
Abstract
Mutations leading to premature termination codons in ATP-Binding Cassette Subfamily A Member 7 (ABCA7) are high penetrant risk factors of Alzheimer’s disease (AD). The influence of other genetic variants in ABCA7 and downstream functional mechanisms, however, is poorly understood. To address this knowledge gap, we investigated tandem repetitive regions in ABCA7 in a Belgian cohort of 1529 AD patients and control individuals and identified an intronic variable number tandem repeat (VNTR). We observed strong association between VNTR length and a genome-wide associated signal for AD in the ABCA7 locus. Expanded VNTR alleles were highly enriched in AD patients [odds ratio = 4.5 (1.3–24.2)], and VNTR length inversely correlated with amyloid β1–42 in cerebrospinal fluid and ABCA7 expression. In addition, we identified three novel ABCA7 alternative splicing events. One isoform in particular—which is formed through exon 19 skipping—lacks the first nucleotide binding domain of ABCA7 and is abundant in brain tissue. We observed a tight correlation between exon 19 skipping and VNTR length. Our findings underline the importance of studying repetitive DNA in complex disorders and expand the contribution of genetic and transcript variation in ABCA7 to AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a highly prevalent, incurable, multifactorial neurodegenerative disease with a long presymptomatic phase of several decades. ATP-Binding Cassette Subfamily A Member 7 (ABCA7) was identified as a risk factor for AD through genome-wide association studies (GWAS) [16, 17, 23, 24, 31, 41]. Subsequently, rare heterozygous premature termination codon (PTC) mutations in ABCA7 were identified and were up to five times more frequent in AD patients. Up to 4% of AD patients in Caucasian populations carry an ABCA7 PTC mutation [2, 10, 15, 22, 38, 40, 46, 48]. In African Americans (AA) a PTC-causing 44 bp exonic deletion (rs142076058) was identified in 15% of patients, which doubled AD risk, and explained an AA-specific GWAS hit [9]. Risk-increasing PTC mutations are not confined to specific ABCA7 protein domains, proposing reduction of overall ABCA7 dosage—due to nonsense mediated mRNA decay (NMD) or inactive truncated proteins—as the most plausible pathomechanism. ABCA7 is deemed to be involved in phagocytosis and/or lipid metabolism and is mainly expressed in hippocampal neurons and microglia in the brain [20]. Knockout of the mouse ortholog Abca7 was shown to reduce phagocytic clearance of amyloid β (Aβ) [12, 21, 39], the main constituent of senile plaques, which together with neurofibrillary tangles and reactive gliosis form the neuropathological hallmarks of AD.
To date, however, a biological variant explaining the GWAS association in Caucasian cohorts remained elusive. None of the PTC mutations are in linkage disequilibrium (LD) with associated GWAS SNPs. Studies have shown association between GWAS SNP rs3764650 and ABCA7 expression, albeit in opposite directions [3, 49]. Methylation of CpG sites in the ABCA7 promoter region was independent of this SNP [54]. Possibly, rs3764650 itself could directly affect AD risk [4], however conditional regression analyses suggest a stronger association with a less frequent SNP rs78117248 [10, 22]. No apparent biological consequences can be linked to rs78117248 (e.g. no overlap with transcription factor motifs), but its stronger association suggests the presence of a yet unknown common genetic variant underlying the discovery of ABCA7 as a susceptibility locus of AD.
We previously sequenced the ABCA7 locus—including coding, intronic, upstream and downstream sequences—in the Belgian AD cohort, which identified PTC mutations, but did not reveal a functionally strong genetic variant that could explain the GWAS signal [10]. Here we report the identification of a variable number of tandem repeats (VNTR) with a 25 bp repeat unit located in intron 18 of ABCA7, immediately flanking exon 18 and including the exon 18 splice donor site (chr19:1049437–1050028 [hg19]). We assessed the role of this ABCA7 VNTR in AD and associated cerebrospinal fluid (CSF) biomarkers, expression, and alternative splicing.
Materials and methods
Study population
Participants were part of a large prospective cohort of Belgian AD patients and healthy elderly control individuals. Patients were ascertained at the Memory Clinic of Middelheim and Hoge Beuken (Hospital Network Antwerp, Belgium), and at the Memory Clinic of the University Hospitals of Leuven, Belgium. Possible, probable, and definite AD diagnosis was based on NINCDS-ADRA [28] and/or NIA-AA [18, 29] criteria. Control individuals were recruited from partners of patients, or were volunteers from the Belgian community. All control individuals scored > 25 on the Montreal Cognitive Assessment (MoCA) test [32], and were negative for subjective memory complaints, neurological or psychiatric antecedents, and family history of neurodegeneration. All participants and/or their legal guardian provided written informed consent before inclusion. The study protocols were approved by the ethics committees of the Antwerp University Hospital and the participating neurological centers at the different hospitals of the BELNEU consortium and by the University of Antwerp. Table S1 provides an overview of individuals for whom DNA, Epstein-Barr virus transformed lymphoblastoid cell lines (LCL), or CSF were included.
VNTR genotyping
Genomic start and end coordinates of the ABCA7 VNTR were delineated with the Tandem Repeat Finder algorithm [6], as implemented in the Tandem Repeats Database (TRDB) [14]. To further assess whether the VNTR length could contribute to AD risk, we used the genotyping methods described below.
