
Abstract
The adenosine triphosphate–binding cassette subfamily A member 7 gene (ABCA7) is associated with Alzheimer’s disease (AD) in large genome-wide association studies. Targeted sequencing of ABCA7 suggests a role for rare premature termination codon (PTC) mutations in AD, with haploinsufficiency through nonsense-mediated mRNA decay as a plausible pathogenic mechanism. Since other classes of rare variants in ABCA7 are poorly understood, we investigated the contribution and pathogenicity of rare missense, indel and splice variants in ABCA7 in Belgian AD patient and control cohorts. We identified 8.36% rare variants in the patient cohort versus 6.05% in the control cohort. For 10 missense mutations identified in the Belgian cohort we analyzed the pathogenetic effect on protein localization in vitro using immunocytochemistry. Our results demonstrate that rare ABCA7 missense mutations can contribute to AD by inducing protein mislocalization, resulting in a lack of functional protein at the plasma membrane. In one pedigree, a mislocalization-inducing missense mutation in ABCA7 (p.G1820S) co-segregated with AD in an autosomal dominant inheritance pattern. Brain autopsy of six patient missense mutation carriers showed typical AD neuropathological characteristics including cerebral amyloid angiopathy type 1. Also, among the rare ABCA7 missense mutations, we observed mutations that affect amino acid residues that are conserved in ABCA1 and ABCA4, of which some correspond to established ABCA1 or ABCA4 disease-causing mutations involved in Tangier or Stargardt disease.
Similar content being viewed by others

Introduction
ABCA7 was identified as a risk gene for late onset (≥ 65 years) AD through genome-wide association studies [1,2,3,4,5,6]. Since then, genetic studies have demonstrated that rare heterozygous premature termination codon (PTC) mutations in ABCA7 are associated with AD risk in both early (< 65 years) and late onset AD patients [7,8,9,10,11,12,13,14,15]. Rare PTC mutations include nonsense, frameshift and canonical splice site mutations as well as the noncanonical c.5570 + 5G > C mutation, which leads to a stop codon due to aberrant splicing of exon 41 [11, 16]. Haploinsufficiency through nonsense-mediated mRNA decay (NMD) is suggested as a plausible downstream mechanism, although incomplete NMD and alternative splicing events, removing the PTC from the transcript, have been observed [11]. In a Belgian cohort, we previously identified PTC mutations in 4.87% of the AD patients compared to 1.84% of the control individuals [9]. In addition, a variable number of tandem repeats (VNTR) is present in intron 18 [17]. Expanded ABCA7 VNTR alleles (> 5720 bp) are highly enriched in AD patients and correlate with reduced ABCA7 expression and with increased exon 19 skipping [17]. However, the contribution to AD of other rare variants in ABCA7, e.g., missense, indels and noncanonical splice mutations, and their potential downstream pathogenic mechanism is less well investigated.
ABCA7 is involved in lipid metabolism and phagocytosis [18]. In vitro and in vivo studies suggest that ABCA7 deficiency leads to a decreased microglial Aβ clearance and elevated amyloid precursor protein (APP) processing, thereby exacerbating Aβ accumulation in the brain, a key pathological feature of AD [19,20,21,22,23]. ABCA7 belongs to the ATP-binding cassette (ABC) transporter family, a superfamily of highly conserved proteins responsible for the active transport of various substrates across cellular membranes [24]. ABC transporters share a characteristic architecture consisting of four core domains: two nucleotide binding domains (NBD) providing energy for substrate transport by ATP binding and hydrolysis, and two transmembrane domains (TMD) providing a pathway across the membrane for the transport of a substrate [25]. The A-subclass of ABC-transporters (ABCA) gained special interest since several members are causatively linked to monogenic diseases.
ABCA1 and ABCA4 show the most extensive sequence identity with ABCA7 (54% and 49%) [24]. Monoallelic mutations in ABCA1 are the cause of familial high-density lipoprotein (HDL) deficiency, whereas homozygous or compound heterozygous mutations represent a more severe phenotype known as Tangier disease [26]. Bi-allelic mutations in ABCA4 are responsible for various forms of recessive retinal dystrophies, including Stargardt disease (STGD1), while heterozygous mutations can increase disease risk or lead to later disease onset compared to bi-allelic mutation carriers [27,28,29]. The genetic spectrum of pathogenic ABCA1 and ABCA4 mutations is extensive, and missense mutations represent the largest group in both genes (http://www.hgmd.cf.ac.uk) [30]. Several studies demonstrated that disease-linked missense mutations in ABCA1 and ABCA4 affect subcellular localization and function, likely by inducing protein misfolding [31,32,33,34,35,36,37]. Also, missense mutations in regions of high sequence similarity between ABCA1 and ABCA4 show a similar impact on the protein in vitro, implying a similar structure–function relationship [35]. How mutations in ABCA7 influence the subcellular protein localization or secretion is not known.
Studies analyzing the genetic contribution of ABCA7 missense mutations to AD are limited, and conflicting results have been reported [11, 14, 16]. Additional studies reported missense mutation frequencies in AD patients and controls [8, 10, 38]. A recent meta-analysis showed a significant enrichment of ABCA7 missense mutations in AD patients [39]. One study reported co-segregation of the ABCA7 missense mutation p.R880Q with autosomal dominant AD [40]. Earlier, we described the frequency and characteristics of ABCA7 PTC mutation carriers in the Belgian AD patient and control cohort [9]. In this study, we investigate the frequency of rare ABCA7 missense mutations, indels, noncanonical splice mutations and compound heterozygous mutations in the Belgian cohort. We analyzed the effect of rare missense mutations in ABCA7 on the subcellular localization of the protein and observed that mutated ABCA7, transiently expressed in HeLa cells, displays a decreased plasma membrane localization and instead is retained in the endoplasmic reticulum (ER), leading to a loss of functional ABCA7.
