
Abstract
Cleome spinosa is widely used as a garden ornamental in many countries. Here we determined the optimal conditions for plant regeneration from different tissue explants grown in vitro. Induction medium containing MS salts, MS vitamins, 3% sucrose, 1 mg l−1 BA, 200 mg l−1 timentin, and 0.8% agar was sufficient for shoot regeneration of all the tissue explants examined, including leaf, hypocotyl, and cotyledon. Subsequently, an Agrobacterium tumefaciens-mediated method was developed to transform the vector pCHS, which carries the transgenes Petunia chalcone synthase (chs) and selection marker neomycin phosphotransferase II (nptII), into C. spinosa. From a total of 368 cotyledon explants, 13 putative transgenic lines were regenerated from selection medium supplemented with 50 mg l−1 kanamycin and 200 mg l−1 timentin, and transferred to the greenhouse. Genomic PCR and Southern blot analyses revealed that the nptII transgene was present in all 13 transgenic plants. Similarly, when the Petunia chs transgene was used as a probe in Southern blot analysis, single or multiple hybridization bands were detected in 12 out of the 13 transgenic plants. In addition, T1 progeny assay from selected transformants showed that the nptII transgene can be transmitted in a Mendelian manner from transgenic parents into their progeny. This is the first report of stable transformation of the C3 dicotyledon C. spinosa, which will facilitate functional comparison of cell-type specific genes with counterpart C4 dicotyledon C. gynandra using transgenic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Cleome, belonging to the family Cleomaceae, includes more than 200 species that are distributed in tropical and subtropical areas worldwide (Voznesenskaya et al. 2007). Many species in this genus are used to treat inflammation and rheumatism; and antioxidant, antimicrobial, and analgesic activities have been reported (Albarello et al. 2006).
Cleome spinosa is a tough-stemmed shrub, sometimes known as “spider flower”, which is widely used as a garden ornamental plant (Albarello et al. 2006). Previous chemical studies have reported the isolation of glucosinolates, which are known as flavor compounds, cancer prevention agents, and biopesticides, from seeds of C. spinosa (Ahmed et al. 1972). Moreover, a rare flavone flindulatin and five cembranes were isolated from aerial portions of C. spinosa; among them, cembranes are known to exhibit anti-HIV (human immunodeficiency virus) activities, neuroprotection, and cytotoxicity against several human cancer cell lines (Collins et al. 2004). In addition, five essential oil extracts, which displayed antimicrobial activities, were isolated from the aerial parts of C. spinosa (McNeil et al. 2010). As yet, no therapeutic uses for the compounds isolated from C. spinosa have been reported.
Besides the ornamental value and chemical properties, the dicotyledonous genus Cleome is also of interest in photosynthesis research as both C3- and C4-type photosynthesis can be found in some species within this genus. C4 photosynthesis is partitioned between mesophyll and bundle sheath cells of the leaves in most C4 plants (such as maize and sugar cane) and corresponds to characteristic leaf anatomy, such as the presence of Kranz anatomy and enzyme activities; this contrasts with photosynthesis of C3 plants (such as rice and Arabidopsis), which occurs in leaf mesophyll cells. The model plant Arabidopsis thaliana is a C3 species from the family Brassicaceae. Based on recent phylogenetic studies, Cleomaceae and Brassicaceae are very closely related (Voznesenskaya et al. 2007). To look for a species with C4-type photosynthesis in the family Cleomaceae, more than 230 samples of Cleomaceae species were screened, and several species of Cleome were selected for further extensive studies: C. gynandra, C. angustifilia, and C. oxalidea were identified as having C4-type photosynthesis; C. paradoxa was identified as a C3–C4 intermediate type; and six species including C. Africana, C. gigantean, C. hassleriana, C. monophylla, C. ornithopodioides, and C. spinosa were identified as C3-type photosynthesis (Voznesenskaya et al. 2007). Thus, Cleome is considered as a useful genus for the study of the genetic basis and regulation of C4 photosynthesis and the evolution from C3 species. Among the six Cleome species carrying C3-type photosynthesis, only C. spinosa has market value as a floricultural plant. To facilitate the functional analysis of C4-type photosynthesis, an Agrobacterium-mediated transformation protocol was recently developed in the C4 species C. gynandra (Newell et al. 2010). More recently, comparative transcriptomics of two closely related C3-type C. spinosa and C4-type C. gynandra have been carried out to examine the differences in gene expression between C3 and C4 species in the genus Cleome (Bräutigam et al. 2011a, b). However, to date, no transformation protocol has been reported in C. spinosa.
