
Abstract
B cells are central for the adaptive immune system to mount successful immune responses not only as antibody producers but also as regulators of cellular immunity. These multifaceted features are also reflected in autoimmunity where autoreactive B cells can fuel disease by production of cytotoxic autoantibodies, presentation of autoantigens to autoreactive T cells, and secretion of cytokines and chemokines that either promote detrimental immune activation or impair regulatory T and B cells. The role of B cells and autoantibodies in autoimmune hepatitis (AIH) have been controversially discussed, with typical autoantibodies and hypergammaglobulinemia indicating a key role, while strong HLA class II association suggests T cells as key players. In this review, we summarize current knowledge on B cells in AIH and how different B cell subpopulations may drive AIH progression beyond autoantibodies. We also discuss recent findings of B cell-directed therapies in AIH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autoimmune hepatitis (AIH) is a severe chronic and relapsing inflammatory liver disease with a female preponderance characterized by an ongoing autoimmune reaction directed against hepatic autoantigens [1,2,3]. Like in other autoimmune diseases, the exact pathogenesis remains uncertain. Both B cell and T cell-mediated autoimmunity and immune dysregulation have been proposed as key mechanisms. While the strong association with distinct HLA-DRB1 alleles strongly supports an important role for CD4+ cells in disease development, the characteristic and specific elevation of immunoglobulin G (IgG) levels and the development of both specific and non-specific autoantibodies, which is also of important diagnostic value, support the role of B cells in AIH pathogenesis [1, 3,4,5]. Furthermore, recently, the establishment of new mouse models and the apparent success of B cell depletion therapies in distinct patient subsets provided further support for the concept of B cells as substantial contributors to AIH immunopathogenesis.
Therefore, in this review, we summarize current knowledge on B cells in AIH and how different B cell subpopulations may drive AIH progression beyond autoantibodies. We also discuss recent findings of B cell depletion in clinical trials as well as further B cell-directed therapeutic approaches beyond depletion.
B cell development and maturation
B cells constitute one of the essential arms of the adaptive immune system [6]. They are produced from hematopoietic precursor cells throughout life starting in the fetal liver to the bone marrow (BM) in adults (Fig. 1) [7]. During a multistep developmental and selection process, their unique characterizing feature, the B cell receptor (BCR), is generated randomly for each single B cell in a complex genomic rearrangement event generating a diverse B cell repertoire with virtually unlimited specificities [8]. B cells with correctly assembled BCRs that pass checkpoints of central tolerance exit the BM as IgM+ immature or transitional B cells and migrate via the bloodstream to the spleen where they complete their maturation process by differentiating into either naïve, follicular, or marginal zone (MZ) B cells after passing peripheral tolerance checkpoints [9, 10]. Based on phenotypic and topographic features, these mature B cells can now circulate in the blood and lymph vessels or populate the secondary lymphoid organs (spleen, lymph nodes, tonsils, and Peyer’s patches) ready for detection of antigens.

B cell development and differentiation. B cell development is a multistep process initiated in hematopoietic stem cells (HSCs) in the bone marrow or fetal liver. During this process, the B cell receptor (BCR), which consists of a heavy and a light chain, is generated in a complex genomic rearrangement event. This rearrangement, termed V(D)J recombination, randomly assembles one of 40 V, 23 D, and 6 J genes with a constant part (CH) in case of the heavy chains. The light chains lack D chains and can have either a λ or κ constant region. Immature B cells with correctly assembled BCRs finalize their maturation after migration to the spleen, where they either differentiate into naïve, follicular, or marginal zone (MZ) B cells. All stages of B cell development are characterized by sets of surface markers from which a selection is depicted. Upon antigen encounter, activated B cells can either rapidly expand in an extrafollicular response into short-lived plasmablast or engage in a germinal center reaction in secondary lymphoid tissues like lymph nodes. The germinal center reaction is a T cell-assisted BCR diversification process, which facilitates class-switch recombination (CSR) and increases BCR affinity via introduction of random mutations (somatic hypermutation, SHM). Germinal center B cells then differentiate into antibody-secreting plasma cells which can become long-lived plasma (LLP) or memory B cells
Mature B cells which encounter cognate antigen and receive additional activation signals from co-stimulatory molecules expand and differentiate either into short-lived plasmablasts or into germinal center (GC) B cells [11]. While plasmablasts rapidly produce and secrete antibodies corresponding to germline-encoded BCR configurations (naïve BCRs), GC B cells engage in the GC reaction. The GC reaction is a T cell-assisted BCR diversification and selection process which increases affinity towards the initial antigen by introducing random mutations into the paratope sequence during clonal expansion [12, 13]. The GC reaction may also facilitate class-switch recombination to further diversify antibody effector functions. B cells that pass the GC reaction become antibody-secreting plasma cells which can differentiate into long-lived plasma (LLP) or memory B cells providing lasting protection against reinfection.
Pathogenic B cells in autoimmune diseases
While B cell antigenic selection and maturation is vital to protect against pathogens, it can have detrimental effects when triggered by self-antigens. This may result in autoreactive B cell populations that target an individual’s own tissues or instruct other cells of the adaptive and innate immune system to do so. B cell dyscrasias driven by autoantigen are well defined for some autoimmune diseases [14]. For example, in diabetes, autoantibodies can target insulin producing β-cells or insulin itself [15, 16], in dilated cardiomyopathy (DCM), which is the leading cause for heart failure and heart transplantation in younger adults, circulating autoantibodies mediate organ-specific tissue damage by targeting different epitopes on cardiac myocytes [17, 18]. In pemphigus vulgaris, IgG autoantibodies directed against desmosomes of keratinocytes cause epithelial acantholysis [19]. The pathogenic potential of several autoantibody classes found in rheumatoid arthritis (RA), especially antibodies targeting post-translational modifications like citrullination (ACPA) and carbamylation (anti-CarP antibodies), was recently substantiated [20, 21]. Another prototypic example for the pathogenic role of B cells is systemic lupus erythematosus (SLE), where ISG15-secreting plasmablast expansions are a hallmark of activity [22] and dysregulated GC reactions mediate the positive selection of high-affinity autoantibodies driving pathogenesis [23]. Notably, SLE also shows ongoing somatic hypermutation (SHM) in extrafollicular responses resulting in affinity matured autoantibodies, a feature also found in other autoimmune settings like RA and Sjögren’s syndrome [24]. While these examples illustrate the direct pathogenic potential of autoreactive B cells in different autoimmune diseases, their role in AIH pathogenesis is much less defined.
The role of B cells as diagnostic markers in AIH
Clinical presentation of AIH is highly variable, ranging from mild and intermittent elevation of liver enzymes to acute and fulminant hepatitis [25]. Since AIH lacks a distinct pathognomonic feature and its etiology is largely unknown, it is diagnosed by exclusion based on clinical, serological, and histopathological features [26,27,28,29]. Key to the diagnostic workup are AIH-specific autoantibodies; selective elevation of polyclonal IgG (hyper-IgG), usually in the absence of an elevation of IgA and IgM; and abnormalities in liver histology.
B cells in AIH liver histology
Liver histology in active AIH is typically characterized by a lympho-plasmacellular infiltrate of the hepatic tissue mediating tissue damage and hepatic necroinflammation. However, the changes in AIH histopathology lack pathognomonic characteristics, as histomorphological changes in chronic AIH mimic findings in chronic viral hepatitis [30,31,32]. Chronic AIH typically shows portal-based lympho-plasmacellular infiltrates with interface hepatitis (formerly called piece-meal-necrosis) [28, 29]. In the inflammatory infiltrate, plasma cells are detectable in two out of three cases; however, their abundance may vary [30, 33, 34]. Histopathological features such as rosette formation and emperipolesis were described to be typical characteristics of AIH. Recently, these histological findings were considered not to be specific for AIH, but rather reflect the hepatic tissue damage and can act as markers of disease severity regardless of the underling disease [30, 32, 35]. However, predominance of plasma cells as well as appearance in clusters (defined as > 5 plasma cells/one focus) in the context of interface hepatitis were reported to be rather specific for AIH [32]. Thus, despite not being necessarily required for diagnosis, plasma cells can be considered typical in AIH [32]. In immunohistochemistry, expression of CD38 or multiple myeloma-1 (MUM-1) can be used to identify plasma cells and determine their distribution and frequency in hepatic tissue [36].
Autoantibodies in AIH
Seropositivity for distinct autoantibody classes represents a key diagnostic feature of AIH. In addition, autoantibodies can serve as biomarkers for grouping different AIH subsets. After exclusion of other autoimmune diseases, ANA and SMA (including anti-F-actin antibody) define AIH type 1. Anti-LKM1 and anti-LC2 are characteristic for the AIH type 2 subset. Antibodies to soluble liver antigen/liver pancreas antigen (anti-SLA/LP) are also regarded by many authors as classifying a third subgroup of AIH, AIH type 3. Further autoantibodies such as pANCA, anti-LM, and anti-ASGPR antibodies have also been discussed as characteristic markers of AIH (Table 1).
Only very few patients lack any type of autoantibody. Autoantibody negativity may, however, occur at acute presentation, but most of these cases develop seropositivity upon further follow-up [37]. While anti-SLA/LP antibodies are highly specific for autoimmune hepatitis, ANA, SMA, and to a lesser extent anti-LKM1 can also be found in other liver diseases or even other autoimmune diseases [38]. Hepatotropic and non-hepatotropic viral infections can lead to a transient development and/or increase of these autoantibodies [38,39,40,41]. This limited degree of specificity thus calls into question a direct pathogenic role of autoantibodies in AIH. On the other hand, a high degree of specificity, as observed for anti-SLA/LP, including a high degree of epitope specificity, speaks strongly in favor of a pathogenic role [42, 43]. Albeit a single case report, transfer of maternal SLA/LP autoantibodies both in utero and via breast feeding did not lead to any hepatitis in the newborn [44].
Since these conventional autoantibodies lack diagnostic sensitivity and accuracy [45], several groups aimed to identify other potential targets for more precise diagnostic differentiation of AIH from other related diseases [46,47,48,49]. While these efforts identified more than 80 potential targets, only one recent publication systematically validated their findings [50]. Taubert et al. report that polyreactive IgG against HIP1R/BSA proved to be more specific than ANA, anti-SMA, anti-LKM1, anti-SLA/LP, and other autoantibodies [50]. HIP1R/BSA reactivity was detected in up to 88% of otherwise seronegative patients and in up to 71% of AIH patients with normal IgG levels [50]. Another class of non-conventional autoantibodies are programmed cell death 1 (PD-1)-targeting antibodies found in type 1 AIH where they correlated with levels of bilirubin and alanine aminotransferase but not with IgG [51]. PD-1 and its ligands PD-L1 and PD-L2 constitute a co-inhibitory signaling axis that limits lymphocyte activation and is key for central and peripheral tolerance [52,53,54]. This essential function is illustrated by several PD-1 knockout mouse models that develop a broad spectrum of autoimmune diseases like arthritis, lupus-like glomerulonephritis, diabetes, or fatal dilated cardiomyopathy [54]. In the cancer context, antibody-mediated blockade of the PD-1/PD-L1 axis helps to overcome cancer immune escape on the other hand favoring adverse autoimmune events including AIH [55]. Since the binding specificities of the anti-PD-1 antibodies from AIH patients and their ability to interfere with the PD-1/PD-L1 axis were not functionally validated, their pathogenic impact in breaking liver tolerance has yet to be determined. However, further evidence for the relevance of this axis derives from studies showing correlation of elevated soluble PD-1 (sPD-1) levels with activity [56, 57]. Secretion of sPD-1 which is generated via alternative splicing [58] might result in the competition of soluble and membrane-bound PD-1 on hepatic B and T cells for ligand interactions within the liver microarchitecture, thus interfering with the tolerogenic functions of PD-L1-expressing hepatocytes, liver sinusoidal epithelial cells (LSECs), hepatic stellate cells, dendritic cells (DCs), and Kupffer cells [59, 60].