Materials and methods
Belgian AD patient and control cohorts
Sequencing data of ABCA7 exons and splice sites of the Belgian AD and control cohort was available from a previous screening effort to investigate the frequency of ABCA7 PTC mutations [9]. Details about in- and exclusion criteria, demographic and patient characteristics were previously described [9]. In short, the patient cohort consists of 1376 individuals (60.0% [826/1376] women) with a mean age at onset (AAO) of 69.3 ± 10.6 years (range 31–96). Genetic screening of APP, PSEN1 and PSEN2 was performed in all AD patients and revealed two pathogenic APP mutations (0.15%) and nine pathogenic PSEN1 mutations (0.65%). The control cohort consists of 976 individuals (63.5% [620/976] women) with a mean age at inclusion (AAI) of 72.4 ± 8.6 years (range 43–100).
Whole exome sequencing
Library preparation and target enrichment was performed using the SeqCap EZ Exome Library v3.0 kit (Nimblegen, Roche, Basel, Switzerland) and sequencing was done on an Illumina® NextSeq platform (Illumina®, San Diego, CA, USA). GenomeComb was used for data processing and variant filtering [41].
Bioinformatics analysis
Read processing, alignment, variant calling, annotation and downstream filtering of resequencing and whole exome sequencing (WES) data was achieved using GenomeComb [41]. Variants located in segmental duplications, repeated sequences or homo-polymer stretches were considered false positives and were excluded from the study. Only variants with a minor allele frequency (MAF) ≤ 1% in the Genome Aggregation database (GnomAD) v2.1.1 [42], the Exome Variant Server (EVS) (NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) and the 1000 Genomes Project [43] were retained.
Sanger sequencing
Validation of all rare (MAF ≤ 1%) non-synonymous coding and splice mutations, identified in the Belgian AD and control cohort’ are performed using Sanger sequencing technology (BigDye Terminator Cycle Sequencing kit v3.1; analysis on an ABI 3730 DNA Analyzer, Thermo Fisher Scientific, MA, US). Sequences were analyzed using NovoSNP [44].
Haplotype sharing analysis
Haplotypes are based on 18 single nucleotide polymorphisms (SNPs) spanning ABCA7 and seven short tandem repeat (STR) markers flanking ABCA7 as described [10]. STR markers were PCR-amplified using fluorescently labelled primers. PCR-products were supplemented with GeneScan™ 500 LIZ™ size standard (Thermo Fisher Scientific, MA, USA) and size-separated using capillary electrophoresis on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific, MA, USA). STR fragment lengths were assigned using the Local Genotype Viewer software (https://www.neuromicssupportfacility.be/) developed by the Neuromics Support Facility, Center for Molecular Neurology, Antwerp, Belgium.
Preparation of cDNA constructs
The coding sequence of WT human ABCA7 was purchased in a Gateway®-adapted entry vector from genomics-online (ABIN3417638). The stop codon was removed from the WT-ABCA7 entry vector with in vitro mutagenesis using KAPA HiFi HotStart DNA polymerase (Kapa Biosystems, Wilmington, MA). Afterwards, ten ABCA7 missense mutations of interest and four likely benign or protective variants were individually introduced in the WT-ABCA7-NoStop entry vector with in vitro mutagenesis. Sequence verified mutant or WT entry clones were subcloned into the in-house developed Gateway®-compatible pCR3 vector with a C-terminal EmGFP-tag to allow the expression of ABCA7-EmGFP fusion proteins. Expression clones were sequence verified. In total, 15 different constructs were generated. Primers used for in vitro mutagenesis are listed in Additional file 1: Table S1.
Cell culture and transfections
Human cervical carcinoma (HeLa) cells were cultured in Modified Eagle’s medium (MEM; Life Technologies), supplemented with 10% fetal calf serum (Sigma Aldrich), 1% penicillin/streptomycin and 1% L-glutamine (Life Technologies) at 37 °C in a humidified 5% CO2 atmosphere.
HeLa cells were transfected using Lipofectamine® 2000 Reagent (Thermofisher) according to the manufacturer’s protocol. Briefly, cells were seeded in a 6-well plate at 2.5 × 105 cells per well, 24 h before transfection. On the day of transfection, cell medium was replaced by medium without antibiotics (Opti-MEM, Life-Technologies). 6 µl Lipofectamine® 2000 Reagent was diluted in 150 µl Opti-MEM and in parallel 2 µg plasmid DNA was added to Opti-MEM in a final volume of 150 µl. The diluted Lipofectamine® 2000 Reagent was added to the diluted DNA and mixed gently by pipetting. After 5 min of incubation at room temperature, the solution was added to the cells in a dropwise manner. 5–6 h after transfection, Opti-MEM was replaced by the original medium.
Immunocytochemistry and microscopy sample preparation
HeLa cells were reseeded 24 h post-transfection on 12 mm glass coverslips (Fisher Scientific). The next day, cells are fixed for 20 min with 4% EM-grade paraformaldehyde (PFA; Electron Microscopy Sciences 15710) and 4% sucrose in PBS at room temperature and washed three times in PBS.
To visualize the plasma membrane, cells were incubated with wheat germ agglutinin (WGA) conjugated with Alexa Fluor 594 (1:200, Invitrogen w11262) for 10 min at room temperature and washed twice with PBS. Afterwards, the nuclei were stained with Hoechst 33342 (1:2,000; Invitrogen H3570) for 10 min at room temperature and washed twice with PBS. Coverslips were mounted using fluorescent mounting medium (Dako S302380) and stored at 4 °C before being subjected to fluorescence microscopy.