As a first step in functional analysis and metabolic engineering in C. spinosa, here we report the set-up conditions for plant regeneration from in vitro plantlets. Subsequently, Agrobacterium-mediated transformation was carried out using the transformation vector pCHS which carries chimeric expression cassettes for chalcone synthase (chs) from Petunia and the selection marker gene neomycin phosphotransferase II (nptII). This paper is the first report of stable transformation in C. spinosa and corresponding analysis of the transgenic plants.
Materials and methods
Plant material and culture conditions
Seeds of Cleome spinosa var. Queen were purchased from Known-You Seed Company, Taiwan, and were grown a greenhouse until maturity. Freshly collected seeds from white flowering plants were sterilized by sequential treatment with 70% ethanol for 0.5 min and 1% sodium hypochloride for 10 min, and were then washed thoroughly with sterile water. Sterilized seeds were placed onto a sterile Petri dish containing moistened filter papers and 200 mg l−1 timentin, stored in a 4°C refrigerator for 2 days, and incubated under a photo cycle of 8-h illumination (100 μmol m−2 s−1) at 25°C and 16-h darkness at 4°C for 2–3 weeks. Seeds producing roots were transferred onto another sterile Petri dish with moistened filter papers and 200 mg l−1 timentin, and placed in a refrigerator at 4°C until sufficient germinating seeds were collected. To obtain synchronous growth, the germinating seeds were then transferred to basal medium, adjusted to a pH value of 5.7, containing MS salts (Murashige and Skoog 1962), 3% sucrose, 200 mg l−1 timentin, and 0.8% Bacto-agar. The cultures were then incubated in a growth chamber at 22°C under a photoperiod of 16-h illumination (100 μmol m−2 s−1) and 8-h darkness. In vitro explants were used to establish the plant regeneration and transformation systems.
For shoot induction, tissue explants were cut into small segments and inoculated on induction medium containing MS salts and MS vitamins, 3% sucrose, and 0.8% Bacto-agar supplemented with 200 mg l−1 timentin (Duchefa, The Netherlands) and various concentrations of 6-benzyladenine (BA). For selection in transformation experiments, kanamycin at 50 mg l−1 was added to the above induction medium. Timentin is an alternative antibiotic for suppression of Agrobacterium tumefaciens in genetic transformation (Cheng et al. 1998), and kanamycin was used for selection in plant tissue culture (Padilla and Burgos 2010).
Agrobacterium strain, binary vector, and transformation
The transformation vector pCHS carrying the expression cassettes for the selection marker gene neomycin phosphotransferase (nptII; 795 bp) under the control of a nopaline synthase (nos) promoter and nos terminator, and for Petunia chalcone synthase cDNA (chs; 1,170 bp) under the control of cauliflower mosaic virus (CaMV) 35S promoter and nos terminator was constructed as previously described (Wang and To 2004). The vector was transformed into the Agrobacterium tumefaciens strain LBA4404 by electroporation (Gene Pulser II, Bio-Rad). A single colony from a YEP plate (1% peptone; 1% yeast extract; 0.5% NaCl; 1.5% Bacto-agar) was picked up and inoculated overnight in 4 ml YEP liquid medium supplemented with 50 μg ml−1 kanamycin at 28°C with shaking at 200 rpm. 35 μl bacterial culture was transferred into 35 ml YEP/kanamycin liquid medium and further incubated overnight at 28°C with shaking until the OD600 reached a value of 1.0. Acetosyringone (AS; Fluka Chemika, Switzerland) and glucose were added to the YEP/kanamycin medium to yield a final concentration of 100 μM and 5%, respectively, and the bacterial culture was further incubated for 2 h at 28°C with shaking to an OD600 of 0.8–1.0. The culture was then centrifuged at 4,000 rpm for 20 min, and the pellet was dissolved in 40 ml simplified induction medium (SIM) (2% glucose; 200 μM AS; 20 mM sodium citrate; pH 5.5). The suspension was then diluted such that the final OD600 reading was 0.8–1.0.
