
Abstract
The syndrome Multiple Congenital Ocular Anomalies (MCOA) is the collective name ascribed to heritable congenital eye defects in horses. Individuals homozygous for the disease allele (MCOA phenotype) have a wide range of eye anomalies, while heterozygous horses (Cyst phenotype) predominantly have cysts that originate from the temporal ciliary body, iris, and/or peripheral retina. MCOA syndrome is highly prevalent in the Rocky Mountain Horse but the disease is not limited to this breed. Affected horses most often have a Silver coat color; however, a pleiotropic link between these phenotypes is yet to be proven. Locating and possibly isolating these traits would provide invaluable knowledge to scientists and breeders. This would favor maintenance of a desirable coat color while addressing the health concerns of the affected breeds, and would also provide insight into the genetic basis of the disease. Identical-by-descent mapping was used to narrow the previous 4.6-Mb region to a 264-kb interval for the MCOA locus. One haplotype common to four breeds showed complete association to the disease (Cyst phenotype, n = 246; MCOA phenotype, n = 83). Candidate genes from the interval, SMARCC2 and IKZF4, were screened for polymorphisms and genotyped, and segregation analysis allowed the MCOA syndrome region to be shortened to 208 kb. This interval also harbors PMEL17, the gene causative for Silver coat color. However, by shortening the MCOA locus by a factor of 20, 176 other genes have been unlinked from the disease and only 15 genes remain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
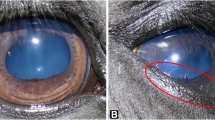
Multiple Congenital Ocular Anomalies (MCOA) syndrome is a congenital nonprogressive syndrome described in horses. The most frequent feature of this disease is fluid-filled cysts of variable sizes (2–20 mm) in the posterior iris and ciliary body epithelium within the eye. Two distinct ocular phenotypes exist: (1) cysts that originate from the posterior iris, temporal ciliary body, and/or peripheral retina (Cyst phenotype), and (2) cysts in combination with additional ocular defects including iridocorneal angle abnormalities, cornea globosa, iris hypoplasia, congenital cataracts, lens subluxation, focal areas of retinal detachment, microphthalmia, and macropalpebral fissures (MCOA phenotype). Horses with MCOA have abnormal pupillary light reflexes and pupils do not dilate after administration of mydriatic drugs (Ramsey et al. 1999a, b). Individual MCOA-affected horses may or may not have the complete set of congenital defects described above. Both the distinct subdivision of phenotypes and the transmission of the disease within our pedigrees are consistent with a mutant allele displaying incomplete dominance. The Cyst horses are heterozygous and have an intermediate phenotype compared to the horses with multiple anomalies that carry two copies of the disease allele (Ewart et al. 2000; Andersson et al. 2008).
Conditions have been favorable for the mutation causing MCOA to be enriched in the Rocky Mountain Horse breed (Ewart et al. 2000). This breed originated from a very limited number of founder horses, which were used extensively to develop the breed. In fact, many Rocky Mountain horses can be traced back to a single foundation stallion. The horses within the breed have been selected for a distinctive four-beat gait and the Silver coat color has been highly favored. The fact that an intensive selection process can lead to amplification of undesirable traits has been demonstrated in several other horse breeds [e.g., hyperkalemic periodic paralysis (Rudolph et al. 1992), hereditary equine regional dermal asthenia (Tryon et al. 2007), severe combined immunodeficiency (Shin et al. 1997), and Overo Lethal White Syndrome (Santschi et al. 1998)]. In the Rocky Mountain Horse breed, selection of horses with the highly desirable Silver coat color has simultaneously increased MCOA syndrome as these traits are linked on horse chromosome 6 (Andersson et al. 2008). The Silver coat color in horses is characterized by dilution of black pigment in the hair and revealed to be associated with a missense mutation in pre-melanosomal protein 17 or PMEL17 (Brunberg et al. 2006). Additional horse breeds that have been diagnosed with MCOA include the Kentucky Mountain Saddle Horse, Mountain Pleasure Horse (both closely related to the Rocky Mountain Horse), Belgian Draft, Morgan Horse, Shetland Pony, American Miniature Horse (Ramsey et al. 1999a; Grahn et al. 2008; Komaromy and Rowlan 2009), and the Icelandic Horse (B. Ekesten, unpublished).
