrc3960
Shay JW, Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33:787–791. https://doi.org/10.1016/S0959-8049(97)00062-2
Shay JW, Pereira-Smith OM, Wright WE (1991) A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 196:33–39
Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY et al (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41:899–904. https://doi.org/10.1038/ng.407
Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, Burrows AE, Anselmo AN, Bredemeyer AL, Li MZ et al (2012) Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science 337:104–109. https://doi.org/10.1126/science.1219580
Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110:4009–4014. https://doi.org/10.1073/pnas.1219747110
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T et al (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15:356–362. https://doi.org/10.1038/ng0497-356
Stone RC, Horvath K, Kark JD, Susser E, Tishkoff SA, Aviv A (2016) Telomere length and the cancer-atherosclerosis trade-off. PLoS Genet 12:e1006144. https://doi.org/10.1371/journal.pgen.1006144
Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF et al (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14:92–107. https://doi.org/10.1038/nrc3655
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV et al (2014) Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157:580–594. https://doi.org/10.1016/j.cell.2014.02.030
Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F et al (2015) Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47:458–468. https://doi.org/10.1038/ng.3273
Tanaka H, Yao MC (2009) Palindromic gene amplification—an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer 9:216–224. https://doi.org/10.1038/nrc2591
Tang YC, Amon A (2013) Gene copy-number alterations: a cost-benefit analysis. Cell 152:394–405. https://doi.org/10.1016/j.cell.2012.11.043
Tarabichi M, Martincorena I, Gerstung M, Markowetz F, Spellman PT, Morris QD, Lingjaerde OC, Wedge DC, Van Loo P (2017) Neutral tumor evolution? bioRxiv: https://doi.org/10.1101/158006
Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. https://doi.org/10.1038/ng.2925
Telomeres Mendelian Randomization C, Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM et al (2017) Association between telomere length and risk of cancer and non-neoplastic diseases: a mendelian randomization study. JAMA Oncol 3:636–651. https://doi.org/10.1001/jamaoncol.2016.5945
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG et al (2016) Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539:309–313. https://doi.org/10.1038/nature20123
Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B (2015) Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci USA 112:118–123. https://doi.org/10.1073/pnas.1421839112
Turcan S, Chan TA (2013) MAPping the genomic landscape of low-grade pediatric gliomas. Nat Genet 45:847–849. https://doi.org/10.1038/ng.2706
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483. https://doi.org/10.1038/nature10866
Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA et al (2017) Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543:122–125. https://doi.org/10.1038/nature21356
Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC (2002) Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Can Res 62:5551–5558
Uhrbom L, Nister M, Westermark B (1997) Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene 15:505–514. https://doi.org/10.1038/sj.onc.1201227
van den Bent MJ (2010) Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol 120:297–304. https://doi.org/10.1007/s00401-010-0725-7
van Thuijl HF, Scheinin I, Sie D, Alentorn A, van Essen HF, Cordes M, Fleischeuer R, Gijtenbeek AM, Beute G, van den Brink WA et al (2014) Spatial and temporal evolution of distal 10q deletion, a prognostically unfavorable event in diffuse low-grade gliomas. Genome Biol 15:471. https://doi.org/10.1186/s13059-014-0471-6
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C et al (2017) Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. https://doi.org/10.1126/science.aai8478
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Vescovi AL, Galli R, Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6:425–436. https://doi.org/10.1038/nrc1889
Vestin A, Mills AA (2013) The tumor suppressor Chd5 is induced during neuronal differentiation in the developing mouse brain. Gene Express Pattern GEP 13:482–489. https://doi.org/10.1016/j.gep.2013.09.003
Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L et al (2013) Frequency of TERT promoter mutations in human cancers. Nat Commun 4:2185. https://doi.org/10.1038/ncomms3185
