
Abstract
Frontotemporal lobar degeneration (FTLD) is the umbrella term encompassing a heterogeneous group of pathological disorders. With recent discoveries, the FTLDs have been show to classify nicely into three main groups based on the major protein deposited in the brain: FTLD-tau, FTLD-TDP and FTLD-FUS. These pathological groups, and their specific pathologies, underlie a number of well-defined clinical syndromes, including three frontotemporal dementia (FTD) variants [behavioral variant frontotemporal dementia (bvFTD), progressive non-fluent aphasia, and semantic dementia (SD)], progressive supranuclear palsy syndrome (PSPS) and corticobasal syndrome (CBS). Understanding the neuropathological background of the phenotypic variability in FTD, PSPS and CBS requires large clinicopathological studies. We review current knowledge on the relationship between the FTLD pathologies and clinical syndromes, and pool data from a number of large clinicopathological studies that collectively provide data on 544 cases. Strong relationships were identified as follows: FTD with motor neuron disease and FTLD-TDP; SD and FTLD-TDP; PSPS and FTLD-tau; and CBS and FTLD-tau. However, the relationship between some of these clinical diagnoses and specific pathologies is not so clear cut. In addition, the clinical diagnosis of bvFTD does not have a strong relationship to any FTLD subtype or specific pathology and therefore remains a diagnostic challenge. Some evidence suggests improved clinicopathological association of bvFTD by further refining clinical characteristics. Unlike FTLD-tau and FTLD-TDP, FTLD-FUS has been less well characterized, with only 69 cases reported. However, there appears to be some associations between clinical phenotypes and FTLD-FUS pathologies. Clinical diagnosis is therefore promising in predicting molecular pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term frontotemporal dementia (FTD) is reserved for a cluster of syndromes that manifest as a result of pathological damage to the frontal and temporal lobes [14, 94]. It encompasses three main clinical syndromes: behavioral variant FTD (bvFTD); progressive non-fluent aphasia (PNFA) and semantic dementia (SD) [94]. The prevalence of FTD has been estimated to be approximately 15 per 100,000 [102] while the incidence ranges from 2.2 to 8.9 per 100,000 depending on the age of onset [70]. In addition, some patients present with features of FTD, as well as motor neuron disease (MND). When these two phenomena co-occur, the syndromic diagnosis rendered is FTD-MND [23, 46]. Two other syndromes, progressive supranuclear palsy syndrome (PSPS) [78] and corticobasal syndrome (CBS) [8] are closely related to the FTDs. These six clinical syndromes are linked to a number of molecular pathologies that target the frontal and temporal lobes, known as the frontotemporal lobar degenerations (FTLDs) [48].
Unlike FTD which is a clinical term, FTLD is a pathological term [87]. Like FTD, the term FTLD encompasses a cluster of diseases, in this instance, a cluster of molecular pathologies [48]. These molecular pathologies are classified according to the major biochemical abnormality identified at post-mortem, i.e. the major protein deposited.
Understanding the relationship between FTLD, FTD, PSPS and CBS has proven challenging. Over the last decade, many researchers have investigated the neuropathological background of the phenotypic variability of FTD, PSPS and CBS. Together these studies have identified variable associations between specific clinical syndromes and molecular pathologies. This review will address the relationship of the FTLD molecular pathologies to the associated clinical syndromes of FTD, PSP and CBS by dissecting each clinical syndrome and molecular pathology. In addition, we pool data from several large clinicopathological studies assessing 544 cases (Table 1) in order to determine associations between clinical syndromes and molecular pathologies. Chi-square tests were performed to assess the strengths of the associations between clinical syndromes and molecular pathologies. These studies were all from different institutions and all studies were reviewed in detail to remove any overlapping cases.
The frontotemporal lobar degenerations
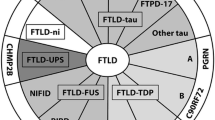
The frontotemporal lobar degenerations (FTLDs) are a heterogeneous group of diseases that overlap in gross and histological features. All are associated with varying degrees of atrophy, neuronal loss and gliosis of the frontal and temporal lobes. However, each disease differs from one another by differences in protein deposition or biochemical signature, and inclusion morphology and distribution [15]. Three major proteins have been identified as leading players in the mechanism of neurodegeneration of the FTLDs. These three proteins are the microtubule-associated protein, tau [43], the transactive response DNA binding protein of 43 kD (TDP-43) [1, 98], and the tumor associated protein fused in sarcoma (FUS) [75]. Therefore, at the highest stratum, the majority of FTLDs can be sub-classified into FTLD-tau, FTLD-TDP and FTLD-FUS [84], based on the biochemical signature of the abnormally deposited protein (Fig. 1). Further sub-classification within each of these three groups is predominantly based on inclusion morphology and lesion distribution [15, 84, 85], although other characteristics may also be utilized, such as, tau isoform dominance, phosphorylation and ubiquitination status, and cleavage sites. Using this classification scheme almost 100% of the FTLDs can be sub-classified, with only a handful of extremely rare cases remaining unclassified (FTLD-other).

Diagram illustrating the current classification scheme for the FTLDs. FTLD-tau pathologies are shown in green, FTLD-TDP in red, FTLD-FUS in yellow and FTLD-other in blue. The genes associated with each pathological group are also shown. FTLD-TDP typing (1–3) is based on the classification by Mackenzie et al.; type 4 is based on the classification of Cairns et al. 9p Linkage identified on chromosome 9P
FTLD-tau
The FTLD-tau group consists of diseases in which the major abnormal protein, identified by immunohistochemistry, is the microtubule-associated protein, tau. This group consists of the following diseases: Pick’s disease (PiD) [28, 29], progressive supranuclear palsy (PSP) [37], corticobasal degeneration (CBD) [30], argyrophilic grains disease (AGD) [11, 12], sporadic multisystem tauopathy with globular inclusions (MST) [7, 53, 73], and diffuse neurofibrillary tangle dementia with calcifications (DNTC) [71, 72] (Fig. 1). Combining data from five large clinicopathological studies [31, 38, 56, 67, 111] of 544 pathologically confirmed FTLD patients, it appears that some FTLD-tau pathologies are much more common than others. The most common pathology was CBD accounting for 35% of cases, followed by PSP with 31%, PiD with 30% and AGD accounting for the remaining 4% of all FTLD-tau (Fig. 2a). No cases of MST or DNTC were reported in any of these large clinicopathological studies. It should be noted that PSP in general is a more common pathology than CBD suggesting a recruitment bias likely driven by the fact that these autopsy cases were mainly from dementia clinics. These studies only recruited patients with FTD-like diagnoses, some even excluded PSPS, and therefore patients with relatively pure motor syndromes without prominent behavioral changes would not have been included in these series; hence it is no surprise that PSP was less common than CBD since cognitive impairment is more likely a feature of CBD than PSP (manuscript in press).

