
Abstract
Ras-associated binding (Rab) proteins and Rab-associated proteins are key regulators of vesicle transport, which is essential for the delivery of proteins to specific intracellular locations. More than 60 human Rab proteins have been identified, and their function has been shown to depend on their interaction with different Rab-associated proteins regulating Rab activation, post-translational modification and intracellular localization. The number of known inherited disorders of vesicle trafficking due to Rab cycle defects has increased substantially during the past decade. This review describes the important role played by Rab proteins in a number of rare monogenic diseases as well as common multifactorial human ones. Although the clinical phenotype in these monogenic inherited diseases is highly variable and dependent on the type of tissue in which the defective Rab or its associated protein is expressed, frequent features are hypopigmentation (Griscelli syndrome), eye defects (Choroideremia, Warburg Micro syndrome and Martsolf syndrome), disturbed immune function (Griscelli syndrome and Charcot–Marie–Tooth disease) and neurological dysfunction (X-linked non-specific mental retardation, Charcot–Marie–Tooth disease, Warburg Micro syndrome and Martsolf syndrome). There is also evidence that alterations in Rab function play an important role in the progression of multifactorial human diseases, such as infectious diseases and type 2 diabetes. Rab proteins must not only be bound to GTP, but they need also to be ‘prenylated’—i.e. bound to the cell membranes by isoprenes, which are intermediaries in the synthesis of cholesterol (e.g. geranyl geranyl or farnesyl compounds). This means that isoprenylation can be influenced by drugs such as statins, which inhibit isoprenylation, or biphosphonates, which inhibit that farnesyl pyrophosphate synthase necessary for Rab GTPase activity. Conclusion: Although protein-trafficking disorders are clinically heterogeneous and represented in almost every subspeciality of pediatrics, the identification of common pathogenic mechanisms may provide a better diagnosis and management of patients with still unknown Rab cycle defects and stimulate the development of therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Germline mutations in the genes involved in the Ras–mitogen-activated protein kinase (MAPK) pathway explain a number of neuro–cardio–facio–cutaneous syndromes, as reported in a paper recently published in this journal [10].
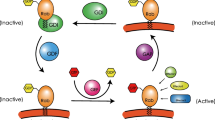
Ras-associated binding (Rab) proteins are small GTPases of the Ras superfamily that continuously cycle between the cytosol and different membranes. In 2002, Pfeffer described 20 different Rab GTPases that have been identified as prenylated proteins localized in distinct membrane-bound compartments [25]. Prenylated proteins are proteins bound to isoprenes, which are intermediaries in the cholesterol synthesis. At the time of the writing this article, more than 60 human Rabs had been identified [26]. This high number of different Rab proteins highlights their importance in the regulation of vesicle trafficking processes, including vesicle formation, motility, tethering and fusion to the acceptor membrane and signaling to other organelles. The ability of Rabs to perform these different tasks in a co-ordinated and regulated manner originates in their highly dynamic conformation and mobility and from their interaction with different Rab-associated proteins, as illustrated in the Rab GTPase cycle (Fig. 1). Rabs can cycle between the active GTP-bound and the non-active GDP-bound forms, assisted by different Rab-associated proteins, such as the GEFs (guanine nucleotide-exchange factors) and GAPs (GTPase-activating proteins), respectively [24, 26]. This conformational cycle is the main driving force for the on/off ‘switch’ mechanism of Rabs and their ability to both regulate binding to downstream effectors and perform the desired function. Rabs are initially synthesized as soluble proteins in the cytosol where they are first recognized by a soluble chaperone-like protein named REP (Rab escort protein) [3]. The REP brings the Rab to the RabGGT (Rab geranylgeranyl transferase) for the addition of—generally—two geranylgeranyl groups. These prenyl groups are added, by covalent thioether bonds, to cysteine residue(s) located at the C-terminus of the Rab protein [7]. This post-translational modification of Rabs is needed to allow for the attachment of the Rab proteins into the lipid bilayer of the organelle; consequently, Rabs are considered to be peripheral membrane proteins (Fig. 1). After performing their function, Rab proteins are extracted from the membranes by RabGDI (GDP dissociation inhibitor), and they remain in the cytosol until they are needed again [7, 24, 37]. Statins are known to inhibit protein isoprenylation. As such, they are able to reduce the production of amyloid-beta and inhibit preamyloid catabolism in the lysosomes, which suggests their possible application in the treatment of Alzheimer [20]. However, the therapeutic role of statins in Rab-related disorders has not yet been established. The potent nitrogen-containing biphosphonates, such as pamidronate, alendronate, ibandronate and zoledronate, inhibit a key enzyme, farnesyl pyrophosphate synthase, which is necessary for Rab activity, hence their inhibitory action on osteoclast activity [29].