Vogelstein B, Kinzler KW (2015) The path to cancer—three strikes and you’re out. New Engl J Med 373:1895–1898. https://doi.org/10.1056/NEJMp1508811
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558. https://doi.org/10.1126/science.1235122
Walsh KM, Codd V, Rice T, Nelson CP, Smirnov IV, McCoy LS, Hansen HM, Elhauge E, Ojha J, Francis SS et al (2015) Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 6:42468–42477. https://doi.org/10.18632/oncotarget.6468
Walsh KM, Codd V, Smirnov IV, Rice T, Decker PA, Hansen HM, Kollmeyer T, Kosel ML, Molinaro AM, McCoy LS et al (2014) Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk. Nat Genet 46:731–735. https://doi.org/10.1038/ng.3004
Walsh KM, Wiencke JK, Lachance DH, Wiemels JL, Molinaro AM, Eckel-Passow JE, Jenkins RB, Wrensch MR (2015) Telomere maintenance and the etiology of adult glioma. Neuro-oncology 17:1445–1452. https://doi.org/10.1093/neuonc/nov082
Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin YJ et al (2016) Clonal evolution of glioblastoma under therapy. Nat Genet 48:768–776. https://doi.org/10.1038/ng.3590
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y et al (2017) Tumor evolution of Glioma-Intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(42–56):e46. https://doi.org/10.1016/j.ccell.2017.06.003
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234. https://doi.org/10.1016/j.ccr.2010.01.020
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153. https://doi.org/10.2353/ajpath.2009.080958
Wesseling P, Capper D (2018) WHO 2016 classification of gliomas. Neuropathol Appl Neurobiol 44:139–150. https://doi.org/10.1111/nan.12432
Wiestler B, Capper D, Hovestadt V, Sill M, Jones DT, Hartmann C, Felsberg J, Platten M, Feiden W, Keyvani K et al (2014) Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro-oncology 16:1630–1638. https://doi.org/10.1093/neuonc/nou138
Wilcox P, Li CC, Lee M, Shivalingam B, Brennan J, Suter CM, Kaufman K, Lum T, Buckland ME (2015) Oligoastrocytomas: throwing the baby out with the bathwater? Acta Neuropathol 129:147–149. https://doi.org/10.1007/s00401-014-1353-4
Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A (2016) Identification of neutral tumor evolution across cancer types. Nat Genet 48:238–244. https://doi.org/10.1038/ng.3489
Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A (2017) Reply: uncertainties in tumor allele frequencies limit power to infer evolutionary pressures. Nat Genet 49:1289–1291. https://doi.org/10.1038/ng.3877
Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S et al (2009) Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 41:905–908. https://doi.org/10.1038/ng.408
Wright WE, Pereira-Smith OM, Shay JW (1989) Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol 9:3088–3092
Wright WE, Shay JW (1992) The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol 27:383–389
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253. https://doi.org/10.1038/ng.1102
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444–450. https://doi.org/10.1038/ng.2938
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30. https://doi.org/10.1016/j.ccr.2010.12.014
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ et al (2009) IDH1 and IDH2 mutations in gliomas. New Engl J Med 360:765–773. https://doi.org/10.1056/NEJMoa0808710
Yates LR, Campbell PJ (2012) Evolution of the cancer genome. Nat Rev Genet 13:795–806. https://doi.org/10.1038/nrg3317
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I et al (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602–612. https://doi.org/10.1038/ng.2611
Zheng S, Fu J, Vegesna R, Mao Y, Heathcock LE, Torres-Garcia W, Ezhilarasan R, Wang S, McKenna A, Chin L et al (2013) A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev 27:1462–1472. https://doi.org/10.1101/gad.213686.113
Acknowledgements
We thank members of the Verhaak laboratory for insightful discussion and valuable input. We thank Zoë Reifsnyder of the Jackson Laboratory creative team for artwork. This work was supported by Grants from the National Institutes of Health R01 CA190121 and P30CA034196, the National Brain Tumor Society Oligo Research Fund and DefeatGBM Initiative, and Grant 11026 from the Dutch Cancer Society KWF. We have no disclosures to make.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Barthel, F.P., Wesseling, P. & Verhaak, R.G.W. Reconstructing the molecular life history of gliomas. Acta Neuropathol 135, 649–670 (2018). https://doi.org/10.1007/s00401-018-1842-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1842-y