Pie charts showing the proportion of specific pathologies observed in FTLD-tau (a) and FTLD-TDP (b), based on pooled data from eight large clinicopathological studies. The number and percentage associated with each pathology is also shown. FTLD-TDP typing is based on the classification by Mackenzie et al.
The characteristic histological features of the FTLD-tau molecular pathologies are shown in Figs. 3, 4. Unlike the other diseases in this category, PiD is a 3 repeat (3R) tauopathy [13], i.e. deposited tau is characterized by 3 binding domains in the carboxyl terminus of the protein, as opposed to 4 binding domains (4R) in PSP [20], CBD [115], AGD [119] and MST [7]. A mixture of 3R and 4R binding domains is observed in intracellular neurofibrillary tangles in DNTC [45], although extracellular tangles (ghost tangles) are predominantly 3R tau (Fig. 4). Additional features differentiating PiD from the other pathologies are the rounded appearance of the inclusions, the fact that inclusions are strongly argyrophilic but do not stain with Gallyas [120, 121], and are localized to the cytoplasm of the neuron, and the absence of features typical for the other diseases [28, 29]. Unlike PiD, neuronal inclusions in PSP are much larger and globose in morphology. In addition, PSP is characterized by the presence of astrocytic lesions, better known as tufted astrocytes. Corticobasal degeneration, like PSP, can show large neuronal inclusions, but they tend to be more rounded (so called cortico-basal bodies) [33]. In addition, CBD is characterized by the presence of a different type of astrocytic lesion, known as the astrocytic plaque [30]. In CBD, unlike in PSP, there is also widespread white matter pathology where numerous thread-like lesions are easily identified. There is also a biochemical difference between PSP and CBD, with PSP showing a 33-kD tau band on immunoblots of sarkosyl-insoluble brain extracts while CBD shows a 37-kD band [2]. Given these differences, many researchers have continued to support splitting PSP and CBD [27]. The other two FTLD-tau entities are characterized by: the presence of globular oligodengroglial inclusions in MST [7, 53, 73]; and a prominence of widespread intracellular neurofibrillary tangles and ghost tangles with intraparenchymal calcification in DNTC [71].

Histological features of the more common 4R FTLD-tau molecular pathologies. PSP features, including globose NFT with H&E (a) and tau (b), and a tufted astrocyte (c) with tau immunohistochemistry. CBD features, including ballooned neurons with H&E (d), and pretangles in basal nucleus (e) and astrocytic plaques in the cortex (f) both with tau immunohistochemistry. Features of a MAPT mutation (P301L) with tau deposition, including ballooned neurons with H&E (g) and threads, ballooned neurons and pretangles in cortex (h), and pretangles in dentate fascia (i) with tau immunohistochemistry

Histological features of the 3R, 3R + 4R and remaining 4R FTLD-tau molecular pathologies. Features of Pick disease, including globose Pick bodies in dentate fascia with H&E (a) and with 3R tau (b), but negative for 4R tau (c). Features of DNTC, including extracellular NFT with H&E (d) and with 3R tau (e), but mostly negative for 4R tau (note some neurites and glial 4R tau immunoreactivity) (f). Features of AGD, including argyrophilic grains with Bielschowsky (g) and sparse pretangles but negative grains with 3R tau (h), and many grains with 4R tau (i)
There are very good clinicopathological associations between FTLD-tau as a group, FTD, PSPS and CBS which are discussed below. However, it is still difficult to differentiate the individual pathologies, such as PSP from CBD, based solely on clinical presentation. The other entities in this group, MST, AGD and DNTC are rare when compared to PiD, PSP and CBD with only a handful of case reports or small case series of each of these pathologies being associated with FTD, PSPS or CBS [7, 26, 44, 45, 53, 71, 73, 93]. Therefore, no strong clinicopathological associations exist for MST, AGD and DNTC.
FTLD-TDP
Similar to FTLD-tau, classification of the subtypes of FTLD-TDP is based on the morphological appearance of the inclusions and the distribution of the lesions. Four subtypes of FTLD-TDP are currently recognized (Fig. 1) [15, 80, 105], although overlap across subtypes have been reported [3]. FTLD-TDP types 1–3 have been described in two different classification schemes by MacKenzie et al. and Sampathu et al. [80, 105], although each classification scheme maps well onto the other. Therefore, FTLD-TDP type 1 in Mackenzie’s scheme maps to FTLD-TDP type 3 in Sampathu’s scheme; Mackenzie’s type 2 maps to Sampathu’s type 1 and Mackenzie’s type 3 maps to Sampathu’s type 2. In this review we use the Mackenzie’s classification scheme since that scheme was initially shown to have good association with clinical phenotype. Based on data from four clinicopathological studies [35, 57, 80, 111], the most common subtype was FTLD-TDP type 1 accounting for 41% of cases followed by FTLD-TDP type 3 with 34% and then FTLD-TDP-type 2 with 25% (Fig. 2b). There were no cases of FTLD-TDP type 4 in any of these large clinicopathological studies.
In Mackenzie’s scheme, FTLD-TDP type 1 is characterized by a combination of neuronal cytoplasmic inclusions (NCI) and short, comma shaped, dystrophic neurites in frontotemporal cortex, as well as the common occurrence of neuronal intranuclear inclusions; FTLD-TDP type 2 by a predominance of long thicker dystrophic neurites and minimal to absent NCI in frontotemporal cortex, and FTLD-TDP type 3 by a predominance of NCI in frontotemporal cortex or the dentate granule cell layer of the hippocampus and absent to minimal dystrophic neurites in cortex (Fig. 5). FTLD-TDP type 4 is characterized by a predominance of intranuclear inclusions, dystrophic neurites and absent to little NCI [95], and is associated with mutations in the valosin containing protein gene [128]. There is good association between FTLD-TDP types and clinical syndromes as discussed below.