Before infection, in vitro explants were cut and incubated in pre-culture medium (MS salts; MS vitamins; 2% sucrose; 0.1 mg l−1 α-naphthalene acetic acid (NAA); 1.0 mg l−1 BA; 0.8% Bacto-agar; pH 5.8) for 2 days in a 25°C growth chamber with a photoperiod of 16-h light (60 μmol m−2 s−1) and 8-h dark cycle. For infection, 20 ml diluted Agrobacterium suspension was added onto each plate, and then immersed for 1 h at room temperature. Excess Agrobacterium was blotted from the explants on filter paper, and then the infected explants were transferred onto co-culture solid medium (MS salts; MS vitamins; 0.1 mg l−1 NAA; 1.0 mg l−1 BA; 200 μM AS; 5% glucose; 0.8% Bacto-agar; pH 5.5) and left to grow for 2 days in a dark growth chamber at 25°C. Afterwards, explants were washed three times by shaking in a washing solution (MS salts; MS vitamins; 2% sucrose; 200 mg l−1 timentin; pH 5.8), transferred to the selection medium containing MS salts and vitamins, 3% sucrose, 1 mg l−1 BA, pH 5.8 and 0.8% agar supplemented with 200 mg l−1 timentin to inhibit Agrobacterium growth and 50 mg l−1 kanamycin for selection, and incubated at 25°C in a growth chamber with a photoperiod of 16-h light (60 μmol m−2 s−1) and 8-h dark cycle. Approximately 2–3 weeks later surviving calli were observed at the edges of tissue explants, and within a period of 1.5 months all shoot buds were excised and transferred into rooting medium (MS salts; 3% sucrose; 200 mg l−1 timentin; 0.8% Bacto-agar; pH 5.8). The culture was incubated in a 25°C growth chamber with a 16-h photoperiod. The rooted plantlets were then transferred into pots and grown in a greenhouse.
Transgenic plant verification by PCR analysis
Transgenic plant verification was performed with putative transformants and wild-type plants by PCR analysis. Total genomic DNA was extracted from green leaves of putative transformants and wild-type plants using the CTAB method (Wilkie 1997). PCR was carried out with the following primer sets: Kan-F (5′-ATGATTGAACAAGATGGA-3′) and Kan-R (5′-TCAGAAGAACTCGTCAAG-3′) for amplification of the 795-bp-long DNA fragment corresponding to the full-length nptII gene (Chen et al. 2003), CHS-F1 (5′-ATGGTGACAGTCGAGGAG-3′) and CHS-R1 (5′-TTAAGTAGCAACACTGTG-3′) for amplification of the 1,170-bp-long DNA fragment corresponding to the full-length Petunia chs cDNA (Wang and To 2004; GenBank accession number AF233638). PCR reactions were performed in a volume of 10 μl under the following conditions: (a) denaturation for 5 min at 94°C; (b) 30 cycles of 0.5 min at 94°C, 0.5 min at 55°C, and 1 min at 72°C, and (c) extension for 15 min at 72°C. Following amplification, PCR products were analyzed on a 1% agarose gel.
Southern blot analysis
For Southern blot analysis, 15 μg of plant DNA was used for overnight digestion at 37°C with restriction enzyme HindIII. After electrophoresis on 0.8% agarose gel in TAE buffer, the gel was washed once by shaking in 0.25 M HCl, followed by twice in denature solution (1.5 M NaCl; 0.5 M NaOH) and twice in transfer solution (1 M CH3COONH4; 0.02 M NaOH), and then the digested DNA fragments were transferred onto Hybond-N+ nylon membrane (Amersham Pharmacia Biotechnol.) in transfer solution overnight. The membrane was washed with distilled water and cross-linked in an UV linker (UV Stratalinker 1800, Stratagene). To prepare the non-radioactive PCR DIG probe (Roche Applied Science, Germany), equal amounts (20 pmol) of the primers CHS-F1 and CHS-R1 were used to amplify the foreign gene Petunia chs (1,170 bp) from the expression vector pCHS (1 μg) and reaction mixture (5 μl of 10 × Taq buffer; 8 μl of 25 mM MgCl2; 1 μl each of 10 mM dATP, 10 mM dGTP, 10 mM dCTG; 0.95 μl of 10 mM dTTP; 0.05 μl of 1 mM DIG-11-dUTP; 0.2 μl of Taq DNA polymerase). The final volume of the mixture was adjusted to 50 μl. PCR mixtures were initially denatured at 94°C for 5 min and then subjected to 30 cycles (1 min at 94°C, 1 min at 55°C, 1 min at 72°C) with a final extension at 72°C for 10 min. For hybridization, the membrane was put onto a hybridization bag, incubated with 10 ml DIG Easy Hyb solution (Roche) at 65°C for 3 h with shaking, and gel-purified PCR product was added to the 10 ml DIG Easy Hyb solution to hybridize the membrane overnight at 65°C with shaking. The membrane was then washed twice in washing solution I (2× SSC; 0.1× SDS) at room temperature for 10 min and then washed twice in washing solution II (0.1× SSC; 0.1× SDS) at 65°C for 20 min. The membrane was incubated at room temperature for 5 min in 50 ml of washing buffer (0.1 M maleic acid, pH 7.5; 0.15 M NaCl); then the membrane was put into a bag, and incubated at room temperature for 30 min in 10 ml of blocking solution (0.1 M maleic acid, pH 7.5; 0.15 M NaCl; 1% blocking reagent). The blocking solution was then discarded, and the membrane was further incubated in 10 ml of anti-dig-AP solution (anti-dig-AP was diluted to 10,000-fold with blocking solution) at room temperature for 30 min. After washing twice with 100 ml washing buffer containing 0.3% Tween-20 for 15 min and washing once with 20 ml detection buffer (0.1 M Tris, pH 9.5; 0.1 M NaCl) for 2 min, the membrane was placed into a bag and 1 ml of diluted CDP-star solution (diluted 4-fold with the detection buffer) was added. The membrane was exposed to X-ray film and then developed.
