
Abstract
Ab initio study of high-pressure phase transition and electronic structure of Fe-doped CeO2 with Fe concentrations of 3.125, 6.25, and 12.5 at% has been reported. At a constant-pressure consideration, the lattice constants and the volume of the supercell were decreased with an increasing concentration of Fe. The average bond length of Fe–O is lower than that of Ce–O. As a result, Fe doping induces the reduced volume of the cell, which is in good agreement with previous experiments. At high pressure (~ 30 GPa), it was found that the transition pressure from the fluorite to the cotunnite orthorhombic phase decreases at a higher concentration of Fe, indicating that the formation energy of the compound is induced by Fe-doping. Furthermore, compression leads to interesting electronic properties too. Under higher pressures, the bandgap increases in the cubic structure under compression and then suddenly plummets after the transition to the orthorhombic phase. The 3d states of Fe mainly induced the impurity states in the bandgap. In both the undoped and Fe-doped systems, the bandgap increased in the cubic phase at high pressure, while the gap and p-d hybridization decrease in the orthorhombic phase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Cerium oxide (CeO2) is an attractive material with various applications, mainly for automobile catalytic converters to decrease pollutants, as well as solid oxide fuel-cell technologies and optical equipment [1,2,3,4,5,6,7,8,9,10]. Due to the importance of the CeO2 properties, such as a high dielectric constant as well as a good epitaxial layer on Si, it is also viewed as a prospective material for future microelectronic applications. Hence, CeO2 has been proposed to be a possible gate dielectric material in metal–oxide–semiconductor devices of the next generation [25]. Several experiments, such as high-pressure Raman spectroscopy, reported that the pressure-induced transition of CeO2 from cubic fluorite structure to orthorhombic PbCl2 structure occurs at around 31 GPa [12]. These results are very close to that investigated by energy-dispersive X-Ray diffraction with phase transition pressure of 31.5 GPa [13]. Later, the properties of CeO2 have been developed by doping transition metals and searching the candidate material, which has great potential for applications in spintronic and multifunctional electronic devices [14,15,16,17,18,19]. The structural and physical properties of the CeO2 doped with Fe have been investigated by previous studies [20,21,22,23,24]. For the crystal structure of Ce1-xFexO2, which was synthesized by various methods, it was shown that Fe-doped CeO2 exhibited only fluorite structure with a small percentage of dopant. This fluorite phase was the only structure without the appearance of another phase of ceria and secondary impurity phase of Fe2O3 until the Fe concentration was increased to 25% [25, 26]. For electronic and optical properties, many previous studies have been obviously revealed the optical bandgap of Fe-doped CeO2 decreases with an increase of Fe concentration [27, 28]. However, there is a lack of clear explanation on, especially, electronic and optical properties of Fe-doped CeO2. A deeper understanding of those will be useful for further applications. In addition, high-pressure phase transformation has also been lacking both experiment and theory. Therefore, the aim of this study is to investigate the effects of Fe-doped CeO2 at concentrations of 3.12%, 6.25%, and 12.50%. The details of structural phase transition and electronic structure of Fe-doped CeO2 based on density functional theory study are discussed.
In this work, the structural relaxation of undoped CeO2 has been carried out using first-principles calculations based on the Kohn–Sham equations to investigate the phase stability as a function of pressure. Further Fe-doped CeO2 has been studied up to 50 GPa. This is because the fluorite phase transforms into the cotunnite phase at a pressure of 29.94 GPa, which is in good agreement with the experimental observations [12, 13]. As a result of this, the structural phase transition of undoped CeO2 is, in a way, similar to that of the Fe-doped case. At this point, it is interesting to note that the percentage of Fe is a key factor for the changing transition pressure. With respect to the dopant, the results demonstrate that the transition pressure depends on the percentage of Fe with respect to the undoped CeO2. Fe-doped CeO2 further reveals that the relative bulk moduli of both the fluorite and cotunnite phases play important roles in the nature of the chemical bonding, hinting at easier compression. Regarding its potential for electronic properties, the concentration of Fe is shown to possibly have an effect on the energy gap. Finally, these findings provide crucial details for fundamentally understanding the structural phase transformation, the changing transition pressure, and the electronic properties of the Fe-doped CeO2 under compressed conditions.
