
Abstract
In 2006 the protein TDP-43 was identified as the major ubiquitinated component deposited in the inclusion bodies found in two human neurodegenerative diseases, amyotrophic lateral sclerosis and frontotemporal lobar degeneration. The pathogenesis of both disorders is unclear, although they are related by having some overlap of symptoms and now by the shared histopathology of TDP-43 deposition. Now, in 2008, several papers have been published in quick succession describing mutations in the TDP-43 gene, showing they can be a primary cause of amyotrophic lateral sclerosis. There are many precedents in neurodegenerative disease in which rare single-gene mutations have given great insight into understanding disease processes, which is why the TDP-43 mutations are potentially very important.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rapidly rising life expectancy is forcing many of the world’s societies to see neurodegenerative diseases as a wider social and economic issue. Such diseases have always been devastating for sufferers and their carers, but aging societies are facing a broader burden resulting from the lack of effective treatments. There is hope for disorders such as Alzheimer disease (AD), where now we have a good understanding of pathogenesis and novel treatments are on the horizon. However, two neurodegenerative disorders that remain in urgent need of attention, and that mainly but not exclusively affect the aging population, are amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD).
ALS is the third most common neurodegenerative cause of adult death after AD and Parkinson disease (http://www.statistics.gov.uk/StatBase) and the lifetime risk of dying from ALS lies between 1/400 and 1/1000 (Boillee et al. 2006; Pasinelli and Brown 2006), and in the UK 1 in 400 death certificates is issued for “motor neuron disease” (J. Stevens, personal communication). In ALS the upper motor neurons that run from the brain into the spinal cord and the lower motor neurons that extend from the spinal cord out to the muscles degenerate, leading inexorably to paralysis and death, typically within 3–5 years of diagnosis (Boillee et al. 2006; Pasinelli and Brown 2006; Schymick et al. 2007; Valdmanis and Rouleau 2008). Intellect usually remains intact and no effective treatments are available. Up to approximately 10% of ALS is familial (FALS), usually autosomal dominant, and mutations in the ubiquitously expressed enzyme superoxide dismutase 1 (SOD1) are causative in less than 20% of FALS (Deng et al. 1993; Rosen et al. 1993) and in approximately 1% of sporadic ALS (SALS) (Pasinelli and Brown 2006). Other rare ALS mutant genes are known, usually associated with variants of ALS rather than the classic typical midlife onset disease (Boillee et al. 2006; Pasinelli and Brown 2006; Schymick et al. 2007; Valdmanis and Rouleau 2008).
FTLD is the most common cause of presenile (below 65 years of age) dementia after AD (Forman et al. 2007; Harvey et al. 2003; Ratnavalli et al. 2002). Affected individuals have a range of characteristic traits that reflect degeneration in the frontal and temporal lobes of the brain—the areas that control behavior, emotions, and language. Early symptoms typically manifest as language difficulties and inappropriate behavior (Neary et al. 1998). Up to 40% of FTLD is thought to be familial with genetically heterogeneous causes. Causative mutations have been identified in several genes, including those encoding tau (Hutton et al. 1998; Poorkaj et al. 1998; Spillantini et al. 1998), charged multivesicular body protein 2B (CHMP2B) (Skibinski et al. 2005), and progranulin (GRN) (Baker et al. 2006; Cruts et al. 2006), and others remain to be found. Mutations have also been identified in the valosin-containing protein (VCP) gene in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia, in which frontotemporal dementia can be, but is not always, a feature (Watts et al. 2004).
At first glance ALS and FTLD appear to be different disorders; however, clinicians have noted for some time that there are overlaps. FTLD symptoms are reported in up to 20% of ALS cases (Valdmanis and Rouleau 2008; Van Deerlin et al. 2008) and there are other intriguing connections such as families that segregate both disorders (Morita et al. 2006; Valdmanis et al. 2007) and imaging studies which show frontal deficits in ALS patients (Kato et al. 1993). A recent study has shown that progranulin is a modifier of ALS disease progression (Sleegers et al. 2008), although this was not replicated in another study (Pickering-Brown et al. 2008).