Cysts are found in most affected horses and are usually bilateral. Horses with cysts usually have normal functional vision irrespective of cyst size since cysts are either translucent or lightly pigmented. A small number of juvenile horses that have cornea globosa as a component of multiple ocular defects have a considerable refractive error and subsequently poor vision. This refractive error is corrected by emmetropization as the juvenile horse eye achieves adult size (Ramsey et al. 2000).
The primary objective of this research was to identify the gene and mutation that regulate the MCOA syndrome in horses. The results would have practical implications such as enable genetic testing for precise diagnosis of carrier horses and horses with the multiple eye defects. We previously mapped the locus for equine MCOA to a 4.9-Mb interval on horse chromosome 6 by linkage mapping (Andersson et al. 2008). In the present study we have significantly limited the interval for the MCOA locus and selected two additional candidate genes in the interval, IKZF4 and SMARCC2, for mutation screening. IKZF4 was selected as a candidate gene because it is expressed in ocular tissue, is a member of the IKAROS family of transcription factors, and interacts with the gene microphthalmia-associated transcription factor, MITF (Hu et al. 2007). MITF influences both pigmentation and ocular development (Hodgkinson et al. 1993). Mutations in MITF can cause Waardenburg syndrome type 2A (Nobukuni et al. 1996), an auditory-pigmentary disorder that affects ocular development. In addition, a mutation in the PAX3 gene causes Waardenburg syndrome type 1, and it has been suggested that it occurs through a failure to regulate MITF (Watanabe et al. 1998). The other candidate gene, SMARCC2, was selected because it encodes a 170-kDa protein that is one of 12 proteins in the SWI/SNF complex (Dechassa et al. 2008). The ATPase component Brm of this complex regulates differentiation of early retinal stem cells (Das et al. 2007). In humans, the SWI/SNF chromatin remodeling complex is involved in the regulation of the CRYAB (αβ-crystallin) gene that is highly expressed in the vertebrate lens where cataracts are commonly associated with crystalline protein deficiencies (Liu et al. 2001; Duncan and Zhao 2007). The present study defines a 208-kb genomic interval for the MCOA locus by identical-by-descent (IBD) mapping of multiple horse breeds.
Materials and methods
Horse material
Four hundred sixty-five horses were genotyped in this study (Table 1), including 362 Rocky Mountain horses, 57 American Miniature horses, 22 Kentucky Mountain Saddle horses, and 24 Icelandic horses. All Rocky Mountain horses evaluated in this study were derived from four half-sibling families and have been described previously (Ewart et al. 2000). The distributions for phenotypes of Rocky Mountain horses were 72 with MCOA, 222 with Cyst phenotype, and 68 unaffected horses. Distribution of phenotypes for the remaining breeds was as follows: American Miniature Horse: 3 MCOA, 14 Cyst, 40 unaffected; Kentucky Mountain Saddle Horse: 4 MCOA, 4 Cyst, 14 unaffected; and Icelandic Horse: 4 MCOA, 14 Cyst, 6 unaffected.
Phenotype assessment
Eye examinations were performed as described previously (Ramsey et al. 1999a). Briefly, direct and indirect pupillary light reflexes were first assessed. Following pharmacologic mydriasis with 1% tropicamide administered topically, a complete ophthalmic examination consisting of slit lamp biomicroscopy, indirect ophthalmoscopy, and applanation tonometry was performed. The study was approved by the Michigan State University Institutional Animal Use and Care Ethics Committee for all breeds except the Icelandic Horse; for that breed, the study was approved by the Ethics Committee for Animal Experiments in Uppsala, Sweden.
Microsatellite genotyping
Fourteen microsatellite markers spanning approximately 12 Mb on ECA6q were used for genotyping (Table 2, Supplementary File 1). Seven of these markers were identified previously (TKY570, TKY412, TKY284, UPP5, UPP6, UPP7, and TKY952), while seven were novel and developed from the horse genome sequence (EquCab2) (UCSC Genome Browser, http://genome.ucsc.edu/) by displaying tracks from simple tandem repeats recognized by Tandem Repeats Finder (Benson 1999). Initially, genotyping was done on a limited number of horses for a wide interval. Subsequently, an increased number of horses were genotyped for a smaller region that was approximately 2–3 Mb wide. After identifying recombinant horses, further marker genotyping focused on these samples. Table 2 summarizes the number of horses that were genotyped for each marker. Primers for novel microsatellites were designed using Primer3 (Rozen and Skaletsky 2000). PCR reactions were performed as described previously (Andersson et al. 2008). Amplified fragments were multiplexed when possible and separated using a MegaBACE™ 1000 instrument (GE Healthcare Bio-Science Corp., Piscataway, NJ) according to the manufacturer’s recommendations. Results were analyzed using Genetic Profiler ver. 2.4 (Amersham Bioscience, GE Healthcare Bio-Science Corp.). Parentage testing was performed according to standard procedures (17 microsatellite markers, Equine Genotypes™ Panel 1.1, Finnzymes Oy, Espoo, Finland) at the Animal Genetics Laboratory, Swedish University of Agricultural Sciences, Uppsala.