Seedling assay for kanamycin resistance
T1 seeds from self-pollinated transgenic plants were sterilized in 70% ethanol for 0.5 min followed by 1% sodium hypochloride for 10 min and washed thoroughly with sterile distilled water. Same treatments of sterilized seeds were described in “Plant material and culture conditions”. The germinating seeds were transferred on a medium containing MS salts, 3% sucrose, 0.8% Bacto-agar, 100 mg l−1 kanamycin sulfate, and 200 mg l−1 timentin. The cultures were incubated at 25°C under 16-h illumination for 2 weeks. Seedlings that displayed restricted growth, pale green color in aerial parts, inhibition of main root extension, and no lateral root development were considered to be kanamycin sensitive, while seedlings with healthy development of leaves and roots were considered to be kanamycin resistant.
Results
Establishment of in vitro regeneration and transformation protocol
Different explants including leaf, cotyledon, and hypocotyl were excised from plantlets grown in vitro, and shoot regeneration was examined under different induction media containing various concentrations of plant growth regulators and various vitamins (Table 1). BA is a kind of cytokinin that promotes shoot formation in plant tissue culture. At the early stage of the optimization of a plant regeneration system from Cleome spinosa, most experiments were carried out with leaf explants as the collection of leaf tissue was easier than other tissues. We found that MS medium supplemented with MS vitamins and different concentrations of BA (0–2 mg l−1; and especially 1 mg l−1) had a higher shoot regeneration rate than other vitamins under the same conditions (Table 1). Then, BA at 1 mg l−1 and a combination of NAA (a kind of auxin that promotes root formation) at 0–0.5 mg l−1 were added to the MS medium supplemented with MS vitamins to examine the shoot formation. We found that addition of NAA was not necessary (Table 1). Finally, leaf, cotyledon, and hypocotyl explants were transferred onto MS medium supplemented with MS vitamins and various concentrations of BA to examine the shoot formation. We found that BA at 1 mg l−1 or 2 mg l−1 significantly improved shoot regeneration (Table 1). Thus, we concluded that induction medium containing MS salts, MS vitamins, 3% sucrose, 1 mg l−1 BA, 200 mg l−1 timentin, and 0.8% agar was sufficient for shoot induction in all the tissue explants we examined. Typical shoot regeneration was observed from different explants, especially from hypocotyl explants (Fig. 1). Roots were easily induced when regenerated shoots were excised and transferred onto rooting medium (MS salts; 3% sucrose; 200 mg l−1 timentin; 0.8% agar; pH 5.8). Timentin is a mixture of ticarcillin and clavulanic acid and has been used for the suppression of Agrobacterium tumefaciens in Agrobacterium-mediated transformation (Cheng et al. 1998). Timentin at concentrations of 200–500 mg l−1 had little or no effect on shoot regeneration in a range of plant species and tissue explants under investigation (Cheng et al. 1998; Wang and To 2004; Slater et al. 2011). In this study, we added 200 mg l−1 timentin to media for seed germination, shoot induction and root regeneration, and found that it had no effect on plant regeneration from different explants of C. spinosa.

Shoot regeneration from different explants of in vitro grown plantlets from Cleome spinosa in induction medium (MS salts; MS vitamins; 3% sucrose; 1 mg l−1 BA; 200 mg l−1 timentin; 0.8% agar; pH 5.8) for 3 or 7 weeks. Three replicates were carried out in this experiment
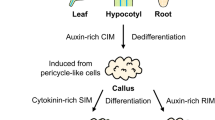
Next, hypocotyl and cotyledon explants were chosen for plant transformation experiments. Agrobacterium infection, co-culture, shoot induction, and plant regeneration were performed as described in “Materials and methods”. Selection was conducted in medium containing MS salts, MS vitamins, 3% sucrose, 1 mg l−1 BA, 200 mg l−1 timentin, 50 mg l−1 kanamycin, 0.8% agar, pH 5.8. A total of 368 cotyledon explants were employed, and 13 putative transgenic lines were obtained and grown in a greenhouse. Schematic representation of in vitro regeneration and Agrobacterium-mediated transformation of C. spinosa from tissue explants is shown in Fig. 2. When 265 hypocotyl explants were used in Agrobacterium-mediated transformation experiment, no transformants were obtained.