Histological features of FTLD-TDP types based on the classification of Mackenzie et al. a Features of FTLD-TDP type 1 (Sampathu, type 3) including short, thin neurites and pleomorphic neuronal cytoplasmic inclusions (NCI) with neuronal intranuclear inclusions (upper inset). b shows features of FTLD-TDP type 2 (Sampathu, type 1) including long, thick neurites in cortex (upper inset) as well as a Pick-body-like NCI in dentate fascia (lower inset). c Features of FTLD-TDP type 3 (Sampathu, type 2) including NCI and granular cytoplasmic staining (pre-inclusions—upper inset) with irregular granular NCI in dentate fascia (lower inset). c, d Features of FTLD-TDP type 4 including NCI, neurites and many neuronal intranuclear inclusions (lower insets). All with TDP-43 immunohistochemistry
FTLD-FUS
This newest FTLD category consists of three relatively rare diseases: neuronal intermediate filament inclusion disease (NIFID) [16, 52], basophilic inclusion body disease (BIBD) [74] and atypical FTLD with ubiquitin-only immunoreactive changes (aFTLD-U) [54, 82, 103] (Fig. 1). These three entities share the fact that they all show striking FUS immunoreactivity [92, 96, 97], but there are differences between each of them that allow each to be considered a separate pathological entity (Fig. 6). The first, NIFID, originally known as neurofilament inclusion body disease [52], is characterized by the presence of variable shaped inclusions that appear more eosinophilic on hematoxylin and eosin (H&E), variable immunoreactivity to ubiquitin, but strikingly immunoreactivity to intermediate filaments, including neurofilament and alpha-internexin [18, 58, 122]. Similar to NIFID, in BIBD, the inclusions are visible on H&E, although in BIBD the inclusions appear basophilic. Unlike in NIFID, antibodies to epitopes of neuronal intermediate filaments do not immunostain the neuronal inclusions, however, like NIFID, ubiquitin staining is variable in BIBD. The third FTLD-FUS entity, aFTLD-U, differs from both NIFID and BIBD as inclusions are not observed on H&E. In addition, aFTLD-U can be further differentiated from NIFID since the inclusions are not immunoreactive to intermediate filaments, and can be further differentiated from BIBD since vermiform intranuclear inclusions present in aFTLD-U are absent or rare in BIBD [83]. Motor neuron degeneration is found in around 50% of NIFID and BIBD cases but has not been reported in aFTLD-U. One feature that appears to be characteristic of the FTLD-FUS group is striking early caudate atrophy [63, 106]; by the time of autopsy, the caudate nucleus usually has a concave appearance.

Histological features of FTLD-FUS molecular pathologies. Features of aFTLD-U, including striking striatal atrophy (a), neuronal cytoplasmic inclusions (NCI) (b) and vermiform neuronal intranuclear inclusions (c) with FUS immunohistochemistry. Features of NIFID, including internexin positive NCI (note weak cytoplasmic staining in pyramidal neurons) (d) and FUS positive NCI in cortex with FUS immunohistochemistry (e). Features of BIBD with juvenile motor neuron disease, including large basophilic cytoplasmic inclusions (f) that are positive with FUS immunohistochemistry (g)
FTLD-other
The FTLD-other category is reserved for diseases in which the major protein associated with the disease entity remains unknown. Currently, there are two rare pathological entities in this category [123]. The first, FTLD without inclusions (FTLD-ni), is diagnosed when there is histological evidence of frontotemporal neuronal loss and gliosis but all routine and immunohistochemical stains fail to reveal the presence of any inclusions. The term FTLD-ni is the preferred terminology, replacing the older terminology of dementia lacking distinctive histology. The second, FTLD with immunohistochemistry against proteins of the ubiquitin proteosomal system (FTLD-UPS), is diagnosed when there are ubiquitin or p62 immunoreactive inclusions, that are negative to tau, alpha-synuclein, alpha-internexin, TDP-43 and FUS. The majority of FTLD-UPS cases are associated with mutations in the charged multivesicular body protein 2B (CHMP2B) gene [40], although a few sporadic cases exist [123]. The FTLD-other category may also be used for extremely rare FTLD cases that do not conform to the pathological criteria discussed above.
The frontotemporal dementias
Three classic syndromes are subsumed under the rubric of frontotemporal dementia (FTD): bvFTD, PNFA and SD [94]. The behavioral variant of FTD is characterized by changes in behavior and personality, resulting in disruption of social interactions. Executive dysfunction is common. Symptoms suggestive of the diagnosis of bvFTD include apathy, disinhibition, poor planning, poor organization, hyperactive behaviors such as wandering and pacing, as well as changes in eating, sleeping and sexual behaviors. The age of onset of patients with bvFTD is typically less than 65 years with an average age of onset around 58 [47]. The syndrome of bvFTD can be associated with many different FTLD pathologies. However, given recent molecular pathological discoveries, some investigators have argued that there may be subtypes of bvFTD that more tightly link to specific molecular pathologies (Fig. 7). Recent clinicopathological studies have identified, for example, a clinical syndrome of behavioral and personality change dominated by hypersexual and hyperphagic behaviors, prominent stereotypy and obsessionality that tightly links to the molecular pathology of aFTLD-U [113, 123]. This finding links two previous reports of (1) striking striatal atrophy in patients with bvFTD and stereotypy [61], and (2) striking striatal atrophy in aFTLD-U [63]. An additional feature common to these patients is a very young age at onset, as patients tend to present with a mean age of onset of around 40 years. Another less well-established association is that of bvFTD with psychosis and FTLD-TDP type 3 with MND [76]. Unlike bvFTD associated with aFTLD-U, bvFTD associated with FTLD-TDP type 3 tends to have a more typical age of onset; patients are also not hypersexual, stereotypic or hyperphagic. This latter bvFTD with psychosis sub-syndrome is reminiscent of the syndrome of dementia with Lewy bodies [22]. One could argue that these ‘sub-syndromes’ are all bvFTD, although there appears to be benefit in separating these sub-syndromes in order to better predict the underlying molecular pathology (Fig. 7).

Diagram showing clinical features that help to predict the underlying FTLD-tau, FTLD-TDP or FTLD-FUS molecular pathology in patients presenting with bvFTD. VSGP vertical supranuclear gaze palsy. FTLD-TDP types based on the classification of Mackenzie et al.