To visualize the ER, fixed cells were permeabilized with 0.5% Triton-X in PBS for 5 min. After washing with PBS, coverslips were blocked with 3% BSA in PBS for 1 h at room temperature. Next, cells were incubated for 2 h with a primary chicken anti-calreticulin antibody as a marker for the ER (1:500, Ab14234, Abcam, USA), followed by three washes with PBS and staining with an Alexa Fluor 594® conjugated goat anti-chicken IgY secondary antibody (1:500, A-11042, Thermo Fisher Scientific, USA) for 1 h. Nuclear staining and mounting were performed as described above.
Image acquisition and analysis
Confocal images are acquired on a Zeiss LSM700 confocal microscope with Zen 2009 software (Carl Zeiss, Zaventem, Belgium), using an EC Plan-Neofluar 40×/1.30 oil objective. Images of ABCA7-EmGFP and plasma membrane or ER co-markers were acquired in different tracks (serial frame scanning) to avoid any possible crosstalk between the channels. Acquisition settings (laser intensities, detector gain and channel settings) of the individual fluorescence channels were kept identical for the different images. For each genotype and co-marker combination (ABCA7-EmGFP with either WGA or Calreticulin), at least 20 images were taken containing random EmGFP positive cells, resulting in more than 700 multi-channel images in total.
Custom ImageJ scripts [45] were used in Fiji [46] to quantify the colocalization of ABCA7-EmGFP with the plasma membrane and the ER. First, individual cells were delineated using automatic segmentation based on intensity thresholding (Huang method) in the green channel, after noise reduction filtering (Median radius 4) and followed by ROI-size filtering (Analyze particles command). In a next step, the resulting ROI’s were manually corrected to precisely correspond with individual cells and stored in ImageJ ROI manager files. To quantify EmGFP colocalization with WGA or calreticulin in individual cells, an ImageJ script was used that extracts the Pearson’s correlation coefficient between the two fluorescent channels in batch for all ROI’s calling the ImageJ Colocalization Test plugin (www.imagej.net). A minimum of 30 individual cells per genotype were analyzed for each co-marker (WGA or calreticulin), resulting in a total of 1224 cell measurements. ImageJ scripts are written to process and analyze all images (and all genotypes) in batch. Means of the independent measurements for each genotype were compared to the WT using Welch ANOVA with Games-Howell post-hoc test.
Results
Rare variants in ABCA7 in Belgian AD patients and control individuals
We observed rare (MAF ≤ 1%) variants i.e., missense, indels, and noncanonical splice mutations in the Belgian AD patient cohort (n = 1376) with a frequency of 8.36% (115/1376) and in the Belgian control cohort (n = 976) with a frequency of 6.05% (59/976) (Fig. 1). Carriers of a rare ABCA7 variant were free from pathogenic mutations in APP, PSEN1 and PSEN2, except for one patient carrying both ABCA7 p.G826R and PSEN1 p.C263F.

Topological model of ABCA7 with all rare missense mutations and deletions identified in the Belgian cohort. Splice mutations are not indicated in the figure because of their location outside the coding sequence. ABCA7 consists of two transmembrane domains (TMD) with large extracellular loops between the first and second transmembrane helix and two nucleotide binding domains (NBD), containing three motifs. Mutations identified exclusively in control individuals are shown in black. Mutations found in both patients and controls are shown in red. Mutations identified exclusively in patients are indicated in bold. Alignment of the ABCA1, ABCA4 and ABCA7 sequence revealed five mutations (underlined) that correspond to positions in ABCA1 and/or ABCA4 on which disease-associated mutations are found (Additional file 1: Table S6). Protein domain and motif information was based on alignment with ABCA1 [24, 70]
Missense mutations represent the largest group of rare variants in both patients and controls, with some individuals carrying two missense mutations (Additional file 1: Table S2). We identified 101 missense mutations in 96 AD patients (7.34%, 101/1376) and 50 in 46 controls (5.12%, 50/976) (Additional file 1: Table S2). Next, splice mutations are identified in 10 AD patients (0.73%, 10/1376) and seven controls (0.72%, 7/976) (Additional file 1: Table S3). The in silico predicted effect of the identified splice mutations on ABCA7 mRNA splicing is listed in Additional file 1: Table S4. Indel mutations are found in four AD patients (0.29%, 4/1376) and two controls (0.20%, 2/976) (Additional file 1: Table S5).
Alignment of the ABCA7, ABCA1 and ABCA4 protein sequences indicates that a subset of the rare ABCA7 missense mutations identified in the Belgian cohort affect amino acid residues that are conserved in ABCA1 and/or ABCA4 and correspond to established disease-causing ABCA1 and ABCA4 mutations (Additional file 1: Table S6).
We did not identify homozygous carriers in the Belgian cohort. Nonetheless, we identified 11 patients (0.80%, 11/1376) and five control individuals (0.51%, 5/976) carrying two rare ABCA7 mutations mutations (p = 0.404), and where possible, cis/trans configuration was determined (Additional file 1: Table S7). Five of the patients carried two missense mutations, four patients combined a missense with a PTC mutation and two patients carried a missense and a splice mutation. Of the control individuals, four carried two missense mutations and one a missense mutation and a PTC mutation (Additional file 1: Table S7). We defined the cis/trans configuration of the mutations in eight of the 11 patients and in all five controls, confirming trans configuration in four patients and in one control (Additional file 1: Table S7). Cis configuration was confirmed in another four patients and four controls (Additional file 1: Table S7). In the remaining three AD patients we could not determine cis/trans configuration. Details of the cis/trans phasing of the compound heterozygous mutations are available in the Additional file 1: methods and results.