The Ras-associated binding (Rab) protein–GTPase cycles. Rab proteins are intrinsically soluble and require a post-translational modification for membrane association. They first associate with a Rab escort protein (REP) and form a stable complex that is the substrate for the subsequent dual prenylation of C-terminal cysteine motifs via Rab geranylgeranyl transferase (RabGGT). RabGGT consists of two different functional subunits (RabGGTα and RabGGTß). After lipid tranfer, REP delivers the prenylated Rab to the donor membrane (the REP cycle is shown with blue arrows). In the absence of REP or RabGGT, Rab proteins remain in the cytosol in an inactive state. The transfer of Rab proteins between acceptor and donor membranes is facilitated by the GDP dissociation inhibitor (GDI) (the GDI cycle is shown with red arrows). Both REP and GDI bind the GDP-bound inactive form of Rab. After REP or GDI dissociate from Rabs at the donor membrane, Rabs can cycle between the inactive (GDP-bound) and active (GTP-bound) states. Rab proteins are activated by guanine nucleotide exchange factors (RabGEFs) and deactivated by GTPase activating proteins (RabGAPs), which accelerate the slow intrinsic rates of nucleotide exchange and GTP hydrolysis. In the active state, Rabs interact with structurally and functionally diverse effectors, including cargo sorting complexes on donor membranes, motor proteins involved in vesicular transport and tethering complexes that regulate vesicle fusion with acceptor membranes
Evolutionarily conserved Rabs are usually expressed in all cell and tissue types and regulate more fundamental vesicle transport pathways whereas the less conserved Rab family members function in more specific pathways, which are often organ-dependent. Defects in intracellular vesicle trafficking underlie a large variety of human diseases, including pathologies associated with defects in Rabs or Rab-associated proteins [14, 32]. In this review, we deal with the role of different Rab and rab-associated proteins in inherited (Table 1) as well as certain multifactorial human diseases in different pediatric disciplines. In addition, the review will also focus briefly on recent advances in understanding human disease through the study of Rab proteins from in vitro and mouse knockout studies.
Rab proteins in skin and hear pigmentation
Griscelli syndrome (GS) is an autosomal recessive disorder that causes partial albinism [15]. There are three variants of this disease: one is a purely hypopigmentation disorder (GS type III), and two, in addition to the pigmentation defect, are also associated with immunological defects (GS type II) or with primary neurological dysfunctions (GS type I). Griscelli syndrome type II with immunological defects is caused by missense mutations in the gene encoding Rab27a [18]. This Rab regulates the movement of melanosomes to the cell periphery of melanocytes and also regulates the secretion of lytic granules in cytotoxic T lymphocytes (CTL) [35]. The lack of Rab27a thus causes pigment anomalies and dysfunctional T lymphocytes, which is in agreement with the defects observed in the patients that present with albinism and hemophagocytic syndrome. The GS type I with neurological symptoms is caused by mutations in the MYO5A gene encoding the motor protein myosin Va [22], a putative Rab27a effector that drives the peripheral distribution of melanosomes along actin filaments [15]. As myosin Va does not participate in the exocytosis of lytic granules of CTL, the inactivation of this protein does not lead to immunological symptoms. Finally, GS type III in patients with typically pigment abnormalities without additional features is caused by a mutation in the MLPH gene encoding melanophilin. A direct interaction exists between the SHD domain of melapholin and Rab27a and between the C-terminal domain of melanophilin and myosin Va [36]. Therefore, although the Rab27a/Mlph/MyoVa complex is crucial for the trafficking of melanosomes, neurodevelopmental abnormalities are specific for the Myo5a deficiency, and Rab27a uses a different effector for its function in the CTL.
Rab proteins in immunology
It has already been mentioned that GS type II patients with Rab27a mutations develop the hemophagocytic syndrome, which is characterized by episodes of life-threatening uncontrolled T lymphocyte and macrophage activation [17]. During these episodes, activated T cells and macrophages infiltrate various organs (including the brain), leading to tissue damage, organ failure and death in the absence of immunosuppressive therapy. In practice, only bone-marrow or cord blood stem cell transplantation can be curative for this condition [33].