Hypergammaglobulinemia
In addition to autoantibodies, polyclonal hypergammaglobulinemia with a selective IgG elevation is another characteristic diagnostic hallmark of AIH [1, 3]. Elevated IgG levels in treated AIH patients mirror an ongoing inflammatory activity [61, 62], and normalization of serum IgG is an accepted treatment goal [37]. However, not all AIH patients have elevated IgG levels, and up to 15% of patients present with normal IgG levels when presenting with acute disease [37, 63]. This observation might be explained with varying baseline immunoglobulin levels due to genetic predisposition [64] which is in line with the observation that immunosuppression leads to a persistent decrease of IgG levels below normal thresholds in patients who present without IgG elevation in acute disease phases [63]. Recent findings have questioned the high selectivity of IgG elevation and suggest that hypergammaglobulinemia in AIH may also extend to IgA [63]. IgA antibodies are predominantly generated on mucosal surfaces [65], and increased serum levels might link alterations in gut microbiota and intestinal permeability to autoimmunity in general or AIH in particular [66,67,68].
Pathological roles of hyper-IgG and autoantibodies
Despite their relevance for diagnosis and therapy monitoring, there is only limited evidence supporting a direct pathogenic role of immunoglobulins in AIH. Hepatocytes isolated from AIH patients are covered with immunoglobulin which may mediate antibody-dependent cellular cytotoxicity (ADCC) [69]. Immunoglobulin coating correlates with biopsy scores and portal but not parenchymal inflammatory activity [69]. In addition, anti-LKM1 can inhibit CYP2D6, which is expressed on hepatocyte plasma membrane, in vitro [70, 71]. However, serum transfer is not able to induce AIH in animal models [72], and fetal or neonatal hepatitis has not been reported in pregnant AIH patients [73, 74].
Other data suggest that autoantibodies and hypergammaglobulinemia might by a by-product reflecting loss of tolerance and/or an overshooting immune response. For example, IL-21 serum levels are elevated in AIH [75] and correlate with immunoglobulin levels [76]. Overexpression of IL-21 has been shown to be sufficient for autoantibody production and initiation of hypergammaglobulinemia in mouse models [77, 78]. IL-21 also triggers class-switching and the plasma cell differentiation program by regulating Blimp-1, Bcl-6, and Pax5 expression [77]. In addition, unpublished data from our group shows elevated IL-10 plasma levels clearly correlating with disease activity. The high plasma IL-10 levels could be seen as a regulatory response, since IL-10 is a dampener of excessive T cell activation while at the same time promoting immunoglobulin class switch recombination and secretion [79,80,81,82]. IL-10 has been described in diverse models of autoimmunity as main driver of autoantibody production (especially IgG) and secretion after initial activation by CD40L [83,84,85].
Cellular functions of B cells in AIH
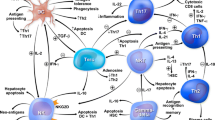
Immune repertoire sequencing can decode complex B and T cell architectures and identify immunogenetic imprints of infection [86], cancer [87], and immune-mediated diseases [88]. In our own recent work, immunosequencing of peripheral blood and liver-infiltrating B and T cell repertoires revealed a strong T cell receptor skewing unaffected by immunosuppression, while the B cell compartment was only marginally affected [89]. This is in line with flow cytometry data showing alterations in the composition of peripheral T cell subsets that persisted upon immunosuppresion, while B cells were not affected [90]. While these findings substantiate that AIH pathogenesis is not fueled by an antigen-driven B cell response, it does not exclude other essential regulatory B cell functions such as antigen presentation or cytokine and chemokine secretion exerted by distinct B cell subsets as important contributors of AIH pathogenesis. Indeed, the presentation of antigen to autoreactive CD4+ cells via B cells might provide a unifying link explaining both the HLA class II association and the various B cell abnormalities including enriched plasma cells in the liver infiltrate (Fig. 2). The regulatory B cell subset may also counteract T cell-mediated immune responses [91, 92].

Potential roles of B cells in the pathogenesis of autoimmune hepatitis. B cells are professional antigen-presenting cells that can take up and present autoantigens processed by the endocytic pathway to naïve CD4+ T cells in the secondary lymphoid tissues. In addition, B cells can be primed by autoantigen activated T cells via interaction with T follicular helper (Tfh) cells in germinal center reactions (T cell help) and differentiate into (auto-)IgG secreting plasma cells and plasmablasts. Activated B and T cells migrate via the blood stream to target tissues and mediate tissue damage. They also secrete pro- and anti-inflammatory cytokines that potentially contribute to inflammation or counteract ongoing pathogenic autoimmune reactions
Antigen presentation
Genetic studies have shown that distinct polymorphisms in the human leukocyte antigen (HLA) region encoding the major histocompatibility complex (MHC) predispose to AIH [1, 3]. MHC class I (MHCI) and II (MHCII) are functionally similar heterodimeric proteins that present processed peptides to T cells [93]. While MHCI proteins are expressed by all nucleated cells and present proteasome-processed antigens of cytosolic and nuclear origin to CD8+ T cells, MHCII molecules are mainly expressed by professional antigen-presenting cells (APCs) like dendritic cells (DCs), macrohages, and B cells and present exogenous peptides processed by the endocytic pathway to CD4+ T cells [93]. Although the AIH-asociated polymorphisms vary between ethnic groups and geographical regions, they are located within the HLA-DR3 and HLA-DR4 loci encoding MHC class II molecules, thus suggesting a disease-driving role for CD4+ T cells [1, 3, 94]. Antigen presentation by B cells is considered a rate-limiting step for diverse autoimmune diseases by inducing activation of autoreactive T cells with the same specificity [95]. Autoreactive B cells that escape self-tolerance mechanisms in the bone marrow become anergic in the periphery and can populate secondary lymphoid tissues where they often locate at T-B cell borders allowing them to interact with antigen-specific T cells [95]. B cells can internalize antigen-bound BCRs via clathrin-mediated endocytosis, proteolytically process and load them on MHCII molecules for presentation [96] (Fig. 2). While peptide-MHCII-mediated T-B cell interactions are necessary for B cell maturation and class switiching (“T cell help”), MHCII antigen presentation by B cells can prime CD4+ T cells in the absence of other APCs and trigger the generation of memory and effector T cells in distinct settings [97, 98] (Fig. 2).
In an AIH mouse model, B cell depletion contributes to AIH by presenting antigens to CD4+ cells and caused a reduction of T follicular helper (Tfh) cell numbers [99]. Tfh cell differentiation is partly dependent on MHCII antigen presentation by B cells [100], and accumulation of Tfh cells has been shown to be necessary for autoimmunity [23]. Although their exact contribution to AIH pathogenesis is not understood, Tfh cell numbers are enriched in AIH patients [101] and mice models [99]. The role of Tfh cell-mediated T and B cell crosstalk for AIH pathogenesis is also substantiated by the finding that SLA-specific autoreactive T cells upregulate transcription programs associated with B cell help [102].
Cytokine production
B cells can produce a broad array of pro- and anti-inflammatory cytokines and chemokines necessary for regulation of different aspects of immunity [103, 104] (Fig. 2). Examples for B cell-derived cytokines that contribute to T cell responses are, among others, IFNγ which promotes TH1 responses, IL‑2 which promotes TH2 memory responses, or TNF-α and CCL3 which regulate Th1 cell responses [104]. Rare subsets of B cells are reported as producers of IL-17 independent of IL-6 and IL-23 signaling and the RORγt transcriptiong factor [105]. In addition, IFN-γ inducible protein 10 (IP-10/CXCL10) which correlates with liver inflammation in AIH [106] is secreted by B cells (and hepatocytes) and mediates hepatic chemoattraction of TH1 and TH17 cells in AIH [107, 108].
Regulatory T cells (Tregs) are crucial for establishing tolerance, especially in the liver [109]. Most Tregs express heterodimeric IL-2 receptors (IL-2Rs) on their surface containing the high-affinitiy alpha chain (IL2RA/CD25) and are thus not highly responsive but also highly dependent on IL-2, a major factor for homeostasis as well as immunosuppressive and cytoprotective Treg function [110]. In a mouse model overexpressing the liver autoantigen FTCD, adoptive transfer of ex vivo IL-2 expanded CXCR3+ Tregs inhibited intrahepatic proliferation of autoreactive FTCD-specific B and T cells and restored peripheral tolerance [111]. AIH patients show decreased IL-2 serum levels and Tregs from AIH patients have been reported to be less responsive to IL-2 [112]. Low-dose IL-2 treatment in two patients with refractory AIH caused an increase in circulating Tregs and reduction of inflammatory liver damage [113].
A well-known driver of autoimmunity is IL-6 [114]. In multiple sclerosis models, B cell-derived IL-6 promotes the activation of pathogenic TH1 and TH17 cells, thereby driving the pathogenesis of this disease [115]. In AIH, IL-17 contributes to AIH pathogenesis by induction of hepatic IL-6 expression [116]. In line with this, several IL-6 polymorphisms are associated with AIH [117], and an effective second-line AIH therapeutic, 6-mercaptopurine, inhibits IL-6 production in B cells [118].
B cells, including plasma cells and plasmablasts, exert immunosuppressive functions by secreting distinct cytokines especially IL-10 and IL-35 [105, 119,120,121]. As mentioned above, our unpublished data shows elevated IL-10 plasma levels that correlated with marker normalization and disease remission. Here, IL-10 is most likely hypersecreted in active AIH to dampen high numbers of activated T cells and later downregulated when T cell activation gets more and more controlled. This provides a mechanistic basis for the finding that the immune system of AIH patients spontaneously attempts to counterregulate T cell autoreactivity in active disease [122] and also explains the observation of normal IgG levels in some patients with acute AIH [37]. Although IL-10 is mainly derived from distinct subsets of regulatory T cells and TH2 cells [81, 84], there is also a subset of regulatory B cells termed B10 solely characterized by intracellular production of IL-10 [123]. B10 cells inhibit inflammation in different model systems and humans but might also elicit proinflammatory functions [123]. However, it needs further investigation, if this subset has a relevant impact on B cell (de-)regulation in AIH. In addition, the immunosuppresive effects of IL-10 are partly mediated through upregulation of the membrane-associated E3 ubiquitin ligase MARCH1 which reduces the half-life of surface MHC-II complexes on antigen-presenting cells [124]. However, this is not exclusive for all B cell subets since IL-10 upregulates MHC-II complexes in follicular B cells [124,125,126].