ABCA7 VNTR identification with NGS data
We analyzed the depth of NGS sequencing reads aligning to the ABCA7 VNTR as a proxy for VNTR length. BAM files from 772 AD patients, and 757 matched healthy elderly control individuals—generated as previously described [10]—were used for this purpose. The total number of reads in the BAM file and the number of reads mapping to the VNTR core sequence (chr19:1049514–1049953 [hg19]), which excludes sequencing reads aligning to the VNTR breakpoints, were calculated with Rsamtools [30]. Individuals with very low coverage (≤ 5 VNTR sequencing reads) were not included. VNTR coverage was then normalized by division by the total number of reads for that sample. Association between normalized coverage and SNP genotypes was calculated with a genotypic Kruskal–Wallis test. D′ and r2 LD values were established previously [10].
To analyze the association between VNTR size and GWAS SNP genotypes in an extended population, we used NGS data of the 1000 Genomes Project [1]. Aligned low coverage whole genome sequencing data from European individuals (i.e. British in England and Scotland, Finnish in Finland, Iberian populations in Spain, Utah residents with Northern and Western European ancestry, and Tuscany in Italy) were processed in an analogous manner as the Belgian NGS dataset described above.
PCR and Sanger sequencing of short VNTR alleles
We designed a PCR protocol to amplify ABCA7 VNTRs up to approximately 3 kb. This upper limit is determined by PCR efficiency, which is inversely correlated with increasing amplicon size and hampered by high GC content. We used the Kapa 2G Robust HotStart PCR Kit (Sigma-Aldrich, Saint Louis, MO, USA), with a custom protocol as described in the Supplementary Methods. The forward and reverse primer sequences, used in this setting, were respectively 5′-GGCTCAGCCTGGACTTCTAC-3′ and 5′-TCCAAAACCCTGTGATAGCC-3′. The shortest VNTRs were Sanger sequenced, to determine the minimal number of tandem repeats. PCR products were purified with ExoSAP-IT (Thermo Fisher Scientific, Waltham, MA, USA), dideoxy terminated with BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), and processed on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific), according to the manufacturer’s protocol. Analysis was done with SeqMan II software (DNASTAR, Madison, WI, USA).
Southern blotting
Southern blotting was used to genotype VNTR lengths in 275 patients and 177 control individuals (Table S1, Fig. S1), including individuals without pathogenic mutations [8] in APP, PSEN1, and PSEN2, or ABCA7 PTC mutations (incl. rs200538373) [10]. Furthermore, we excluded individuals for whom only a single band on Southern blotting was observed, since this could reflect failed detection of the second VNTR allele. The Southern blotting protocol was adapted from the DIG Application Manual For Filter Hybridization (Roche, Basel, Switzerland) (details provided in Supplementary Methods).
Statistical analyses on Southern blotted VNTR alleles were performed in R3.3.2 [35], unless mentioned otherwise. VNTR allele distribution was compared to genotypes of rs3764650 and rs78117248, which were determined previously [10, 45]. Association between SNP genotypes and phenotype was assessed with allelic Fisher exact tests. VNTR length association testing per SNP was done with a Kruskal–Wallis Rank Sum test. Optimal VNTR size cutoff to distinguish expanded and wild-type VNTR lengths was determined as the largest VNTR allele length observed in control individuals without an rs3764650 risk allele. Association testing and calculation of odds ratio (OR) with 95% confidence interval (95% CI) between expanded and wild-type allele carriers in patients and controls was calculated with Fisher exact testing. Calculations of heritability attributable to an ABCA7 VNTR expansion were performed with INDI-V [52], assuming AD heritability of liability of 79% [13], a 13% risk of AD after age 65 years [42], and a multiplicative model. Association of VNTR length with age at onset (AAO) within patients was calculated with linear regression, using the sum of VNTR alleles or longest VNTR allele, APOE ε4 dosage, and gender as independent variables. AAO difference between patients with and without an expanded VNTR allele was assessed using a Mann–Whitney U test.
To test whether VNTR allele lengths share a haplotype with ABCA7 PTC mutations, we used Southern blot to genotype VNTR length in an additional seventeen individuals carrying a previously identified PTC mutation [10]. These individuals were not included in any statistical analyses.
Correlation between VNTR and CSF biomarkers
CSF concentrations of amyloid β1–42 (Aβ1–42), phosphorylated tau181P (P-tau181P), and total tau (T-tau) were determined for a subset of 168 patients from the Southern blotting cohort (Table S1) as part of their diagnostic work-up with commercially available single parameter ELISA kits (Fujirebio Europe, Ghent, Belgium). Linear regression with the sum of VNTR alleles or longest VNTR allele as the independent variable and log2 transformed biomarker levels as the dependent variable was calculated in R [35]. Results are reported as β-regression coefficients with standard error (SE) and p value.
Expression studies
Procedures for RNA extraction, cDNA generation and characteristics of the LCL cohort are given in Supplementary Methods.
Quantitative real time PCR
Quantitative real time PCR (qRT-PCR) was performed with the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific), according to the manufacturer’s protocol in a 384-well plate setting on LCL RNA. Each sample-gene pair was conducted in triplicate and on two different cDNA batches from separate RNA extractions. ABCA7 transcripts were targeted using the Hs01105117_m1 Taqman assay, with primers located in exon 45 and 46 of ABCA7 (Fig. 1), and three reference genes were used: HPRT1 (Hs02800695_m1), GAPDH (Hs99999905_m1), and YWHAZ (Hs00237047_m1). The PCR protocol (2 min at 50 °C, then 20 s at 95 °C, followed by 40 cycles of 1 s at 95 °C, and 20 s at 60 °C), and fluorescence detection was performed on a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). Ct values were exported to Qbase+ (Biogazelle, Gent, Belgium) to calculate relative expression.