2 Computational details
Ab initio study based on density functional theory (DFT) within the CASTEP code [29] was employed to investigate high-pressure phase transition and electronic structure of Fe-doped and undoped CeO2. Generalized gradient approximation (GGA) functional [30] based on Perdew–Burke–Ernzerhof (PBE) [31] exchange–correlation functional was performed. The results of the high-pressure structural phase transition and electronic properties of CeO2 are in good agreement with experimental results by previous studies [12, 13]. The ultrasoft pseudopotential was used with electronic configurations of Fe: 3d64s2, Ce: 4d15s25p65d16s2 and O: 2s22p4. The plane wave basis was set as Ecut = 500 eV, and the k-point was sampled using a mesh size of 0.04 2 \(\pi\) /Å−1. For all the structures, the total energy convergence has been tested within a tolerance level of 0.005 meV/atom. The unit cell parameters and atomic positions were optimized using the BFGS algorithm [32] with the convergence tolerance as 0.01 eV/Å, within the pressure range that corresponds to experiments [12, 13]. For the Fe-doped CeO2, the supercells that have been constructed for 3.125, 6.25, and 12.50% doping of Fe are 2 × 2 × 2, 2 × 2 × 1, and 2 × 1 × 1 of both phases, respectively. Fe atom was replaced on a Ce site at the center of the supercell. The Birch–Murnaghan equation of state [33] was used to calculate the pressure values. Since the DFT method performs at absolute zero temperature, minimum Gibbs free energy of the state leads to its enthalpy. The stable phase at each pressure was compared by considering the minimum enthalpy. For band structure calculation, the k-point mesh that has been used is 0.03 2 \(\pi\) /Å−1 for all structures. Spin setup in GGA-PBE was added for splitting spin types up and down on electronic band and density of states (DOS) calculations. It was performed to clarify the effect of Fe atom on the bandgap reduction, which has been reported by previous studies [27, 28].
3 Results and discussion
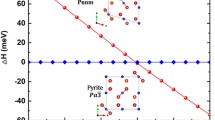
The atomic structures of 3.125% Fe-doped CeO2 in cubic (Fm-3 m) and orthorhombic (Pnma) phases are presented as shown in Fig. 1. In Table 1, it is shown that the undoped CeO2 lattice constant is 5.4634 Å which agrees within 1–2% with the experiment results of 5.411 Å [34] and 5.406 Å [13]. The effect of the Fe atom obviously influences the crystal volume because of the different atomic sizes between Ce and Fe. The decrement of a0 with the increment of Fe is due to the smaller size of Fe atoms. Values of electron affinity and electronegativity in the periodic table of Fe are higher than that of Ce. This also corresponds to the experimental results [20, 21, 28]. At high-pressure consideration, a phase transition from fluorite phase to cotunnite phase is calculated at 28.0 GPa with a volume reduction of 6.45% for undoped CeO2. The relationship between enthalpy versus pressure of four compositions (pristine and three doped cases) is presented in Fig. 2. The decrement of the transition pressure trend depends on the percentage of Fe. This is because the free energy of CeO2 systems is decreased at a high concentration of Fe-doping. Therefore, Fe-doping reduces free energy in the high-pressure phase more than the ambient pressure phase.