Importantly, in common with several other neurodegenerative disorders, ALS and FTLD both present with aggregates of misfolded proteins in the cytoplasm and/or nucleus of neurons. Ubiquitinated inclusion bodies have been reported in the cytoplasm of neurons of both SALS and FALS cases and in mutant SOD1 transgenic mice that model FALS (for further discussion see Boillee et al. 2006; Pasinelli and Brown 2006). FTLD is subdivided into two classes based on the content of the inclusion bodies: (1) those with tau-positive and ubiquitin-negative inclusions (tauopathies) and (2) more common forms with ubiquitinated but tau-negative inclusions known as FTLD-U, including PGRN, VCP, and CHMP2B mutations (reviewed in Mackenzie and Rademakers 2007). ALS and FTLD-U cases both present with cytoplasmic ubiquitin-positive, tau-negative inclusions indicating that there are at least some common pathway(s) involved in the pathogenesis of these diseases.
TDP-43—the new kid on the block
Research into ALS and FTLD-U was radically redirected by the appearance of a new player in 2006. In that year, Neumann, Trojanowski, Lee, and colleagues provided a molecular connection between these disorders by finding that a protein called the TAR DNA-binding protein (TDP-43) is the major protein in the inclusion bodies in both disorders (Neumann et al. 2006), which was quickly confirmed (Arai et al. 2006). Some authors now refer to the TDP-43 inclusion positive types of ALS and FTLD-U as different forms of the same neurodegenerative disorder: TDP-43 proteinopathy (Cairns et al. 2007; Kwong et al. 2008; Winton et al. 2008). TDP-43 positive inclusions have now also been reported in cases of Alzheimer disease, Pick disease, dementia with Lewy bodies, and other neurodegenerative disorders, and are seen in glia as well as neurons (Amador-Ortiz et al. 2007; Freeman et al. 2008; Geser et al. 2008; Hasegawa et al. 2007; Higashi et al. 2007; Nakashima-Yasuda et al. 2007; Neumann et al. 2007).
TDP-43 is a 414-residue, 43-kDa protein, first identified as a binding partner of the TAR DNA element of the human immunodeficiency virus (Ou et al. 1995). Currently we know that TDP-43 is a ubiquitously expressed, highly conserved nuclear protein encoded by a 6-exon gene (TARDBP on human chromosome 1p36.2). The protein consists of two RNA recognition motifs and a glycine-rich domain (Fig. 1). It is found in nuclear bodies, colocalized with SMN and gemin proteins, and may function as a transcriptional repressor and as an activator of exon skipping, or in other roles such as in miRNA biogenesis, apoptosis, and cell division (Ayala et al. 2005, 2008; Buratti et al. 2001; Johnson et al. 2008; Winton et al. 2008). In the TDP-43 proteinopathies TDP-43 is depleted from the nucleus and is sequestered as hyperphosphorylated insoluble aggregates in the nucleus, perikarya, and dystrophic neurites (Neumann et al. 2006). Perturbation of the trafficking of TDP-43 between the nucleus and cytoplasm is thought to lead to the formation of these aggregates; brain samples from both ALS and FTLD-U are enriched for a smaller (∼25 kDa) phosphorylated C-terminal fragment and high-molecular-weight ubiquitinated species (Neumann et al. 2006; Winton et al. 2008). Inhibition of autophagy can also lead to the relocalization of TDP-43 from the nucleus to the cytoplasm in vitro (Filimonenko et al. 2007).

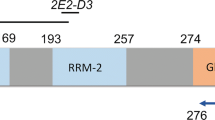
TDP-43 mutations in ALS. TDP-43 is encoded by a 6-exon gene, of which exons 2-6 are protein coding (top). The TDP-43 protein contains four known functional domains: a nuclear localization sequence, two central RNA Recognition Motifs (RRM1 and RRM2), and a C-terminal glycine-rich domain, predicted to mediate protein-protein interactions. All disease mutations so far are found in the glycine-rich domain (encoded by exon 6), with the exception of one mutation in RRM1 (encoded by exon 4). A mutation in the nuclear localization domain has been reported in two unaffected controls (data compiled from Gitcho et al. 2008; Kabashi et al. 2008; Sreedharan et al. 2008; Van Deerlin et al. 2008; Yokoseki et al. 2008)
Intriguingly, while TDP-43 is deposited in sporadic and familial FTLD-U and in sporadic and non-SOD1 familial ALS (Kwong et al. 2008; Neumann et al. 2006), it is not found in inclusions in SOD1 FALS (Mackenzie et al. 2007; Tan et al. 2007) or in the SOD1 G93A, SOD1 G37R, and SOD1 G85R transgenic mouse models of FALS (Robertson et al. 2007).