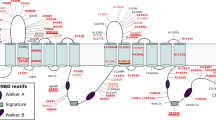
Genomic location of the ABCA7 VNTR and its effect on ABCA7 splicing. a The ABCA7 genomic conformation (NM_019112.3) and positions of rs3764650, the VNTR, and rs78117248 are shown. Pairwise D′ and r2 between rs3764650 and rs78117248 is noted on top. Black arrows superimposed on exons 45 and 46 indicate the primers used for qPCR. Below, ABCA7 exons 17–20 (blue rectangles), introns (black line), VNTR units (black rectangles), and primers in exons 17 and 20 for isoform quantification (black arrows) are represented. Twelve VNTR units are depicted, corresponding to the smallest observed VNTR length. The 25 bp VNTR unit sequence is shown in more detail, with the first two bases corresponding to a splice donor site. Caret-like exon connecting lines depict canonical (green), exon 18 cryptic splice acceptor (orange), retention of a single VNTR unit (purple) and exon 19 skipping (red) splicing. b Canonical spicing of the entire ABCA7 gene leads to a full length ABCA7 protein, containing two nucleotide binding domains (NBD). Usage of a cryptic splice acceptor in exon 18 and retention of a single VNTR unit lead to a frameshift, which causes a premature termination codon (PTC) that leads to either a truncated protein or degradation of the mRNA via nonsense mediated mRNA decay (NMD). Complete skipping of exon 19 causes a deletion of 44 amino acids embedded in the first ABCA7 NBD
Identification of novel alternatively spliced isoforms
Splicing events in proximity to the VNTR were identified using cDNA PCR amplification followed by Sanger sequencing in both LCL and brain cDNA. Forward primers were positioned in exon 17 (5′-TTTCGGAGGAGCTACTGGTG-3′), or on the exon18-cryptic-acceptor-splice-site-breakpoint (5′-GACCCAAAGGCCTGGAGA-3′), and paired with a reverse primer in exon 20 (5′-GTGCTCGTCCACGGTCAG-3′). Amplification was performed with Titanium Taq (Clontech Laboratories, Mountain View, CA, USA) in a 1 M betain solution using the following thermal cycler protocol: 2 min at 95 °C, followed by 35 cycles of 30 s at 95 °C and 1.5 min at 68 °C, and finished by 3 min at 68 °C. The Sanger sequencing reaction was then carried out with BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), and subsequently processed on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific). Analysis was performed with Seqman software (DNASTAR, Madison, WI, USA). All splicing events were further confirmed by analyzing in-house RNAseq data (described in Verheijen et al. [50]) with the use of Integrative Genomics Viewer (IGV) software [37].
Isoform quantification
Capillary fragment analysis was used to quantify eight observed isoform combinations. PCR amplification of cDNA was analogous to the isoform identification method described above, using a 5′ FAM-labeled forward primer in exon 17 (5′-TTTCGGAGGAGCTACTGGTG-3′), and an exon 20 reverse primer (5′-GTGCTCGTCCACGGTCAG-3′). The PCR amplicons were then supplemented with formamide and GeneScan 600 LIZ Size Standard (Thermo Fisher Scientific) and size separated on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific). Analysis was performed with MAQ-S v1.5.0 software (Agilent). Manual inspection was performed to confirm that fluorescence signals corresponded to the expected size, and signal heights were within quantifiable range. Ratios for canonical, exon 18 cryptic splice acceptor, and exon 19 skipping isoforms were calculated as the signal area of the corresponding isoform peaks divided by the total area of these peaks.
Allele-specific isoform expression
To examine the effect of individual VNTR alleles on ABCA7 isoform abundancy we performed allele-specific isoform expression on an individual with a small (837 bp) and large (9711 bp) VNTR allele, and heterozygosis (A/G) for nearby exonic polymorphism rs3752240. VNTR alleles were phased with rs3752240 on genomic DNA according to the “short VNTR allele PCR protocol” (Supplementary Methods) using primers 5′-CAAGACCACCACCCTGTGA-3′ and 5′-AGAGATGGGGAAGGGACCTC-3′. Since the largest allele is too large for PCR amplification, only the shortest allele will be observed, allowing phasing. RNA extraction and cDNA synthesis was performed as described above. Subsequently the “Isoform quantification PCR protocol” was used for which the forward and reverse primers were 5′ supplemented with Illumina Nextera Adapters (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′ respectively). The amplicon was then PCR barcoded with i5 and i7 adapters and 300 bp paired end sequenced on MiSeq with v3 chemistry (Illumina, San Diego, CA, USA). GMAP was used to align and output splicing events of sequencing reads [53]. Genotypes for rs3752240 per read were determined with samtools [25].
Statistical analysis of ABCA7 overall and isoform quantifications
The qRT-PCR and isoform quantifications were compared to the sum of the VNTR alleles. Since the difference between two VNTR alleles within an LCL sample was kept to a minimum (Supplementary Methods), the VNTR sum value corresponds well to the underlying individual alleles. Association between expression and VNTR sum was calculated with linear mixed-effects models, using lme4 in R [5, 35], correcting for gender and batch in which LCL were grown. The full model was compared to the null model (without VNTR sum) using a Likelihood Ratio test. Linear regression was used to calculate the expression fold change between different VNTR sizes. The analyses were additionally performed using the longest VNTR allele as independent variable.
