
Abstract
Forming the outer body barrier, our skin is permanently exposed to pathogens and environmental hazards. Therefore, skin diseases are among the most common disorders. In many of them, the immune system plays a crucial pathogenetic role. For didactic and therapeutic reasons, classification of such immune-mediated skin diseases according to the underlying dominant immune mechanism rather than to their clinical manifestation appears to be reasonable. Immune-mediated skin diseases may be mediated mainly by T cells, by the humoral immune system, or by uncontrolled unspecific inflammation. According to the involved T cell subpopulation, T cell–mediated diseases may be further subdivided into T1 cell–dominated (e.g., vitiligo), T2 cell–dominated (e.g., acute atopic dermatitis), T17/T22 cell–dominated (e.g., psoriasis), and Treg cell–dominated (e.g., melanoma) responses. Moreover, T cell–dependent and -independent responses may occur simultaneously in selected diseases (e.g., hidradenitis suppurativa). The effector mechanisms of the respective T cell subpopulations determine the molecular changes in the local tissue cells, leading to specific microscopic and macroscopic skin alterations. In this article, we show how the increasing knowledge of the T cell biology has been comprehensively translated into the pathogenetic understanding of respective model skin diseases and, based thereon, has revolutionized their daily clinical management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
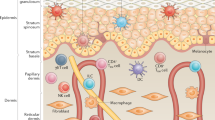
As the barrier between the organism and the environment, the skin protects against external hazards, noxious substances, and pathogens. It is the largest and heaviest human organ and is built up by three distinct layers: the outermost epidermis, the dermis, and the subcutis [1,2,3]. The numerically dominant cell population of the continuously renewing epidermis is that of keratinocytes. Proliferation of these cells usually takes place within the stratum basale (attached to the basement membrane). Here, the epidermal stem cells are situated, which, upon their rare divisions, deliver the so-called transit amplifying cells (TA cells), a frequently proliferating population [1, 4]. Each division of an individual TA cell delivers a daughter cell able to leave the basement membrane and to start terminal differentiation in the suprabasal stratum spinosum. During terminal differentiation, these cells move upward, becoming granular (stratum granulosum) and undergoing a particular apoptotic process to become horny constructs. Tightly covering the skin surface, these so-called corneocytes form the uppermost layer of the epidermis, the stratum corneum [1,2,3]. Under normal conditions, there is a balance between stem cell proliferation, TA cells, terminal differentiation, and the continuous desquamation of corneocytes from the skin surface (about 50 billion daily). This equilibrium is markedly disrupted in some chronic immune-mediated skin diseases [5]. Besides keratinocytes, Merkel cells, melanocytes, and immune cells, including Langerhans cells and resident memory CD8+ T cells, are also present in the epidermis [6, 7]. The dermis, lying under the epidermis, consists of connective tissue containing collagenous, elastic, and reticular fibers as well as fibroblasts and hosts immune cells like macrophages, immature dendritic cells (DCs), mast cells, and some resident memory CD4+ T cells.
The permanent contact of the skin with exogenous stimuli and antigens frequently leads to activation of the resident immune cells. The cutaneous persistence of the stimulus/antigen and/or a relative deficiency of counter-regulatory mechanisms, particularly in the context of a genetic predisposition, results in local immune cell infiltration and chronic activation, which also involves the cutaneous tissue cells. Hence, it is not surprising that chronic immune-mediated skin diseases are some of the most common disorders in humans. For the affected patients, these diseases induce not only physical but also psychological burdens due to the visibility of the symptoms and the frequent association with itching, pain, and burning [8,9,10]. They may be primarily mediated by the uncontrolled activation of T cells, the humoral immune system, or unspecific inflammation (innate immunity). Disorders dominated by pathogenic CD4+ and/or CD8+ T cells comprise the largest group within the chronic immune-mediated skin diseases [11]. A deeper understanding of the molecular and cellular mechanisms underlying these disorders might lead to the identification of novel target molecules and, as a consequence, to the development of innovative therapeutic strategies. In this review, we will discuss the mechanisms of development and maintenance of specialized T cell subtypes and refer to representative diseases, in which the specific T cell subtypes play a crucial pathogenic role.