Agrobacterium-mediated transformation in the floricultural plant Cleome spinosa. a Tissue explants were excised from in vitro grown plantlets, infected with Agrobacterium tumefaciens, and incubated on selection plates containing induction medium supplemented with 50 mg l−1 kanamycin and 200 mg l−1 timentin. The arrow shows a regenerated shoot on the edge of cotyledon explant, b regenerated shoots were excised and transferred onto rooting medium supplemented with 50 mg l−1 kanamycin and 200 mg l−1 timentin. Roots were observed after incubation for 1 week, c healthy plantlet grown in vitro, d the rooted plantlet was transferred to potting soil, e transgenic plant. A mature flower at the later flowering stage is attached
Confirmation of transgenic plants
To verify the transformants obtained, genomic DNA from leaves of plantlets was isolated and PCR analysis was carried out using gene-specific primers for nptII and Petunia chs sequences. All the 13 putative transformants carried the nptII selection marker gene, as revealed by the presence of a unique amplicon of 0.8 bp (Fig. 3a). The transformation efficiency was, thus, 3.5%. Interestingly, only 11 out of 13 transgenic lines carried the transgene Petunia chs, as revealed by the presence of a unique amplicon of 1.2 kb (Fig. 3b). No PCR product was detected in transgenic lines 1 and 13, or in the wild type (Fig. 3b).

Confirmation of transgenes in transgenic plants of Cleome spinosa by genomic PCR analysis. Genomic DNA was isolated from wild-type (WT) and 13 putative transgenic lines, and 500 ng of each DNA sample was used for PCR analysis using nptII-specific primers (a) or chs-specific primers (b). Lanes M in each panel contain a DNA size marker
Furthermore, genomic DNA from the wild-type and 13 transgenic plants was digested with HindIII and probed with the nptII or Petunia chs to estimate the copy number of the transgenes. For the transformation vector pCHS, the unique HindIII restriction site was found between expression cassettes of nptII and Petunia chs in the T-DNA region (Wang and To 2004). No hybridization band was detected in the wild-type sample in either Southern blot (Fig. 4). The hybridization bands in all samples were different, indicating that all of the 13 transgenic plants obtained in this study were independent integration events. One to three copies of the nptII selection marker gene were integrated into the genome of each transgenic plant (Fig. 4a). For the Southern blot probed with Petunia chs cDNA, no hybridization band was detected in transgenic plant #1 (Fig. 4b). This is consistent with the result obtained for this line when chs-specific primers were employed in PCR analysis (Fig. 3b). Two hybridization bands were detected in transgenic plant #13; however, no PCR product was detected when PCR analysis was conducted in this line (Fig. 3b). Single or multiple copies of transgene Petunia chs were integrated into the genomes of the other transgenic plants (Fig. 4b).

Southern blot analysis of all transgenic plants. For each sample, 15 μg of genomic DNA was digested with HindIII and probed with DIG-labeled PCR product of nptII (a) or Petunia chs (b)
Inheritance of the nptII selection marker gene in T1 progeny assay
To demonstrate the stability and inheritance of the transgenes, three transgenic plants (#1, #2, #7), each harboring single copy of nptII (Fig. 4a), were selected for T1 progeny assay. Seeds collected from selfed T0 plants as well as from the wild type were screened for kanamycin resistance. As shown in Fig. 5, the growth of wild-type seedlings was inhibited, the main roots were short, and no lateral roots developed in the medium containing 100 mg l−1 kanamycin. Instead of the lower dosage (i.e., 50 mg l−1 kanamycin) at the selection stage from tissue explants, a higher concentration of kanamycin (i.e., 100 mg l−1) needed to be added to suppress the growth and development of wild-type seedlings in C. spinosa. Both resistant seedlings (that grew healthily with leaves and roots normally developed, and a normal height) and sensitive seedlings were found in the three transgenic plants we examined (Fig. 5). Among them, single insertion of the nptII transgene was determined in transgenic plants #1 and #7, since the segregation ratio of resistant and sensitive seedlings in these transgenic plants was 3:1 (Table 2), which fits the Mendelian rule of progeny segregation for single insertion into the nuclear chromosome of the plant genome. Unexpectedly, kanamycin-resistant and kanamycin-sensitive seedlings segregated in a 15:1 ratio in transgenic plant #2, suggesting the presence of two copies of the nptII transgene in the nuclear genome of this transformant (Table 2). In summary, the three transgenic plants we examined were considered to be kanamycin resistant, and the nptII transgene can be stably transmitted into their progeny in a classical Mendelian manner.