The syndrome of PNFA is characterized by ‘non-fluent’ speech output with agrammatic and telegraphic speech [94]. Frequently accompanying the aphasia is a motor speech disorder known as apraxia of speech (AOS) [51, 99]. Many researchers do not separate AOS from PNFA and hence the term PNFA typically implies the presence of aphasia and AOS [34]. Detailed studies of PNFA, however, have demonstrated that the presence of AOS is tightly associated with FTLD-tau, especially PSP and CBD [26, 51]. Neuroanatomic studies have also demonstrated that the presence of AOS correlates with atrophy of the premotor and supplemental motor cortices [51], regions that are prominently and focally affected in PSP and CBD. It is therefore not surprising that the most common pathologies underling AOS, and hence PNFA, are PSP and CBD, as discussed below. Predicting PSP over CBD, and vice versa, is difficult in PNFA, although there is some evidence to suggest that pure or dominant AOS is more suggestive of PSP, while AOS plus prominent aphasia is more suggestive of CBD [50]. This hypothesis would be supported by the fact that CBD pathology is associated with slightly less focal atrophy than PSP [59], and tends to also involve the inferior posterior lateral frontal lobe or Broca’s area [129]. It remains to be determined whether aphasia without AOS is linked to a specific molecular pathology but evidence exists to suggest that it is linked to FTLD-TDP [26], particularly FTLD-TDP type 1 pathology [111], as discussed below.
The term SD is reserved for a clinical syndrome characterized by fluent speech with prominent anomia, loss of word and object meaning, and poor single word comprehension [110, 127]. The clinical syndrome of SD has been found to highly associate with FTLD-TDP type 2 pathology [39, 57, 80, 111]. Patients with SD commonly present with aphasia that is associated with left anterior medial temporal lobe atrophy [21, 91]. The right temporal lobe is typically also affected in SD, and in some instances is even more atrophic than the left [118]. In such cases of right greater than left temporal lobe atrophy, the clinical presentation of SD is dominated, not by aphasia, but by prosopagnosia and behavioral dyscontrol [64, 107, 118]. Not surprisingly, therefore, behavioral changes have been shown to be prominent in SD [112]. Unlike the anomic presentation of SD that has been strongly associated with FTLD-TDP type 2 pathology, the behavioral/prosopagnostic presentation of SD is more easily confused with bvFTD. Detailed studies on SD, however, have shown that the behavioral/prosopagnostic presentation of SD also links to FTLD-TDP type 2 pathology. One study suggested that the presence or absence of prosopagnosia is helpful in differentiating SD with greater right temporal lobe atrophy and behavioral features from bvFTD with prominent right temporal lobe atrophy [62]. This is important since it appears that bvFTD with prominent right temporal lobe atrophy is associated with FTLD-tau pathology [62].
Some patients present with symptoms suggestive of a diagnosis of FTD, however, clinical and electrophysiological examination confirms coexisting MND (FTD-MND) [23, 79]. In FTD-MND, the FTD syndrome is typically characterized by behavioral change, although cases with prominent aphasia have been described [19]. The MND domain can be characterized by bulbar problems such as swallowing or speech difficulties [42, 76], although limb weakness associated with fasciculations in the weak limb is also common. Signs of upper motor neuron disease or pyramidal tract signs, such as spasticity, hyper-reflexia, Babinski and clonus, are less common, but have been described [55]. A clinical diagnosis of FTD-MND has almost perfect association with molecular pathology as revealed below.
Progressive supranuclear palsy and corticobasal syndromes
The terms PSPS and CBS are reserved for clinical diagnosis, while the terms PSP and CBD are now reserved for pathological diagnosis. Progressive supranuclear palsy syndrome is characterized by a symmetric akinetic syndrome with prominent axial rigidity, vertical supranuclear gaze palsy (VSGP) and early falls [131]. Apathy is common in PSPS, while other behavioral and personality changes are less common [4, 10, 60]. A diagnosis of classic PSPS, i.e. without prominent behavioral dyscontrol, is highly predictive of PSP pathology [56]. Recent evidence suggests, however, that patients with VSGP, as in PSPS, but prominent behavioral dyscontrol and dementia as in bvFTD, may actually more likely be CBD, instead of PSP (manuscript in press) (Fig. 7). Corticobasal syndrome is characterized by asymmetric cortical and extrapyramidal signs occurring together [8]. Cortical signs include limb apraxia, action-induced myoclonus and alien limb phenomenon, while limb rigidity and dystonia are relatively common extrapyramidal features. Unfortunately, CBS shows poor association to any single molecular pathology [9], although CBD and PSP, both FTLD-tau, account for the majority of cases of CBS.
Clinicopathological associations
Four large studies representing work from six different centers in the United States, Canada, United Kingdom and Australia have been published on clinicopathological associations between FTLD, and specific FTD, PSPS and CBS clinical diagnoses [38, 56, 67, 111]. When data from all four studies were pooled, variable clinicopathological associations were observed. The observed proportions of FTLD-tau to FTLD-TDP differed significantly across the different clinical syndromes (p < 0.0001) (Fig. 8). In addition, the observed proportions of FTLD-tau to FTLD-TDP differed from that expected by chance in all clinical groups (p < 0.05), except for bvFTD (p = 0.22). Almost all cases of PSPS and CBS had FTLD-tau pathology, while 100% of FTD-MND cases had FTLD-TDP pathology. Semantic dementia was associated with FTLD-TDP, with 83% of cases showing FTLD-TDP pathology, and PNFA was associated predominantly with FTLD-tau (70%). The bvFTD syndrome showed almost equal proportions of FTLD-tau and FTLD-TDP.

Bar chart showing the proportion of patients with FTLD-tau or FTLD-TDP within each clinical diagnosis based on pooled data from large clinicopathological studies. Actual number of subjects with each clinical diagnosis is also shown
Associations between clinical syndromes and specific FTLD-tau pathologies are shown in Fig. 9a. The observed proportions of FTLD-tau pathologies (PiD, CBD, PSP, AGD) differed significantly across the different clinical syndromes (p < 0.0001). In addition, the observed proportions of FTLD-tau pathologies differed from that expected by chance in all clinical groups (p < 0.05), except for PNFA (p = 0.10). Of the patients diagnosed with SD and having FTLD-tau pathology (n = 3), all showed PiD, suggesting that PSP and CBD are extremely unlikely to be associated with the SD syndrome. Another excellent association was noted between PSPS and PSP pathology, where over 90% of patients with PSPS had PSP, and the remainder had CBD. Weaker associations were found between the clinical diagnosis of bvFTD and pathology, although the data showed that if bvFTD was associated with FTLD-tau there was almost a 70% chance that the pathology would be PiD. The diagnosis of PNFA was not associated with any specific FTLD-tau pathology, and was split almost equally between PiD, CBD and PSP. The most common tau pathology associated with CBS was CBD, followed by PSP and PiD.