Results
VNTR identification and correlation with the GWAS index SNP at ABCA7
We examined repetitive regions in ABCA7, which were poorly assessed by conventional NGS data analysis, in our previously generated NGS dataset [10]. This revealed many sequencing reads with low mapping quality in intron 18, close to exon 18, aligning to a low complexity C-rich region. Further inspection indicated a 25 bp tandem repeating pattern. The VNTR comprises 592 bp of the human reference genome (chr19:1049437–1050028 [hg19]), corresponding to 23.7 repetitions of the 25 bp VNTR core repeat motif (Fig. 1). The first two nucleotides (GT) of the VNTR correspond to the canonical splice donor site of exon 18. The sequence between VNTR repeat units can vary slightly (Fig. S2).
First, we investigated the depth of NGS sequencing reads aligning to the VNTR (chr19:1049514–1049953 [hg19]) as a proxy for VNTR length, in relation to the genotype of ABCA7 GWAS index SNP rs3764650. Carriers of the risk increasing rs3764650[G]-allele had increased read depth in the Belgian cohort (n = 1529; Fig. S3, p = 2.2 × 10−16) [10], as well as in low coverage whole genome sequencing data in European subjects derived from the 1000 Genomes Project (n = 403; Fig. S4, p = 0.01) [1].
To directly genotype VNTR length, we performed Southern blotting on 452 individuals. For each individual, two size-separated bands were imaged corresponding to the VNTR alleles; no signs of mosaicism were present (Fig. S1). We observed high variability in VNTR lengths between individuals. The smallest allele was 298 bp (approx. 12 repeat units), which could be confirmed with Sanger sequencing. The largest observed allele size was 10678 bp (approx. 427 tandem repeats). Relative to the ABCA7 VNTR in the human reference genome (chr19:1049437–1050028 [hg19], 592 bp), the longest VNTR length causes an eightfold size increase of intron 18 and a 39% extension of ABCA7 pre-mRNA. Increasing VNTR lengths were associated with risk alleles of rs3764650 (p = 6.7 × 10−8) and rs78117248 (p = 6.3 × 10−4) (Fig. 2a and Fig. S5), confirming our observations based on NGS-based approximation.

Association of ABCA7 VNTR lengths with AD and AD-associated SNPs. a The distribution of largest and smallest Southern blotted VNTR alleles is shown in relation to the genotype of rs3764650 and rs78117248 in three categories: homozygous reference allele (blue), heterozygous (green), and homozygous risk allele (red) carriers. These distributions are shown per phenotype in Fig. S5. b The main panel depicts patients (red) and healthy elderly control individuals (blue) according to their largest (x-axis) and smallest (y-axis) VNTR allele, as determined by Southern blotting. The upper and right panel show the Q–Q distance, respectively corresponding to the largest and smallest VNTR allele. A positive distance reflects enrichment in AD patients, while a negative distance corresponds to enrichment in controls
We assessed normal variability of VNTR lengths (median = 2388 bp, interquartile range = 2130 bp) within healthy elderly without an rs3764650 risk allele (n = 133) and observed a maximum VNTR length of 5720 bp (Fig. S6).
ABCA7 VNTR expansion is associated with AD
For individuals with two alleles within the normal range (< 5720 bp, “wild-type”), distribution was similar between patients and controls (Fig. 2b). However, individuals carrying an allele > 5720 bp (“expanded”), were mostly patients [OR 4.5 (95% CI 1.3–24.2), p = 0.008, Fig. 2b]. Twenty patients (7.3%) carried an expanded VNTR allele in contrast to three controls (1.7%) (Table S2). In this cohort, the heritability of liability attributable to ABCA7 VNTR expansion is estimated at 3.1%. Among carriers of an expanded allele, the second (smaller) allele was longer in patients (2751 ± 1164 bp) than controls (1001 ± 652 bp). In the group with Southern blot data, 25% of patients and 22% of controls carried at least one rs3764650 risk allele. The latter is not significantly associated with AD in this cohort (p = 0.6), precluding conditional regression analysis. For expanded VNTR allele carriers, however, the proportion of rs3764650 risk alleles increased to 63 and 100% for patients and controls, respectively (Table S2). We observed no shared haplotype between expanded VNTR alleles and ABCA7 PTC mutations (Fig. S7). Patients with an expanded VNTR allele had an onset age ranging from 44 to 90 years. The age at inclusion for all three control individuals falls within this range (Table S2). We observed no association between VNTR length and onset age in patients (p = 1, Table S3, Table S4), nor for expanded VNTR allele carriers compared to other patients (p = 0.9). We analyzed the correlation of VNTR length with CSF biomarkers for AD (Fig. 3a–c) in 168 AD patients. A decrease in Aβ1–42 was observed with increasing VNTR length (βlog(Aβ) = − 4.6 × 10−5, SE = 2.1 × 10−5, p = 0.03 for sum of VNTR alleles). For P-tau181P, a directly proportional trend was observed, albeit not significant (βlog(p-tau) = − 4.0 × 10−5, SE = 2.3 × 10−5, p = 0.09 for sum of VNTR alleles). We did not detect an association of VNTR length on T-tau (Table S3).

Effect of the ABCA7 VNTR on CSF biomarkers of AD. Logarithmic levels (y-axis) of CSF Aβ1–42 (a), P-tau181P (b), and T-tau (c) in AD patients corresponding to the sum of VNTR allele lengths (x-axis) within those patients. A trendline (blue) is shown with standard error (shaded area)
VNTR length is associated with decreased ABCA7 expression
We observed a decrease in overall ABCA7 expression with increasing VNTR length (β = − 4.7 × 10−5, SE = 1.9 × 10−5, p = 0.01 for sum of VNTR alleles) (Fig. 4a, Table S3). This trend was observed for both patients and controls. The average expression for the sample with the largest diploid combination of VNTR alleles (5399 + 4575 bp) was 33% lower than the expression at the smallest diploid VNTR combination (557 + 315 bp).