The important function of Rab proteins in intracellular traffic, endocytosis and exocytosis explains their role in counteracting bacterial and viral infections. Phagocytosis by macrophages leads to the destruction of bacteria. Endocytosis leads to the formation of phagosomes, but phagosomes fuse with lysosomes (phagolysosomes), resulting in the release of toxic products that kill the bacterium as well as the release of the degradation products by exocytosis [19]. However, some bacteria survive this process and avoid bactericide by escaping from the phagosomes into the cytoplasm [19]. Listeria monocytogenes is a paramount example. It has been shown that the stimulation of macrophages with interferon (IFN)-γ, a cytokine that is secreted by natural killer and T helper cells, results in the expression of at least 200 proteins, including Rab5a [11]. In L. monocytogenes-infected macrophages, overexpression of Rab5a stimulates the intracellular degradation of the pathogen [6]. It has been suggested that Rab5a participates in the active recruitment of Rac2 to phagolysosomes, which is important for the destruction of the bacterium [27]. Other microbes develop antiphagocytosis (e.g. Yersinia enterocolitica) or inhibit the formation of phagosomes (e.g. Salmonella typhimurium). In Salmonella infections, there is evidence that Rab5a is stabilized on the phagosome by the Salmonella type III secretory protein SopE [23]. SopE has GEF activity for Rab5a, stimulating activated GTP-bound Rab5a and preventing its membrane extraction by RabGDI. A general conclusion that can be drawn is that parasites recruit endocytic Rabs to prevent or delay the formation of the degradative phagolysosome in the host organism.
Viruses enter host cells by receptor-mediated endocytosis, but they usually escape from endosomal vesicles to enter either the cytosol or the nucleus where they replicate. The binding of hepatitis C virus (HCV) non-structural protein (NS) with Rab1 GTPase-activating protein suggests that viruses are able to subvert host cell machinery in this way because this binding is necessary for HCV replication.
Rab1 depletion decreases HCV-RNA levels, which could lead to therapeutic applications [34].
Rab proteins in vision
Choroideremia is an X-linked disease that involves the degeneration of the retinal pigment epithelium and the adjacent choroid and retinal photoreceptor cell layers, leading to blindness. The mutated gene in the choroideremia is one of the two REP isoforms, REP1 [3, 30]. Although the other isoform, REP-2, seems to be sufficient for the geranylgeranylation of all Rab GTPases in all tissues, the retinal pigment epithelium seems to be the exception. A role for REP-2 in human disease has not yet been discovered. REP-1 is essential for the efficient geranylgeranylation of Rab27a in the retinal pigment epithelium. Thus, a lack of REP1 leads to a lack of functional Rab27a specifically in the retinal pigment epithelium [30, 31]. The degeneration of this epithelium and its adjacent layers may be due to deficient melanosome transport and, consequently, a lack of protection against harmful light exposure.
Rab proteins in neurology
A subgroup of patients with X-linked non-specific mental retardation have mutations in the GDI1 gene for one of the GDI isoforms, GDI-α [9]. This isoform is particularly abundant in the brain, and dysfunctional membrane recycling of one or more Rab GTPases in brain synapses, leading to aberrant neurotransmission, is likely to underlie the symptoms in this disease.
Hereditary sensory and autonomic neuropathies (HSAN), known for many years as familial dysautonomia in Jewish patients, are characterized by the loss of sensation for pain and temperature, alacrima, excessive sweating and the absence of fungiform tongue papillae. The onset is congenital, and the transmission is autosomal recessive. The clinical symptoms of HSAN have been classified into five types [21]. HSAN type II encompasses patients with Charcot–Marie–Tooth disease type II. This is an axonal neuropathy with a median motor conduction velocity of >38 m/s. This peripheral neuropathy starts in the second or third decade of life and presents as mixed motor and sensory involvement associated with ulcers. Four mutations in the Rab7 gene have been identified in four families with Charcot–Marie–Tooth disease type 2B (CMT2B), with symptoms leading to ulcerations and amputations [38, 39]. However, a hallmark feature of patients with HSAN type 1 is lancinating pain, which is not commonly observed in CMT2B. Moreover, HSAN type 1 is associated with a mutation in serine palmitoyl transferase (SPT), a rate-limiting step in the sphingo-lipid synthesis. This overlapping of the two syndromes is possibly explained by Rab7 being involved in both the endocytosis and transport of sphingolipids [39].