SNP genotyping
Six single nucleotide polymorphism (SNP) markers were selected in regions lacking microsatellite markers. Custom TaqMan SNP Genotyping assays [Applied Biosystems (ABI), Foster City, CA] were used for genotyping five of the SNPs. Probe and primer designs were obtained from the ABI web page (http://www5.appliedbiosystems.com/tools/cadt/) using the custom genotyping assays order option. The ABI PRISM 7900 HT sequence detection system for 384-well format (ABI) was used for the analysis. The six selected SNP markers were genotyped in 123–465 horses (Table 2). One additional SNP was investigated in 33 horses by traditional Sanger sequencing (see the next subsection). The PMEL17 SNP in exon 11 was interrogated using pyrosequencing as described previously (Andersson et al. 2008).
Sanger sequencing of SMARCC2 and IKZF4
Two candidate genes, SMARCC2 and IKZF4, within the identified genomic interval were sequenced using the Sanger method. Three horses with MCOA and two unaffected controls were used to sequence SMARCC2, while two MCOA horses and one unaffected control were used for sequencing IKZF4. The generated DNA sequences were also compared with the horse reference genome (Wade et al. 2009). Since none of the genes are well annotated in the horse genome, exon number, size, and position were deduced by displaying the track “None-Horse mRNA” from GenBank. Sequencing primers were designed to amplify 500–700-bp fragments using Primer3 (Rozen and Skaletsky 2000). Primer sequences are listed in Supplementary File 1. All exons, as well as some introns, the untranslated regions (UTRs), and certain evolutionary conserved elements were sequenced (Supplementary File 1). Sequencing reactions were carried out as described previously (Brunberg et al. 2006) and results were analyzed using CodonCode software (CodonCode Aligner ver. 1.6.3, CodonCode Corporation, Dedham, MA). Consensus sequences were deposited in GenBank (accession Nos. HQ331540 and HQ331541 for SMARCC2 and IKZF4, respectively).
Results
Haplotype analysis
Genotyping of 14 microsatellite and 7 SNP markers firmly identified a 264-kb genomic interval for the MCOA locus (Tables 2, 3 haplotype A, B, C1, and D1). SNP markers N1 and 8453 represented the 5′ and 3′ borders of the interval, respectively. Four of the microsatellite markers (MS14, MS3, TKY284, and MS13) and three SNPs (PMEL17ex11, 8345, and 8377) reside within the interval. Table 2 displays the results from the genotyping of each marker. With the exception of markers PMEL17 and TKY284, disease-associated alleles are also common in unaffected horses. Phased data of chromosomes that carry the mutation is depicted in Table 3. Forty-eight percent of the individual genotypes were inferred based on surrounding marker information. This was performed only when the haplotype could be assessed with certainty. In total, a combination of 457 affected and unaffected horses of four breeds were used to define the interval (see schematic figure in Supplementary File 2). This corresponds to 409 analyzed chromosomes that carry the MCOA mutation (including horses with both the MCOA and the Cyst phenotype). Of 83 MCOA horses included in this study, 81 were homozygous for the disease-associated haplotype; the remaining two horses were diagnosed clinically with MCOA but carried only one copy of the disease haplotype. Of the 254 horses with the Cyst phenotype, 245 were heterozygous for the disease-associated haplotype and one horse had two copies of the disease haplotype. The remaining eight horses had individually unique haplotypes (see Supplementary File 2). These horses were parentage-tested to determine whether DNA and/or blood sample error had occurred. However, parentage results were accurate. Our genetic data also revealed 16 horses in which the Cyst genotype showed nonpentetrance. These horses represent only 6% of all genetically defined heterozygous horses.