Quantification of ABCA7 VNTR effects on ABCA7 expression and splicing. a–c Quantification of ABCA7 expression in LCL according to the sum of the two VNTR alleles within an individual (x-axis). a Relative RT-PCR determined ABCA7 expression targeting all known isoforms of ABCA7 for patients (dots) and controls (triangles). A trendline (blue) is shown with standard error (shaded area). b The ratio of canonical (green), alternative exon 18 splicing (orange), VNTR unit retention (purple), and exon 19 skipping (red) shown for individuals (dots) and with a trendline per isoform. c Exon 19 skipping shown in more detail for patients (dots), and controls (triangles) with a trendline and standard error (shaded area). d–f Distribution of the four isoforms (x-axis) across three different tissues: LCL (d), hippocampus (e), and Brodmann area (BA) 10 (f)
ABCA7 alternative splicing is affected by VNTR length
We observed three separate isoform changes in exon 18 and exon 19 (exons flanking the VNTR), currently unknown to public databases (Fig. 1). These alternative splicing events include usage of a cryptic splice acceptor site in exon 18 (chr19:1049314–1049316 [hg19]), usage of a cryptic splice donor site in intron 18 (chr19:1049461–1049463 [hg19]), and exon 19 skipping. The first two alternative splicing events respectively cause a 52 bp loss of coding sequence, and a 25 bp intron retention (corresponding to one VNTR repeat unit), hence both resulting in an out-of-frame transcript. The third splicing event leads to complete skipping of exon 19, which causes in-frame loss of coding sequence, corresponding to 44 amino acids that are part of the first nucleotide binding domain (NBD) of ABCA7 (Fig. 1). We analyzed the effect of VNTR length on abundance of these splicing events in LCL (Fig. 4b). Exon 18 alternative splicing and intron 18 retention events were not affected by different VNTR sizes (Table S3). Exon 19 skipping, however, strongly increased with expanding VNTR size (p = 3.24 × 10−13 for sum of VNTR alleles; Table S3). This trend was present in patients and controls (Fig. 4c). Exon 19 skipping was 3.2 times more abundant in the LCL sample with the largest diploid combination of VNTR alleles (5399 + 4575 bp) compared to smallest VNTR combination (557 + 315 bp). To determine the effect of individual alleles on exon 19 skipping, we examined allele-specific expression in LCL of an AD patient with an expanded VNTR allele (9711 bp) and a small wild-type allele (837 bp) (Table S2)—which were respectively phased with the rs3752240[G] and rs3752240[A] allele—and observed a 4.1-fold increase in exon 19 skipping for the expanded allele (Fig. S8).
In addition to quantification in LCL (Fig. 4d), we quantified the three alternative splicing events in the hippocampus, originating from six AD patients and six controls, as well as in Brodmann area 10 (BA10) brain tissue from four patients and six controls (Fig. 4e, f). For each brain tissue, three individuals had VNTR Southern blotted lengths and none carried an expanded VNTR allele. Splicing isoforms causing a frameshift had approximately similar expression level in all tissues: 14 ± 6 and 2 ± 1%, respectively. Exon 19 skipping, however, varied strongly between different brain regions and between brain and LCL: 45 ± 10% in BA10, 29 ± 12% in hippocampus, and 12 ± 6% in LCL. In 90% of BA10 cases and 31% of hippocampal tissue, the cumulative abundance of the alternative splicing transcripts exceeded the abundance of the canonically spliced transcript (Fig. 4e, f).
Since exon 18 alternative splicing and intron 18 retention introduce PTCs, they are subject to NMD. To evaluate whether the VNTR has an effect on NMD efficiency, we analyzed LCL treated with cycloheximide (CHX). Addition of CHX substantially increased the abundance of PTC introducing transcripts (Fig. S9), but no linear relationship was observed between the VNTR sum and the change in transcript abundance (p = 0.7 for exon 18 alternative splicing) (Fig. S10).
Discussion
We report a VNTR in ABCA7 that increases risk of AD up to 4.5-fold. VNTR length was tightly correlated with a GWAS index SNP for AD in ABCA7, with expansions predominantly occurring on the risk allele haplotype. VNTR length was negatively correlated with ABCA7 expression, in line with the mode of action of rare PTC mutations in ABCA7 that increase risk of AD [2, 9, 10, 15, 22, 38, 40, 46, 48]. Interestingly, VNTR expansion resulted in an increase in exon 19 skipping, a splicing event that has not previously been reported. This splicing event was most prominent in brain tissue.
A genetic variant with downstream biological consequences underlying the GWAS association in ABCA7 in Caucasian AD cohorts had not been identified yet. Here, we show that individuals with the risk haplotype of rs3764650 and rs78117248 have longer VNTR lengths on average. Furthermore, the majority of carriers with an expanded VNTR allele have at least one rs3764650 risk allele. Within this study population, rs3764650 is not associated with AD due to a comparable risk allele frequency in patients (MAF = 13.4%) and controls (MAF = 12.1%), therefore restricting the use of conditional regression. Nevertheless, the number of patients with an expanded VNTR allele (7.3%) is much higher than controls (1.7%), rendering this ABCA7 VNTR a common high-penetrant [OR 4.5 (95% CI 1.3–24.2)] risk factor for AD. Expanded VNTR alleles were not in LD with ABCA7 PTC mutations, and independently contribute to AD risk. Within this discovery cohort, the heritability of liability attributable to expanded ABCA7 VNTRs equals 3.1%. Compared to other genetic AD risk factors, this estimate is second to only APOE ε4 [11].