The importance of Rab proteins in brain development and function is even more emphasized by the recent discoveries of the involvement of Rab3-associated proteins in human disease [4, 5]. Rab3A is the most abundantly expressed protein in the brain, where it is present in all synapses and involved in calcium-dependent neurotransmitter release [13]. The activity of Rab3 proteins is tightly regulated by Rab3GAP, which specifically converts active Rab3-GTP to the inactive-GDP form. Germline-inactivating mutations in the catalytic subunit of Rab3GAP (Rab3GAP1) cause Warburg Micro syndrome, an autosomal recessive disorder characterized by ocular defects (microphthalmos, microcornea, congenital cataracts and optic atrophy), neurodevelopmental defects (microcephaly, cortical gyral abnormalities, such as pachygyria and polymicrogyria, hypoplasia of the corpus callosum, severe mental retardation, and spastic cerebral palsy) and hypothalamic hypogenitalism [4]. Mutations in the non-catalytic subunit of Rab3GAP (Rab3GAP2) have been described in patients with Martsolf syndrome, an autosomal recessive disorder with a similar but milder phenotype than the Warburg Micro syndrome, including congenital cataracts, hypogonadism and mild mental retardation [5]. The precise mechanisms whereby the Rab3GAP1 and Rab3GAP2 mutations cause the human phenotype is still unclear.
Rab proteins in endocrinology
Abnormal trafficking of the insulin-sensitive glucose transporter GLUT4 has been described in patients with diabetes type 2. Glucose transporter 4 accumulates in the dense membrane compartments, suggesting that defects in membrane trafficking may be involved in insulin resistance [12]. Rab4 has been found to be implicated in GLUT4 biogenesis, sorting and exocytosolic movement but more studies are needed to define the exact role of this and other Rabs and Rab-associated proteins in GLUT4 trafficking [16]. Rab4 mutations in humans or mice do not yet exist.
Rab27a is necessary for insulin secretion [2]. Diabetes mellitus is characterized by both low insulin and high glucagon levels. These conditions have been shown recently to be due to the expression of an inducible cAMP early repressor (ICER) [1], resulting in a repression of the transcription of the insulin gene, and associated with a diminished level of granuphilin, a Rab27a-dependent protein. However, patients with GS type II-homozygous deficiency in Rab27a do not have diabetes mellitus.
The control of thyroid hormone production depends on endocytic catalysts and the tandem regulators Rab5a and Rab7, which have been found in excess in thyroid adenomas [8].
Rab proteins in nephrology
Nephrogenic diabetes insipidus (NDI) is characterized by the inability to concentrate the urine in response to the antidiuretic hormone vasopressin (AVP). Under normal conditions, binding of AVP to the vasopressin receptor (V2R) leads to the insertion of aquaporin 2 (AQP2) water channels in the apical membrane of the renal collecting duct [28] (Fig. 2). The defect can be inherited and is either X-linked due to mutations in the genes encoding the V2R or autosomal recessive or dominant in mutations in the AQP2 gene. The trafficking of AQP2 in the cell and its internalization are cAMP and especially Rab11-dependent [40]. However, Rab11 mutations in humans or mice are not yet described.

Regulation of aquaporin-2 (AQP2)-mediated water transport by vasopressin (AVP). Shown is a nephron with a magnified principal cell. In this cell, the vasopressin V2 receptor (V2R), stimulatory GTP binding protein (Gs), adenylate cyclase (AC), ATP, cAMP, and phosphorylated proteins (O-P) are indicated. This figure has been used with permission of the American Journal of Physiology and Renal Physiology
Conclusion
The Rab GTPases are a large family of proteins with a variety of regulatory functions in membrane trafficking. The central role of these proteins has become clear during the past decade, as part of the progress that has been made in understanding in detail the mechanistic principles of transport vesicle formation, movement, and fusion. Sequencing of the human genome has enabled researchers to realize the diversity of the Rab gene family, although the functions of the majority of the gene products are still unknown. The availability of complete genomic sequences as well as the important advances in molecular and cell biological methods that have already been made hold the promise of significant progress being booked in our understanding of Rab function in the near future. The identification of further genes involved not only in monogenic but also in common multifactorial human vesicle-trafficking disorders will result in a better understanding of this complex transport pathway but, more importantly, will also lead to opportunities to develop novel treatments.