IL-35 belongs to the IL-12 family and is a heterodimeric cytokine formed by p35 and EBI3 with immunosuppressive functions via increasing numbers of Tregs and regulatory B cells [127]. IL-35 fosters the generation of Tregs and inhibits CD4+ effector T cells like TH1 and TH17 cells [127, 128]. In AIH, the hepatic expression of the IL-35 subunits p35 and EBI3 is elevated and correlates with liver inflammation and fibrosis, the level of p35 also with age and serum levels of IgG and transaminases [129]. Notably, p35 is also a subunit of IL-12 (together with p40) and EBI3 of IL-27 (together with p28) [130]. Although p40 and p28 did not show equivalent correlations in immunohistochemical staining of liver tissue from AIH patients [129], higher expression of p35 and EBI3 might also hint towards production of IL-27 or IL-12; the latter of is also produced by B cells [131, 132]. Interestingly, transient hepatic overexpression of IL-12 in mice causes loss of tolerance to hepatocellular antigens leading to an AIH-like disease with hypergammaglobulinemia, autoantibodies, persistent immune cell infiltration of the liver, and hepatic fibrosis [133].
Evidence for the pathogenic role of B cells in AIH from treatment studies
Standard of care and impact on B cell function
Current therapies block pathogenic immune responses without reliably reestablishing immune tolerance [1, 3, 26, 134]. Standard of care for the induction of remission are the glucocorticoid derivatives prednisone or prednisolone, while azathioprine is given for the maintenance of remission with or without low levels of corticosteroids. In case of intolerance to azathioprine, its metabolite 6-mercaptopurine or mycophenolate mofetil (MMF) may be alternatives that alleviate adverse effects [135]. Functionally, these therapeutics represent nonspecific systemic immunosuppressants that have varying effects on immune cell types including B lineage cells.
Glucocorticoids bind the ubiquitously expressed cytosolic glucocorticoid receptor (GR) which acts as a ligand-inducible transcription factor after nuclear translocation [136,137,138,139]. In primary human B cells, GR bound prednisolone impairs BCR (downregulation of CR2/CD21, CD19, SYK, BTK, BLNK, and CD79B but not CD79A) and Toll-like receptor (TLR) 7 signaling, while immunosuppressive IL-10 and the marker for terminal plasma cell differentiation, PRDM1/Blimp-1, are upregulated [140]. Other reports showed that prednisolone inhibits proliferation, plasma cell differentiation, and IgG secretion in a dose-dependent manner and also reduces IL-10 and IL-21 cytokine levels, while low levels of prednisolone increase IgG secretion when added to peripheral blood mononuclear cells (PBMCs) [141,142,143]. In AIH patients, prednisolone therapy suppresses intrahepatic B and Treg cell proliferation and portal B and T cell densities [143]. Decline of B and Treg densities was proportional with similar Treg/B cell ratios before and under therapy [143]. In addition, portal CD79A+ B cell infiltrate density significantly correlated with serum IgG levels suggesting these cells as source of elevated IgG levels [143]. Given that mature B cells are more resistant as immature B cells towards long-term prednisolone administration although GR is expressed throughout all stages of B cell development [144, 145] and PRDM1/Blimp-1 is essential for maintenance of long-lived plasma cells (LLPCs) in the bone marrow [146], these findings might provide a mechanistic explanation for the observation that IgG normalizes upon glucocorticoid treatment, while autoantibody titers do not correlate with remission [37]. In this notion, glucocorticoid treatment would select an autoreactive B cell memory which persistently secretes autoantibodies, supports chronic inflammation, or might contribute to the fluctuating course of AIH including flares as also suggested for other autoimmune diseases [147].
Azathioprine is a pro-drug of 6-mercaptopurine that is converted by hypoxanthine–guanine phosphoribosyltransferase (HPRT1) to cytotoxic thioguanine nucleotides which are incorporated into newly synthesized nucleic acids and also reduce nucleotide synthesis by inhibiting enzymes of the purine metabolism [148]. The main immunosuppressive effect of azathioprine and its metabolites is attributed to blockade of DNA synthesis and proliferation of leukocytes by incorporation of cytotoxic purine analoga [148]. In addition, azathioprine-derived 6-mercaptopurine can also directly induce apoptosis in T cells by blocking the activity of the RAS-related C3 botulinum toxin substrate 1 (Rac1) GTPase [149]. It is reasonable to postulate the same mechanism for B cells which are highly sensitive to azathioprine [150] and dependent on Rac1 as mediator of BCR proliferation and survival signals [151]. Interestingly, low doses of azathioprine selectively reduce B cell numbers [152]. This property is used to effectively minimize the immunogenicity of anti-TNF antibodies and thus increase therapeutic efficacy [153, 154].
Mycophenolic acid (MPA) is the pharmacological active metabolite of MMF and is long known for its anti-inflammatory properties [148]. MPA reversibly inhibits inosine-5´-monophosphate dehydrogenase (IMPDH) and thus the formation of guanosine nucleotides with a high preference for T and B cells [155]. IMPDH has two isoforms, IMPDH1 and 2, from which IMPDH2 is more susceptible to MPA inhibition and also more abundant in lymphocytes [156]. MPA has a lower impact on B cell survival as compared to azathioprine, especially on antigen-naïve and resting memory B cells, but selectively inhibits B cell activation and plasma cell formation, while T cells appear not affected in SLE patients [157, 158]. MPA arrests B cells in the G0/G1 phase of the cell cycle and blocks immunoglobulin production from activated primary cells but not from terminally differentiated plasma cells expressing low levels of IMPDH2 [159]. Inhibition of immunoglobulin production of primary human B cells after CD40 ligation is independent of dose [160]. In addition, MPA reduces IL-6 production by B cells [118], which is linked to IL-17-driven AIH pathogenesis [116].
B cell depletion in clinical AIH trials
Given the pivotal role of B cells for the development and outcome of many autoimmune diseases including AIH, therapeutic approaches targeting the B lineage emerge as promising therapeutic options. The majority of available treatment options are conceived as antibody-mediated B cell depletion therapies which either target the B cell-specific surface markers CD19 and CD20 or essential survival factors like the B cell activation factor (BAFF) as well as its homolog A proliferation-inducing ligand (APRIL) [161]. B cells express the CD20 molecule from the late pro-B cells to the development of memory cells, but lost during plasmablast/plasma cell differentiation [92]. Anti-CD20 antibody therapy with the monoclonal antibody rituximab has shown efficacy in the treatment of a range of autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, pemphigus vulgaris, immune thrombocytopenia, or systemic lupus erythematosus by B lymphocyte depletion and decreased production of autoantibodies as reviewed [161,162,163,164]. However, since CD20 expression is lost on long-lived plasma cells, autoantibody production is not abrogated in all cases and might contribute to persistent inflammation or flares [147, 165].
In AIH, B cell depletion is so far only used as third-line therapy in small cohorts of difficult to treat patients showing promising results without safety concerns [166, 167]. A single center open label study of rituximab in 6 AIH patients and a retrospective multi-center cohort of 22 patients demonstrated significant improvements in serum IgG and liver transaminases sustained for up to 24 months after treatment and reported no significant adverse events [166, 167]. In paired liver biopsies of AIH before and after rituximab therapy, inflammation grade that correlated with CD4 regulatory T cells improved with treatment [166]. This suggests B cell depletion in AIH might work therapeutically through an indirect reduction in liver infiltrating CD4 T cells. However, prospective studies are yet to be obtained to validate the use of B cell depletion therapies in AIH. This is especially true for the long-term perspective of B cell depletion since anti-CD20 treatment of an AIH mouse model showed reduction in serum IgG but no histopathological normalization [168].
The success of rituximab led to the development of a second generation of humanized or full-humanized anti-CD20 (ocrelizumab, ofatumumab, ublituximab, obinutuzumab) and anti-CD19 (inebilizumab, obexelimab) antibodies [161] (Table 2). Currently, usage of next generation antibodies is evaluated in different autoimmune conditions; however, to our knowledge, no data is available about safety and efficiency in AIH [161].
Targeting of B cell regulating cytokines in AIH
BAFF and APRIL are crucial for survival and proliferation of B cells and plasma cells [169, 170]. These cytokines belong to the tumor necrosis factor family and are mainly provided by T cells and dendritic cells [171]. Both factors are known for their modulation of autoimmunity [161, 172, 173]. Self-tolerance can be achieved by an inactivation mechanism (anergy) which renders autoreactive B cells unresponsive to self-antigens. However, depending on the antigen, the anergic threshold necessary to stimulate B cells via their BCR can be overcome by BAFF-mediated signaling, thus activating autoreactive B cell clones [95]. Since BCR-coupled BAFF signaling defines a clone-specific threshold to rescue autoreactive B cells, low BAFF level maintain peripheral tolerance [174]. In AIH, BAFF levels are reported to correlate with liver inflammation [106, 175].
Ianalumab (VAY736) is an engineered, humanized, defucosylated, IgG1κ monoclonal antibody designed to block the BAFF receptor (BAFF-R/TNFRSF13C) and induce antibody-dependent cellular cytotoxicity (ADCC) of activated B cells. In primary Sjögren’s syndrome, treatment with ianalumab yielded improvements in salivary gland function, reduced tissue inflammation, sustained B cell depletion, and absence of major side effects [176, 177]. The anti-BAFF antibody belimumab has demonstrated promising results in different trials in SLE [178] and is therefore the first approved monoclonal antibody for treatment of SLE for patients intolerant or unresponsive for standard treatment [179]. First data in AIH show complete response in two AIH patients with refractory and advanced liver-related fibrosis who remained in remission while receiving low-dose corticosteroids. No adverse events related to belimumab and/or disease decompensation were observed [180]. Currently, the use of anti-BAFF receptor antibodies is evaluated in a randomized, placebo-controlled, double-blind dose range study in patients refractory or intolerant for standard treatment (NCT03217422).
Targeting of B cell in their role as co-stimulators
Abatacept is a fusion protein comprising the extracellular domain of human CTLA-4. Therefore, it specifically inhibits the proliferation and activation of T cells by binding the surface markers CD80 and CD86 [181]. On B cells, abatacept binds CD80/CD86, thereby abrogating B cell-mediated co-stimulation of T cells. Studies in RA patients showed efficient decrease in symptoms, disease activity, and structural damage upon intravenous or subcutaneous administration [181]. Treatment of RA patients with abatacept showed CD80/86 downregulation on peripheral B cells. This was associated with decreased numbers of plasma cells as well as serum IgG levels [182]. Abatacept, which was successfully used in a RA case with adalimumab-induced hepatitis [183] and to treat graft-vs-host disease upon liver transplantation [184, 185], is currently investigated in treatment of recurrent or de novo AIH in liver transplanted patients (NCT04203875).