Typical growth pattern in T1 seedlings from self-crossed T0 transgenic plants and wild type (WT) in MS basal medium supplemented with 100 mg l−1 kanamycin and 200 mg l−1 timentin for 14 days. Only kanamycin-sensitive seedlings were observed in WT, whilst both kanamycin-resistant and kanamycin-sensitive seedlings were found in each transgenic plant we examined
Discussion
In order to obtain in vitro plantlets of C. spinosa for plant regeneration and transformation, at the beginning of this study, seeds were sterilized with standard protocol using 70% ethanol followed by 1% sodium hypochloride, and then cultured in MS basal medium for seed germination. Unexpectedly, no germination or a very low germination rate (<5%) with non-uniform growth was observed. Poor and delayed seed germination due to dormancy has been previously reported as one of the major problems in the propagation of Cleome species (Ekpong 2009; Raboteaux and Anderson 2010). To overcome the problem of dormancy, several treatments including chilling, heating, soaking, washing, adding gibberellic acid GA3 (Ekpong 2009), stratification, changing photoperiod, temperature, and storage time (Raboteaux and Anderson 2010) have been attempted in Cleome seeds. Seeds of C. gynandra from fresh intact pods were sown directly (without standard sterilization) onto sterile Petri dishes with wet filter papers, resulting in high rates (80–90%) of seed germination (Newell et al. 2010). In this study, fresh collection of seeds from C. spinosa, standard sterilization, chilling, and photoperiod, as described in “Materials and methods“, were crucial to achieving high rates of seed germination (more than 90% were regularly obtained) and low levels of contamination.
Previously, in vitro propagation of C. spinosa using different explants has been reported, and it has been found that the use of BA alone at 4.4 μM (equivalent to 1 mg l−1) could induce maximal shoot production (10.1 ± 2.3 shoots) through direct organogenesis from hypocotyl explants (Albarello et al. 2006). These findings are consistent with our study that 1 mg l−1 BA is sufficient to induce shoot formation from different explants of C. spinosa, and multiple shoots per hypocotyl explant were observed (Fig. 1). Although hypocotyl explants had a better regeneration potential than other tissues (Table 1; Fig. 1), no transgenic lines were produced in Agrobacterium-mediated transformation experiments. We noticed that several tiny regenerated shoots from hypocotyl explants could also be induced in selection medium; however, these tiny shoots ceased growing, did not turn green, and finally died.
The transformation vector pCHS contains the expression cassettes of Petunia chs cDNA and kanamycin resistance gene nptII in the T-DNA region (Wang and To 2004). Thus, the copy number of these two transgenes should be the same in each transgenic plant. To confirm the transgenic plants we obtained, and to validate the integrity of the T-DNA in each transgenic plant, gene-specific primers for nptII and Petunia chs were used for genomic PCR and Southern blot analyses (Figs. 3, 4). Presence of a single copy of nptII but absence of Petunia chs was detected in transgenic plant #1, suggesting incomplete integration of T-DNA into the plant chromosome during the transformation process. Loss of one of the two transgenes within the same T-DNA has been clearly demonstrated in transgenic wheat (Wu et al. 2006) and rice (Kim et al. 2003). In addition, no PCR product was obtained using chs-specific primers in transgenic plant #13; however, two hybridization bands for each transgene were detected by Southern blot analysis in this plant. This may be because the partial 5′- or 3′-end sequence of Petunia chs was deleted, but the DNA hybridization bands could still be detected since the truncated T-DNA in plant #13 may contain most of the sequence of Petunia chs. Truncated T-DNA inserts, which may be caused by partial T-DNA transfer or recombination, have been reported elsewhere (Yin and Wang 2000; Wu et al. 2006). Single T-DNA insertion, with 1 copy of nptII and Petunia chs transgenes, was found in transgenic plants #2, #7, and #11. Two or more T-DNA inserts, with the same or different copy numbers of both transgenes, were also detected. Unequal copy numbers of the transgenes in the same T-DNA can be caused by deletion, recombination, rearrangement, direct or inverted repeats (Yin and Wang 2000; Kim et al. 2003; Wu et al. 2006; Rai et al. 2007). Transmission of the nptII selection marker gene from selected transgenic plants into its progeny was clearly demonstrated (Table 2).