Supercell structures of a the cubic fluorite phase (Fm-3 m) and b the orthorhombic phase (Pnma) of 3.125% Fe-doped CeO2

The relationship between relative enthalpy versus pressure of 0.00%, 3.125%, 6.25% and 12.50% of Fe concentration for a, b, c, and d, respectively
The evidence from fluorite to cotunnite phase can be clearly given through an explanation of the changing transition pressure with Fe concentration. It can be clarified from the bond lengths of Fe and O (Fe atom is surrounded by 8 atoms of O), and the distribution of Ce–O bond lengths. Figure 3a shows the bond length evolution of Fe–O with pressure. At ambient pressure, the bond lengths of Fe–O are absolutely the same for all Fe concentrations, which is equal to 2.195 Å. This causes the bond lengths of Ce–O to deviate from that of the undoped CeO2, which is equal to 2.366 Å. The equivalent value of Ce–O bond lengths (\(l_{Ce - O}\)) is proposed here and directly calculated from the equation as; \(l_{Ce - O} = \left( {\sqrt 3 a_{x} - 4xl_{Fe - O} } \right)/4\left( {1 - x} \right)\); where \({l}_{Fe-O}\) and \({a}_{x}\) are Fe–O bond length and lattice constant of fluorite at Fe concentration of \(x\) (\(x\) = 0.03125, 0.0625, and 0.1250). At ambient, \({l}_{Ce-O}\) is 2.367, 2.363, and 2.355 Å, for \(x\) equal to 0.03125, 0.0625, and 0.1250, respectively. These support each other with the changing bulk modulus of Fe-doped CeO2 compared to undoped CeO2. In the case of 3.12% of Fe, \({l}_{Ce-O}\) is slightly higher than undoped CeO2. This means that it could be easier to compress because of the weaker bonds, the majority of the Ce–O pairs are more than 2.366 Å. Therefore, a larger pressure range could be needed in this case in order to drive it to structural phase transition. At higher Fe concentrations, not only the same phenomena in the previous discussion are the effect associated with the decreasing transition pressure, but the enthalpy barriers of cotunnite and fluorite phase at ambient pressure also are the cause as well, as shown in Table 2. The calculation reveals the enthalpy barrier oppositely changes with the amount of doping Fe. Consequently, these obviously imply the decreasing transition pressure in 6.25% and 12.50% of doping Fe in CeO2.

The relationship between a bond length of Fe–O of fluorite phase and pressure, b the GGA-PBE’s energy gap and Fe concentration at ambient pressure
Following the remarkable result, as shown in Table 2, it is worthy to note that the transition pressure depends on the percentage of Fe, resulting in the dramatic decrease in the transition pressure with the percentage of Fe. This implies that the computational results might be considered to be consistent with the increase of Fe concentrations. Besides, it is interesting to note the bulk moduli because the percentage of Fe plays a critical role in the mechanical properties. The result of the relative bulk moduli, therefore, has been carefully investigated by comparing to Fe-doped CeO2 of both fluorite and cotunnite phases. Subsequently, the relative bulk modulus of fluorite is smaller than that of cotunnite with the increasing percentage of Fe, supporting the transition pressure behavior. A possible cause for this could be the nature of the chemical bonding that is the Fe–O being the weaker bonds, and thus, it is easier to compress.
For Fe-doped CeO2, it has two naturally occurring energy bandgaps. The first one, the optical bandgap, is the gap between the maximum of the valence band, situated under the Fermi level, which are mainly O-2p states, and the narrow upper bands mainly contributing by Ce-4f states. The electronic bandgap is due to the unoccupied conduction bands formed by Ce-5d states hybridizing with O-2p states. The two energy bandgaps of undoped fluorite CeO2 and Fe-doped CeO2 at ambient pressure are shown in Fig. 3b. The optical and electronic gap is 5.82 eV and 2.24 eV, respectively, for undoped CeO2. These values are slightly underestimated compared to experimental results (\({E}_{g}\)= 6 eV and 3 eV) [36]. At 3.125% of Fe, the optical and electronic gaps dramatically decrease to 1.06 and 4.76 eV, respectively. They further slightly reduce when the concentration of Fe increases to 6.25% and 12.50%.
To further understand the electronic property of Fe-doped CeO2, energy dispersion of Ce0.9688Fe0.0312O2 along with the high symmetry directions in the Brillouin zone, the total density of states (DOS) and partial density of states (PDOS) are shown in Fig. 4. The finding obviously discloses the effect of Fe-doped CeO2. There are two pieces of evidence consisting of: (1) states of Fe-3d that are situated around the Fermi level. These consist of two energy bands and are represented by the solid line in PDOS. The first one, exhibiting occupied states, is slightly higher than O-2p states, which regularly are the valence band maximum of undoped CeO2. Therefore, the effect of Fe results in enhancing the Fermi level compared to the case of undoped Fe. The Fermi levels of the two cases are shown in the DOS panel of Fig. 4. The Fe-3d states above the Fermi level are located close to the bottom of the narrow states of Ce-4f. As a result, this makes a lower tail in the total DOS of narrow states of Ce-4f. This is the cause of the decrement of the optical bandgap. (2) A splitting of unoccupied Ce-5d states emerges at the lowest band. It is approximately at an energy of 5 eV denoted by the cross symbols (x) in the energy dispersion panel. This could occur due to the long bond lengths of Ce–O pairs comparing to their equivalent value.