Remarks and current research gaps/outlook
In summary, B cell-directed therapies such as blockade of BAFF and B cell-depletion have shown first evidence to be safe and efficient in treatment of AIH in patients unresponsive or intolerant to standard treatments. However, the experience of B cell-targeted therapies is limited, as only few cases or case series are reported, all of them were evaluated retrospectively. Therefore, special interest comes to the results of the first prospective phase 2 and 3 studies of ianalumab (NCT05124925, NCT05126277, NCT03656562, NCT02962895), one of which recently reported first data on safety and efficacy in patients with primary Sjögren’s syndrome [186]. Further, there is currently no data about the use of second generation of anti-CD20 antibodies in AIH as well as the use of antibodies targeting B cells in a broader spectrum of development, as it would be possible by the use of anti-CD19-targeted therapies. In addition, we lack information about targeting specifically plasma cells in AIH, which would be possible by use of a small molecule proteasome inhibitor bortezomib promoting plasma cell apoptosis. Bortezomib was shown to be efficient in various models of autoimmune diseases [187,188,189], and data from other autoimmune-mediated diseases provides evidence for efficiency in patients refractory to standard treatment [147, 190,191,192,193,194,195,196,197,198,199,200]. However, the potential side effects of, e.g., peripheral neuropathy, may limit its use. For evaluation of safety and efficiency, further studies are required.
The success of B cell-targeted therapies in AIH points out that B cells should not be considered innocent bystanders in AIH liver inflammation but rather substantial contributors to and mediators of pathogenic inflammatory processes. This can be facilitated either by being source of proinflammatory cytokines and (auto)antibodies, but also to provide help in sustaining the inflammatory state in AIH by supporting ongoing inflammation as B cells can also act as APC in secondary in chronic immune responses [97, 98, 201]. Irrespective of their substantial contribution to disease maintenance and progression, T cells still seem to play the pivotal role in disease onset in AIH [1,2,3,4, 202], and the use of B cell-targeted therapies as primary treatment in AIH remains questionable.
Since placebo-controlled trails of B cell-depletion therapies resulted in highly variable responses in other autoimmune diseases such as SLE [203], the multifaceted roles of B cells become clear. Also in RA B cells as producers of naturally arising antibodies (Nabs) are reported to be protective in the development of disease complications and might alter the disease burden [204]. Induction of regulatory B cells as potential mediators of regulatory functions with anti-inflammatory capacities is discussed as a novel treatment in autoimmunity. Regulatory B cells are reported to repopulate upon B cell-directed therapies, and their abundance are described to correlate with responsiveness to immunosuppressive treatment [162]. These hints to a more diverse role of B cells in autoimmune diseases, which to this point is only insufficiently understood in AIH.
References
Mieli-Vergani G, Vergani D, Czaja AJ, Manns MP, Krawitt EL, Vierling JM et al (2018) Autoimmune hepatitis Nat Rev Dis Primers 4:18017. https://doi.org/10.1038/nrdp.2018.17
Krawitt EL (2006) Autoimmune hepatitis. N Engl J Med 354(1):54–66. https://doi.org/10.1056/NEJMra050408
Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D (2021) Autoimmmune hepatitis. Cell Mol Immunol. https://doi.org/10.1038/s41423-021-00768-8
Lohse AW, Mieli-Vergani G (2011) Autoimmune hepatitis. J Hepatol 55(1):171–182. https://doi.org/10.1016/j.jhep.2010.12.012
Lohse AW, Weiler-Normann C, Tiegs G (2010) Immune-mediated liver injury. J Hepatol 52(1):136–144. https://doi.org/10.1016/j.jhep.2009.10.016
Cooper MD (2015) The early history of B cells. Nat Rev Immunol 15(3):191–197. https://doi.org/10.1038/nri3801
Hardy RR, Hayakawa K (2001) B cell development pathways. Annu Rev Immunol 19:595–621. https://doi.org/10.1146/annurev.immunol.19.1.595
Herzog S, Reth M, Jumaa H (2009) Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol 9(3):195–205. https://doi.org/10.1038/nri2491
Melchers F (2015) Checkpoints that control B cell development. J Clin Invest 125(6):2203–2210. https://doi.org/10.1172/JCI78083
Pieper K, Grimbacher B, Eibel H (2013) B-cell biology and development. J Allergy Clin Immunol 131(4):959–971. https://doi.org/10.1016/j.jaci.2013.01.046
Cyster JG, Allen CDC (2019) B Cell responses: cell interaction dynamics and decisions. Cell 177(3):524–540. https://doi.org/10.1016/j.cell.2019.03.016
De Silva NS, Klein U (2015) Dynamics of B cells in germinal centres. Nat Rev Immunol 15(3):137–148. https://doi.org/10.1038/nri3804
Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L (2018) Regulation of the germinal center response. Front Immunol 9:2469. https://doi.org/10.3389/fimmu.2018.02469
Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM et al (2017) Mechanisms of autoantibody-induced pathology. Front Immunol 8:603. https://doi.org/10.3389/fimmu.2017.00603
Robles DT, Eisenbarth GS, Dailey NJ, Peterson LB, Wicker LS (2003) Insulin autoantibodies are associated with islet inflammation but not always related to diabetes progression in NOD congenic mice. Diabetes 52(3):882–886. https://doi.org/10.2337/diabetes.52.3.882
Taplin CE, Barker JM (2008) Autoantibodies in type 1 diabetes. Autoimmunity 41(1):11–18. https://doi.org/10.1080/08916930701619169
Jahns R, Boivin V, Schwarzbach V, Ertl G, Lohse MJ (2008) Pathological autoantibodies in cardiomyopathy. Autoimmunity 41(6):454–461. https://doi.org/10.1080/08916930802031603
Caforio AL, Tona F, Bottaro S, Vinci A, Dequal G, Daliento L et al (2008) Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity 41(1):35–45. https://doi.org/10.1080/08916930701619235
Didona D, Maglie R, Eming R, Hertl M (2019) Pemphigus: current and future therapeutic strategies. Front Immunol 10:1418. https://doi.org/10.3389/fimmu.2019.01418
Bax M, Huizinga TW, Toes RE (2014) The pathogenic potential of autoreactive antibodies in rheumatoid arthritis. Semin Immunopathol 36(3):313–325. https://doi.org/10.1007/s00281-014-0429-5
van Delft MAM, Huizinga TWJ (2020) An overview of autoantibodies in rheumatoid arthritis. J Autoimmun 110:102392. https://doi.org/10.1016/j.jaut.2019.102392
Care MA, Stephenson SJ, Barnes NA, Fan I, Zougman A, El-Sherbiny YM et al (2016) Network analysis identifies proinflammatory plasma cell polarization for secretion of ISG15 in human autoimmunity. J Immunol 197(4):1447–1459. https://doi.org/10.4049/jimmunol.1600624
Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL et al (2009) Follicular helper T cells are required for systemic autoimmunity. J Exp Med 206(3):561–576. https://doi.org/10.1084/jem.20081886
Elsner RA, Shlomchik MJ (2020) Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity 53(6):1136–1150. https://doi.org/10.1016/j.immuni.2020.11.006
Rahim MN, Miquel R, Heneghan MA (2020) Approach to the patient with acute severe autoimmune hepatitis. JHEP Rep 2(6):100149. https://doi.org/10.1016/j.jhepr.2020.100149
Weiler-Normann C, Lohse AW (2021) Autoimmune hepatitis: from immunopathogenesis to diagnostic and therapeutic innovation. Curr Opin Gastroenterol 37(2):86–90. https://doi.org/10.1097/MOG.0000000000000701
Herkel J, Carambia A, Lohse AW (2020) Autoimmune hepatitis: possible triggers, potential treatments. J Hepatol 73(2):446–448. https://doi.org/10.1016/j.jhep.2020.03.015
Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL et al (2008) Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 48(1):169–176. https://doi.org/10.1002/hep.22322
Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL et al (1999) International autoimmune hepatitis group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 31(5):929–938. https://doi.org/10.1016/s0168-8278(99)80297-9
de Boer YS, van Nieuwkerk CM, Witte BI, Mulder CJ, Bouma G, Bloemena E (2015) Assessment of the histopathological key features in autoimmune hepatitis. Histopathology 66(3):351–362. https://doi.org/10.1111/his.12558
Balitzer D, Shafizadeh N, Peters MG, Ferrell LD, Alshak N, Kakar S (2017) Autoimmune hepatitis: review of histologic features included in the simplified criteria proposed by the international autoimmune hepatitis group and proposal for new histologic criteria. Mod Pathol 30(5):773–783. https://doi.org/10.1038/modpathol.2016.267
Gurung A, Assis DN, McCarty TR, Mitchell KA, Boyer JL, Jain D (2018) Histologic features of autoimmune hepatitis: a critical appraisal. Hum Pathol 82:51–60. https://doi.org/10.1016/j.humpath.2018.07.014
Bach N, Thung SN, Schaffner F (1992) The histological features of chronic hepatitis C and autoimmune chronic hepatitis: a comparative analysis. Hepatology 15(4):572–577. https://doi.org/10.1002/hep.1840150403
Czaja AJ, Carpenter HA (1993) Sensitivity, specificity, and predictability of biopsy interpretations in chronic hepatitis. Gastroenterology 105(6):1824–1832. https://doi.org/10.1016/0016-5085(93)91081-r
Crawford AR, Lin XZ, Crawford JM (1998) The normal adult human liver biopsy: a quantitative reference standard. Hepatology 28(2):323–331. https://doi.org/10.1002/hep.510280206
Covelli C, Sacchi D, Sarcognato S, Cazzagon N, Grillo F, Baciorri F et al (2021) Pathology of autoimmune hepatitis. Pathologica 113(3):185–93. https://doi.org/10.32074/1591-951X-241
European Association for the Study of the Liver (2015) EASL clinical practice guidelines: autoimmune hepatitis. J Hepatol 63(4):971–1004. https://doi.org/10.1016/j.jhep.2015.06.030
Schultheiss C, Paschold L, Willscher E, Simnica D, Wostemeier A, Muscate F et al (2021) Maturation trajectories and transcriptional landscape of plasmablasts and autoreactive B cells in COVID-19. IScience 24(11):103325. https://doi.org/10.1016/j.isci.2021.103325