As mentioned earlier, the transgene Petunia chs cDNA in the pCHS vector encodes chalcone synthase. Chalcone synthase catalyzes condensation of one molecule of p-coumaroyl-CoA and three molecules of malonyl-CoA, resulting in one molecule of 4′,2′,4′,6′-tetrahydroxychalcone (chalcone or naringenin chalcone), which is a key intermediate in the formation of flavonoids. Flavonoids (mainly anthocyanins) are the most common flower pigments contributing to a range of colors from yellow to orange to purple. By using the transgenic approach of overexpressing of sense or anti-sense chs constructs, floral color can be partially or evenly reduced, or completely altered into white color in a range of plant species (To and Wang 2006). Previously, we obtained seven transgenic tobacco plants using the Agrobacterium-mediated method and the same pCHS vector, four of the transgenic plants produced white flowers, and three produced pink flowers similar to the wild-type plants (Wang et al. 2006). In this study, no petal coloration was altered in any of the 13 transgenic plants we obtained, because the petal color in wild-type C. spinosa was white. Besides providing colorful pigments in plant tissues, expression of chalcone synthase is also involved in plant resistance such as UV irradiation, bacterial, and fungal infection (Forkmann and Martens 2001; Winkel-Shirley 2001; Dao et al. 2011). Further experiments examining the relationship between the expression of chalcone synthase and the plant resistance system in selected transgenic plants we obtained will be useful in molecular understanding of defense mechanisms and application in horticultural biotechnology.
In this study, we have established Agrobacterium tumefaciens-mediated transformation and regeneration of transgenic plants of C. spinosa. To the best of our knowledge, this is the first report regarding stable transformation in C. spinosa, a C3 plant in the genus Cleome. Since transformation of C. gynandra (a C4 plant in the genus Cleome) has been reported recently (Newell et al. 2010), our protocol will facilitate the functional comparison between C3 plant C. spinosa and C4 plant C. gynandra using transgenic approaches.
References
Ahmed ZF, Rizk AM, Hammouda FM, Seif El-Nasr MM (1972) Naturally occurring glucosinolates with special reference to those family Capparidaceae. Planta Med 21:35–60. doi:10.1055/s-0028-1099522
Albarello N, Simões C, Rosas PFG, de Castro TC, Gianfaldoni MG, Callado CH, Mansur E (2006) In vitro propagation of Cleome spinosa (Capparaceae) using explants from nursery-grown seedlings and axenic plants. In Vitro Cell Dev Biol-Plant 42:601–606. doi:10.1079/IVP2006828
Bräutigam A, Kajala K, Wullenweber J, Sommer M, Gagneul D, Weber KL, Carr KM, Gowik U, Maβ J, Lercher MJ, Westhoff P, Hibberb JM, Weber APM (2011a) An mRNA blueprint for C4 photosynthesis derived from comparative transcriptomics of closely related C3 and C4 species. Plant Physiol 155:142–156. doi:10.1104/pp.110.159442
Bräutigam A, Mullick T, Schliesky S, Weber APM (2011b) Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C3 and C4 species. J Exp Bot 62:3093–3102. doi:10.1093/jxb/err029
Chen PY, Wang CK, Soong SC, To KY (2003) Complete sequence of the binary vector pBI121 and its application in cloning T-DNA insertion from transgenic plants. Mol Breed 11:287–293. doi:10.1023/A:1023475710642
Cheng ZM, Schnurr JA, Kapaun JA (1998) Timentin as an alternative antibiotic for suppression of Agrobacterium tumefaciens in genetic transformation. Plant Cell Rep 17:646–649. doi:10.1007/s002990050458
Collins DO, Reynolds WF, Reese PB (2004) New cembranes from Cleome spinosa. J Nat Prod 67:179–183. doi:10.1021/np0303299
Dao TTH, Linthorst HJM, Verpoorte R (2011) Chalcone synthase and its functions in plant resistance. Phytochem Rev 10:397–412. doi:10.1007/s11101-011-9211-7
Ekpong B (2009) Effects of seed maturity, seed storage and pre-germination treatments on seed germination of cleome (Cleome gynandra L.). Scientia Hort 119:236–240. doi:10.1016/j.scienta.2008.08.003
Forkmann G, Martens S (2001) Metabolic engineering and applications of flavonoids. Curr Opin Biotechnol 12:155–160. doi:10.1016/S0958-1669(00)00192-0