Characteristics, development, and functions of T cell subpopulations
T cells, a central component of the adaptive immunity, play a pivotal role in the defense against pathogens and tumors, while their dysregulation contributes to the development and maintenance of various diseases. T cells mature in the thymus, where they undergo somatic gene rearrangement resulting in the expression of a unique T cell receptor (TCR) [12]. During the positive selection process, detection of antigens presented on major histocompatibility complex class 1 (MHCI) or class 2 (MHCII) by the rearranged TCR implements either a CD8+ or CD4+ T cell lineage fate, respectively [13]. Presentation of autoantigens in the thymic medulla ensures the elimination of autoreactive T cells [14], and remaining T cells egress into circulation where they patrol blood and lymph as CD45RA+CCR7+ naïve T cells [15].
When T cells bind their cognate antigen by the TCR accompanied by a sufficient co-stimulatory signal, they become activated, start proliferating, and contribute to pathogen clearance as effector cells [16]. After pathogen clearance, 95% of the effector cells undergo apoptosis; the remaining T cells give rise to a highly specialized set of memory cells that have lost CD45RA expression and instead express CD45RO [17]. The memory compartment can be subdivided into CCR7+ central memory (TCM), CCR7- effector memory (TEM), and CCR7- effector memory T cells re-expressing CD45RA (TEMRA) [15]. While TCM migrate through lymphatic tissue and were described to be less responsive, TEM patrol peripheral tissues and provide rapid effector function upon reactivation [15]. Single-cell-based experiments suggest a progressive maturation of T cells from naive via TCM and TEM to TEMRA cells that are associated with chronic activation and display features of exhaustion such as impaired cytokine secretion and the expression of exhaustion markers such as programmed death-1 (PD-1) and TIM3 [18,19,20]. Upon migration into the different lymphoid and non-lymphoid tissues, some memory T cells gain tissue residency characterized by the upregulation of CD69 and CD103, the hallmarks of tissue-resident memory T cells (TRM), as well as expression of Hobit and Blimp1, which together suppress the re-egress into circulation [21]. Those cells exhibit an outstanding long-term maintenance and act as sentinels that protect against re-infections.
Antigen detection by CD4+ T cells is restricted to MHCII expressed on professional antigen-presenting cells (APCs) including dendritic cells (DCs), B cells, and macrophages [22]. These APCs continuously sample proteins and present peptides derived thereof on their surface. In the presence of conserved pathogenic structures or danger signals that are released by distressed cells, they become activated and migrate to the secondary lymphoid organs, where they in turn activate CD4+ T cells [23, 24]. A central role of CD4+ T cells is to migrate into B cell follicles upon activation, where they mediate B cell help by CD40L:CD40 interaction. This CD40L-mediated help is indispensable for the induction of germinal center formation, antibody class switch, and somatic hypermutation [25]. During T cell activation, the cytokine milieu at the site of infection moreover modulates the differentiation and subsequent specialization of the T cells, allowing pathogen-tailored responses. The cytokines IL-12 and IFN-γ induce the expression of the transcription factor T-bet in T cells, resulting into IFN-γ-producing type 1 helper (Th1) T cells that contribute to the clearance of virus-infected cells and intracellular pathogens [26]. Induction of Gata3 expression by IL-4 gives rise to type 2 helper (Th2) cells secreting IL-4, IL-5, and IL-13, which are critical mediators of extracellular parasite expulsion and mediate B cell class switch [27]. In recent years, the spectrum of CD4+ T cell subsets rapidly broadened by the identification of type 17 and type 22 helper (Th17, Th22), T follicular helper (Tfh) and regulatory (Treg) T cells. Th17 cells differentiate upon RORγt expression and produce IL-17 that induces epithelial antimicrobial defense and leads to recruitment and activation of neutrophils [28]. The work of Acosta-Rodriguez et al. suggests that IL-17-producing cells are a heterogeneous population consisting of Th17 cells dominating anti-fungal responses, whereas Th17+1 cells additionally secrete IFN-γ and are the main responders in the defense of extracellular bacteria [29]. Th22 cells differentiate upon aryl hydrocarbon receptor expression, act on epithelial cells like keratinocytes by IL-22 secretion, and promote wound healing and tissue protection against damage [30]. Upon activation, some naïve CD4+ T cells upregulate the transcription factor Bcl6 and migrate into B cell follicles where they become resident Tfh cells contributing to germinal center formation [31, 32]. In contrast, FoxP3+ Treg cells do not contribute to pathogen defense but instead prevent autoimmune disorders by suppressing unwanted immune responses [33]. The major population of CD4+ Treg cells was found to be characterized by high expression of the IL-2 receptor alpha chain (CD25), and the transcription factor FoxP3, the latter being indispensable for the development and suppressive function of Treg cells [34, 35].