TDP-43 is a criminal, not just a bystander
The identification of TDP-43 in ALS and FTLD-U inclusion bodies immediately led to surveys of patient cohorts to find either association with or mutations in the gene. All returned with negative results (e.g., see Gijselinck et al. 2007; Rollinson et al. 2007; Schumacher et al. 2007), leading to the reasonable conclusion that TDP-43 deposition may simply be a consequence of disease and therefore possibly of less interest in providing novel insight into the cause of these diseases. However, in 2008 all this has changed with the publication of five articles, all of which report rare TDP-43 mutations in sporadic and familial ALS in patients of different ethnicities (mainly but not exclusively Caucasian) (Gitcho et al. 2008; Kabashi et al. 2008; Sreedharan et al. 2008; Van Deerlin et al. 2008; Yokoseki et al. 2008). In summary (Fig. 1, Table 1), all the mutations lie in exon 6 except for one in exon 4. The FALS cases are autosomal dominant with some variability in presentation, bulbar and limb onset cases, some with more or less lower motor neuron loss and different ages of onset and rates of progression, but all fairly classic FALS. No dementia is found, although Kabashi et al. (2008) report apathy, major anxiety, and agitation in two individuals. For cases in which postmortems have been carried out, TDP-43 deposition has been found in the brains of affected individuals (Van Deerlin et al. 2008; Yokoseki et al. 2008).
The mutations are already starting to give some insight into pathogenesis (Gitcho et al. 2008; Kabashi et al. 2008; Sreedharan et al. 2008; Van Deerlin et al. 2008; Yokoseki et al. 2008). Disruption of protein interactions is one possible disease mechanism. Most mutations lie in the C-terminal, a glycine-rich region that may mediate interactions with proteins, including heterogeneous ribonucleoproteins. Also, some of the mutations found in this region could increase phosphorylation by substituting threonine or serine residues or through the creation of a new protein kinase A site (Sreedharan et al. 2008). This may result in disruption of protein interactions and/or disruption of transport through the nuclear pore complex. In transfected cell lines and patient lymphocytes some variants also show a clearly increased propensity to aggregate and to produce a lower-molecular–weight, detergent-insoluble protein product (Kabashi et al. 2008; Sreedharan et al. 2008; Yokoseki et al. 2008). One mutation, D169G, lies in the first RNA-binding motif and may affect RNA binding (Kabashi et al. 2008). When mutant TDP-43 is electroporated into the neural tube of developing chick embryos, two different mutations showed a reduction in rate of maturation of the neural tube with an increase in apoptotic nuclei, suggesting a toxic gain of function or dominant negative effect (Sreedharan et al. 2008).
It is noteworthy that just one mutation lies in exon 4, within the first RNA recognition motif (RRM 1), whereas the other 13 mutations identified so far occur in exon 6, in the putative protein interaction domain. This raises the question of whether the exon 4 mutation (D169G) is a genuine pathogenic mutation and, if so, if it leads to disease via a different effect on TDP-43 function than the other mutations. D169 is a highly conserved amino acid (Kabashi et al. 2008) and the mutation was absent in 360 ethnically matched controls from France (Kabashi et al. 2008), as well as in 872 nonethnically matched controls (700 British, 172 Australian) for which all exons of TDP-43 were sequenced in the study of Sreedharan et al. (2008). The absence of the mutation in a large number of controls and the evolutionary conservation of the amino acid argue for a pathogenic role; however, absence in a larger number of ethnically matched controls and functional data or the identification of further mutations in RRM 1 will be required to fully resolve this issue. If D169G is pathogenic, then it suggests that mutations in the C-terminal domain, and the RRM1 domain, which has been shown to be essential for the RNA-binding ability of TDP-43 (Buratti and Baralle. 2001), affect the function of TDP-43 in a similar way, such as a general loss of function, or that there are different ways to disrupt TDP-43 function and still lead to disease.
Rare single-gene defects, protein aggregates, and neurodegeneration—sounds familiar
The identification of the major protein in neurodegenerative disease inclusion bodies, followed by finding rare mutations in the gene encoding this protein, is a powerful route to understanding pathogenesis and is turning into a surprisingly common approach in neurodegenerative diseases. It is exemplified by the classic example of finding mutations in the amyloid precursor protein (APP) gene that encodes the Abeta peptide deposited in the plaques of Alzheimer disease; it revolutionized our understanding of AD (Hardy and Selkoe 2002). Similar rare dominant single-gene mutations have also been found in the proteins that aggregate in Parkinson disease, prion diseases, and tauopathies, for example, also giving us great insight into the pathogenesis of these disorders. The articles that reported on TDP-43 mutations in ALS have shifted the focus of attention on this protein from being deposited as a by-product of disease processes to actually being a causative agent that triggers the processes resulting in neuronal death. From this point investigations will proceed using transgenic and knockout mice and a variety of different cellular systems to understand the link between mutation and disease.