Wucherpfennig KW (2001) Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest 108(8):1097–1104. https://doi.org/10.1172/JCI14235
Chakravarty EF (2008) Viral infection and reactivation in autoimmune disease. Arthritis Rheum 58(10):2949–2957. https://doi.org/10.1002/art.23883
Gilman AJ, Le AK, Zhao C, Hoang J, Yasukawa LA, Weber SC et al (2018) Autoantibodies in chronic hepatitis C virus infection: impact on clinical outcomes and extrahepatic manifestations. BMJ Open Gastroenterol 5(1):e000203. https://doi.org/10.1136/bmjgast-2018-000203
Baeres M, Herkel J, Czaja AJ, Wies I, Kanzler S, Cancado EL et al (2002) Establishment of standardised SLA/LP immunoassays: specificity for autoimmune hepatitis, worldwide occurrence, and clinical characteristics. Gut 51(2):259–264. https://doi.org/10.1136/gut.51.2.259
Herkel J, Heidrich B, Nieraad N, Wies I, Rother M, Lohse AW (2002) Fine specificity of autoantibodies to soluble liver antigen and liver/pancreas. Hepatology 35(2):403–408. https://doi.org/10.1053/jhep.2002.30699
Lohse AW, Gerken G, Altes U, Mayet WJ, Meyer zumBuschenfelde KH (1993) Transmission of maternal IgG autoantibodies via cord blood and breastmilk without transmission of hepatitis. Lancet 341(8854):1216–7. https://doi.org/10.1016/0140-6736(93)91046-o
Zhang WC, Zhao FR, Chen J, Chen WX (2014) Meta-analysis: diagnostic accuracy of antinuclear antibodies, smooth muscle antibodies and antibodies to a soluble liver antigen/liver pancreas in autoimmune hepatitis. PLoS ONE 9(3):e92267. https://doi.org/10.1371/journal.pone.0092267
Zingaretti C, Arigo M, Cardaci A, Moro M, Crosti M, Sinisi A et al (2012) Identification of new autoantigens by protein array indicates a role for IL4 neutralization in autoimmune hepatitis. Mol Cell Proteomics 11(12):1885–1897. https://doi.org/10.1074/mcp.M112.018713
Song Q, Liu G, Hu S, Zhang Y, Tao Y, Han Y et al (2010) Novel autoimmune hepatitis-specific autoantigens identified using protein microarray technology. J Proteome Res 9(1):30–39. https://doi.org/10.1021/pr900131e
Lammert C, Zhu C, Lian Y, Raman I, Eckert G, Li QZ et al (2020) Exploratory study of autoantibody profiling in drug-induced liver injury with an autoimmune phenotype. Hepatol Commun 4(11):1651–1663. https://doi.org/10.1002/hep4.1582
Zhang W, Rho JH, Roehrl MH, Wang JY (2019) A comprehensive autoantigen-ome of autoimmune liver diseases identified from dermatan sulfate affinity enrichment of liver tissue proteins. BMC Immunol 20(1):21. https://doi.org/10.1186/s12865-019-0304-1
Taubert R, Engel B, Diestelhorst J, Hupa-Breier KL, Behrendt P, Baerleckeen NT et al (2021) Quantification of polyreactive immunoglobulin G facilitates the diagnosis of autoimmune hepatitis. Hepatology 75(1):13–27. https://doi.org/10.1002/hep.32134
Matsumoto K, Miyake Y, Matsushita H, Ohnishi A, Ikeda F, Shiraha H et al (2014) Anti-programmed cell death-1 antibody as a new serological marker for type 1 autoimmune hepatitis. J Gastroenterol Hepatol 29(1):110–115. https://doi.org/10.1111/jgh.12340
Francisco LM, Sage PT, Sharpe AH (2010) The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 236:219–242. https://doi.org/10.1111/j.1600-065X.2010.00923.x
Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z et al (2019) The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol 10:2298. https://doi.org/10.3389/fimmu.2019.02298
Okazaki T, Honjo T (2006) The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 27(4):195–201. https://doi.org/10.1016/j.it.2006.02.001
Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M et al (2019) Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 16(9):563–580. https://doi.org/10.1038/s41571-019-0218-0
Aarslev K, Dige A, Greisen SR, Kreutzfeldt M, Jessen N, Vilstrup H et al (2017) Soluble programmed death-1 levels are associated with disease activity and treatment response in patients with autoimmune hepatitis. Scand J Gastroenterol 52(1):93–99. https://doi.org/10.1080/00365521.2016.1233576
Hadley T, Gillespie S, Espinoza H, Prince J, Gronbaek H, Chandrakasan S et al (2020) Soluble PD1 levels are increased with disease activity in paediatric onset autoimmune hepatitis and inflammatory bowel disease. Autoimmunity 53(5):253–260. https://doi.org/10.1080/08916934.2020.1755964
Gu D, Ao X, Yang Y, Chen Z, Xu X (2018) Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer 6(1):132. https://doi.org/10.1186/s40425-018-0449-0
Curran CS, Sharon E (2017) PD-1 immunobiology in autoimmune hepatitis and hepatocellular carcinoma. Semin Oncol 44(6):428–432. https://doi.org/10.1053/j.seminoncol.2017.12.001
Thomson AW, Knolle PA (2010) Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 10(11):753–766. https://doi.org/10.1038/nri2858
Luth S, Herkel J, Kanzler S, Frenzel C, Galle PR, Dienes HP et al (2008) Serologic markers compared with liver biopsy for monitoring disease activity in autoimmune hepatitis. J Clin Gastroenterol 42(8):926–930. https://doi.org/10.1097/MCG.0b013e318154af74
Hartl J, Ehlken H, Sebode M, Peiseler M, Krech T, Zenouzi R et al (2018) Usefulness of biochemical remission and transient elastography in monitoring disease course in autoimmune hepatitis. J Hepatol 68(4):754–763. https://doi.org/10.1016/j.jhep.2017.11.020
Hartl J, Miquel R, Zachou K, Wong GW, Asghar A, Pape S et al (2020) Features and outcome of AIH patients without elevation of IgG. JHEP Rep 2(3):100094. https://doi.org/10.1016/j.jhepr.2020.100094
Jonsson S, Sveinbjornsson G, de Lapuente Portilla AL, Swaminathan B, Plomp R, Dekkers G et al (2017) Identification of sequence variants influencing immunoglobulin levels. Nat Genet 49(8):1182–1191. https://doi.org/10.1038/ng.3897
Abokor AA, McDaniel GH, Golonka RM, Campbell C, Brahmandam S, Yeoh BS et al (2021) Immunoglobulin A, an active liaison for host-microbiota homeostasis. Microorganisms 9(10):2117. https://doi.org/10.3390/microorganisms9102117
Yuksel M, Wang Y, Tai N, Peng J, Guo J, Beland K et al (2015) A novel “humanized mouse” model for autoimmune hepatitis and the association of gut microbiota with liver inflammation. Hepatology 62(5):1536–1550. https://doi.org/10.1002/hep.27998
Lin R, Zhou L, Zhang J, Wang B (2015) Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int J Clin Exp Pathol 8(5):5153–5160
Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N et al (2018) Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359(6380):1156–1161. https://doi.org/10.1126/science.aar7201
Vergani D, Mieli-Vergani G, Mondelli M, Portmann B, Eddleston AL (1987) Immunoglobulin on the surface of isolated hepatocytes is associated with antibody-dependent cell-mediated cytotoxicity and liver damage. Liver 7(6):307–315. https://doi.org/10.1111/j.1600-0676.1987.tb00361.x
Manns M, Zanger U, Gerken G, Sullivan KF, Meyer zumBuschenfelde KH, Meyer UA et al (1990) Patients with type II autoimmune hepatitis express functionally intact cytochrome P-450 db1 that is inhibited by LKM-1 autoantibodies in vitro but not in vivo. Hepatology 12(1):127–32. https://doi.org/10.1002/hep.1840120120
Muratori L, Parola M, Ripalti A, Robino G, Muratori P, Bellomo G et al (2000) Liver/kidney microsomal antibody type 1 targets CYP2D6 on hepatocyte plasma membrane. Gut 46(4):553–561. https://doi.org/10.1136/gut.46.4.553
Hardtke-Wolenski M, Fischer K, Noyan F, Schlue J, Falk CS, Stahlhut M et al (2013) Genetic predisposition and environmental danger signals initiate chronic autoimmune hepatitis driven by CD4+ T cells. Hepatology 58(2):718–728. https://doi.org/10.1002/hep.26380
Schramm C, Herkel J, Beuers U, Kanzler S, Galle PR, Lohse AW (2006) Pregnancy in autoimmune hepatitis: outcome and risk factors. Am J Gastroenterol 101(3):556–560. https://doi.org/10.1111/j.1572-0241.2006.00479.x
Wang CW, Grab J, Tana MM, Irani RA, Sarkar M (2021) Outcomes of pregnancy in autoimmune hepatitis: a population-based study. Hepatology 75(1):5–12. https://doi.org/10.1002/hep.32132
Abe K, Takahashi A, Imaizumi H, Hayashi M, Okai K, Kanno Y et al (2016) Interleukin-21 plays a critical role in the pathogenesis and severity of type I autoimmune hepatitis. Springerplus 5(1):777. https://doi.org/10.1186/s40064-016-2512-y
Ma L, Qin J, Ji H, Zhao P, Jiang Y (2014) Tfh and plasma cells are correlated with hypergammaglobulinaemia in patients with autoimmune hepatitis. Liver Int 34(3):405–415. https://doi.org/10.1111/liv.12245
Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF et al (2004) Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol 173(9):5361–5371. https://doi.org/10.4049/jimmunol.173.9.5361
Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A et al (2002) A critical role for IL-21 in regulating immunoglobulin production. Science 298(5598):1630–1634. https://doi.org/10.1126/science.1077002
Wang X, Wong K, Ouyang W, Rutz S (2019) Targeting IL-10 family cytokines for the treatment of human diseases. Cold Spring Harb Perspect Biol 11(2). https://doi.org/10.1101/cshperspect.a028548
Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM et al (2011) Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34(4):566–578. https://doi.org/10.1016/j.immuni.2011.03.018
Abe M, Hiasa Y, Onji M (2013) T helper 17 cells in autoimmune liver diseases. Clin Dev Immunol 2013:607073. https://doi.org/10.1155/2013/607073
Malisan F, Briere F, Bridon JM, Harindranath N, Mills FC, Max EE et al (1996) Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. J Exp Med 183(3):937–947. https://doi.org/10.1084/jem.183.3.937
Llorente L, Zou W, Levy Y, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J et al (1995) Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J Exp Med 181(3):839–844. https://doi.org/10.1084/jem.181.3.839
Geginat J, Larghi P, Paroni M, Nizzoli G, Penatti A, Pagani M et al (2016) The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev 30:87–93. https://doi.org/10.1016/j.cytogfr.2016.02.003
Laman JD, Claassen E, Noelle RJ (1996) Functions of CD40 and its ligand, gp39 (CD40L). Crit Rev Immunol 16(1):59–108. https://doi.org/10.1615/critrevimmunol.v16.i1.40