Extensive analyses of the CD4+ T cell subsets revealed differing migration abilities, which are reflected by the expression of unique sets of chemokine receptors that mediate migration along a chemokine gradient. Combinations of the chemokine receptors CCR4, CCR6, CCR10, and CXCR3 were identified as separators of Th1 (CCR6-CCR4-CXCR3+), Th2 (CCR6-CCR4+CXCR3-), Th17+1 (CCR6+CCR4-CXCR3+), Th17 (CCR6+CCR4+CXCR3-CCR10-), and Th22 (CCR6+CCR4+CXCR3-CCR10+) cells [29, 36, 37]. CCR6 expression is—together with CD161—a common feature of IL-17-secreting cells [29, 38, 39]. The ligand for CCR6 is CCL20, which is predominantly produced by epithelial cells, organ-associated lymphoid tissues, and liver, allowing a broad migration pattern that is specified by the co-expression of further chemokine receptors [40]. In contrast, CCR4 and CCR10 expression is implemented by DCs in skin-draining lymph nodes and allows the chemotactic migration along CCL17/CCL22 and CCL27/28, respectively [41, 42]. CCR4- and CCR10-expressing T cells co-express the so-called cutaneous leukocyte-antigen (CLA) and altogether mediate homing into the skin [43]. CXCR3 binds to CXCL9, CXCL10, and CXCL11, which are secreted in the presence of IFN-γ and recruit CXCR3+ cells to sites of inflammation (reviewed in [44]). Beyond, the expression of CXCR5 is characteristic of Tfh cells, which binds the chemokine CXCL13 secreted by the follicular stroma, allowing the recruitment into the B cell follicle zones [45]. This concerted differentiation of T cells orchestrated by DCs ensures the right response at the right place in the body. CD4+ T cells possess a broad flexibility regarding the subset they differentiate into. By comparison of the TCR clone repertoire in Mycobacterium tuberculosis and Candida albicans infection, Becattini et al. could demonstrate overlaps in the clones found in the different CD4+ helper subsets, suggesting that priming of a single naïve CD4+ T cell can give rise to multiple fates [46].