Co-segregation of p.G1820S with AD in a Belgian family
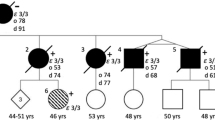
For all patient rare variant carriers with a positive familial history, we checked whether DNA of relatives was available for genetic testing. Only one family was informative with several patients and segregation of disease over multiple generations (Fig. 2). The disease pattern in this family mimics autosomal dominant inheritance (Fig. 2). The index patient (II.1) was diagnosed with probable AD at the age of 63 and carries the ABCA7 p.G1820S mutation. Cerebrospinal fluid analysis shows a pathological biomarker profile compatible with AD. The index patient (II.1) has five siblings of whom two, II.2 and II.4, are diagnosed with probable AD and another sibling, II.3, with AD by hearsay.

Co-segregation of ABCA7 p.G1820S with AD. To respect the privacy of the participants, the sex of each person is masked, and the pedigree is scrambled. The black arrow indicates the proband. Slashed symbols indicate deceased individuals. Black diamonds indicate individuals diagnosed with AD. Diamonds with question marks represent individuals with hearsay AD. An asterisk represents participants of whom genomic DNA is available. A red ‘c’ represents (obligate) carriers of the ABCA7 p.G1820S mutation. The shared haplotype of the p.G1820S carriers is indicated in blue. AAO, age at onset; AAI, age at inclusion
In generation II, DNA was available from two affected siblings (II.1 and II.2) and two healthy siblings (II.5 and II.6). Genetic screening disclosed the presence of the ABCA7 p.G1820S mutation in II.1 and II.2. The II.5 and II.6 siblings tested negative and are cognitively healthy at the advanced ages of 85 and 80 years. In generation III, DNA was available of seven offspring of the three affected and two non-affected individuals of generation II. None of the offspring are affected but III.1 and III.5 are carriers of the ABCA7 p.G1820S mutation, although they are considerably younger than their affected parents, II.1 and II.4, and therefore possibly presymptomatic. Also, since III.5 is an offspring carrier of II.4, we can consider II.4 an obligate carrier. In addition, whole exome sequencing was performed in affected individuals II.1 and II.2 and unaffected individuals II.5 and II.6 and confirms the absence of co-segregation of other rare variants in Mendelian AD genes and AD-associated genes.
Haplotyping reveals a common haplotype of at least 2.63 Mb shared by all mutation carriers in the family. Within our Belgian AD patient cohort, one additional patient (DR1744.1) with an onset age of 69 years carries the p.G1820S mutation. Haplotyping indicates at least 1.36 Mb sharing with the family demonstrating a genetic relationship due to a distant common ancestor (Additional file 1: Fig. S1).
ABCA7 missense mutations affect protein localization in HeLa cells
To investigate whether ABCA7 missense mutations affect the subcellular localization of ABCA7, immunocytochemistry experiments are performed on HeLa cells transiently transfected with EmGFP-tagged wild type ABCA7 and 10 constructs containing a predicted pathogenic ABCA7 missense mutation. In addition, three constructs containing a likely benign ABCA7 variant and one construct containing a protective variant are used as positive controls.
The 10 different ABCA7 missense mutations used in the protein localization experiment are selected from the Belgian cohort (Table 1). We selected mutations with an in silico predicted pathogenic effect, present throughout the full length and the different domains of the protein (Table 1; Fig. 1). In addition, we included mutations that are of particular interest because of their segregation with AD in families (p.R880Q, p.G1820S), because they correspond to Tangier or Stargardt disease mutations in respectively ABCA1 (p.A845V) or ABCA4 (p.R1932C) or because they show enrichment in patients versus controls (p.L620P) (Table 1; Fig. 1). The p.G826R mutation is observed with equal frequency in patients and controls. Of note, all selected mutations affect amino acids that are conserved between ABCA7, ABCA1 and, except for p.G826R, ABCA4. As a positive control, we included in the experiment likely benign variants which are highly frequent in the GnomAD database (p.E188G, p.R1349Q and p.G1527A), and we also included p.G215S, previously shown to have a protective effect against AD in GWAS [38] (Table 1). The minor ‘G’ allele coding for the Glycine at position 1527 is significantly associated with late onset AD in GWAS [5].
The EmGFP signal of cells transfected with WT ABCA7 is present intracellularly with signal accumulations at cell borders, corresponding to the expected location of ABCA7 at the plasma membrane and at intracellular membrane bound organelles e.g., Golgi, endosomes, ER [47]. Co-staining with a marker for the plasma membrane (WGA) demonstrated an obvious overlap with WT ABCA7-EmGFP (Fig. 3). Similarly, HeLa cells transfected with the four ABCA7-EmGFP constructs, containing a likely benign or protective variant (p.E188G, G215S, R1349Q and G1527A), show a similar localization and overlap with the plasma membrane (Additional file 1: Fig. S9). However, hardly any overlap with the plasma membrane is detected when HeLa cells are transfected with any of the mutant ABCA7 constructs (Fig. 3; Additional file 1: Fig. S9). Quantification of the amount of ABCA7 at the plasma membrane, by measuring the Pearson’s correlation coefficient between the WGA and ABCA7-EmGFP fluorescent signals in a high number of cells, confirm the absence of mutant ABCA7 at the plasma membrane (Fig. 4). Instead, mutants generally show an intense intracellular EmGFP signal, in which typical morphological reticular ER-like membrane structures can be distinguished (Fig. 3; Additional file 1: Fig. S9). As expected, co-staining with a marker for the ER (calreticulin) shows a strong colocalization with mutant ABCA7-EmGFP (Fig. 5, Additional file 1: Fig. S10). While WT ABCA7 and benign ABCA7 variants also show some overlap with the ER in addition to their presence on the plasma membrane (Fig. 5; Additional file 1: Fig. S10), quantification by fluorescence colocalization analysis verifies that mutant ABCA7 is retained in the ER (Fig. 6). Confocal microscopy images for the remaining constructs under study are found in the Additional file 1: Data. Overall, the results show a retention of the selected ABCA7 mutants within the ER and indicate aberrant exocytotic processing resulting in an inability to reach the correct cellular destiny at the plasma membrane.