Increasing VNTR length was associated with an overall reduction of ABCA7 expression. This points towards a similar pathomechanism as observed in ABCA7 PTC mutation carriers. We observed no effect of VNTR length on NMD, hence ABCA7 expression reduction is not caused by differential proportions of alternatively spliced exon 18 and VNTR retention transcripts. The sequence of the ABCA7 VNTR contains recurring transcription factor binding motifs for ZNF263, PLAG1, and RREB1 [27]. ZNF263 has the strongest expression in brain (GTEx Consortium, accessed November 2017) and contains a Krüppel associated box (KRAB) domain which is involved in transcriptional repression [26]. Hence, increased binding of ZNF263 to longer VNTRs could decrease ABCA7 expression. Other factors contributing to the effect of the VNTR on ABCA7 expression could be chromatin state in the VNTR region, and stability of pre-mRNA with varying VNTR lengths, warranting further investigation.
VNTR expansion resulted in increased skipping of exon 19, which contains 132 highly conserved nucleotides (55% GERP-score > 2) and encodes 44 amino acids that are part of the first nucleotide binding domain of ABCA7. This domain is required for ATP hydrolysis and export of phospholipids [34]. Skipping of this exon can therefore cause reduction of functional ABCA7. Due to high abundance of this isoform in hippocampus and frontal cortex, it has a strong impact on ABCA7 dosage in brain tissue. Analysis of the VNTR repeat unit sequence with SpliceAid [33] revealed binding motifs for three splice factors in particular: SRp30c, YB-1, and Nova-1. SRp30c—in conjunction with YB-1 [36]—represses the use of a downstream splice acceptor site [44], and was shown to regulate exon skipping in MAPT, which is associated with frontotemporal dementia [51]. Increase of SRp30c binding motifs due to ABCA7 VNTR length could therefore influence skipping of downstream exon 19. Nova-1 is only expressed in neurons where it affects alternative splicing [19] and could potentially explain higher exon 19 skipping levels in brain tissue.
AD patients carrying an ABCA7 PTC mutation display a wide variability in onset age (45–90 years) [7, 10, 15, 38], which may in part be explained by reduced penetrance due to variation in NMD efficiency and alternative splicing events [38]. In line with this, AD carriers of an expanded VNTR allele had onset ages ranging from 44 to 90 years. We observed no association between onset age and VNTR length. Of note, the age at inclusion for the three control individuals with an expanded VNTR allele varied from 63 to 79 years, which is still within the onset age range. Therefore, the current disease penetrance estimate of this ABCA7 VNTR could be underestimated; clinical follow-up of these individuals is warranted. We observed nominal significant association between increasing ABCA7 VNTR length and decrease of CSF Aβ1–42, which is one of the hallmarks of AD [29]. This observation is in line with a previous report showing association between the risk allele of rs3764650 and increased neuritic plaque load [43], and exacerbation of plaque load in Abca7 knockout mice [12, 21, 39].
Due to technical limitations related to long GC-rich tandem repeats, Southern blotting is currently the only method for size determination of the two VNTR alleles within an individual. Southern blotting is a low-throughput method, however, and requires highly concentrated and high molecular weight DNA, which limited the sample size for whom bi-allelic data were available. Nevertheless, due to the strong effect of VNTR expansion on AD risk, we were able to detect association between expanded VNTR alleles and AD in this study cohort. Furthermore, approximation of VNTR length based on NGS data of our full cohort as well as the 1000 Genomes Project supported the association between VNTR and AD risk. Techniques are emerging for target enrichment of GC-rich regions [47], which may in the near future enable a more in-depth characterization of this VNTR at higher throughput. Outstanding questions that should then be explored include studying the effect of intermediate and smaller VNTR lengths on AD and its endophenotypes, epistasis between the two alleles within an individual, sequence differences between VNTR repeat units, nucleotide modifications, and phenotypic heterogeneity due to varying VNTR length.
In conclusion, we identified high penetrant expanded ABCA7 VNTR alleles in more than 7% of AD patients which increase abundance of a novel NBD-null ABCA7 isoform. With this study, we emphasize the need for the analysis of tandem repeats and their biological consequences to resolve indirect GWAS signals. Expanded VNTR alleles as well as PTC mutations in ABCA7 have a strong contribution to AD and both converge to a disease mechanism involving decreased ABCA7 expression. Future research will be paramount to study this AD subtype with potential benefit from ABCA7-specific therapies for AD.