Abbreviations
- AQP2:
-
aquaporin 2
- AVP:
-
antidiuretic hormone vasopressin
- CHM:
-
choroideremia
- CMT:
-
Charcot–Marie–Tooth
- CTL:
-
cytotoxic T lymphocytes
- ER:
-
endoplasmic reticulum
- GAP:
-
GTPase-activating protein
- GDI:
-
GDP-dissociation inhibitor
- GEF:
-
guanine nucleotide-exchange factor
- GLUT4:
-
glucose transporter 4
- GS:
-
Griscelli syndrome
- HSAN:
-
hereditary sensory and autonomic neuropathies
- IFNγ:
-
interferon gamma
- MAPK:
-
mitogen-activated protein kinase
- NDI:
-
nephrogenic diabetes insipidus
- Rab:
-
Ras-associated binding protein
- RabGGT:
-
Rab geranylgeranyl transferase
- Ras:
-
Rat sarcoma viral oncogene homologue
- REP:
-
Rab escort protein
References
Abderrahmani A, Cheviet S, Ferdaoussi M, Coppola T, Waeber G, Regazzi R (2006) ICER induced by hyperglycemia represses the expression of genes essential for insulin exocytosis. EMBO J 25:977–986
Aizawa T, Komatsu M (2005) Rab27a: a new face in beta cell metabolism-secretion coupling. J Clin Invest 115:227–230
Alexandrov K, Horiuchi H, Steele-Mortimer O, Seabra MC, Zerial M (1994) Rab escort protein-1 is a multifunctional protein that accompanies newly prenylated Rab proteins to their target membranes. EMBO J 13:5262−5273
Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, Maina EN, Morgan NV, Tee L, Morton J, Ainsworth JR, Horn D, Rosser E, Cole TR, Stolte-Dijkstra I, Fieggen K, Clayton-Smith J, Mégarbané A, Shield JP, Newbury-Ecob R, Dobyns WB, Graham JM Jr, Kjaer KW, Warburg M, Bond J, Trembath RC, Harris LW, Takai Y, Mundlos S, Tannahill D, Woods CG, Maher ER (2005) Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 37:221–223
Aligianis IA, Morgan NV, Mione M, Johnson CA, Rosser E, Hennekam RC, Adams G, Trembath RC, Pilz DT, Stoodley N, Moore AT, Wilson S, Maher ER (2006) Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum Genet 78:702–707
Alvarez-Dominguez C, Madrazo-Toca F, Fernandez-Prieto L, Vandekerckhove J, Pareja E, Tobes R, Gomez-Lopez MT, Del Cerro-Vadillo E, Fresno M, Leyva-Cobián F, Carrasco-Marín E (2008) Characterization of a Listeria monocytogenes protein Interfering with Rab5a. Traffic 9:325–337
Anant JS, Desnoyers L, Machius M, Demeler B, Hansen JC, Westover KD, Deisenhofer J, Seabra MC (1998) Mechanism of Rab geranylgeranylation: formation of the catalytic ternary complex. Biochemistry 37:12559–12568
Croizet-Berger K, Daumerie C, Couvreur M, Courtoy PJ, van den Hove MF (2002) The endocytic catalysts, Rab5a and Rab7, are tandem regulators of thyroid hormone production. Proc Natl Acad Sci USA 99:8277–8282
D’Adamo P, Menegon A, Lo Nigro C, Grasso M, Gulisano M, Tamanini F, Bienvenu T, Gedeon AK, Oostra B, Wu SK, Tandon A, Valtorta F, Balch WE, Chelly J, Toniolo D (1998) Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat Genet 19:134–139
Denayer E, Legius E (2007) What’s new in neuro-cardio-facial-cutaneous syndromes? Eur J Pediatr 166:1091–1098
Der SD, Zhou A, Williams BR, Silverman RH (1998) Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA 95:15623–156628
Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD (1998) Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest 101:2377–2386
Geppert M, Südhof TC (1998) RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci 21:75–95
Gissen P, Maher ER (2007) Cargos and genes: insights into vesicular transport from inherited human disease. J Med Genet 44:545–555
Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC (2001) Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J Cell Biol 152:795–808
Imamura T, Huang J, Usui I, Satoh H, Bever J, Olefsky JM (2003) Insulin-induced GLUT4 translocation involves protein kinase C-lambda-mediated functional coupling between Rab4 and the motor protein kinesin. Mol Cell Biol 23:4892–4900
Janka GE (2007) Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr 166:95–109
Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulfraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G (2000) Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25:173–176