Band strucure, DOS, and PDOS of 3.125% Fe-doped CeO2 from GGA-PBE calculation
The spin-up and down on GGA-PBE functional are also analyzed for pure and doped CeO2 at 0 and 25 GPa for the cubic and 35 GPa for the orthorhombic structures, as presented in Fig. 5. It was first verified for pure CeO2 and found that spin-up gave a lower energy level compared with spin down, which relates to basic idea in quantum chemistry. The edge of the conduction band minimum is due to the spin-up state of the f-state in Ce atom. Under high pressure in the cubic phase up to 25 GPa, it was found that the bandgap of pure CeO2 increased. The CBM gets pushed to a higher energy level with pressure. This supports the idea that the decreasing bond length and cell volume are mainly influenced by the electronic states of the nearly free electrons in CeO2. After phase transition to the orthorhombic phase, the electronic DOS was calculated at 35 GPa. The gap between CBM and VBM is decreased in the orthorhombic phase. Reconstruction of the new stable phase increases the average bond length of Fe–O, although cell volume reduces under compression. The new stable phase (Pnma) results in the reduction of the bandgap and energy states of CBM in pure CeO2, as shown in Fig. 5a. The same trend is seen for the Fe-doped system between 0 and 35 GPa, and the DOS of 3.125%Fe is depicted in Fig. 5b. Fe atom gave contribute to impurity states between VBM and CBM. Figures 5c and d shows that for 3.125 and 6.25% of Fe, the DOS is not significantly different. Figure 5e presents the DOS in the orthorhombic phase and shows that the p-d hybridization decreases when compared with the cubic phase.

DOS and PDOS in a DOS of pure CeO2, b DOS of 3.125% Fe-doped CeO2, c PDOS of 6.25% Fe-doped CeO2 at 0 GPa, d and e PDOS of 3.125% Fe-doped CeO2 at 0 and 35 GPa, respectively
In addition, the two bandgaps slowly decrease for Fe concentrations at 6.25% and 12.50%. This can be clearly explained because of the saturation of Fe electronic states, which cannot be much more extensive over the edge of the bands at high Fe concentrations. The decrease of volume at high concentrations of Fe affects the decrease in bond length of Ce–O, and it directly leads to the increasing bandgaps as well. However, these results are useful to clarify the background phenomena of doping Fe in CeO2; on the other hands, these also need evidence from the further experiment, which is still lacking. Furthermore, effects of Fe concentrations that are investigated in this study would be beneficial to material engineering for designing modern electronic devices.
4 Conclusions
CeO2 doped with Fe has been investigated using first-principles computational techniques. The effect of Fe doping with concentrations of 3.12, 6.25, and 12.5% has been considered. The GGA-PBE functional was employed to calculate the stability of electronic-crystal structures under high pressure based on the fluorite to cotunnite phase transition, as well as the change of the optical and electronic bandgaps. The calculated lattice constant and density of the cubic fluorite structure at ambient pressure decrease with increasing concentration of Fe dopants because the substituting Fe atom is smaller and lighter than the Ce atom. The two bandgaps dramatically decrease at 3.12% of doping of Fe, and they further sluggishly reduce at two higher Fe concentrations and 6.25% and 12.50%. The 3d states of Fe atom are the main cause of impurity states within the intrinsic bandgap. In both the undoped and Fe-doped systems, the bandgap increases for the cubic phase under pressure, while the gap and p-d hybridization suddenly decrease after the phase transition. We believe that these results of the effects of Fe will be beneficial to bandgap engineering in materials for designing modern electronic devices.