References
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Allen M, Lincoln SJ, Corda M, Watzlawik JO, Carrasquillo MM, Reddy JS et al (2017) ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol Genet 3:e126. https://doi.org/10.1212/NXG.0000000000000126
Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS et al (2012) Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 79:221–228. https://doi.org/10.1212/WNL.0b013e3182605801
Bamji-Mirza M, Li Y, Najem D, Liu QY, Walker D, Lue L-F et al (2016) Genetic variations in ABCA7 can increase secreted levels of amyloid-β40 and amyloid-β42 peptides and ABCA7 transcription in cell culture models. J Alzheimer’s Dis 53:875–892. https://doi.org/10.3233/JAD-150965
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw. https://doi.org/10.18637/jss.v067.i01
Benson G (1999) Tandem repeats: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Van den Bossche T, Sleegers K, Cuyvers E, Engelborghs S, Sieben A, De Roeck A et al (2016) Phenotypic characteristics of Alzheimer patients carrying an ABCA7 mutation. Neurology 86:2126–2133. https://doi.org/10.1212/WNL.0000000000002628
Cruts M, Theuns J, Van Broeckhoven C (2012) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33:1340–1344. https://doi.org/10.1002/humu.22117
Cukier HN, Kunkle BW, Vardarajan BN, Rolati S, Hamilton-Nelson KL, Kohli MA et al (2016) ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet 2:e79. https://doi.org/10.1212/NXG.0000000000000079
Cuyvers E, De Roeck A, Van den Bossche T, Van Cauwenberghe C, Bettens K, Vermeulen S et al (2015) Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol 14:814–822. https://doi.org/10.1016/S1474-4422(15)00133-7
Cuyvers E, Sleegers K (2016) Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol 15:857–868. https://doi.org/10.1016/S1474-4422(16)00127-7
Fu Y, Hsiao J-HT, Paxinos G, Halliday GM, Kim WS (2016) ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis 54:569–584. https://doi.org/10.3233/JAD-160456
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S et al (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63:168–174. https://doi.org/10.1001/archpsyc.63.2.168
Gelfand Y, Rodriguez A, Benson G (2007) TRDB—the tandem repeats database. Nucleic Acids Res 35:D80–D87. https://doi.org/10.1093/nar/gkl1013
Le Guennec K, Nicolas G, Quenez O, Charbonnier C, Wallon D, Bellenguez C et al (2016) ABCA7 rare variants and Alzheimer disease risk. Neurology 86:2134–2137. https://doi.org/10.1212/WNL.0000000000002627
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088–1093. https://doi.org/10.1038/ng.440
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43:429–435. https://doi.org/10.1038/ng.803
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement 8:1–13. https://doi.org/10.1016/j.jalz.2011.10.007
Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ et al (2000) Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25:359–371. https://doi.org/10.1016/S0896-6273(00)80900-9
Kim WS, Guillemin GJ, Glaros EN, Lim CK, Garner B (2006) Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. NeuroReport 17:891–896. https://doi.org/10.1097/01.wnr.0000221833.41340.cd
Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK et al (2013) Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci 33:4387–4394. https://doi.org/10.1523/JNEUROSCI.4165-12.2013
Kunkle BW, Carney RM, Kohli MA, Naj AC, Hamilton-Nelson KL, Whitehead PL et al (2017) Targeted sequencing of ABCA7 identifies splicing, stop-gain and intronic risk variants for Alzheimer disease. Neurosci Lett 649:124–129. https://doi.org/10.1016/j.neulet.2017.04.014
Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41:1094–1099. https://doi.org/10.1038/ng.439
Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. https://doi.org/10.1038/ng.2802
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen HJ, Rauscher FJ (1994) Kruppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci 91:4509–4513. https://doi.org/10.1073/pnas.91.10.4509
Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J et al (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44:D110–D115. https://doi.org/10.1093/nar/gkv1176
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34:939–944
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263–269. https://doi.org/10.1016/j.jalz.2011.03.005
Morgan M, Pagès H, Obenchain V, Hayden N (2017) Rsamtools: binary alignment (BAM), FASTA, variant call (BCF), and tabix file import, R package version 1.30.0. http://bioconductor.org/packages/release/bioc/html/Rsamtools.html
Naj AC, Jun G, Beecham GW, Wang L, Vardarajan BN, Buros J et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441. https://doi.org/10.1038/ng.801
Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I et al (2005) The montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53:695–699. https://doi.org/10.1111/j.1532-5415.2005.53221.x
Piva F, Giulietti M, Nocchi L, Principato G (2009) SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics 25:1211–1213. https://doi.org/10.1093/bioinformatics/btp124
Quazi F, Molday RS (2013) Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem 288:34414–34426. https://doi.org/10.1074/jbc.M113.508812
R Core Team (2017) R: a language and environment for statistical computing. https://www.R-project.org/
Raffetseder U, Frye B, Rauen T, Jürchott K, Royer H-D, Jansen PL et al (2003) Splicing factor SRp30c interaction with Y-box protein-1 confers nuclear YB-1 shuttling and alternative splice site selection. J Biol Chem 278:18241–18248. https://doi.org/10.1074/jbc.M212518200
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G et al (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26. https://doi.org/10.1038/nbt.1754
De Roeck A, Van den Bossche T, van der Zee J, Verheijen J, De Coster W, Van Dongen J et al (2017) Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol 134:475–487. https://doi.org/10.1007/s00401-017-1714-x