In contrast to CD4+ T cells, CD8+ T cells were described cytotoxic T lymphocytes (Tc) that directly kill malign or infected cells. They detect antigens presented by MHCI, which is expressed by almost every cell in the body, to either eliminate the cell by the secretion of cytolytic molecules including perforin and granzymes or to induce Fas-mediated apoptosis (reviewed in [47]). In the memory stage, most (Tc1) cytotoxic CD8+ T cells express the transcription factor T-bet and secrete high levels of IFN-γ. However, some CD8+ T cells were identified that express Gata3 and display a type 2 cytotoxic (Tc2) T cell phenotype with secretion of IL-4, IL-5, and IL-13. Comparable with their CD4+ Th2 counterparts, they possess a CCR4+ and CRTH2+ phenotype [48]. Effector profiles of CD8+ T cells in multiple diseases such as psoriasis vulgaris demonstrated that, among memory CD8+ T cells, also IL-17, IL-22, and IL-17/IFN-γ producers exist [49]. We could demonstrate that the CD8+ T cell subsets Tc1, Tc2, Tc17, Tc1+1, and Tc22 express the same set of chemokine receptors and utilize the same differentiation programs based on T-bet, Gata3, RORγt, and aryl hydrocarbon receptor as do CD4+ T cells (Loyal et al., manuscript under review). While Tc1 and Tc17+1 CD8+ T cells display a classical cytotoxic phenotype, Tc2, Tc17, and Tc22 lack the capability to kill target cells and express the Th cell–typical molecule CD40L instead (Loyal et al., manuscript under review; [50]). In contrast to CD4+ T cells, the differentiation flexibility is restricted among CD8+ T cells, with a certain flexibility to gain Tc1 or Tc17+1 phenotype on the one side or to gain Tc2, Tc17, or Tc22 phenotype on the other side, but with very little clonal overlap between these two groups (Loyal et al., manuscript under review). They share the ability to secrete IL-13 and provide CD40L-dependent help (Loyal et al., manuscript under review; [50]). This striking effect might be caused by the site of priming, the involved APC, the priming conditions, and especially the type of antigen that gives rise to non-cytotoxic, “helper-type” CD8+ T cells. Their chemokine receptor expression, effector profile, and lack of cytotoxicity suggest a tissue homeostasis–maintaining function instead of contribution to the elimination of infected/malign/distorted cells. Cheuk et al. demonstrated that, in human skin, a significant fraction of CD8+ T cells lack cytotoxic features including the expression of CD49a and instead produce IL-17 [51]. In a murine model, those skin Tc17 cells were shown to contribute to wound healing by IL-13 release upon recognition of non-classical MHCI (H2-M3)-presented peptides derived from commensal bacteria [52, 53]. Altogether, CD4+ and CD8+ T cells provide a broad repertoire of highly specific features and functions adapted to the diverse spectrum of challenges such as infections but also tissue homeostasis, wound healing, and tolerance as summarized in Fig. 1.

Phenotype and function of T cell subsets
Besides their role in pathogen defense, the activation of skin-directed T cells can lead to chronic T cell–mediated skin diseases, whose characteristics correspond to the specific effector mechanisms of the different T cell subpopulations. In fact, those diseases may be dominated by a Th/c1-specific (e.g., vitiligo), Th/c2-specific (e.g., acute stage of atopic dermatitis), or Th/c17-/Th/c22-specific (e.g., psoriasis) pattern. Moreover, the activation of Treg cells is associated with skin tumors like melanoma, a malignant condition derived from melanocytes. Finally, T cell responses may be paralled by non-T cell responses, as in the case of hidradenitis suppurativa, where T17 cell activation [54, 55], a relative deficiency in Treg cells [56], and strong unspecific inflammation [57] have been found.
Since, as described above, the classical division of T cells into CD4+ helper T cells and CD8+ cytotoxic T cells has become out of date, the following sections will use the terms T1, T2, T17, T22, and Treg cells. The role of specialized T cells in representative skin diseases will be discussed in the following.
Common T cell–related skin disorders
T1 cells and vitiligo
Epidemiology, clinical, and histological characteristics
Vitiligo is a chronic disease characterized by the appearance of pigment-free patches of the skin and rarely of the mucosa. In Europe, about 0.5–2% of people suffer from vitiligo. However, the prevalence of vitiligo in, e.g., India and Arabic countries, is appreciably higher [58, 59]. Both sexes are equally affected, and a quarter of the cases concern children [60]. Typically, the vitiligo patches are sharply demarcated and differ in shape and size [58, 61]. However, apart from the depigmentation, they are macroscopically very similar to intact skin. Periorbital, perioral, and acral regions of the body are often affected. During the course of the disease, the number and size of the depigmented patches can increase, and patches can coalesce. As expected from the loss of pigment, the histological examination of vitiligo patches shows lack of melanocytes and melanin-containing keratinocytes in the stratum basale [61, 62]. During the early stage, a perivascular lymphocyte infiltration is observed in the dermis. In the late stage, though, the lymphocytes are mainly present at the edge of the patches [62].