Sanger sequencing of IKZF4 and SMARCC2
In total, 13.6 kb of IKZF4 (87%) were evaluated for polymorphisms, including all exons and conserved sequences within introns and 3.6 kb upstream of the gene (Supplementary File 1). Comparison among MCOA horses and unaffected control horses revealed three intronic polymorphic sites (Supplementary File 3). Two of these were SNPs (introns 3 and 4) and not completely associated with the disease. The third polymorphism was a homozygous adenine insertion (intron 5) in both MCOA horses that was not present in either of the unaffected controls or the reference genome.
Resequencing of the SMARCC2 gene covered 12.6 kb (76%) and included all 29 exons (Supplementary File 1) and intronic conserved elements. Ten SNPs were detected and two of them were positioned within coding regions (Supplementary File 3). Only two SNPs, in intron 19 and 24, respectively, matched the correct mode of inheritance in the horses selected for resequencing. Since SMARCC2 is positioned close to the 3′ downstream border of the 264-kb IBD interval, the intron 19 SNP was genotyped in recombinant horses. These are the horses with the shortest IBD region compared with the majority of horses (Table 3, haplotypes B-D). The disease-associated allele for this SNP is G, but genotyping revealed nine genetically well-defined Cyst horses as being TT (Table 3, haplotypes C2 and D2). Two additional American Miniature horses displaying the Cyst phenotype can be phased as either C1 or C2 but have been labeled as C2 due to a close relatedness to the other horses carrying the C2 haplotype. Furthermore, a subset of horses, including the newly identified recombinants, was also genotyped for the SNP in intron 24. SMARCC2 is positioned on the minus strand and hence intron 24 is upstream of intron 19 according to the depicted interval. The disease-associated allele for this SNP is also guanine but the C2 haplotype holds an adenine at this position. These horses thus shortened the genetic interval by 57 kb, providing a minimum shared haplotype of 208 kb.
Discussion
Identical-by-descent mapping was used to firmly define a 208-kb genomic interval on horse chromosome 6 for the MCOA locus. This has shortened the previous 4.9-Mb MCOA genetic interval by a factor of 20, which has unlinked 176 other genes from the disease. In addition, a large number of horses were utilized to verify that the mutation causing MCOA syndrome does have a clear additive effect and that the two distinct phenotypic categories, Cyst and MCOA, are caused by one and two copies of the mutant allele, respectively (p = 2.2 × 10−16). Results of the study reported here are based on analysis of 21 genetic markers in 465 horses from four different breeds. The identified interval is a gene-dense area of the genome and includes 15 genes (see Supplementary File 4). Based on information on gene function from studies in other species, two candidate genes within the interval, SMARCC2 and IKZF4, were evaluated for DNA polymorphisms using Sanger sequencing. Two intronic SNPs that segregated with the disease phenotype were detected in SMARCC2. After analysis of these SNPs in recombinant horses, the interval for the MCOA syndrome was shortened by 57 kb (from 265 to 208 kb). Sequencing also revealed one disease-associated insertion in intron 5 of IKZF4. However, since this polymorphism resides within a noncoding region, it is less likely than the PMEL17 mutation to have a phenotypic consequence. Nevertheless, this variant will be tested in a larger number of horses.
Another candidate gene within this interval is PMEL17. A missense mutation in this gene is most likely causative of the Silver coat color and this mutation may have pleiotropic effects. The protein PMEL17 is involved in the production of eumelanin, but its precise function remains unknown (Theos et al. 2005). Mutations within this gene are associated with dilution of eumelanin in several species, including mouse, chicken, zebrafish, dog, cow, and horse (Kwon et al. 1995; Kerje et al. 2004; Schonthaler et al. 2005; Clark et al. 2006; Brunberg et al. 2006; Kuehn and Weikard 2007). The different species vary in the type and the location of the mutation they exhibit, although several mutations reside near the C-terminal portion of the protein.
To date, ocular defects associated with known PMEL17 mutants have been reported in zebrafish and dog (Schonthaler et al. 2005; Clark et al. 2006), which supports PMEL17 as a candidate gene for MCOA syndrome in horses. Conversely, eye defects in these species appear dissimilar, although they are not so easily compared. In zebrafish fdv mutant larvae, the retinal pigment epithelium (RPE) melanosome biogenesis is impaired, leading to photoreceptors with fewer, shorter, and misaligned outer segments. The retina is partially recovered in the adult fdv zebrafish. The zebrafish RPE melanosomes have an aberrant shape and contain less melanin, giving the eye and the body a lighter appearance. Merle dogs that are homozygous for the proposed SILV mutation exhibit several different aberrant eye phenotypes such as starburst/irregular dropped pupils, heterochromia, and microphthalmia. Like the zebrafish, affected dogs have defects in the pigmentation of skin, coat, and RPE.