Schultheiss C, Paschold L, Simnica D, Mohme M, Willscher E, von Wenserski L et al (2020) Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity 53(2):442–55 e4. https://doi.org/10.1016/j.immuni.2020.06.024
Simnica D, Smits M, Willscher E, Fanchi LF, Kloots ISH, Iv Oort et al (2020) Responsiveness to immune checkpoint inhibitors is associated with a peripheral blood T-cell signature in metastatic castration-resistant prostate cancer. JCO Precision Oncology 4:1374–85. https://doi.org/10.1200/po.20.00209
Bashford-Rogers RJM, Bergamaschi L, McKinney EF, Pombal DC, Mescia F, Lee JC et al (2019) Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574(7776):122–126. https://doi.org/10.1038/s41586-019-1595-3
Schultheiss C, Simnica D, Willscher E, Oberle A, Fanchi L, Bonzanni N et al (2021) Next-generation immunosequencing reveals pathological T-cell architecture in autoimmune hepatitis. Hepatology 73(4):1436–1448. https://doi.org/10.1002/hep.31473
Renand A, Habes S, Mosnier JF, Auble H, Judor JP, Vince N et al (2018) Immune alterations in patients with type 1 autoimmune hepatitis persist upon standard immunosuppressive treatment. Hepatol Commun 2(8):968–981. https://doi.org/10.1002/hep4.1202
Taylor SA, Assis DN, Mack CL (2019) The contribution of B cells in autoimmune liver diseases. Semin Liver Dis 39(4):422–431. https://doi.org/10.1055/s-0039-1688751
Czaja AJ (2021) Review article: targeting the B cell activation system in autoimmune hepatitis. Aliment Pharmacol Ther 54(7):902–922. https://doi.org/10.1111/apt.16574
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11(12):823–836. https://doi.org/10.1038/nri3084
Fasano R, Malerba E, Prete M, Solimando AG, Buonavoglia A, Silvestris N et al (2021) Impact of antigen presentation mechanisms on immune response in autoimmune hepatitis. Front Immunol 12:814155. https://doi.org/10.3389/fimmu.2021.814155
Shlomchik MJ (2008) Sites and stages of autoreactive B cell activation and regulation. Immunity 28(1):18–28. https://doi.org/10.1016/j.immuni.2007.12.004
Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung SC et al (2017) The other function: class II-restricted antigen presentation by B cells. Front Immunol 8:319. https://doi.org/10.3389/fimmu.2017.00319
Rodriguez-Pinto D, Moreno J (2005) B cells can prime naive CD4+ T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur J Immunol 35(4):1097–1105. https://doi.org/10.1002/eji.200425732
Crawford A, Macleod M, Schumacher T, Corlett L, Gray D (2006) Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol 176(6):3498–3506. https://doi.org/10.4049/jimmunol.176.6.3498
Beland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F (2015) Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology 62(5):1511–1523. https://doi.org/10.1002/hep.27991
Barnett LG, Simkins HM, Barnett BE, Korn LL, Johnson AL, Wherry EJ et al (2014) B cell antigen presentation in the initiation of follicular helper T cell and germinal center differentiation. J Immunol 192(8):3607–3617. https://doi.org/10.4049/jimmunol.1301284
Wang L, Sun Y, Zhang Z, Jia Y, Zou Z, Ding J et al (2015) CXCR5+ CD4+ T follicular helper cells participate in the pathogenesis of primary biliary cirrhosis. Hepatology 61(2):627–638. https://doi.org/10.1002/hep.27306
Renand A, Cervera-Marzal I, Gil L, Dong C, Garcia A, Kervagoret E et al (2020) Integrative molecular profiling of autoreactive CD4 T cells in autoimmune hepatitis. J Hepatol 73(6):1379–1390. https://doi.org/10.1016/j.jhep.2020.05.053
Lino AC, Dorner T, Bar-Or A, Fillatreau S (2016) Cytokine-producing B cells: a translational view on their roles in human and mouse autoimmune diseases. Immunol Rev 269(1):130–144. https://doi.org/10.1111/imr.12374
Shen P, Fillatreau S (2015) Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol 15(7):441–451. https://doi.org/10.1038/nri3857
Wang AA, Gommerman JL, Rojas OL (2021) Plasma cells: from cytokine production to regulation in experimental autoimmune encephalomyelitis. J Mol Biol 433(1):166655. https://doi.org/10.1016/j.jmb.2020.09.014
Nishikawa H, Enomoto H, Iwata Y, Kishino K, Shimono Y, Hasegawa K et al (2016) B-cell activating factor belonging to the tumor necrosis factor family and interferon-gamma-inducible protein-10 in autoimmune hepaTitis. Medicine (Baltimore) 95(12):e3194. https://doi.org/10.1097/MD.0000000000003194
Czaja AJ (2022) Advancing biologic therapy for refractory autoimmune hepatitis. Dig Dis Sci 2022. https://doi.org/10.1007/s10620-021-07378-4
Nishioji K, Okanoue T, Itoh Y, Narumi S, Sakamoto M, Nakamura H et al (2001) Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol 123(2):271–279. https://doi.org/10.1046/j.1365-2249.2001.01391.x
Herkel J (2015) Regulatory T cells in hepatic immune tolerance and autoimmune liver diseases. Dig Dis 33(Suppl 2):70–74. https://doi.org/10.1159/000440750
Ye C, Brand D, Zheng SG (2018) Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther 3:2. https://doi.org/10.1038/s41392-017-0002-5
Lapierre P, Beland K, Yang R, Alvarez F (2013) Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology 57(1):217–227. https://doi.org/10.1002/hep.26023
Czaja AJ (2021) Exploring the pathogenic role and therapeutic implications of interleukin 2 in autoimmune hepatitis. Dig Dis Sci 66(8):2493–2512. https://doi.org/10.1007/s10620-020-06562-2
Lim TY, Martinez-Llordella M, Kodela E, Gray E, Heneghan MA, Sanchez-Fueyo A (2018) Low-dose interleukin-2 for refractory autoimmune hepatitis. Hepatology 68(4):1649–1652. https://doi.org/10.1002/hep.30059
Jones BE, Maerz MD, Buckner JH (2018) IL-6: a cytokine at the crossroads of autoimmunity. Curr Opin Immunol 55:9–14. https://doi.org/10.1016/j.coi.2018.09.002
Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S et al (2012) B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med 209(5):1001–1010. https://doi.org/10.1084/jem.20111675
Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X et al (2011) Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE 6(4):e18909. https://doi.org/10.1371/journal.pone.0018909
Yousefi A, Najafi M, Motamed F, Mahmoudi E, Bidoki AZ, Sadr M et al (2018) Association of interleukin-6 and interleukin-1 family gene polymorphisms in autoimmune hepatitis. Ann Hepatol 17(6):1021–1025. https://doi.org/10.5604/01.3001.0012.7202
von Borstel A, Abdulahad WH, Dekkema G, Rutgers A, Stegeman CA, Veldman J et al (2020) Mycophenolic acid and 6-mercaptopurine both inhibit B-cell proliferation in granulomatosis with polyangiitis patients, whereas only mycophenolic acid inhibits B-cell IL-6 production. PLoS ONE 15(7):e0235743. https://doi.org/10.1371/journal.pone.0235743
Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E et al (2014) IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507(7492):366–370. https://doi.org/10.1038/nature12979
Dang VD, Hilgenberg E, Ries S, Shen P, Fillatreau S (2014) From the regulatory functions of B cells to the identification of cytokine-producing plasma cell subsets. Curr Opin Immunol 28:77–83. https://doi.org/10.1016/j.coi.2014.02.009
Anderton SM, Fillatreau S (2008) Activated B cells in autoimmune diseases: the case for a regulatory role. Nat Clin Pract Rheumatol 4(12):657–666. https://doi.org/10.1038/ncprheum0950
Lohse AW, Kogel M, Meyer zumBuschenfelde KH (1995) Evidence for spontaneous immunosuppression in autoimmune hepatitis. Hepatology 22(2):381–8
Tedder TF (2015) B10 cells: a functionally defined regulatory B cell subset. J Immunol 194(4):1395–1401. https://doi.org/10.4049/jimmunol.1401329
Thibodeau J, Bourgeois-Daigneault MC, Huppe G, Tremblay J, Aumont A, Houde M et al (2008) Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol 38(5):1225–1230. https://doi.org/10.1002/eji.200737902
Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M et al (2007) Novel regulation of MHC class II function in B cells. EMBO J 26(3):846–854. https://doi.org/10.1038/sj.emboj.7601556
Galbas T, Steimle V, Lapointe R, Ishido S, Thibodeau J (2012) MARCH1 down-regulation in IL-10-activated B cells increases MHC class II expression. Cytokine 59(1):27–30. https://doi.org/10.1016/j.cyto.2012.03.015
Choi J, Leung PS, Bowlus C, Gershwin ME (2015) IL-35 and Autoimmunity: a comprehensive perspective. Clin Rev Allergy Immunol 49(3):327–332. https://doi.org/10.1007/s12016-015-8468-9
Hu S, Lian PP, Hu Y, Zhu XY, Jiang SW, Ma Q et al (2020) The role of IL-35 in the pathophysiological processes of liver disease. Front Pharmacol 11:569575. https://doi.org/10.3389/fphar.2020.569575
Lian M, Zhang J, Zhao L, Chen X, Peng Y, Wang Q et al (2019) Interleukin-35 regulates immune microenvironment of autoimmune hepatitis through inducing the expansion of myeloid-derived suppressor cells. Front Immunol 10:2577. https://doi.org/10.3389/fimmu.2019.02577
Vignali DA, Kuchroo VK (2012) IL-12 family cytokines: immunological playmakers. Nat Immunol 13(8):722–728. https://doi.org/10.1038/ni.2366
Sartori A, Ma X, Gri G, Showe L, Benjamin D, Trinchieri G (1997) Interleukin-12: an immunoregulatory cytokine produced by B cells and antigen-presenting cells. Methods 11(1):116–127. https://doi.org/10.1006/meth.1996.0395
Sugimoto K, Ogawa A, Shimomura Y, Nagahama K, Mizoguchi A, Bhan AK (2007) Inducible IL-12-producing B cells regulate Th2-mediated intestinal inflammation. Gastroenterology 133(1):124–136. https://doi.org/10.1053/j.gastro.2007.03.112
Gil-Farina I, Di Scala M, Salido E, Lopez-Franco E, Rodriguez-Garcia E, Blasi M et al (2016) Transient expression of transgenic IL-12 in mouse liver triggers unremitting inflammation mimicking human autoimmune hepatitis. J Immunol 197(6):2145–2156. https://doi.org/10.4049/jimmunol.1600228
Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D et al (2010) Diagnosis and management of autoimmune hepatitis. Hepatology 51(6):2193–2213. https://doi.org/10.1002/hep.23584