Immunopathophysiology of vitiligo
Every tenth cell in the stratum basale of healthy skin is a melanocyte, and - under physiological conditions - melanocytes are not attacked by the immune system. However, melanocytes can be targeted or even destroyed by T cell–mediated immune responses initiated by autoimmune processes or therapeutic intervention. Like in other autoimmune diseases, genetic predisposition is also present in individuals with vitiligo. Besides certain HLA genotypes, patients with vitiligo can show single nucleotide polymorphisms (SNPs) within genes that are implicated in T cell signaling or activation (NLRP1, TICAM1, FOXP3, BACH2, PTPN22, CD80) or genes associated with cytotoxic T cell responses (GZMB, IL2RA) [60]. Nonetheless, the role of genetic predisposition in vitiligo etiology seems to be less important than that in other chronic T cell–mediated diseases like psoriasis or atopic dermatitis. In fact, only 1–10% of vitiligo patients have a positive family history for vitiligo in contrast to about 30% in the case of psoriasis.
It is generally accepted that the first step in vitiligo pathogenesis is a slight damage of melanocytes, e.g., by ultraviolet (UV) radiation or chemical substances. Such damage leads to an increase of reactive oxygen species (ROS), in particular when low levels of enzymatic and non-enzymatic antioxidants are present [63]. In fact, the impairment of the nuclear factor E2-related factor 2 (Nrf2), a protein important for protection against oxidative stress, seems to be critical for the increased sensitivity of vitiligo melanocytes to oxidative stress [64] as observed in lesional and non-lesional skin of patients. ROS and respective chemical substances provoke alteration of the folding machinery of the endoplasmic reticulum, leading to accumulation of immature proteins and finally to autophagy or apoptosis [60]. The increase of ROS is associated with the release of melanocyte-specific antigens and molecules like heat-shock proteins (HSPs) and self RNA/DNA, which activate pathogen recognition receptors on macrophages and DCs [63]. As reported, inducible HSP70 promotes an inflammatory DC phenotype and accelerates disease progression in a murine model of vitiligo [65].
The described events induce generation of T1 cells in lymph nodes that are specific for melanocyte antigens (Fig. 2). The infiltration of such T cells into the skin seems to depend on the chemokine receptor CXCR3 expressed by T1 cells and its ligands CXCL9, CXCL10, and CXCL11 produced by cutaneous tissue cells like keratinocytes [66]. Interestingly, vitiligo mouse models suggest that CXCL9 promotes Tc1 recruitment into the skin but not their effector function, whereas CXCL10 is required for effector function [67]. In the progressive phase of the disease, the immigrated T1 cells, in particular Tc1 cells, destroy melanocytes through the production of IFN-γ and TNF-α as well as cytotoxic molecules like granzyme B and perforin [51] (Fig. 2). In fact, Tc1 cells isolated from the edges of patches induced apoptosis in autologous melanocytes in co-cultures in vitro [68]. Furthermore, IFN-γ induces CXCL9 and CXCL10 in cutaneous tissue cells [69]. In contrast, the Treg cell response in the skin of patients with vitiligo seems to be limited [70], so that Treg cells are not able to prevent the cytotoxic IFN-γ-dominated T1 cell response [71]. Of note, individuals with vitiligo have a lower risk for developing malignant melanoma (see below). This observation shows that immune activation directed against melanocytic antigens can be of benefit in the setting of carcinogenesis.

Immunopathophysiology of vitiligo
Besides IFN-γ, IL-17 expression is also increased in perilesional skin of vitiligo patients, where T cells appear as the main source of this cytokine [72]. Since depigmentation is not a typical finding in psoriasis, a disease with high IL-17 expression in the skin, it is questionable, whether IL-17 significantly contributes to vitiligo pathogenesis.