References
S. Rossignol, Y. Madier, D. Duprez, Catal, Preparation of zirconia–ceria materials by soft chemistry. Today 50, 261 (1999)
M. Shiono, K. Kobayashi, T.L. Nguyen, K. Hosoda, T. Kato, K. Ota, M. Dokiya, Effect of CeO2 interlayer on ZrO2 electrolyte/La(Sr)CoO3 cathode for low-temperature SOFCs. Solid State Ionic 170, 1 (2004)
A. Trovarelli, C.D. Leitenburg, G. Dolcetti, Design better cerium-based oxidation catalysts. Chem. Tech. 27, 32 (1997)
M. Akbar, Z. Tu, B. Jin, N. Mushtaq, Z. He, W. Dong, B. Wang, X. Wang, C. Xia, Demonstrating the dual functionalities of CeO2–CuO composites in solid oxide fuel cells. Int. J. Hydrog. Energy. 46, 9938 (2021)
Z. Qiao, C. Xia, Y. Cai, M. Afzal, H. Wang, J. Qiao, B. Zhu, Electrochemical and electrical properties of doped CeO2-ZnO composite for low-temperature solid oxide fuel cell applications. J. Power Sour. 392, 33 (2018)
J.W. Shin, S. Oh, S. Lee, J.G. Yu, J. Park, D. Go, B.C. Yang, H.J. Kim, J. An, Ultrathin atomic layer- deposited CeO2 overlayer for high-performance fuel cell electrodes. ACS Appl. Mater. Interfaces 11, 46651 (2019)
B.C. Yang, D. Go, S. Oh, J.W. Shin, H.J. Kim, J. An, Atomic-layer-deposited ZrO2-doped CeO2 thin film for facilitating oxygen reduction reaction in solid oxide fuel cell. Appl. Surf. Sci 473, 102 (2019)
A. Haghighatzadeh, Enhanced third-order optical susceptibility in Ag-doped CeO2 nanostructures under pulsed Nd-YVO4 laser. Opt Laser Technol 126, 106114 (2020)
B. Soni, S. Makkar, S. Biswas, Defects induced tailored optical and magnetic properties of Zn-doped CeO2 nanoparticles synthesized by a facile sol-gel type process. J. Alloys Compd 879, 160149 (2021)
J.S.P. Cresi, L.D. Mario, D. Catone, F. Martelli, A. Paladini, S. Turchini, S. D’Addato, P. Luches, P. O’Keeffe, Ultrafast formation of small polarons and the optical gap in CeO2. J. Phys. Chem. Lett. 11, 5686 (2020)
S. Mehrotra, P. Sharma, M. Rajagopalan, A.K. Bandyopadhyay, High pressure phase transition and band structures of different phases in CeO2. Solid State Commun. 140, 313 (2006)
G.A. Kourouklis, A. Jayaraman, G.P. Espinosa, High-pressure Raman study of CeO2 to 35 GPa and pressure-induced phase transformation from the fluorite structure. Phys. Rev. B 37, 8 (1988)
S.J. Duclos, Y.K. Vohra, A.L. Ruoff, A. Jayaraman, G.P. Espinosa, High-pressure x-ray diffraction study of CeO2 to 70 GPa and pressure-induced phase transformation from the fluorite structure. Phys. Rev. B 38, 7755 (1988)
A. Fert, Nobel lecture: origin, development, and future of spintronics. Rev. Mod. Phys. 80, 17 (2008)
S.A. Wolf, D.D. Awschalom, R.A. Buhrman, J.M. Daughton, S. von Molna, M.L. Roukes, A.Y. Chtchelkanova, D.M. Treger, Spintronics: a spin-based electronics vision for the future. Science 294, 1488 (2001)
S. Zhao, D. Kang, Y. Liu, Y. Wen, X. Xie, H. Yi, X. Tang, Spontaneous formation of asymmetric oxygen vacancies in transition-metal-doped CeO2 nanorods with improved activity for carbonyl sulfide hydrolysis. ACS Catal. 10, 11739 (2020)
K. Subramanyam, N. Sreelekha, D.A. Reddy, M. Ramanadha, B. Poornaprakash, K.C. Reddy, R.P. Vijayalakshmi, Influence of transition metals co-doping on CeO2 magnetic and photocatalytic activities. Ceram. Int. 46, 5086 (2020)
R.N. Bharathi, S. Sankar, Effects of transition metal element (Co, Fe, Ni) codoping on structural, optical and magnetic properties of CeO2: Er nanoparticles. Superlattices Microstruct. 123, 37 (2018)
B. Poornaprakash, K. Subramanyam, M. Kumar, Y.L. Kim, M.S.P. Reddy, Enhanced photoluminescence characteristics and intrinsic ferromagnetism in Co-substituted CeO2 nanoparticles. Mater Sci Semicond Process 123, 105566 (2021)