Lohse AW, Sebode M, Jorgensen MH, Ytting H, Karlsen TH, Kelly D et al (2020) Second-line and third-line therapy for autoimmune hepatitis: a position statement from the European Reference Network on Hepatological Diseases and the International Autoimmune Hepatitis Group. J Hepatol 73(6):1496–1506. https://doi.org/10.1016/j.jhep.2020.07.023
Hardy RS, Raza K, Cooper MS (2020) Therapeutic glucocorticoids: mechanisms of actions in rheumatic diseases. Nat Rev Rheumatol 16(3):133–144. https://doi.org/10.1038/s41584-020-0371-y
Timmermans S, Souffriau J, Libert C (2019) A General introduction to glucocorticoid biology. Front Immunol 10:1545. https://doi.org/10.3389/fimmu.2019.01545
Quatrini L, Ugolini S (2021) New insights into the cell- and tissue-specificity of glucocorticoid actions. Cell Mol Immunol 18(2):269–278. https://doi.org/10.1038/s41423-020-00526-2
Cain DW, Cidlowski JA (2017) Immune regulation by glucocorticoids. Nat Rev Immunol 17(4):233–247. https://doi.org/10.1038/nri.2017.1
Franco LM, Gadkari M, Howe KN, Sun J, Kardava L, Kumar P et al (2019) Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J Exp Med 216(2):384–406. https://doi.org/10.1084/jem.20180595
Haneda M, Owaki M, Kuzuya T, Iwasaki K, Miwa Y, Kobayashi T (2014) Comparative analysis of drug action on B-cell proliferation and differentiation for mycophenolic acid, everolimus, and prednisolone. Transplantation 97(4):405–412. https://doi.org/10.1097/01.TP.0000441826.70687.f6
Yan SX, Deng XM, Wang QT, Sun XJ, Wei W (2015) Prednisone treatment inhibits the differentiation of B lymphocytes into plasma cells in MRL/MpSlac-lpr mice. Acta Pharmacol Sin 36(11):1367–1376. https://doi.org/10.1038/aps.2015.76
Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J et al (2014) Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J Hepatol 61(5):1106–1114. https://doi.org/10.1016/j.jhep.2014.05.034
Voetberg BJ, Garvy BA, Mayer HK, King LE, Fraker PJ (1994) Apoptosis accompanies a change in the phenotypic distribution and functional capacity of murine bone marrow B-cells chronically exposed to prednisolone. Clin Immunol Immunopathol 71(2):190–198. https://doi.org/10.1006/clin.1994.1071
Gruver-Yates AL, Quinn MA, Cidlowski JA (2014) Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology 155(2):463–474. https://doi.org/10.1210/en.2013-1473
Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K (2005) Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med 202(11):1471–1476. https://doi.org/10.1084/jem.20051611
Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A (2011) Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol 7(3):170–178. https://doi.org/10.1038/nrrheum.2011.1
Broen JCA, van Laar JM (2020) Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol 16(3):167–78. https://doi.org/10.1038/s41584-020-0374-8
Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D et al (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest 111(8):1133–1145. https://doi.org/10.1172/JCI16432
Dimitriu A, Fauci AS (1978) Activation of human B lymphocytes XI Differential effects of azathioprine on B lymphocytes and lymphocyte subpopulations regulating B cell function. J Immunol 121(6):2335–9
Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A et al (2003) Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science 302(5644):459–462. https://doi.org/10.1126/science.1089709
Tareyeva IE, Shilov EM, Gordovskaya NB (1980) The effects of azathioprine and prednisolone on T- and B-lymphocytes in patients with lupus nephritis and chronic glomerulonephritis. Clin Nephrol 14(5):233–237
Krieckaert CL, Bartelds GM, Lems WF, Wolbink GJ (2010) The effect of immunomodulators on the immunogenicity of TNF-blocking therapeutic monoclonal antibodies: a review. Arthritis Res Ther 12(5):217. https://doi.org/10.1186/ar3147
Garces S, Demengeot J, Benito-Garcia E (2013) The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis 72(12):1947–1955. https://doi.org/10.1136/annrheumdis-2012-202220
Allison AC (2005) Mechanisms of action of mycophenolate mofetil. Lupus 14(Suppl 1):s2-8. https://doi.org/10.1191/0961203305lu2109oa
Carr SF, Papp E, Wu JC, Natsumeda Y (1993) Characterization of human type I and type II IMP dehydrogenases. J Biol Chem 268(36):27286–27290
Eickenberg S, Mickholz E, Jung E, Nofer JR, Pavenstadt HJ, Jacobi AM (2012) Mycophenolic acid counteracts B cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res Ther 14(3):R110. https://doi.org/10.1186/ar3835
Bijl M, Horst G, Bootsma H, Limburg PC, Kallenberg CG (2003) Mycophenolate mofetil prevents a clinical relapse in patients with systemic lupus erythematosus at risk. Ann Rheum Dis 62(6):534–539. https://doi.org/10.1136/ard.62.6.534
Karnell JL, Karnell FG 3rd, Stephens GL, Rajan B, Morehouse C, Li Y et al (2011) Mycophenolic acid differentially impacts B cell function depending on the stage of differentiation. J Immunol 187(7):3603–3612. https://doi.org/10.4049/jimmunol.1003319
Heidt S, Roelen DL, Eijsink C, van Kooten C, Claas FH, Mulder A (2008) Effects of immunosuppressive drugs on purified human B cells: evidence supporting the use of MMF and rapamycin. Transplantation 86(9):1292–1300. https://doi.org/10.1097/TP.0b013e3181874a36
Lee DSW, Rojas OL, Gommerman JL (2021) B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov 20(3):179–199. https://doi.org/10.1038/s41573-020-00092-2
Hofmann K, Clauder AK, Manz RA (2018) Targeting B cells and plasma cells in autoimmune diseases. Front Immunol 9:835. https://doi.org/10.3389/fimmu.2018.00835
Frampton JE (2020) Rituximab: a review in pemphigus vulgaris. Am J Clin Dermatol 21(1):149–156. https://doi.org/10.1007/s40257-019-00497-9
Chen DM, Odueyungbo A, Csinady E, Gearhart L, Lehane P, Cheu M et al (2020) Rituximab is an effective treatment in patients with pemphigus vulgaris and demonstrates a steroid-sparing effect. Br J Dermatol 182(5):1111–1119. https://doi.org/10.1111/bjd.18482
Favas C, Isenberg DA (2009) B-cell-depletion therapy in SLE–what are the current prospects for its acceptance? Nat Rev Rheumatol 5(12):711–716. https://doi.org/10.1038/nrrheum.2009.218
Burak KW, Swain MG, Santodomingo-Garzon T, Lee SS, Urbanski SJ, Aspinall AI et al (2013) Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol 27(5):273–280. https://doi.org/10.1155/2013/512624
Than NN, Hodson J, Schmidt-Martin D, Taubert R, Wawman RE, Botter M et al (2019) Efficacy of rituximab in difficult-to-manage autoimmune hepatitis: results from the international autoimmune hepatitis group. JHEP Rep 1(6):437–445. https://doi.org/10.1016/j.jhepr.2019.10.005
Buitrago-Molina LE, Dywicki J, Noyan F, Schepergerdes L, Pietrek J, Lieber M et al (2021) Anti-CD20 therapy alters the protein signature in experimental murine AIH, but not exclusively towards regeneration. Cells 10(6). https://doi.org/10.3390/cells10061471
Mackay F, Schneider P, Rennert P, Browning J (2003) BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol 21:231–264. https://doi.org/10.1146/annurev.immunol.21.120601.141152
Bossen C, Schneider P (2006) BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol 18(5):263–275. https://doi.org/10.1016/j.smim.2006.04.006
Mackay F, Schneider P (2009) Cracking the BAFF code. Nat Rev Immunol 9(7):491–502. https://doi.org/10.1038/nri2572
Vincent FB, Morand EF, Schneider P, Mackay F (2014) The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol 10(6):365–373
Nakayamada S, Tanaka Y (2016) BAFF- and APRIL-targeted therapy in systemic autoimmune diseases. Inflamm Regen 36:6. https://doi.org/10.1186/s41232-016-0015-4
Stadanlick JE, Cancro MP (2008) BAFF and the plasticity of peripheral B cell tolerance. Curr Opin Immunol 20(2):158–161. https://doi.org/10.1016/j.coi.2008.03.015
Migita K, Abiru S, Maeda Y, Nakamura M, Komori A, Ito M et al (2007) Elevated serum BAFF levels in patients with autoimmune hepatitis. Hum Immunol 68(7):586–591. https://doi.org/10.1016/j.humimm.2007.03.010
McWilliams EM, Lucas CR, Chen T, Harrington BK, Wasmuth R, Campbell A et al (2019) Anti-BAFF-R antibody VAY-736 demonstrates promising preclinical activity in CLL and enhances effectiveness of ibrutinib. Blood Adv 3(3):447–460. https://doi.org/10.1182/bloodadvances.2018025684
Dorner T, Posch MG, Li Y, Petricoul O, Cabanski M, Milojevic JM et al (2019) Treatment of primary Sjogren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann Rheum Dis 78(5):641–647. https://doi.org/10.1136/annrheumdis-2018-214720
Wise LM, Stohl W (2020) Belimumab and rituximab in systemic lupus erythematosus: a tale of two B cell-targeting agents. Front Med (Lausanne) 7:303. https://doi.org/10.3389/fmed.2020.00303
Blair HA, Duggan ST (2018) Belimumab: a review in systemic lupus erythematosus. Drugs 78(3):355–366. https://doi.org/10.1007/s40265-018-0872-z
Arvaniti P, Giannoulis G, Gabeta S, Zachou K, Koukoulis GK, Dalekos GN (2020) Belimumab is a promising third-line treatment option for refractory autoimmune hepatitis. JHEP Rep 2(4):100123. https://doi.org/10.1016/j.jhepr.2020.100123
Blair HA, Deeks ED (2017) Abatacept: a review in rheumatoid arthritis. Drugs 77(11):1221–1233. https://doi.org/10.1007/s40265-017-0775-4
Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L et al (2019) Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun 101:145–152. https://doi.org/10.1016/j.jaut.2019.04.016
Grasland A, Sterpu R, Boussoukaya S, Mahe I (2012) Autoimmune hepatitis induced by adalimumab with successful switch to abatacept. Eur J Clin Pharmacol 68(5):895–898. https://doi.org/10.1007/s00228-011-1191-4
Elfeki MA, Genco PV, Pungpapong S, Nakhleh RE, Nguyen JH, Harnois DM (2014) Abatacept use in graft-versus-host disease after orthotopic liver transplantation: a case report. Transplant Proc 46(7):2422–2425. https://doi.org/10.1016/j.transproceed.2014.06.061
Watkins B, Qayed M, McCracken C, Bratrude B, Betz K, Suessmuth Y et al (2021) Phase II trial of costimulation blockade with abatacept for prevention of acute GVHD. J Clin Oncol 39(17):1865–1877. https://doi.org/10.1200/JCO.20.01086