Immunopathology-based therapy of vitiligo
Treating vitiligo is a challenge, since no systemic therapies are yet available. Understanding the exact pathogenetic processes in vitiligo could help in developing successful therapeutic strategies (Table 1). The dominant role of IFN-γ in the depigmentation in mouse models of vitiligo [73] suggests that neutralizing this cytokine, inhibiting its production or signaling pathway, may help to stop the disease. More recently, two case reports described that patches rapidly repigmented in vitiligo patients treated with JAK inhibitors like tofacitinib or ruxolitinib that interfere with IFN-γ signaling [74]. Since cumulating reports show that pathogenic T1 cells in vitiligo are tissue-resident memory T cells, interventions focused on IFN-γ neutralization or hindrance of the effect of this cytokine should be periodically repeated [51]. Besides IFN-γ as cytokine factor, T cell–based cytotoxic mechanisms are involved in melanocyte destruction. Thus, the depletion of T1 cells or the inhibition of their migration into the skin may result in promising approaches. The minimization of skin infiltration by T1 cells might be achieved by inhibiting CXCR3 function, as demonstrated in experimental mice [67]. Targeting the CXCR3 chemokine receptor to deplete T1 cells from skin is another alternative approach, as also recently demonstrated in mice [75]. Interestingly, this latter approach did not only prevent depigmentation but also lead to perifollicular re-pigmentation.
T2 cells and atopic dermatitis
Clinical and histological picture of atopic dermatitis
A more frequent T cell–mediated skin disease than vitiligo is atopic dermatitis. It usually begins in infancy. Its prevalence is very high in the Western population, with 15–20% of children and 3–4% of adults being affected [10]. The clinical manifestation of atopic dermatitis is age- and stage-dependent. While, in infants, skin lesions occur especially in the face and on the scalp, at later age, the flexural surfaces of the elbows and knees, the hands, feet, and the neck are increasingly affected. Acute lesions present as strongly itchy with red papules, serous exudation, and crusting. Histologically, edemas, vesiculation, and moderate hypogranularity and hyperkeratosis can be observed in the epidermis. Immune infiltration of the skin includes T cells, mast cells and eosinophilic granulocytes, macrophages, and DCs. Chronic lesions show increased collagen deposition in the dermis resulting in skin lichenification. Microscopically, acanthosis and more macrophage-dominated dermal infiltrations are visible at this stage. In contrast to psoriasis, lesions are less clearly demarcated [76]. In addition to the cutaneous alterations, 80% of patients suffer from allergies and often develop allergic asthma and rhinitis (extrinsic disease) [77].
Immunopathophysiology of atopic dermatitis
Atopic dermatitis has a multifactorial nature with a genetic component and environmental factors being involved (Fig. 3). A positive family history has been reported in 40–60% of patients [78, 79]. The strongest genetic association concerns the gene encoding the skin-barrier molecule filaggrin (FLG). In fact, 20–30% of patients carry a FLG null mutation [80]. This matches the fact that the impaired skin barrier is an essential factor in the pathogenesis and correlates with the severity of this disease [81]. Atopic dermatitis has also been linked to variants within the genes encoding the T2 pathway-associated cytokines/cytokine receptors IL-4, IL-13, IL-4RA, and IL-31 and associated downstream molecules like STAT6 and GATA3 [82]. A characteristic MHC variant reported in some patient populations with atopic dermatitis is HLA-DRB1 [83]. Exogenous triggers of the disease include allergens, microbial antigens/superantigens, mental stress, and scratching of the skin [76].

Immunopathophysiology of atopic dermatitis
T2 cell mediators are crucial for the pathogenesis of atopic dermatitis [80] (Fig. 3). At the chronic disease stage, the T22 mediator IL-22 is also of relevance [84, 85]. IL-4, IL-13, IL-31, and IL-22 seem to interfere with keratinocyte terminal differentiation [86,87,88,89]. This may explain the decreased epidermal expression of filaggrin and other molecules necessary for skin differentiation and barrier function even in patients without FLG mutation. IL-31 is involved in the pathophysiology of itching, a characteristic finding patients with atopic dermatitis suffer from. IL-5 is known to activate eosinophilic granulocytes that are found histologically in skin lesions from the patients. IL-4 also promotes production of IgE, which shows elevated serum levels in the majority of patients with atopic dermatitis [80]. Chronic lesions also express moderate levels of the T1 cell cytokine IFN-γ, while IL-17 can hardly be detected [76, 90].