Sakae N, Liu C-C, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y et al (2016) ABCA7 deficiency accelerates amyloid-beta generation and Alzheimer’s neuronal pathology. J Neurosci 36:3848–3859. https://doi.org/10.1523/JNEUROSCI.3757-15.2016
Sassi C, Nalls MA, Ridge PG, Gibbs JR, Ding J, Lupton MK et al (2016) ABCA7 p. G215S as potential protective factor for Alzheimer’s disease. Neurobiol Aging 46:235.e1–235.e9. https://doi.org/10.1016/j.neurobiolaging.2016.04.004
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M et al (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303:1832–1840. https://doi.org/10.1001/jama.2010.574
Seshadri S, Wolf PA (2007) Lifetime risk of stroke and dementia: current concepts, and estimates from the Framingham Study. Lancet Neurol 6:1106–1114. https://doi.org/10.1016/S1474-4422(07)70291-0
Shulman JM, Chen K, Keenan BT, Chibnik LB, Fleisher A, Thiyyagura P et al (2013) Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol 70:1150–1157. https://doi.org/10.1001/jamaneurol.2013.2815
Simard MJ, Chabot B (2002) SRp30c is a repressor of 3′ splice site utilization. Mol Cell Biol 22:4001–4010. https://doi.org/10.1128/MCB.22.12.4001-4010.2002
Sleegers K, Bettens K, De Roeck A, Van Cauwenberghe C, Cuyvers E, Verheijen J et al (2015) A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Aβ 42. Alzheimer’s Dement 11:1452–1460. https://doi.org/10.1016/j.jalz.2015.02.013
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H et al (2015) Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 47:445–447. https://doi.org/10.1038/ng.3246
Tsai Y-C, Greenberg D, Powell J, Hoijer I, Ameur A, Strahl M et al (2017) Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. bioarxiv 203919. https://doi.org/10.1101/203919
Vardarajan BN, Ghani M, Kahn A, Sheikh S, Sato C, Barral S et al (2015) Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 78:487–498. https://doi.org/10.1002/ana.24466
Vasquez JB, Fardo DW, Estus S (2013) ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci Lett 556:58–62. https://doi.org/10.1016/j.neulet.2013.09.058
Verheijen J, Van den Bossche T, van der Zee J, Engelborghs S, Sanchez-Valle R, Lladó A et al (2016) A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol 132:213–224. https://doi.org/10.1007/s00401-016-1566-9
Wang Y, Wang J, Gao L, Lafyatis R, Stamm S, Andreadis A (2005) Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c.SRp55 complex that either recruits or antagonizes htra2beta1. J Biol Chem 280:14230–14239. https://doi.org/10.1074/jbc.M413846200
Witte JS, Visscher PM, Wray NR (2014) The contribution of genetic variants to disease depends on the ruler. Nat Publ Gr 15:765–776. https://doi.org/10.1038/nrg3786
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21:1859–1875. https://doi.org/10.1093/bioinformatics/bti310
Yamazaki K, Yoshino Y, Mori T, Yoshida T, Ozaki Y, Sao T et al (2017) Gene expression and methylation analysis of ABCA7 in patients with Alzheimer’s disease. J Alzheimers Dis 57:171–181. https://doi.org/10.3233/JAD-161195
Acknowledgements
The authors thank the personnel of the VIB Neuromics Support Facility of the VIB-UAntwerp Center for Molecular Neurology, the Diagnostic Service Facility and the Antwerp Biobank of the Institute Born-Bunge, and the neurological centers of the BELNEU consortium partners. The research was funded in part by the Alzheimer Research Foundation (SAO-FRA), the Belgian Science Policy Office Interuniversity Attraction Poles program, the Flemish government-initiated Flanders Impulse Program on Networks for Dementia Research (VIND), Flemish government-initiated Methusalem excellence program, the VIB Technology Fund, The Research Foundation Flanders (FWO, Grant nos. G030718N, 1S4421216N), the University of Antwerp Research Fund, Belgium; ADR is recipient of a PhD fellowship of FWO, and a Hope in Head award from the Belgian Rotary. RC is recipient of a Postdoc fellowship of FWO.
Belgian Neurology (BELNEU) Consortium: the following members of the BELNEU Consortium have contributed to the clinical and pathological phenotyping and follow-up of the Belgian patient cohorts: Johan Goeman, Roeland Crols (Hospital Network Antwerp (ZNA) Middelheim and Hoge Beuken, Antwerp, Belgium); Dirk Nuytten (Hospital Network Antwerp (ZNA) Stuivenberg, Antwerp, Belgium); Rudy Mercelis (Antwerp University Hospital, Edegem); Mathieu Vandenbulcke (University of Leuven and University Hospitals Leuven, Leuven, Belgium); Anne Sieben, Jan L. De Bleecker, Patrick Santens (University Hospital Ghent, Ghent, Belgium); Jan Versijpt, Alex Michotte (University Hospital Brussels, Brussels, Belgium); Olivier Deryck, Ludo Vanopdenbosch, Bruno Bergmans (AZ Sint-Jan Brugge, Bruges, Belgium); Christiana Willems, Nina De Klippel (Jessa Hospital, Hasselt, Belgium); Jean Delbeck (General Hospital Sint-Maria, Halle); Adrian Ivanoiu (Saint-Luc University Hospital, Université Catholique de Louvain, Louvain-la-Neuve, Belgium); and Eric Salmon (University of Liege and Memory Clinic, CHU Liege, Liege, Belgium).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Informed consent
All participants and/or their legal guardian gave written informed consent for participation in clinical and genetic studies. Autopsied patients or their legal guardian gave written informed consent for inclusion in neuropathological studies. Clinical study protocol and the informed consent forms for patient ascertainment were approved by the ethic committee of the respective hospitals at the cohort sampling sites. The genetic study protocols and informed consent forms were approved by the Ethics Committees of the University of Antwerp and the University Hospital of Antwerp, Belgium.
Conflict of interest
The authors declare no conflict of interest.
Additional information
The members of BELNEU Consortium are listed in Acknowledgements.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
De Roeck, A., Duchateau, L., Van Dongen, J. et al. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer’s disease. Acta Neuropathol 135, 827–837 (2018). https://doi.org/10.1007/s00401-018-1841-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1841-z