Subcellular localization of WT ABCA7 and mutant ABCA7. Confocal microscopy images of HeLa cells transiently expressing WT or mutant ABCA7-EmGFP. Cells were labelled with WGA (magenta) as a plasma membrane marker. WT ABCA7 locates to the plasma membrane and intracellularly, whereas for the mutants the colocalization with WGA is lost. Instead, the EmGFP signal is concentrated intracellularly and shows a reticulate pattern characteristic for the ER. Scale bars represent 20 µm

Quantification of the colocalization of ABCA7-EmGFP and the plasma membrane. The Pearson’s correlation coefficient between the ABCA7-EmGFP channel and the WGA channel was measured with ImageJ on a minimum of 30 cells per genotype. Data are represented as median values, with lower and upper quartiles and the value ranges (whiskers). (*p < 0.05; **p < 0.01; ***p < 0.001 after Games-Howell correction)

Subcellular localization of WT ABCA7 and mutant ABCA7. Confocal microscopy images of HeLa cells transiently expressing WT or mutant ABCA7-EmGFP. Calreticulin (magenta) was used as a marker for the ER. WT ABCA7 is present inside the ER as well as outside the ER, whereas the expression of mutant ABCA7 is predominantly seen in the ER, as shown by the near perfect colocalization between EmGFP and calreticulin. Scale bars represent 20 µm

Quantification of the colocalization of ABCA7-EmGFP and the ER. The Pearson’s correlation coefficient between the ABCA7-EmGFP channel and the Calreticulin channel was measured with ImageJ on a minimum of 30 cells per genotype. Data are represented as median values, with lower and upper quartiles and the value ranges (whiskers). (*p < 0.05; **p < 0.01; ***p < 0.001 after Games-Howell correction). Presence of neurofibrillary tangles (arrow) and dystrophic neurites (asterisk) in the hippocampus (A). Presence of diffuse plaques and capillary amyloid angiopathy (arrow) in the hippocampus (B). Scale bars represent 50 µm. Details on the protocol of the neuropathologic assessment can be found in the Additional file 1: Materials and Method section
Clinicopathological phenotype of missense mutation carriers
Mean AAO of missense mutation carriers (n = 96) is 67.9 ± 11.0 years, with a wide range of 37–92 years. Mean age at death (AAD) is 78.3 ± 10.4 years with a mean disease duration (DD) of 8.6 ± 4.3 years. Information on familial history of dementia is available for 73/96 (76.0%) carriers and a positive familial history is noted in 43/73 (58.9%) carriers (Additional file 1: Table S8). The fraction of patients with a missense mutation carrying at least one APOE ε4 allele (66/96, 68.8%) is significantly higher compared to the full AD cohort (790/1350, 58.5%) (p = 0.002) (Additional file 1: Table S8) [9]. Brain autopsy is performed in six missense mutation carriers (Table 2). High AD neuropathological changes are noted in five carriers, and intermediate changes in one carrier (Table 2, Fig. 7). Meningeal and parenchymal blood vessels present with cerebral amyloid angiopathy (CAA) in all carriers although in different degrees. Capillary CAA (i.e., CAA type 1) is present in all individuals (Table 2).

Neuropathological examination of a patient carrying the p.G1731S missense mutation. Presence of neurofibrillary tangles (arrow) and dystrophic neurites (asterisk) in the hippocampus (A). Presence of diffuse plaques and capillary amyloid angiopathy (arrow) in the hippocampus (B). Scale bars represent 50 µm. Details on the protocol of the neuropathologic assessment can be found in the Additional file 1: Materials and Method section
Discussion
We investigated the presence of rare (MAF ≤ 1%) ABCA7 missense, indel and noncanonical splice mutations in the Belgian cohort and identified 115 mutations (8.36%, 115/1376) in the patient cohort In Addition in both patients (7.34%) and controls (5.12%) and are present throughout the full length of the protein, like pathogenic missense mutations in ABCA1 and ABCA4 (http://www.hgmd.cf.ac.uk) (Fig. 1). In fact, several of the identified missense mutations in the Belgian AD cohort correspond to pathogenic ABCA1 or ABCA4 mutations, indicating a functional role of these amino acids (Additional file 1: Table S6). Taken all rare missense mutations together, in our study we do not observe an enrichment in patients versus controls (SKAT-O p = 0.348, Additional file 1: Methods). This might be due to the presence of a high level of benign and protective missense variants. However, the carefully selected mutations included in the immunocytochemistry experiments show an effect on protein localization. Therefore, subgrouping the mutations based on e.g., homology to pathogenic mutations in other ABCA proteins might capture mutations that are likely pathogenic. Nonetheless, functional investigation of single rare missense mutations is required for classification purposes and to estimate their precise contribution to AD.