Bar charts showing the proportion of patients with specific FTLD-tau (a) or FTLD-TDP (b) pathologies within each clinical diagnosis, based on pooled data from large clinicopathological studies. Actual number of subjects with each clinical diagnosis is also shown. FTLD-TDP types based on the classification of Mackenzie et al.
For the FTLD-TDP group, when we pooled data we also found variable clinicopathological associations [25, 35, 57, 80] (Fig. 9b). The observed proportions of FTLD-TDP types differed significantly across the different clinical syndromes (p < 0.0001) and differed from that expected by chance in all clinical groups (p < 0.05). The strongest associations were identified between FTD-MND and FTLD-TDP type 3 pathology, between SD and FTLD-TDP type 2 pathology, and between PNFA and FTLD-TDP type 1 pathology. These clinicopathological associations support the sub-typing of FTLD-TDP. Once again, bvFTD was not as strongly associated with any one type, although the most common was FTLD-TDP type 1. As mentioned earlier, one has to wonder whether the syndrome of bvFTD is too loose and hence needs to be revised. One has to also wonder whether bvFTD associated with FTLD-TDP type 2 represents misdiagnosed cases of SD.
There are no large clinicopathological studies that take into account FTLD-tau, FTLD-TDP and FTLD-FUS. Therefore, less is known about associations between FTLD-FUS, FTD, PSPS and CBS. With that said, there are many small case series and a few large ones that allow us to pool data on 69 FTLD-FUS cases and get a glimpse of possible associations between clinical syndromes and FTLD-FUS [16, 54, 58, 90, 92, 96, 97, 123] (Fig. 10). It appears that NIFID is associated with three syndromes; the most common of which is bvFTD (Fig. 10a). It can also be associated with FTD-MND. A third syndrome associated with NIFID is a CBS/primary lateral sclerosis (PLS) hybrid which not only shows features of CBS but also prominent upper motor neuron signs such as spasticity, Babinski and clonus. On the flip side, one could say that this CBS/PLS syndrome is characterized by asymmetric upper motor neuron disease plus parkinsonism. Many syndromes can underlie the pathology of BIBD including bvFTD, FTD-MND, PSPS, CBS/PLS and others, such as, pure MND (Fig. 10b) or juvenile amyotrophic lateral sclerosis. An excellent association was, however, identified between aFTLD-U and bvFTD (Fig. 10c), as discussed earlier.

Bar charts showing the proportion of patients with each clinical diagnosis in the FTLD-FUS variants, NIFID (a), BIBD (b), and aFTLD-U (c). Data is pooled from small case series on FTLD-FUS variants. Actual number of subjects with each clinical diagnosis is also shown
There are no clinicopathological studies on FTLD-UPS or FTLD-ni. Furthermore, less than a handful of such cases exists [123] with the exception of the CHMP2B subtype of FTLD-UPS which is discussed below.
Clinicopathological associations of genetic variants of FTLD
Although the majority of FTLD cases are sporadic, a small familial subset is associated with gene mutations. In many instances, these gene mutations result in a syndrome presenting similar to one of the syndromes described above. Six genes are currently associated with FTLD. Mutations in the microtubule-associated tau gene, MAPT [43], are typically associated with neuronal and glial tau deposition (Fig. 3), and although there is no consistent pattern to the tau deposition [125] it can sometimes resemble other sporadic tauopathies such as PiD, PSP and CBD [32]. Most commonly, patients with MAPT mutations present with a bvFTD-like phenotype [100] in which extrapyramidal features may also be present. Not surprisingly, however, given the prominent anterior medial temporal lobe atrophy identified in patients with a MAPT mutation [104, 130], an SD-like presentation associated with features of bvFTD (or bvFTD with semantic impairment), in particular disinhibition, has also been associated with MAPT mutations [62, 100, 111] (Fig. 7). Another gene associated with familial FTLD is progranulin (GRN) [5, 24]. Unlike MAPT that is associated with tau, GRN mutations are associated with TDP-43 deposition; specifically FTLD-TDP type 1 pathology with neuronal intranuclear inclusions [17, 49, 81]. It is now well recognized that familial GRN is strongly associated with a CBS-like presentation [66, 86, 116], as well as a PNFA-like presentation [88, 111, 114] and a bvFTD-like presentation [66], especially with apathy [101] (Fig. 7). Mutations in other genes associated with FTLD pathology are extremely rare and include mutations in the VCP gene [128], the CHMP2B gene [109], the TDP-43 (TARDBP) gene [6] and the FUS gene [132]. While mutations in the VCP gene are associated with a syndrome combining FTD, Paget’s disease of the bone and myopathy [128], mutations in CHMP2B are typically associated with a bvFTD-like syndrome [36] and has only been identified in two families to date [36, 124]. Mutations in the TARDBP and FUS genes are predominantly associated with familial MND [65, 75, 117, 126], but rarely with an FTD-like syndrome.
These genetic clinicopathological associations differ somewhat from the clinicopathological associations identified in sporadic FTD and related disorders. For example, clinical diagnoses of PNFA and CBS are more likely to be associated with FTLD-tau in sporadic cases while in familial cases they are more likely to be associated with FTLD-TDP, particularly type 1, pathology. However, a diagnosis of sporadic or familial bvFTD could be associated with either FTLD-tau or FTLD-TDP. Semantic dementia and PSP are almost always sporadic.
Overlap with Alzheimer’s disease
Distinguishing FTLD from Alzheimer’s disease (AD) is also of paramount importance. AD, like DNTC, is characterized by 3R + 4R tau deposition. Alzheimer’s disease is usually associated with loss of episodic memory. However, there have been reports of AD being associated with some variants of FTD, commonly PNFA, and CBS which would not be captured in clinicopathological studies of FTLD pathology [41, 67, 68, 77, 108]. Recent work has revealed that AD is more likely present when the aphasia syndrome is dominated by phonological errors, i.e. logopenic progressive aphasia [89], a syndrome that can be mistaken for PNFA. A diagnosis of frontal variant AD is given when the ante-mortem clinical diagnosis is bvFTD and the pathological diagnosis is AD. Such cases occur but are rare, with AD pathology being reported in only 5–6% of bvFTD cases [67, 69].