Kim SR, Lee J, Jun SH, Park S, Kang HG, Kwon S, An G (2003) Transgene structures in T-DNA-inserted rice plants. Plant Mol Biol 52:761–773. doi:10.1023/A:1025093101021
McNeil MJ, Porter RBR, Williams LAD, Rainford L (2010) Chemical composition and antimicrobial activity of the essential oils from Cleome spinosa. Nat Prod Commun 5:1301–1306
Murashige T, Skoog F (1962) A revised medium for the rapid growth and bioassays with tobacco cultures. Physiol Plant 15:473–497. doi:10.1111/j.1399-3054.1962.tb08052.x
Newell CA, Brown NJ, Liu Z, Pflug A, Gowik U, Westhoff P, Hibberd JM (2010) Agrobacterium tumefaciens-mediated transformation of Cleome gynandra L., a C4 dicotyledon that is closely related to Arabidopsis thaliana. J Exp Bot 61:1311–1319. doi:10.1093/jxb/erg009
Padilla IMG, Burgos L (2010) Aminoglycoside antibiotics: structure, functions and effects on in vitro plant culture and genetic transformation protocols. Plant Cell Rep 29:1203–1213. doi:10.1007/s00299-010-0900-2
Raboteaux NNG, Anderson NO (2010) Germination of Cleome hassleriana and Polanisia dodecandra seed lots in response to light, temperature and stratification. Res J Seed Sci 3:1–17. doi:10.3923/rjss.2010.1.17
Rai M, Datta K, Parkhi V, Tan J, Oliva N, Chawla HS, Datta SK (2007) Variable T-DNA linkage configuration affects inheritance of carotenogenic transgenes and carotenoid accumulation in transgenic indica rice. Plant Cell Rep 26:1221–1231. doi:10.1007/s00299-007-0333-8
Slater SMH, Keller WA, Scoles G (2011) Agrobacterium-mediated transformation in Eruca sativa. Plant Cell Tiss Organ Cult 106:253–260. doi:10.1007/s11240-010-9915-1
To KY, Wang CK (2006) Molecular breeding of flower color. In: Teixeira da Silva JA (ed) Floriculture, ornamental and plant biotechnology, vol. 1. Global Science Books, UK, pp 300–310
Voznesenskaya EV, Koteyeva NK, Chuong SDX, Ivanova AN, Barroca J, Craven LA, Edwards GE (2007) Physiological, anatomical and biochemical characterisation of photosynthetic types in genus Cleome (Cleomaceae). Functional Plant Biol 34:247–267. doi:10.1071/FP06287
Wang HM, To KY (2004) Agrobacterium-mediated transformation in the high-value medicinal plant Echinacea purpurea. Plant Sci 166:1087–1096. doi:10.1016/j.plantsci.2003.12.035
Wang CK, Chen PY, Wang HM, To KY (2006) Cosuppression of tobacco chalcone synthase using Petunia chalcone synthase construct results in white flowers. Bot Stud 47:71–82
Wilkie S (1997) Isolation of total genomic DNA. In: Clark MS (ed) Plant molecular biology—a laboratory manual. Springer, Berlin, pp 3–15
Winkel-Shirley B (2001) Flavonoid biosynthesis: a colorful model for genetic, biochemistry, cell biology, and biotechnology. Plant Physiol 126:485–493. doi:10.1104/pp.126.2.485
Wu H, Sparks CA, Jones HD (2006) Characterisation of T-DNA loci and vector backbone sequences in transgenic wheat produced by Agrobactium-mediated transformation. Mol Breed 18:195–208. doi:10.1007/s11032-006-9027-0
Yin Z, Wang GL (2000) Evidence of multiple complex patterns of T-DNA integration into the rice genome. Theor Appl Genet 100:461–470. doi:10.1007/s001220050060
Acknowledgments
The authors thank Miranda Loney for critical reading and editing of the manuscript, Director Wen-Hsiung Li (Biodiversity Research Center, Academia Sinica) for initiating and supporting the research project, Director Ming-Che Shih (Agricultural Biotechnology Research Center, Academia Sinica), Professor Maurice S. B. Ku (Department of BioAgricultural Science, National Chaiyi University, Taiwan) and colleagues in this research project for valuable suggestions. We also thank Show-Jane Sun (Genomics Research Center, Academia Sinica) for providing and maintaining the greenhouse facility. This work was financially supported by Academia Sinica, Republic of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Kumar.
Rights and permissions
About this article
Cite this article
Tsai, YT., Chen, PY. & To, KY. Plant regeneration and stable transformation in the floricultural plant Cleome spinosa, a C3 plant closely related to the C4 plant C. gynandra . Plant Cell Rep 31, 1189–1198 (2012). https://doi.org/10.1007/s00299-012-1240-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-012-1240-1