Missense mutations can affect structure, stability and folding of the protein [48]. Computational modeling can be used to predict the consequence of amino acid changes on protein stability, however, a crystallographic structure of ABCA7 is currently not available. To assess the effect of rare missense mutations on the ABCA7 protein, we investigated the subcellular localization of 10 predicted deleterious missense mutations identified in the Belgian cohort. Previous literature described the presence of a short splicing variant in ABCA7 (type II) in addition to full-length ABCA7 (type I) [47]. In vitro studies with HEK293 cells revealed that both proteins have a different subcellular localization and function [47]. Type I ABCA7 is mainly present at the plasma membrane and at sites corresponding to the intracellular route of exocytosis, while type II ABCA7 is predominantly restricted to the ER and is incapable of ApoA1-mediated lipid release unlike type I ABCA7 [47]. We transiently expressed wild type ABCA7 in HeLa cells and confirm its localization on the plasma membrane and intracellular compartments. We reveal that the 10 mutant ABCA7 constructs under study lead to a significant decrease in ABCA7 plasma membrane expression and are largely retained in the ER, likely impacting their cellular lipid release capacities, while predicted benign variants show the same localization pattern as wild type ABCA7. Our results are in line with data from ABCA1 and ABCA4 studies investigating the effect of missense mutations on protein localization and functional properties. Disease-causing mutations in these genes were found to cause mislocalization and/or to impair the substrate binding or ATPase activity of the transporter [32,33,34, 36, 49]. Our findings highlight the cellular localization of ABCA7 as a potential therapeutic target for AD, although validation of our results in induced pluripotent stem cell (iPSC) derived brain cells is needed to confirm the pathogenic effect of the studied missense mutations on endogenous ABCA7.
Two of the studied ABCA7 mutants, p.A845V and p.R1932C, are in conserved amino acid residues and mutations at these residues in ABCA1 or ABCA4 are known to cause Tangier disease or Stargardt disease respectively. The p.A845V mutation resides in the Walker A motif of NBD1. The Walker A motif is characterized by the consensus sequence GXNGAGKT/S, where X can be any amino acid. This motif is conserved in NBD2 of all members of the ABCA subfamily (ABCA1-13) and in NBD1 of the ABCA subgroup consisting of ABCA1, 2, 3, 4, 7, 12 and 13 [34, 50]. Importantly, the corresponding amino acid substitution of the conserved Alanine to Valine in ABCA1 (p.A937V) is responsible for Tangier disease and highlights the importance of this residue [51]. Interestingly, the ABCA1 p.A937V mutation was found to segregate with AD in a family [52]. In contrast to our results from ABCA7 p.A845V, studies investigating the subcellular location and functional characteristics of ABCA1 p.A937V did not show mislocalization, although cholesterol efflux was abolished, leaving the mutant dysfunctional [53, 54]. p.R1932C is present in the signature motif of NBD2. Substitution of the corresponding basic Arginine to the hydrophobic Tryptophan in ABCA4 (p.R2077W) is identified in a patient with Stargardt disease [32]. Characterization of this variant showed a significantly reduced functional activity as well as mislocalization of ABCA4 indicative of ER-retention [32]. In the ABCA7 p.R1932C, the basic Arginine is replaced for a hydrophobic Cysteine, and we observe a similar pattern of mislocalization.
We also identified patients carrying two rare ABCA7 alleles, either in trans or cis configuration. Allen et al. described a remarkably lower brain ABCA7 expression in two c.5570 + 5G > C PTC carriers who caried an additional predicted ABCA7 deleterious missense mutation compared to two c.5570 + 5G > C patient carriers with normal WT protein expression [7]. A higher mutational burden in ABCA7 might have a modifying effect on penetrance and pathogenicity. Mutational profiling of ABCA4 revealed that common variants contribute to increased risk for developing ABCA4-related disease and might act as penetrance modifiers when they are combined with a second ABCA4 mutation [55,56,57]. In addition, pathogenic deep-intronic variants in ABCA4 are found to contribute to the recessive inheritance of Stargardt disease [58, 59]. However, in our study we only considered rare (MAF ≤ 1%) coding and splice variants, while additional common, noncoding, or structural variants might as well contribute to the mutational spectrum.
Familial load of missense mutation carriers (58.9%) was lower compared to PTC mutation carriers (77.3%), but still higher than the overall AD cohort (50.0%) [9]. The difference in familial load between PTC and missense carriers might be explained by the bidirectional effect of missense mutations, which is reflected in the high numbers of missense mutations in control individuals. Highly variable onset ages are noted between missense mutation carriers (range 37–92 years old), highlighting once more the need to determine pathogenicity of individual missense mutations as well as the contribution of other genetic factors, e.g., APOE genotype. Since the inclusion age of control carriers (range 54–92) falls within the onset age range of patient carriers within our study, we cannot exclude that control carriers may develop AD later in life. Nonetheless, co-segregation of the mislocalizing p.G1820S mutation with AD on a shared haplotype of at least 2.63 Mb in a Belgian AD family with an autosomal dominant inheritance pattern is strengthening our findings on the potential role of ABCA7 missense mutations in AD. Since individuals II.1 and II.2 both have the APOE ε4/ε4 genotype, which is associated with a higher risk for AD [60], we cannot exclude that both p.G1820S and the APOE genotype contribute to the disease. The p.G1820S mutation affects the second NBD of ABCA7 and is predicted deleterious by Mutation Taster, Polyphen2 and SIFT and has a CADD score of 32. In our previous screening of ABCA7 in a European cohort of 928 EOAD patients, we observed the p.G1820S mutation in a Swedish AD-patient of 64 years old (APOE ε33) while it was absent in 980 control individuals [11]. The p.G1820S mutation is also absent in 478 healthy control individuals age 60 or older without neurodegenerative disease from the healthy exome (HEX) database (https://www.alzforum.org/exomes/hex). One earlier study reported co-segregation of the ABCA7 missense mutation p.R880Q with AD [40]. p.R880Q affects the first NBD of ABCA7 and has a CADD score of 28.7. This mutation is present in three patients and one control individual (0.22% versus 0.10%) within our Belgian cohorts and was found to affect protein localization. All patient carriers reported a positive familial history of dementia. Segregation analysis was not performed since DNA of relatives was not available.