Summary
In this review, we have discussed the neuropathological background of phenotypic variability of FTD. Some clinical syndromes have good–excellent associations with the FTLD pathological group, and even with specific molecular pathologies. These syndromes allow for better prediction of pathology than those syndromes with average-poor clinicopathological associations. One such syndrome with excellent prediction of underlying pathology is FTD-MND. Other syndromes, however, show little association with a single FTLD pathological group and even less association with any single FTLD pathology. This latter finding, implores us, as researchers, to continue to dissect the complex labyrinth of FTLD.
Abbreviations
- FTLD:
-
Frontotemporal lobar degeneration
- TDP-43:
-
Transactive response DNA binding protein of 43 kD
- FUS:
-
Fused in sarcoma
- AD:
-
Alzheimer’s disease
- 3R:
-
Three repeat
- 4R:
-
Four repeat
- FTLD-tau:
-
FTLD characterized by tau immunoreactive inclusions
- FTLD-TDP:
-
FTLD characterized by TDP-43 immunoreactive inclusions
- FTLD-FUS:
-
FTLD characterized by FUS immunoreactive inclusions
- FTLD-other:
-
Unclassified FTLD (not characterized by tau, TDP-43 or FUS immunoreactive inclusions)
- FTLD-MND:
-
FTLD with motor neuron degeneration
- FTLD-U:
-
FTLD with ubiquitin-only immunoreactive inclusions
- PiD:
-
Pick’s disease
- PSP:
-
Progressive supranuclear palsy
- CBD:
-
Corticobasal degeneration
- AGD:
-
Argyrophilic grains disease
- MST:
-
Sporadic multisystem tauopathy with globular inclusions
- DNTC:
-
Diffuse neurofibrillary tangle dementia with calcifications
- NIFID:
-
Neuronal intermediate filament inclusion disease
- BIBD:
-
Basophilic inclusion body disease
- aFTLD-U:
-
Atypical FTLD with ubiquitin-only immunoreactive changes
- FTLD-ni:
-
FTLD without inclusions
- FTLD-UPS:
-
FTLD with immunohistochemistry against proteins of the ubiquitin proteosomal system
- NCI:
-
Neuronal cytoplasmic inclusions
- H&E:
-
Hematoxylin and eosin
- NFT:
-
Neurofibrillary tangle
- FTD:
-
Frontotemporal dementia
- bvFTD:
-
Behavioral variant of FTD
- MND:
-
Motor neuron disease
- FTD-MND:
-
Frontotemporal dementia with motor neuron disease
- SD:
-
Semantic dementia
- PNFA:
-
Progressive non-fluent aphasia
- AOS:
-
Apraxia of speech
- PSPS:
-
Progressive supranuclear palsy syndrome
- CBS:
-
Corticobasal syndrome
- PLS:
-
Primary lateral sclerosis
- VSGP:
-
Vertical supranuclear gaze palsy
- CHMP2B :
-
Charged multivesicular body protein 2B
- MAPT :
-
Microtubule associated protein tau
- GRN :
-
Progranulin
- VCP :
-
Valosin containing protein
- TARDBP :
-
Transactive response DNA binding protein
References
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K, Iritani S, Tsuchiya K, Iseki E, Yagishita S, Oda T, Mochizuki A (2004) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79
Armstrong RA, Ellis W, Hamilton RL, Mackenzie IR, Hedreen J, Gearing M, Montine T, Vonsattel JP, Head E, Lieberman AP, Cairns NJ (2010) Neuropathological heterogeneity in frontotemporal lobar degeneration with TDP-43 proteinopathy: a quantitative study of 94 cases using principal components analysis. J Neural Transm 117:227–239
Bak TH, Crawford LM, Berrios G, Hodges JR (2010) Behavioural symptoms in progressive supranuclear palsy and frontotemporal dementia. J Neurol Neurosurg Psychiatry 81:1057–1059
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65:470–473
Bigio EH, Lipton AM, Yen SH, Hutton ML, Baker M, Nacharaju P, White CL 3rd, Davies P, Lin W, Dickson DW (2001) Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol 60:328–341
Boeve BF, Lang AE, Litvan I (2003) Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 54(Suppl 5):S15–S19
Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, Dickson DW, Kokmen E, Petersen RC (1999) Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 53:795–800
Borroni B, Alberici A, Agosti C, Cosseddu M, Padovani A (2009) Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int Psychogeriatr 21:463–468
Braak H, Braak E (1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76:124–127
Braak H, Braak E (1989) Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 15:13–26
Bronner IF, ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC (2005) Hereditary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain 128:2645–2653
Brun A (1994) Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 57:416–418
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22
Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, Duyckaerts C, Stankoff B, Pillon B, Skullerud K, Cruz-Sanchez FF, Bigio EH, Mackenzie IR, Gearing M, Juncos JL, Glass JD, Yokoo H, Nakazato Y, Mosaheb S, Thorpe JR, Uryu K, Lee VM, Trojanowski JQ (2004) Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 63:1376–1384
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR, Gearing M, Duyckaerts C, Yokoo H, Nakazato Y, Jaros E, Perry RH, Lee VM, Trojanowski JQ (2004) Alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol 164:2153–2161
Caselli RJ, Windebank AJ, Petersen RC, Komori T, Parisi JE, Okazaki H, Kokmen E, Iverson R, Dinapoli RP, Graff-Radford NR et al (1993) Rapidly progressive aphasic dementia and motor neuron disease. Ann Neurol 33:200–207
Chambers CB, Lee JM, Troncoso JC, Reich S, Muma NA (1999) Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer’s disease. Ann Neurol 46:325–332
Chan D, Fox NC, Scahill RI, Crum WR, Whitwell JL, Leschziner G, Rossor AM, Stevens JM, Cipolotti L, Rossor MN (2001) Patterns of temporal lobe atrophy in semantic dementia and Alzheimer’s disease. Ann Neurol 49:433–442
Claassen DO, Parisi JE, Giannini C, Boeve BF, Dickson DW, Josephs KA (2008) Frontotemporal dementia mimicking dementia with Lewy bodies. Cogn Behav Neurol 21:157–163
Cooper PN, Jackson M, Lennox G, Lowe J, Mann DM (1995) Tau, ubiquitin, and alpha B-crystallin immunohistochemistry define the principal causes of degenerative frontotemporal dementia. Arch Neurol 52:1011–1015
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113:521–533
Deramecourt V, Lebert F, Debachy B, Mackowiak-Cordoliani MA, Bombois S, Kerdraon O, Buee L, Maurage CA, Pasquier F (2010) Prediction of pathology in primary progressive language and speech disorders. Neurology 74:42–49
Dickson DW (1999) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246(Suppl 2):II6–II15
Dickson DW (2001) Neuropathology of Pick’s disease. Neurology 56:S16–S20
Dickson DW (1998) Pick’s disease: a modern approach. Brain Pathol 8:339–354
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee VM, Miller BL, Trojanowski JQ, Grossman M (2006) Frontotemporal dementia: clinicopathological correlations. Ann Neurol 59:952–962
Ghetti B, Hutton M, Wszolek Z (2003) Frontotemporal dementia and parkinsonism linked to chromosome 17 associated with tau gene mutations (FTDP-17t). In: Dickson D (ed) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 86–102
Gibb WR, Luthert PJ, Marsden CD (1989) Corticobasal degeneration. Brain 112(Pt 5):1171–1192