Recently, chickens homozygous for the Dominant white mutation, a 6-bp insertion (ins723–725) within the transmembrane region of PMEL17, were found to be free from ocular abnormalities and visual impairment (Karlsson et al. 2009). Dominant white mutant chickens appeared less pigmented than wild-type chickens in the outer layers of the retina, but the RPE seemed to be unaltered. Further investigations of eyes of chickens that carry the Dun allele would be helpful since they have the exact same missense mutation (R740C) as the horse and also have a 12-bp deletion (del731–735).
The degree of ocular pigmentation and MCOA are poorly correlated. All but eight horses, diagnosed with MCOA syndrome, carried the Silver mutation, however their RPE appears to be unaltered (Ramsey et al. 1999a; B. Ekesten, unpublished). Pigmented uveal tissues (iris, ciliary body, choroid) may also be darkly pigmented or lightly pigmented in both MCOA-affected and normal horses. Nevertheless, the fact that the MCOA syndrome has been diagnosed in all breeds where the Silver coat color is present favors PMEL17 as a candidate gene.
Several other possible candidate genes exist in the interval evaluated. According to AceView, a database that gathers all cDNA sequences available in public databases, 14 of 15 genes in the associated interval are variably expressed in ocular tissues (Thierry-Mieg and Thierry-Mieg 2006). A similar search in the Mouse Gene Expression Database (http://www.informatics.jax.org/expression.shtml) showed that six of the genes were known to be expressed in ocular tissues. To date, the remaining genes were either not found (six genes) or there was no evidence of expression (three genes). In addition to PMEL17, IKZF4, and SMARCC2, a literature search identified four other genes within the interval, SUOX, CDK2, ErbB3, and RAB5B, that have documented connection to eye development or function. Mutations in SUOX can cause Isolated sulfite oxidase deficiency, a severe autosomal recessive neurological disorder. One symptom of this disease is dislocation of the ocular lenses (Johnson et al. 2002). The role of the cell cycle protein CDK2 has been studied extensively during cell differentiation and proliferation in the lens (Wang et al. 2005). ErbB3 is a member of the epidermal growth factor receptor family and activates a pathway leading to cell proliferation and differentiation. Expression of ErbB3 in conjunctiva has been shown to be higher in patients suffering from keratoconjunctivitis sicca (dry eye syndrome) compared with normal subjects (Liu et al. 2000). Although direct evidence for a role of RAB5B in ocular development was not established, several other Rab and Rab-associated proteins are known to be involved. Mutations in some of these genes are responsible for the heritable eye defects choroideremia, Warburg Micro syndrome, and Martsolf syndrome (Corbeel and Freson 2008).
In the present study, 16 horses had clinically normal eyes but were heterozygous for the disease haplotype (including the PMEL17 mutation). Twelve of these had the Silver coat color and four were chestnuts. This nonpenetrance could be explained by genetic background or, more likely, that very small cysts located on the posterior iris or peripheral ciliary body may not have been detected. Conversely, eight horses that had the Cyst phenotype but not the disease-associated haplotype were also identified. However, it is conceivable that these cysts may be attributable to postinflammatory ocular disease or other nonhereditary causes.
Results of this study are of great importance for veterinarians and horse breeders since genetic markers within our identified interval (e.g., PMEL17 mutation) can be used for genetic testing of horses. Genetic information could be used for diagnosis of MCOA and to advise horse breeders regarding selection of horses for breeding. Horses that are homozygous for the disease-causing alleles exhibit the most severe clinical signs. Therefore, breeding an affected horse (MCOA or Cyst phenotype) with another affected horse (MCOA or Cyst phenotype) should be avoided as this could produce a horse with MCOA. Genetic testing for PMEL17 is of particular use since the Silver coat color can be confused with similar coat colors and because it is not possible to detect the mutation on coat colors that do not express any black pigment or have coat color genes such as Gray that could mask the Silver phenotype. So far it remains speculative whether the PMEL17 mutation causes both the Silver coat color and MCOA syndrome. However, linkage disequilibrium between the two is complete; if the phenotypes are regulated by separate mutations, they are a maximum of 183 kb apart. Because of this, it may be practically impossible to find horses that have recombined between these loci (i.e., carrying a haplotype with the Silver mutation but devoid of the mutation causing MCOA syndrome). On both sides of the MCOA interval, it is less than 20 kb between the marker defining the interval border and the markers within the IBD region (18 and 13 kb, respectively). Thus, additional fine mapping with our present horse material will not substantially change the size of the interval. Therefore, future aims include sequencing the entire genetic interval by next generation, deep sequencing to identify all candidate mutations in conserved sequences, and subsequently permitting functional assay evaluations.