Neuropathological examination in six carriers revealed the hallmarks of AD i.e., senile plaques and neurofibrillary tangles, in combination with (capillary) CAA in all patients. These findings are like the ABCA7 PTC carriers, in whom extensive levels of CAA and capillary CAA were noted as well (E. Hendrickx Van de Craen et al., personal communication) [61]. Several studies using in vitro and in vivo models have shown that ABCA7 deficiency may lead to a decreased microglial Aβ clearance and an increased APP processing, therefore exacerbating Aβ accumulation in the brain [62]. ABCA1 also plays a role in the pathogenesis of AD and CAA, as increased Aβ deposition as well as increased levels of CAA and CAA-related microhemorrhages were observed in AD mouse models lacking ABCA1 [63, 64].
In conclusion, we describe the frequency of rare ABCA7 indel, missense and splice mutations in the Belgian AD and control cohorts and explore the pathogenic nature of missense mutations on protein localization in vitro using immunocytochemistry. Our data support a role for rare ABCA7 missense mutations in the pathogenesis of AD and propose loss of functional ABCA7 at the plasma membrane due to impaired protein localization as the downstream pathogenic mechanism.
Availability of data and material
All data relevant to this study are included in the research paper or added to the Additional file 1. The corresponding author will share additional information upon reasonable request.
Abbreviations
- AAD:
-
Age at death
- AAI:
-
Age at inclusion
- AAO:
-
Age at onset age
- ABC:
-
ATP-binding cassette
- ABCA:
-
ATP-binding cassette subfamily A
- AD:
-
Alzheimer’s disease
- CAA:
-
Cerebral amyloid angiopathy
- CADD:
-
Combined annotation dependent depletion
- DD:
-
Disease duration
- ER:
-
Endoplasmic reticulum
- MAF:
-
Minor allele frequency
- NBD:
-
Nucleotide binding domain
- NMD:
-
Nonsense-mediated mRNA decay
- PTC:
-
Premature termination codon
- SNP:
-
Single nucleotide polymorphism
- STR:
-
Short tandem repeat
- TMD:
-
Transmembrane domain
- VNTR:
-
Variable number of tandem repeats
- WES:
-
Whole exome sequencing data
- WGA:
-
Wheat germ agglutinin
- WT:
-
Wild type
References
Cukier HN et al (2016) ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet 2(3):e79–e79
Hollingworth P et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43(5):429–435
Lambert J-C et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45(12):1452–1458
Logue MW et al (2011) A comprehensive genetic association study of Alzheimer Disease in African Americans. Arch Neurol 68(12):1569–1579
Naj AC et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43(5):436–441
Reitz C et al (2013) Variants in the ATP-binding cassette transporter (ABCA7), Apolipoprotein E ϵ4, and the risk of late-onset Alzheimer Disease in African Americans. JAMA 309(14):1483–1492
Allen M et al (2017) ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol Genet 3(1):e126–e126
Bellenguez C et al (2017) Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging 59:220.e1-220.e9
Bossaerts L et al (2021) Premature termination codon mutations in ABCA7 contribute to Alzheimer’s disease risk in Belgian patients. Neurobiol Aging 106:307-e1
Cuyvers E et al (2015) Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol 14(8):814–822
De Roeck A et al (2017) Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol 134(3):475–487
Del-Aguila JL et al (2015) Role of ABCA7 loss-of-function variant in Alzheimer’s disease: a replication study in European-Americans. Alzheimer’s Res Therapy 7(1):73–73
Kunkle BW et al (2017) Targeted sequencing of ABCA7 identifies splicing, stop-gain and intronic risk variants for Alzheimer disease. Neurosci Lett 649:124–129
Le Guennec K et al (2016) ABCA7 rare variants and Alzheimer disease risk. Neurology 86(23):2134
Vardarajan BN et al (2015) Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 78(3):487–498
Steinberg S et al (2015) Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 47(5):445–447
De Roeck A et al (2018) An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer’s disease. Acta Neuropathol 135(6):827–837
Jehle AW et al (2006) ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 174(4):547–556
Satoh K et al (2015) ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem 290(40):24152–24165
Sakae N et al (2016) ABCA7 deficiency accelerates amyloid-β generation and Alzheimer’s neuronal pathology. J Neurosci: Off J Soc Neurosci 36(13):3848–3859
Li M et al (2017) Study on lentivirus-mediated ABCA7 improves neurocognitive function and related mechanisms in the C57BL/6 mouse model of Alzheimer’s disease. J Mol Neurosci 61(4):489–497
Kim WS et al (2013) Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci: Off J Soc Neurosci 33(10):4387–4394
Fu Y et al (2016) ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis 54:569–584
Kaminski WE et al (2000) Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem Biophys Res Commun 273(2):532–538
Dean M, Rzhetsky A, Allikmets R (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 11(7):1156–1166
Brooks-Wilson A et al (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 22(4):336–345
Allikmets R (2000) Further evidence for an association of ABCR alleles with age-related macular degeneration. the International ABCR Screening Consortium. Am J Hum Genet 67(2):487–491
Allikmets R et al (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat Genet 15(3):236–246
Westeneng-van Haaften SC et al (2012) Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 119(6):1199–1210
Landrum MJ et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D1067
Brunham LR et al (2015) Clinical, biochemical, and molecular characterization of novel mutations in ABCA1 in families with Tangier disease. JIMD Rep 18:51–62
Garces F et al (2018) Correlating the expression and functional activity of ABCA4 disease variants with the phenotype of patients With Stargardt disease. Investig Ophthalmol Vis Sci 59(6):2305–2315
Garces FA, Scortecci JF, Molday RS (2020) Functional characterization of ABCA4 missense variants linked to Stargardt macular degeneration. Int J Mol Sci 22(1):185
Molday LL et al (2018) Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum Mol Genet 27(2):295–306
Quazi F, Molday RS (2013) Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem 288(48):34414–34426
Singaraja RR et al (2006) Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circ Res 99(4):389–397