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Grossman M, Wood EM, Moore P, Neumann M, Kwong L, Forman MS, Clark CM, McCluskey LF, Miller BL, Lee VM, Trojanowski JQ (2007) TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol 64:1449–1454
Gydesen S, Brown JM, Brun A, Chakrabarti L, Gade A, Johannsen P, Rossor M, Thusgaard T, Grove A, Yancopoulou D, Spillantini MG, Fisher EM, Collinge J, Sorensen SA (2002) Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 59:1585–1594
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I (1994) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2019
Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406
Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, Nestor PJ, Patterson K (2010) Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 133:300–306
Holm IE, Isaacs AM, Mackenzie IR (2009) Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol 118:719–720
Hu WT, Rippon GW, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Josephs KA (2009) Alzheimer’s disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 24:1375–1379
Hu WT, Seelaar H, Josephs KA, Knopman DS, Boeve BF, Sorenson EJ, McCluskey L, Elman L, Schelhaas HJ, Parisi JE, Kuesters B, Lee VM, Trojanowski JQ, Petersen RC, van Swieten JC, Grossman M (2009) Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol 66:1359–1364
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
Ishihara K, Araki S, Ihori N, Shiota J, Kawamura M, Yoshida M, Hashizume Y, Nakano I (2005) Argyrophilic grain disease presenting with frontotemporal dementia: a neuropsychological and pathological study of an autopsied case with presenile onset. Neuropathology 25:165–170
Iwasaki Y, Ito M, Mori K, Deguchi A, Nagaoka M, Yoshida M, Hashizume Y (2009) An autopsy case of diffuse neurofibrillary tangles with calcification: early stage pathologic findings. Neuropathology 29:697–703
Jackson M, Lennox G, Lowe J (1996) Motor neurone disease-inclusion dementia. Neurodegeneration 5:339–350
Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, Chute DJ, Roberson ED, Pace-Savitsky C, Neumann M, Chow TW, Rosen HJ, Forstl H, Kurz A, Miller BL (2005) Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 62:925–930
Josephs KA (2008) Frontotemporal dementia and related disorders: deciphering the enigma. Ann Neurol 64:4–14
Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151
Josephs KA, Duffy JR (2008) Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 21:688–692
Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, Hauser MF, Witte RJ, Boeve BF, Knopman DS, Dickson DW, Jack CR Jr, Petersen RC (2006) Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 129:1385–1398
Josephs KA, Holton JL, Rossor MN, Braendgaard H, Ozawa T, Fox NC, Petersen RC, Pearl GS, Ganguly M, Rosa P, Laursen H, Parisi JE, Waldemar G, Quinn NP, Dickson DW, Revesz T (2003) Neurofilament inclusion body disease: a new proteinopathy? Brain 126:2291–2303
Josephs KA, Katsuse O, Beccano-Kelly DA, Lin WL, Uitti RJ, Fujino Y, Boeve BF, Hutton ML, Baker MC, Dickson DW (2006) Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 65:396–405
Josephs KA, Lin WL, Ahmed Z, Stroh DA, Graff-Radford NR, Dickson DW (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116:159–167
Josephs KA, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW (2006) Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol 63:506–512
Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW (2006) Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66:41–48
Josephs KA, Stroh A, Dugger B, Dickson DW (2009) Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol 118:349–358
Josephs KA, Uchikado H, McComb RD, Bashir R, Wszolek Z, Swanson J, Matsumoto J, Shaw G, Dickson DW (2005) Extending the clinicopathological spectrum of neurofilament inclusion disease. Acta Neuropathol 109:427–432
Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Jack CR Jr (2008) Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 29:280–289
Josephs KA, Whitwell JL, Eggers SD, Senjem ML, Jack CR Jr (2011) Gray matter correlates of behavioral severity in progressive supranuclear palsy. Mov Disord 26:493–498
Josephs KA, Whitwell JL, Jack CR Jr (2008) Anatomic correlates of stereotypies in frontotemporal lobar degeneration. Neurobiol Aging 29:1859–1863
Josephs KA, Whitwell JL, Knopman DS, Boeve BF, Vemuri P, Senjem ML, Parisi JE, Ivnik RJ, Dickson DW, Petersen RC, Jack CR Jr (2009) Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology 73:1443–1450
Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack CR Jr, Dickson DW (2010) Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol 17:969–975
Josephs KA, Whitwell JL, Vemuri P, Senjem ML, Boeve BF, Knopman DS, Smith GE, Ivnik RJ, Petersen RC, Jack CR Jr (2008) The anatomic correlate of prosopagnosia in semantic dementia. Neurology 71:1628–1633
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Van de Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, Frank AR, Jack CR Jr, Shiung M, Knopman DS, Josephs KA, Parashos SA, Rademakers R, Hutton M, Pickering-Brown S, Adamson J, Kuntz KM, Dickson DW, Parisi JE, Smith GE, Ivnik RJ, Petersen RC (2009) Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging 30:739–751
Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG (2005) The evolution and pathology of frontotemporal dementia. Brain 128:1996–2005
Knibb JA, Xuereb JH, Patterson K, Hodges JR (2006) Clinical and pathological characterization of progressive aphasia. Ann Neurol 59:156–165
Knopman DS, Boeve BF, Parisi JE, Dickson DW, Smith GE, Ivnik RJ, Josephs KA, Petersen RC (2005) Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol 57:480–488
Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA (2004) The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology 62:506–508
Kosaka K (1994) Diffuse neurofibrillary tangles with calcification: a new presenile dementia. J Neurol Neurosurg Psychiatry 57:594–596
Kosaka K, Shibayama H, Kobayashi H, Hoshino T, Iwase S (1973) An autopsy case of unclassifiable presenile dementia. Seishin Shinkeigaku Zasshi 75:18–34
Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, Fraser G, Crowther RA, Goedert M, Budka H, Ghetti B (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lillo P, Garcin B, Hornberger M, Bak TH, Hodges JR (2010) Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol 67:826–830
Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Paviour DC, Lees AJ (2010) Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 133:2045–2057
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47:1–9
Lowe JS, Rossor MN (2003) Frontotemporal lobar degeneration. In: Dickson DW (ed) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 342–348
Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM (2006) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 112:539–549
Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH (2006) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129:3081–3090