Bowman SJ, Fox R, Dorner T, Mariette X, Papas A, Grader-Beck T et al (2022) Safety and efficacy of subcutaneous ianalumab (VAY736) in patients with primary Sjogren’s syndrome: a randomised, double-blind, placebo-controlled, phase 2b dose-finding trial. Lancet 399(10320):161–171. https://doi.org/10.1016/S0140-6736(21)02251-0
Bontscho J, Schreiber A, Manz RA, Schneider W, Luft FC, Kettritz R (2011) Myeloperoxidase-specific plasma cell depletion by bortezomib protects from anti-neutrophil cytoplasmic autoantibodies-induced glomerulonephritis. J Am Soc Nephrol 22(2):336–348. https://doi.org/10.1681/ASN.2010010034
Gomez AM, Vrolix K, Martinez-Martinez P, Molenaar PC, Phernambucq M, van der Esch E et al (2011) Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol 186(4):2503–2513. https://doi.org/10.4049/jimmunol.1002539
Taddeo A, Khodadadi L, Voigt C, Mumtaz IM, Cheng Q, Moser K et al (2015) Long-lived plasma cells are early and constantly generated in New Zealand Black/New Zealand white F1 mice and their therapeutic depletion requires a combined targeting of autoreactive plasma cells and their precursors. Arthritis Res Ther 17:39. https://doi.org/10.1186/s13075-015-0551-3
Rosenberg AS, Pariser AR, Diamond B, Yao L, Turka LA, Lacana E et al (2016) A role for plasma cell targeting agents in immune tolerance induction in autoimmune disease and antibody responses to therapeutic proteins. Clin Immunol 165:55–59. https://doi.org/10.1016/j.clim.2016.02.009
Mehta B, Mahadeo K, Zaw R, Tang S, Kapoor N, Abdel-Azim H (2014) Bortezomib for effective treatment of a child with refractory autoimmune hemolytic anemia post allogeneic hematopoietic stem cell transplant. Pediatr Blood Cancer 61(12):2324–2325. https://doi.org/10.1002/pbc.25172
Jakez-Ocampo J, Atisha-Fregoso Y, Llorente L (2015) Refractory primary Sjogren syndrome successfully treated with bortezomib. J Clin Rheumatol 21(1):31–32. https://doi.org/10.1097/RHU.0000000000000210
Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, Jansen G (2015) Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res Ther 17:17. https://doi.org/10.1186/s13075-015-0529-1
Khandelwal P, Davies SM, Grimley MS, Jordan MB, Curtis BR, Jodele S et al (2014) Bortezomib for refractory autoimmunity in pediatrics. Biol Blood Marrow Transplant 20(10):1654–1659. https://doi.org/10.1016/j.bbmt.2014.06.032
Yates S, Matevosyan K, Rutherford C, Shen YM, Sarode R (2014) Bortezomib for chronic relapsing thrombotic thrombocytopenic purpura: a case report. Transfusion 54(8):2064–2067. https://doi.org/10.1111/trf.12614
van Balen T, Schreuder MF, de Jong H, van de Kar NC (2014) Refractory thrombotic thrombocytopenic purpura in a 16-year-old girl: successful treatment with bortezomib. Eur J Haematol 92(1):80–82. https://doi.org/10.1111/ejh.12206
Gomez AM, Willcox N, Molenaar PC, Buurman W, Martinez-Martinez P, De Baets MH et al (2012) Targeting plasma cells with proteasome inhibitors: possible roles in treating myasthenia gravis? Ann N Y Acad Sci 1274:48–59. https://doi.org/10.1111/j.1749-6632.2012.06824.x
Danchaivijitr P, Yared J, Rapoport AP (2011) Successful treatment of IgG and complement-mediated autoimmune hemolytic anemia with bortezomib and low-dose cyclophosphamide. Am J Hematol 86(3):331–332. https://doi.org/10.1002/ajh.21950
Hiepe F, Radbruch A (2016) Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat Rev Nephrol 12(4):232–240. https://doi.org/10.1038/nrneph.2016.20
Mauri C, Menon M (2015) The expanding family of regulatory B cells. Int Immunol 27(10):479–486. https://doi.org/10.1093/intimm/dxv038
Barr TA, Brown S, Mastroeni P, Gray D (2010) TLR and B cell receptor signals to B cells differentially program primary and memory Th1 responses to Salmonella enterica. J Immunol 185(5):2783–2789. https://doi.org/10.4049/jimmunol.1001431
Muscate F, Woestemeier A, Gagliani N (2021) Functional heterogeneity of CD4(+) T cells in liver inflammation. Semin Immunopathol 43(4):549–561. https://doi.org/10.1007/s00281-021-00881-w
Parodis I, Stockfelt M, Sjowall C (2020) B Cell therapy in systemic lupus erythematosus: from rationale to clinical practice. Front Med (Lausanne) 7:316. https://doi.org/10.3389/fmed.2020.00316
Wu F, Gao J, Kang J, Wang X, Niu Q, Liu J et al (2021) B cells in rheumatoid arthritispathogenic mechanisms and treatment prospects. Front Immunol 12:750753. https://doi.org/10.3389/fimmu.2021.750753
Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D (2021) Autoimmune hepatitis: serum autoantibodies in clinical practice. Clin Rev Allergy Immunol. https://doi.org/10.1007/s12016-021-08888-9
Villalta D, Girolami E, Alessio MG, Sorrentino MC, Tampoia M, Brusca I et al (2016) Autoantibody profiling in a cohort of pediatric and adult patients with autoimmune hepatitis. J Clin Lab Anal 30(1):41–46. https://doi.org/10.1002/jcla.21813
Strassburg CP, Obermayer-Straub P, Alex B, Durazzo M, Rizzetto M, Tukey RH et al (1996) Autoantibodies against glucuronosyltransferases differ between viral hepatitis and autoimmune hepatitis. Gastroenterology 111(6):1576–1586. https://doi.org/10.1016/s0016-5085(96)70020-3
Martini E, Abuaf N, Cavalli F, Durand V, Johanet C, Homberg JC (1988) Antibody to liver cytosol (anti-LC1) in patients with autoimmune chronic active hepatitis type 2. Hepatology 8(6):1662–1666. https://doi.org/10.1002/hep.1840080632
Kanzler S, Weidemann C, Gerken G, Lohr HF, Galle PR, Meyer zumBuschenfelde KH et al (1999) Clinical significance of autoantibodies to soluble liver antigen in autoimmune hepatitis. J Hepatol 31(4):635–40
Ballot E, Homberg JC, Johanet C (2000) Antibodies to soluble liver antigen: an additional marker in type 1 auto-immune hepatitis. J Hepatol 33(2):208–215. https://doi.org/10.1016/s0168-8278(00)80361-x
Czaja AJ, Donaldson PT, Lohse AW (2002) Antibodies to soluble liver antigen/liver pancreas and HLA risk factors for type 1 autoimmune hepatitis. Am J Gastroenterol 97(2):413–419. https://doi.org/10.1111/j.1572-0241.2002.05479.x
Kirstein MM, Metzler F, Geiger E, Heinrich E, Hallensleben M, Manns MP et al (2015) Prediction of short- and long-term outcome in patients with autoimmune hepatitis. Hepatology 62(5):1524–1535. https://doi.org/10.1002/hep.27983
Rigopoulou EI, Roggenbuck D, Smyk DS, Liaskos C, Mytilinaiou MG, Feist E et al (2012) Asialoglycoprotein receptor (ASGPR) as target autoantigen in liver autoimmunity: lost and found. Autoimmun Rev 12(2):260–269. https://doi.org/10.1016/j.autrev.2012.04.005
Manns MP, Strassburg CP (2001) Autoimmune hepatitis: clinical challenges. Gastroenterology 120(6):1502–1517. https://doi.org/10.1053/gast.2001.24227
Zauli D, Ghetti S, Grassi A, Descovich C, Cassani F, Ballardini G et al (1997) Anti-neutrophil cytoplasmic antibodies in type 1 and 2 autoimmune hepatitis. Hepatology 25(5):1105–1107. https://doi.org/10.1002/hep.510250510
Terjung B, Bogsch F, Klein R, Sohne J, Reichel C, Wasmuth JC et al (2004) Diagnostic accuracy of atypical p-ANCA in autoimmune hepatitis using ROC- and multivariate regression analysis. Eur J Med Res 9(9):439–448
Sonthalia N, Rathi PM, Jain SS, Surude RG, Mohite AR, Pawar SV et al (2017) Natural history and treatment outcomes of severe autoimmune hepatitis. J Clin Gastroenterol 51(6):548–556. https://doi.org/10.1097/MCG.0000000000000805
Bergantini L, d’Alessandro M, Cameli P, Vietri L, Vagaggini C, Perrone A et al (2020) Effects of rituximab therapy on B cell differentiation and depletion. Clin Rheumatol 39(5):1415–1421. https://doi.org/10.1007/s10067-020-04996-7
Myhr KM, Torkildsen O, Lossius A, Bo L, Holmoy T (2019) B cell depletion in the treatment of multiple sclerosis. Expert Opin Biol Ther 19(3):261–271. https://doi.org/10.1080/14712598.2019.1568407
Babiker HM, Glode AE, Cooke LS, Mahadevan D (2018) Ublituximab for the treatment of CD20 positive B-cell malignancies. Expert Opin Investig Drugs 27(4):407–412. https://doi.org/10.1080/13543784.2018.1459560
Freeman CL, Sehn LH (2018) A tale of two antibodies: obinutuzumab versus rituximab. Br J Haematol 182(1):29–45. https://doi.org/10.1111/bjh.15232
Cartron G, Watier H (2017) Obinutuzumab: what is there to learn from clinical trials? Blood 130(5):581–589. https://doi.org/10.1182/blood-2017-03-771832
Chen D, Gallagher S, Monson NL, Herbst R, Wang Y (2016) Inebilizumab, a B cell-depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med 5(12). https://doi.org/10.3390/jcm5120107
Zhao Q (2020) Bispecific antibodies for autoimmune and inflammatory diseases: clinical progress to date. BioDrugs 34(2):111–119. https://doi.org/10.1007/s40259-019-00400-2
Chu SY, Pong E, Bonzon C, Yu N, Jacob CO, Chalmers SA et al (2021) Inhibition of B cell activation following in vivo co-engagement of B cell antigen receptor and Fcgamma receptor IIb in non-autoimmune-prone and SLE-prone mice. J Transl Autoimmun 4:100075. https://doi.org/10.1016/j.jtauto.2020.100075
Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A et al (2012) Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum 64(7):2328–2337. https://doi.org/10.1002/art.34400
Ramskold D, Parodis I, Lakshmikanth T, Sippl N, Khademi M, Chen Y et al (2019) B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine 40:517–527. https://doi.org/10.1016/j.ebiom.2018.12.035
Bonelli M, Scheinecker C (2018) How does abatacept really work in rheumatoid arthritis? Curr Opin Rheumatol 30(3):295–300. https://doi.org/10.1097/BOR.0000000000000491
Acknowledgements
All figures were generated using BioRender. Figure 1 was adapted from “B-1 and B-2 Cell Development,” by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study is funded by DFG SFB 841 and YAEL foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on: Tolerance and autoimmunity in the liver - Guest Editors: Christoph Schramm, Ansgar Lohse & Ye Oo
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schultheiß, C., Steinmann, S., Lohse, A.W. et al. B cells in autoimmune hepatitis: bystanders or central players?. Semin Immunopathol 44, 411–427 (2022). https://doi.org/10.1007/s00281-022-00937-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00937-5