Tanaka AR et al (2003) Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis*. J Biol Chem 278(10):8815–8819
Sassi C et al (2016) ABCA7 p.G215S as potential protective factor for Alzheimer’s disease. Neurobiol Aging 46:235.e1-235.e2359
De Roeck A, Van Broeckhoven C, Sleegers K (2019) The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol 138(2):201–220
May P et al (2018) Rare ABCA7 variants in 2 German families with Alzheimer disease. Neurol Genet 4(2):e224
Reumers J et al (2012) Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol 30(1):61–68
Lek M et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291
Auton A et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74
Weckx S et al (2005) novoSNP, a novel computational tool for sequence variation discovery. Genome Res 15(3):436–442
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675
Schindelin J et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682
Ikeda Y et al (2003) Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem Biophys Res Commun 311(2):313–318
Stein A et al (2019) Biophysical and mechanistic models for disease-causing protein variants. Trends Biochem Sci 44(7):575–588
Wiszniewski W et al (2005) ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet 14(19):2769–2778
Dean M, Hamon Y, Chimini G (2001) The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 42(7):1007–1017
Bodzioch M et al (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 22(4):347–351
Beecham GW et al (2018) Rare genetic variation implicated in non-Hispanic white families with Alzheimer disease. Neurol Genet 4(6):e286–e286
Landry YD et al (2006) ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions*. J Biol Chem 281(47):36091–36101
Nandi S et al (2009) ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J Lipid Res 50(3):456–466
Schulz HL et al (2017) Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter German cohort-impact of selected deep intronic variants and common SNPs. Investig Ophthalmol Vis Sci 58(1):394–403
Zernant J et al (2017) Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet 54(6):404–412
Runhart EH et al (2018) The common ABCA4 variant p.Asn1868Ile shows nonpenetrance and variable expression of Stargardt disease when present in trans with severe variants. Investig Ophthalmol Vis Sci 59(8):3220–3231
Sangermano R et al (2019) Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med 21(8):1751–1760
Khan M et al (2020) Detailed phenotyping and therapeutic strategies for intronic ABCA4 variants in stargardt disease. Mol Therapy - Nucleic Acids 21:412–427
Roses AD (1996) Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med 47:387–400
Van den Bossche T et al (2016) Phenotypic characteristics of Alzheimer patients carrying an ABCA7 mutation. Neurology 86(23):2126–2133
Aikawa T, Holm M-L, Kanekiyo T (2018) ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci 8(2):27
Koldamova R, Staufenbiel M, Lefterov I (2005) Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice*. J Biol Chem 280(52):43224–43235
Wahrle SE et al (2005) Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease*. J Biol Chem 280(52):43236–43242
Adzhubei IA et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Sim N-L et al (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40(W1):W452–W457
Kircher M et al (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315
Montine TJ et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11
Love S et al (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 3(1):19–32
Le My LT et al (2021) Cryo-EM structure of lipid embedded human ABCA7 at 3.6Å resolution. bioRxiv. https://doi.org/10.1101/2021.03.01.433448
Acknowledgements
The following members of the Belgian Neurology (BELNEU) consortium contributed by diagnosing the Alzheimer patients and selection patients to include in the Belgian AD cohort for this study: Johan Goeman, Roeland Crols, Peter P. De Deyn (General Hospital Network Antwerp, Antwerp); Anne Sieben, Patrick Cras (University Hospital Antwerp, Edegem); Tim Van Langenhove, Bart Dermaut (University Hospital Ghent, Ghent); Olivier Deryck, Bruno Bergmans (General Hospital Sint-Jan Brugge, Bruges); Jan Versijpt (University Hospital Brussels, Brussels); Bernard Hanseeuw (University Hospital Saint-Luc, Brussels); Boudewijn Michielsens (General Hospital Sint-Jozef, Malle). Anne Sieben (University Hospital Antwerp, Edegem) provided the autopsy neuropathological data. The authors are thankful for the support of the personnel of the VIB CMN Neuromics Support Facility, the DNA Screening Facility and the Human Biobank of the NBD group.
Funding
The research was in part supported by the Flemish Government initiated Methusalem excellence program and the Flanders Impulse Program on Networks for Dementia Research.
Author information
Authors and Affiliations
Contributions
LB conceived part of the design of the study, performed experiments, analyzed the data, created the figures, and drafted the manuscript; EH(VdC) conceived part the design of the study, delivered phenotype data of the patients, selected patients for the study, wrote part of the drafted manuscript; RC conceived part of the design of the study, provided training and advised cell experiments, assisted in data interpretation, revised the manuscript; BA assisted designing the cell work and immunohistochemistry studies, analyzed confocal microscopy images, assisted in creating the respective figures and revised the manuscript; CVB conceived and oversighted the project and the design of the study, provided funding for the research, contributed to the writing of the paper and the critical revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants and/or their legal guardian provided written informed consent before inclusion in the study. The ethic committee of the University Hospital of Antwerp and University of Antwerp approved the informed consent form and the clinical, pathological and genetic protocols used in this study.
Consent for publication
Not applicable.
Competing interests
None of the authors reported personal competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary material and methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bossaerts, L., Hendrickx Van de Craen, E., Cacace, R. et al. Rare missense mutations in ABCA7 might increase Alzheimer’s disease risk by plasma membrane exclusion. acta neuropathol commun 10, 43 (2022). https://doi.org/10.1186/s40478-022-01346-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01346-3