S.K. Sharma, M. Knobel, C.T. Meneses et al., Ferromagnetic properties of bulk Fe-doped CeO2 dilute magnetic semiconductors. J Korean Phys. Soc. 55, 1018 (2009)
S. Kumar, G.W. Kim, B.H. Koo, S.K. Sharma, M. Knobel, C.G. Lee, Structural and magnetic study of a diluted dagnetic semiconductor: Fe-doped CeO2 nanoparticles. J Nanosci. Nanotechnol. 11, 555 (2011)
M. El-Hagary, E.R. Shaaban, S.H. Moustafa, G.M.A. Gad, The particle size-dependent optical band gap and magnetic properties of Fe-doped CeO2 nanoparticles. Solid State Sci. 91, 15 (2019)
Y. Xue, D. Tian, C. Zeng, Y. Fu, K. Li, Effect of Fe doping concentration on photocatalytic performance of CeO2 from DFT insight into analysis. AIP Adv. 9, 125341 (2019)
H. Bao, K. Qian, J. Fang, W. Huang, Fe-doped CeO2 solid solutions: Substituting-site doping versus interstitial-site doping, bulk doping versus surface doping. Appl. Surf. Sci 414, 131 (2017)
L. Li, X. Li, Strain-engineered cube nanocrystals Ce1–yFeyO2 that brought forth abnormal structural and magnetic properties. J. Phys. Chem. C 117, 15383 (2013)
P. Singh, M.S. Hegde, Sonochemical synthesis Ce1-xFexO2-δ (0 ≤ x ≤ 0.45) and Ce0.65Fe0.33Pd0.02O2-δ of nanocrystallites: oxygen storage material, CO oxidation and water gas shift catalyst. Dalton Trans 39, 10768 (2010)
T. Dhannia, S. Jayalekshmi, M.C.S. Kumar, T.P. Rao, A.C. Bose, Effect of iron doping and annealing on structural and optical properties of cerium oxide nanocrystals. J. Phys. Chem. Solids 71, 1020 (2010)
P.C.A. Brito, D.A.A. Santos, J.G.S. Duque, M.A. Macêdo, Structural and magnetic study of Fe-doped CeO2. Physica B 405, 1821 (2010)
M.D. Segall, P.L.D. Lindan, M.J. Probert, C.J. Pickard, P.J. Hasnip, S.J. Clark, M.C. Payne, First-principles simulation: ideas illustrations and the CASTEP code. J. Phys Condens. Matter 14, 2717 (2002)
J.P. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson, M.R. Pederson, D.J. Singh, Atoms molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671 (1991)
J.P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996)
B.G. Pfrommer, M. Cote, S.G. Louie, M.L. Cohen, Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 131, 133 (1997)
F. Birch, Finite elastic strain of cubic crystals. Phys. Rev. B 71, 809 (1947)
W. F. NcClune, Powder Diffraction File, edited by W. F. NcClune (JCPDS International Centre for Diffraction Data, Swarthmore, PA, (1980).
A. Bendjedid et al., Structural, electronic, bonding and thermo-elastic properties of orthorhombic and cubic CeO2 compound. Chin. J.Phys. 54, 1 (2016)
E. Wuilloud, B. Delley, W.-D. Schneider, Y. Baer, Spectroscopic evidence for localized and extended f-symmetry states in CeO2. Phys. Rev. Lett. 53, 202 (1984)
Acknowledgements
The authors would like to express their gratitude to the 90th Year Chulalongkorn Scholarship and Thailand Center of Excellence in Physics. K. K. and T. B. would like to express their gratitude to the Thailand Research Fund for financial support through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0277/2552). T.B. acknowledges financial support from Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University; Grant for Research. Computing facilities have been partially supported by Sci-Super IV research grant.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sathupun, K., Kotmool, K., Tsuppayakorn-aek, P. et al. Fe-doped effects on phase transition and electronic structure of CeO2 under compressed conditions from ab initio calculations. Appl. Phys. A 127, 784 (2021). https://doi.org/10.1007/s00339-021-04911-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-021-04911-0