The aforementioned cytokine pattern is also the reason for the deficient epidermal production of anti-microbial proteins (AMPs) and antiviral proteins (AVPs) atopic dermatitis patients show (Fig. 3). In fact, high T2 cytokine/low IL-17 levels result in low AMP production by keratinocytes [91,92,93], while lacking expression of the T17 cytokine IL-29 is associated with impaired keratinocyte AVP expression [94] (see also “Common T cell–related disorder”). Low AMP levels in the barrier-disturbed skin of atopic dermatitis patients predestinate for atypical cutaneous colonization with Staphylococcus aureus, penetration of microbial pathogens and their immunostimulating constituents into the skin, and infections with this pathogen [95,96,97,98,99]. Interestingly, subclinical S. aureus colonization also occurs in non-lesional skin of patients, correlating with disturbed skin barrier function and disease extent [99, 100]. Atopic dermatitis patients also show an increased risk of developing skin infections with viral pathogens, including human papillomavirus, herpes simplex virus (HSV), and molluscum contagiosum virus [95, 98]. In rare cases, HSV infection may spread and cause Ekzema herpeticatum [101]. Importantly, the impaired skin barrier function also promotes epicutaneous sensitization to allergens and may explain the high allergy frequency in affected patients.
Immunopathology-based therapy of atopic dermatitis
Classically, topical corticosteroids and, for more severe disease, systemic immunosuppressive agents are used. Since 2016, topical phosphodiesterase inhibitors such as crisaborole [102] (for the treatment of mild to moderate disease) are approved. More recently, the first biologic for treating atopic dermatitis, namely dupilumab, was introduced [103]. This antibody targets the IL-4/IL-13 receptor and was approved in 2017 for the treatment of moderate to severe disease (Table 2). A large number of other biologics for the treatment of atopic dermatitis are under development (Table 3).
T17 cells, T22 cells, and psoriasis
Clinical and histological picture of psoriasis
With a prevalence of 2–3% in Western countries, psoriasis is another very common T cell–mediated skin disease [104]. Psoriasis manifests with sharply demarcated, raised, erythematous plaques covered by silvery scales. Lesions preferentially develop in mechanically stressed areas such as the extensor sides of the arms and legs, the sacral region, and the head [105].
Microscopically, psoriatic skin lesions show a massively thickened epidermis. This is the result of a substantial elongation of the epidermal rete ridges and an increased stratum corneum (hyperkeratosis). Furthermore, a reduced stratum granulosum and presence of nuclear remnants in the stratum corneum (parakeratosis) are typical features. Mechanistically, these changes are based on excessive proliferation of basal keratinocytes (TA cells) and an impaired cornification process of the keratinocytes of the upper epidermal layers [106]. In the dermis, dilatated blood capillaries greatly extend between epidermal rete ridges toward the skin surface [107]. The massive immune cell infiltration, which is most prominent in the dermis but not restricted to it, predominantly consists of monocytes/macrophages, dendritic cells, and T cells [108]. There are also accumulations of partially netose-forming neutrophilic granulocytes in the stratum corneum, called Munro’s microabscesses [109].
Interestingly, inflammation in psoriasis is not restricted to the skin. More than 20% of patients show involvement of the joints [110]. In addition, the prevalence of colitis is increased, and metabolic and cardiovascular alterations lead to a shortened life expectancy in the patients [111, 112].
Immunopathophysiology of psoriasis
Both genetic and extern/lifestyle factors are involved in the development of psoriatic skin alterations (Fig. 4). Approximately 75% percent of patients report a positive family history [113]. A great proportion of patients carry the MHC haplotype HLA-Cw6 [113, 114], which has been correlated with certain clinical characteristics and therapeutical outcome in patients with psoriasis [115]. In addition, proposed autoantigens in psoriasis like the cathelicidin-derived peptide LL-37 and the melanocytic protein ADAMTSL5 were demonstrated to have T cell–stimulatory activity in HLA-Cw6-carrying patients [115]. Furthermore, there are associations with genes related to the keratinocyte terminal differentiation, antimicrobial defense, and the T17 cell pathway [116]. Regarding the latter (see also below), psoriasis has been linked for example to polymorphisms within IL12B, IL23A, IL23R, and, in some patients with a special psoriasis subtype, pustular psoriasis, also IL36RN. Moreover, there are associations with variants in REL, TYK2, RUNX3, STAT3, and TRAF3IP2 [115]. Exogenous triggering factor for psoriasis involves mechanical skin trauma, streptococcal infections, and certain drugs [115].