References
Andersson LS, Juras R, Ramsey DT, Eason-Butler J, Ewart S, Cothran G, Lindgren G (2008) Equine Multiple Congenital Ocular Anomalies maps to a 4.9 megabase interval on horse chromosome 6. BMC Genet 9:88
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580
Brunberg E, Andersson L, Cothran G, Sandberg K, Mikko S, Lindgren G (2006) A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet 7:46
Clark LA, Wahl JM, Rees CA, Murphy KE (2006) Retrotransposon insertion in SILV is responsible for Merle patterning of the domestic dog. Proc Natl Acad Sci USA 103(5):1376–1381
Corbeel L, Freson K (2008) Rab proteins and Rab-associated proteins: major actors in the mechanism of protein-trafficking disorders. Eur J Pediatr 167(7):723–729
Das AV, James J, Bhattacharya S, Imbalzano AN, Antony ML, Hegde G, Zhao X, Mallya K, Ahmad F, Knudsen E, Ahmad I (2007) SWI/SNF chromatin remodeling ATPase Brm regulates the differentiation of early retinal stem cells/progenitors by influencing Brn3b expression and Notch signaling. J Biol Chem 282(48):35187–35201
Dechassa ML, Zhang B, Horowitz-Scherer R, Persinger J, Woodcock CL, Peterson CL, Bartholomew B (2008) Architecture of the SWI/SNF–nucleosome complex. Mol Cell Biol 28(19):6010–6021
Duncan B, Zhao K (2007) HMGA1 mediates the activation of the CRYAB promoter by BRG1. DNA Cell Biol 26(10):745–752
Ewart SL, Ramsey DT, Xu J, Meyers D (2000) The horse homolog of congenital aniridia conforms to codominant inheritance. J Hered 91(2):93–98
Grahn BH, Pinard C, Archer S, Bellone R, Forsyth G, Sandmeyer LS (2008) Congenital ocular anomalies in purebred and crossbred Rocky and Kentucky Mountain horses in Canada. Can Vet J 49(7):675–681
Hodgkinson CA, Moore KJ, Nakayama A, Steingrímsson E, Copeland NG, Jenkins NA, Arnheiter H (1993) Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 74(2):395–404
Hu R, Sharma SM, Bronisz A, Srinivasan R, Sankar U, Ostrowski MC (2007) Eos, MITF, and PU.1 recruit corepressors to osteoclast-specific genes in committed myeloid progenitors. Mol Cell Biol 27(11):4018–4027
Johnson JL, Rajagopalan KV, Renier WO, Van der Burgt I, Ruitenbeek W (2002) Isolated sulfite oxidase deficiency: mutation analysis and DNA-based prenatal diagnosis. Prenat Diagn 22(5):433–436
Karlsson AC, Kerje S, Hallbook F, Jensen P (2009) The Dominant white mutation in the PMEL17 gene does not cause visual impairment in chickens. Vet Ophthalmol 12(5):292–298
Kerje S, Sharma P, Gunnarsson U, Kim H, Bagchi S, Fredriksson R, Schutz K, Jensen P, von Heijne G, Okimoto R, Andersson L (2004) The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 168(3):1507–1518
Komaromy A, Rowlan JS (2009) Multiple congenital ocular abnormalities (MCOA) syndrome in two silver coat colored ponies. Proc Am Coll Vet Ophthalmol 40:113
Kuehn C, Weikard R (2007) Multiple splice variants within the bovine silver homologue (SILV) gene affecting coat color in cattle indicate a function additional to fibril formation in melanophores. BMC Genomics 8:335
Kwon BS, Halaban R, Ponnazhagan S, Kim K, Chintamaneni C, Bennett D, Pickard RT (1995) Mouse silver mutation is caused by a single base insertion in the putative cytoplasmic domain of Pmel17. Nucleic Acids Res 23(1):154–158
Liu Z, Carvajal M, Carothers Carraway CA, Carraway KL, Pflugfelder SC (2000) Increased expression of the type 1 growth factor receptor family in the conjunctival epithelium of patients with keratoconjunctivitis sicca. Am J Ophthalmol 129(4):472–480
Liu R, Liu H, Chen X, Kirby M, Brown PO, Zhao K (2001) Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106(3):309–318