Many questions need to be addressed, such as does TDP-43 take on a toxic gain of function when mutated in ALS, as happens with SOD1 mutations in ALS? Will TDP-43 mutations be found in cases of FTLD-U, and if not, why not? How important is timing? Does having a germline TDP-43 mutation give rise to ALS which might develop into FTLD-U if individuals lived long enough? Another question is: What separates TDP-43 ALS from SOD1 ALS? These two diseases are very similar at a clinical level, but clearly different pathways are affected for at least some of the pathogenesis. We assume that there are many parallel cellular pathways, which, if disrupted can lead to the same outcome. TDP-43 ALS and SOD1 ALS may allow the investigation of this phenomenon.
The new mutations found in TDP-43 are a breakthrough for ALS and FTLD-U research and we are looking forward to seeing what new discoveries they herald to help us treat and ultimately cure these terrible neurodegenerative diseases.
References
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Ayala YM, Pantano S, D’Ambrogio A, Buratti E, Brindisi A et al (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348:575–588
Ayala YM, Misteli T, Baralle FE (2008) TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A 105:3785–3789
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Boillee S, Vande VC, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36
Buratti E, Dork T, Zuccato E, Pagani F, Romano M et al (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774–1784
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Cruts M, Gijselinck I, van der ZJ, Engelborghs S, Wils H et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A et al (1993) Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261:1047–1051
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500
Forman MS, Trojanowski JQ, Lee VM (2007) TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol 17:548–555
Freeman SH, Spires-Jones T, Hyman BT, Growdon JH, Frosch MP (2008) TAR-DNA binding protein 43 in Pick disease. J Neuropathol Exp Neurol 67:62–67
Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX et al (2008) Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115:133–145
Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin JJ et al (2007) Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging
Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB et al (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Harvey RJ, Skelton-Robinson M, Rossor MN (2003) The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry 74:1206–1209
Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H et al (2007) TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 130:1386–1394
Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H et al (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184:284–294
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S et al (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
Johnson BS, McCaffery JM, Lindquist S, Gitler AD (2008) A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 105:6439–6444
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kato S, Hayashi H, Yagishita A (1993) Involvement of the frontotemporal lobe and limbic system in amyotrophic lateral sclerosis: as assessed by serial computed tomography and magnetic resonance imaging. J Neurol Sci 116:52–58
Kwong LK, Uryu K, Trojanowski JQ, Lee VM (2008) TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals 16:41-51
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Mackenzie IR, Rademakers R (2007) The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8:237–248
Morita M, Al Chalabi A, Andersen PM, Hosler B, Sapp P et al (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66:839–844
Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H et al (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114:221–229
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA et al (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183
Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723
Pickering-Brown SM, Rollinson S, Du PD, Morrison KE, Varma A et al (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131:721–731
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM et al (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825
Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621
Robertson J, Sanelli T, Xiao S, Yang W, Horne P et al (2007) Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420:128–132
Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DM et al (2007) TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett 419:1–4
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Schumacher A, Friedrich P, Diehl-Schmid J, Ibach B, Perneczky R et al (2007) No association of TDP-43 with sporadic frontotemporal dementia. Neurobiol Aging
Schymick JC, Talbot K, Traynor BJ (2007) Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 16 Spec No. 2:R233–R242
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL et al (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808
Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P et al (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A et al (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95:7737–7741
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T et al (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542
Valdmanis PN, Dupre N, Bouchard JP, Camu W, Salachas F et al (2007) Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol 64:240–245
Valdmanis PN, Rouleau GA (2008) Genetics of familial amyotrophic lateral sclerosis. Neurology 70:144–152
Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7:409–416
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ et al (2008) Disturbance of nuclear and cytoplasmic Tar DNA binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J Biol Chem 283:13302-13309
Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H et al (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63:538–542
Acknowledgments
For brevity we have cited recent review articles rather than original literature. Also, we have used the most frequently used abbreviation for the TDP-43 protein, which does not conform to human or mouse gene nomenclature rules. We thank Simon Mead and James Stevens for comments and Ray Young for graphics. These authors are supported by the Wellcome Trust, the ENDOCYTE Research and Training Network funded by the European Union, and the UK Medical Research Council.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Banks, G.T., Kuta, A., Isaacs, A.M. et al. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome 19, 299–305 (2008). https://doi.org/10.1007/s00335-008-9117-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-008-9117-x