Mackenzie IR, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M (2011) Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121:207–218
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Masellis M, Momeni P, Meschino W, Heffner R Jr, Elder J, Sato C, Liang Y, St George-Hyslop P, Hardy J, Bilbao J, Black S, Rogaeva E (2006) Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 129:3115–3123
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58:1803–1809
Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL, Bigio EH, Weintraub S, Dickson DW, Hutton ML, Graff-Radford NR (2007) Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch Neurol 64:43–47
Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, Weintraub S, Bigio EH (2008) Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 63:709–719
Molina-Porcel L, Llado A, Rey MJ, Molinuevo JL, Martinez-Lage M, Esteve FX, Ferrer I, Tolosa E, Blesa R (2008) Clinical and pathological heterogeneity of neuronal intermediate filament inclusion disease. Arch Neurol 65:272–275
Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges JR (2000) A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol 47:36–45
Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, Mackenzie IR (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Nanda S, Bhatt SP, Pamula J, Woodruff WW, Fowler M, Miller D (2007) Diffuse neurofibrillary tangles with calcification (DNTC): Kosaka-Shibayama disease in America. Am J Alzheimers Dis Other Dement 22:535–537
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 66:152–157
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Ogar J, Slama H, Dronkers N, Amici S, Gorno-Tempini ML (2005) Apraxia of speech: an overview. Neurocase 11:427–432
Pickering-Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DM (2002) Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 125:732–751
Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AM, Neary D, Snowden JS, Mann DM (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131:721–731
Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, Rossor MN, Warren JD (2010) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53:1070–1076
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352
Seelaar H, Klijnsma KY, de Koning I, van der Lugt A, Chiu WZ, Azmani A, Rozemuller AJ, van Swieten JC (2010) Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol 257:747–753
Seeley WW, Bauer AM, Miller BL, Gorno-Tempini ML, Kramer JH, Weiner M, Rosen HJ (2005) The natural history of temporal variant frontotemporal dementia. Neurology 64:1384–1390
Shelley BP, Hodges JR, Kipps CM, Xuereb JH, Bak TH (2009) Is the pathology of corticobasal syndrome predictable in life? Mov Disord 24:1593–1599
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808
Snowden J, Goulding P, Neary D (1989) Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol 2:167–182
Snowden J, Neary D, Mann D (2007) Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 114:31–38
Snowden JS, Bathgate D, Varma A, Blackshaw A, Gibbons ZC, Neary D (2001) Distinct behavioural profiles in frontotemporal dementia and semantic dementia. J Neurol Neurosurg Psychiatry 70:323–332
Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, Momeni P, Baborie A, Griffiths TD, Jaros E, Perry RH, Richardson A, Pickering-Brown SM, Neary D, Mann DM (2011) The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta neuropathologica. doi:10.1007/s00401-011-0816-0
Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, Mann DM (2006) Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129:3091–3102
Spillantini MG, Bird TD, Ghetti B (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8:387–402
Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Grafman J, Ghetti B (2007) Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol 66:892–900
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Thompson SA, Patterson K, Hodges JR (2003) Left/right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology 61:1196–1203
Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, de Silva R, Lees A, Dickson DW (2002) Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 61:547–556
Uchihara T, Shibuya K, Nakamura A, Yagishita S (2005) Silver stains distinguish tau-positive structures in corticobasal degeneration/progressive supranuclear palsy and in Alzheimer’s disease–comparison between Gallyas and Campbell-Switzer methods. Acta Neuropathol 109:299–305
Uchihara T, Tsuchiya K, Nakamura A, Akiyama H (2005) Silver staining profiles distinguish Pick bodies from neurofibrillary tangles of Alzheimer type: comparison between Gallyas and Campbell-Switzer methods. Acta Neuropathol 109:483–489
Uchikado H, Shaw G, Wang DS, Dickson DW (2005) Screening for neurofilament inclusion disease using alpha-internexin immunohistochemistry. Neurology 64:1658–1659
Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Seelaar H, Van Swieten JC, Brown JM, Johannsen P, Nielsen JE, Holm IE, Dickson DW, Rademakers R, Graff-Radford NR, Parisi JE, Petersen RC, Hatanpaa KJ, White CL 3rd, Weiner MF, Geser F, Van Deerlin VM, Trojanowski JQ, Miller BL, Seeley WW, van der Zee J, Kumar-Singh S, Engelborghs S, De Deyn PP, Van Broeckhoven C, Bigio EH, Deng HX, Halliday GM, Kril JJ, Munoz DG, Mann DM, Pickering-Brown SM, Doodeman V, Adamson G, Ghazi-Noori S, Fisher EM, Holton JL, Revesz T, Rossor MN, Collinge J, Mead S, Isaacs AM (2010) FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120:33–41
van der Zee J, Urwin H, Engelborghs S, Bruyland M, Vandenberghe R, Dermaut B, De Pooter T, Peeters K, Santens P, De Deyn PP, Fisher EM, Collinge J, Isaacs AM, Van Broeckhoven C (2008) CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet 17:313–322
van Swieten J, Spillantini MG (2007) Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol 17:63–73
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Warrington EK (1975) Selective impairment of semantic memory. Q J Exp Psychol 27:635–657
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381
Whitwell JL, Jack CR Jr, Boeve BF, Parisi JE, Ahlskog JE, Drubach DA, Senjem ML, Knopman DS, Petersen RC, Dickson DW, Josephs KA (2010) Imaging correlates of pathology in corticobasal syndrome. Neurology 75:1879–1887
Whitwell JL, Jack CR Jr, Boeve BF, Senjem ML, Baker M, Rademakers R, Ivnik RJ, Knopman DS, Wszolek ZK, Petersen RC, Josephs KA (2009) Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 72:813–820
Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ (2005) Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 128:1247–1258
Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, Liu E, Donkervoort S, Zheng JG, Shi Y, Ahmeti KB, Brooks B, Engel WK, Siddique T (2010) Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75:807–814
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Josephs, K.A., Hodges, J.R., Snowden, J.S. et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122, 137–153 (2011). https://doi.org/10.1007/s00401-011-0839-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0839-6