Nobukuni Y, Watanabe A, Takeda K, Skarka H, Tachibana M (1996) Analyses of loss-of-function mutations of the MITF gene suggest that haploinsufficiency is a cause of Waardenburg syndrome type 2A. Am J Hum Genet 59(1):76–83
Ramsey DT, Ewart SL, Render JA, Cook CS, Latimer CA (1999a) Congenital ocular abnormalities of Rocky Mountain horses. Vet Ophthalmol 2(1):47–59
Ramsey DT, Hauptman JG, Petersen-Jones SM (1999b) Corneal thickness, intraocular pressure, and optical corneal diameter in Rocky Mountain Horses with cornea globosa or clinically normal corneas. Am J Vet Res 60(10):1317–1321
Ramsey DT, Mutti DO, Zadnik C, Bullimore MA, Murphy CJ (2000) Refractive error in Rocky Mountain horses with cornea globosa and with normal corneas. Invest Ophthalmol Vis Sci 41(4):S135
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Rudolph JA, Spier SJ, Byrns G, Hoffman EP (1992) Linkage of hyperkalaemic periodic paralysis in quarter horses to the horse adult skeletal muscle sodium channel gene. Anim Genet 23(3):241–250
Santschi EM, Purdy AK, Valberg SJ, Vrotsos PD, Kaese H, Mickelson JR (1998) Endothelin receptor B polymorphism associated with lethal white foal syndrome in horses. Mamm Genome 9(4):306–309
Schonthaler HB, Lampert JM, von Lintig J, Schwarz H, Geisler R, Neuhauss SC (2005) A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev Biol 284(2):421–436
Shin EK, Perryman LE, Meek K (1997) A kinase-negative mutation of DNA-PK(CS) in equine SCID results in defective coding and signal joint formation. J Immunol 158(8):3565–3569
Theos AC, Truschel ST, Raposo G, Marks MS (2005) The Silver locus product Pmel17/gp100/Silv/ME20: controversial in name and in function. Pigment Cell Res 18(5):322–336
Thierry-Mieg D, Thierry-Mieg J (2006) AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7 Suppl 1:S12.1–14
Tryon RC, White SD, Bannasch DL (2007) Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse. Genomics 90(1):93–102
Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR, Blocker H, Distl O, Edgar RC, Garber M, Leeb T, Mauceli E, MacLeod JN, Penedo MC, Raison JM, Sharpe T, Vogel J, Andersson L, Antczak DF, Biagi T, Binns MM, Chowdhary BP, Coleman SJ, Della Valle G, Fryc S, Guerin G, Hasegawa T, Hill EW, Jurka J, Kiialainen A, Lindgren G, Liu J, Magnani E, Mickelson JR, Murray J, Nergadze SG, Onofrio R, Pedroni S, Piras MF, Raudsepp T, Rocchi M, Roed KH, Ryder OA, Searle S, Skow L, Swinburne JE, Syvanen AC, Tozaki T, Valberg SJ, Vaudin M, White JR, Zody MC, Lander ES, Lindblad-Toh K (2009) Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 326(5954):865–867
Wang L, Wormstone IM, Reddan JR, Duncan G (2005) Growth factor receptor signalling in human lens cells: role of the calcium store. Exp Eye Res 80(6):885–895
Watanabe A, Takeda K, Ploplis B, Tachibana M (1998) Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet 18(3):283–286
Acknowledgments
We thank the personnel at the Animal Genetics Laboratory of the Department of Animal Breeding and Genetics (SLU, Uppsala) for their work with paternity testing of horses. We are grateful to anonymous horse breeders who have provided DNA samples. Financial support was obtained from the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) and the Swedish Foundation for Equine Research (SSH) to GL.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Andersson, L.S., Lyberg, K., Cothran, G. et al. Targeted analysis of four breeds narrows equine Multiple Congenital Ocular Anomalies locus to 208 kilobases. Mamm Genome 22, 353–360 (2011). https://doi.org/10.1007/s00335-011-9325-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-011-9325-7