Méresse S, Steele-Mortimer O, Moreno E, Desjardins M, Finlay B, Gorvel JP (1999) Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat Cell Biol 1:E183–E188
Ostrowski SM, Wilkinson BL, Golde TE, Landreth G (2007) Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem 282:26832–26844
Parman Y (2007) Hereditary neuropathies. Curr Opin Neurol 20:542–547
Pastural E, Barrat FJ, Dufourcq-Lagelouse R, Certain S, Sanal O, Jabado N, Seger R, Griscelli C, Fischer A, De Saint-Basile G (1997) Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 16:289–292
Patel JC, Galán JE (2006) Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol 175:453–463
Pereira-Leal JB, Seabra MC (2000) The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol 301:1077–1087
Pfeffer SR (1992) GTP-binding proteins in intracellular transport. Trends Cell Biol 2:41–46
Pfeffer SR (2007) Unsolved mysteries in membrane traffic. Annu Rev Biochem 76:629–645
Prada-Delgado A, Carrasco-Marín E, Peña-Macarro C, Del Cerro-Vadillo E, Fresno-Escudero M, Leyva-Cobián F, Alvarez-Dominguez C (2005) Inhibition of Rab5a exchange activity is a key step for Listeria monocytogenes survival. Traffic 6:252–265
Robben JH, Knoers NV, Deen PM (2006) Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 291:F257–F270
Russell RG (2007) Bisphosphonates: mode of action and pharmacology. Pediatrics 119[Suppl 2]:S150–S162
Seabra MC, Brown MS, Goldstein JL (1993) Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science 259:377–381
Seabra MC, Ho YK, Anant JS (1995) Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 270:24420–24427
Seabra MC, Mules EH, Hume AN (2002) Rab GTPases, intracellular traffic and disease. Trends Mol Med 8:23–30
Schuster F, Stachel DK, Schmid I, Baumeister FA, Graubner UB, Weiss M, Haas RJ, Belohradsky BH (2001) Griscelli syndrome: report of the first peripheral blood stem cell transplant and the role of mutations in the RAB27A gene as an indication for BMT. Bone Marrow Transplant 28:409–412
Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS (2007) TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J Biol Chem 282:d36354–61
Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, Seabra MC, Griffiths GM (2001) Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol 152:825–834
Strom M, Hume AN, Tarafder AK, Barkagianni E, Seabra MC, A family of Rab27-binding proteins (2002) Melanophilin links Rab27a and myosin Va function in melanosome transport. J Biol Chem 277:25423–25430
Ullrich O, Stenmark H, Alexandrov K, Huber LA, Kaibuchi K, Sasaki T, Takai Y, Zerial M (1993) Rab GDP dissociation inhibitor as a general regulator for the membrane association of rab proteins. J Biol Chem 268:18143–18150
Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, Van Gerwen V, Wagner K, Hartung HP, Timmerman V (2003) Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72:722–727
Verhoeven K, Timmerman V, Mauko B, Pieber TR, De Jonghe P, Auer-Grumbach M (2006) Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol 19:474–480
Vossenkämper A, Nedvetsky PI, Wiesner B, Furkert J, Rosenthal W, Klussmann E (2007) Microtubules are needed for the perinuclear positioning of aquaporin-2 after its endocytic retrieval in renal principal cells. Am J Physiol Cell Physiol 293:C1129–C1138
Acknowledgments
This research was supported by the ‘Excellentie financiering KULeuven’ (EF/05/013), by research grants G.0453.05 and G.0124.02 from the FWO-Vlaanderen (Belgium) and by grant GOA/2004/09 from the Research Council of the University of Leuven (Onderzoeksraad K.U.Leuven, Belgium). We thank Doctor Chris Van Geet for helpful and valuable advice and cooperation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Corbeel, L., Freson, K. Rab proteins and Rab-associated proteins: major actors in the mechanism of protein-trafficking disorders. Eur J Pediatr 167, 723–729 (2008). https://doi.org/10.1007/s00431-008-0740-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-008-0740-z