Immunopathophysiology of psoriasis
The central pathways crucial to psoriasis pathogenesis involve T17 and T22 cells, whose mediators and upstream and downstream molecules are highly present in the lesions (Fig. 4). In addition to T cells (CD4+ and CD8+), type 3 innate lymphoid cells play a role as producers of IL-17 and IL-22 [117, 118]. One of the most relevant cytokines promoting IL-17 and IL-22 expression by immune cells is IL-23. This heterodimeric cytokine is highly expressed in psoriatic skin. IL-23 inhibits IL-10 production by T17 cells and instead induces an inflammatory T17 phenotype [119]. Moreover, TNF-α, primarily secreted by T17 cells, T22 cells, and macrophages, as well as the T1 cell mediator IFN-γ are abundant in the psoriatic skin [120]. In sharp contrast, IL-4 is not found in psoriatic lesions.
Main target cells of IL-17A, IL-17F, and IL-22 in the skin are keratinocytes, although IL-17 effects were also described for immune cells and other tissue cells. In keratinocytes, IL-17 induces the production of selected chemokines (such as CCL20, which attracts T17 cells, T22 cells, and DCs, as well as CXCL1, CXCL2, CXCL5, and CXCL8, which all attract neutrophilic granulocytes) and other cytokines (such as IL-6, the granulocyte-activating cytokine G-CSF, and IL-19) in the skin. Presumably, IL-17 alone causes only moderate cellular responses while mainly synergizing with TNF-α, and IL-22 [121,122,123,124,125,126,127]. Together with IL-22, IL-17 induces the production of AMPs and therefore plays an essential role in the remarkable immune defense of the psoriatic plaque against extracellular bacteria and fungi [54, 126, 128,129,130]. In fact, it is a peculiarity of psoriasis patients that the impairment of the skin barrier function is not associated with an increased skin infection risk [95, 96]. Apart from its function in antibacterial defense, IL-22 is the main mediator of the impaired keratinocyte cornification process in psoriasis. IL-22 reduces the expression of molecules like filaggrin required for the terminal differentiation of keratinocytes [89]. The consequences of the IL-22-mediated inhibition of the keratinocyte terminal differentiation are reflected by a marked epidermal thickening and hypogranulosis of reconstituted three-dimensional human epidermis models and IL-22-transgenic mice [88]. Apart from the direct effects of IL-22, this mediator also acts via the induction of IL-20 in keratinocytes, which, by partially using the same receptor (IL-22R1), can exert IL-22-like effects [131]. It should be noted that the activity of IL-22 is regulated by IL-22 binding protein [132]. In psoriasis patients, the expression of this natural inhibitor is downregulated in non-lesional skin, and such downregulation is associated with an increased sensitivity of the skin to the pathogenetic action of IL-22 [133]. Increased levels of the T1 cell cytokine IFN-γ may support the activation of dermal endothelia to allow infiltration of immune cells from the bloodstream into the psoriatic lesion [134]. IFN-γ further induces chemokines attracting T1 cells including CXCR10 [135] and upregulates the expression of MHC molecules on both tissue and antigen-presenting immune cells [134]. The pleiotropic and highly inflammatory cytokine TNF-α induces a wide range of immune cell–attracting chemokines and contributes to endothelial activation, two functions necessary for immune cell infiltration [136]. Many effects of cytokines are enhanced in the presence of TNF-α [121, 122, 125], arguing for a central role of this cytokine in skin inflammation.
Another mediator, which can be produced by T17 cells in psoriasis, is IL-29. IL-29 is able to inhibit the replication of viruses via the induction of AVPs [137] and seems to be responsible for the high resistance of psoriatic epidermis toward viral superinfections [94].
Immunopathology-based therapy of psoriasis
The excellent knowledge about the specific cytokine pathways involved in psoriasis pathogenesis has allowed the tremendous success in the development of innovative drugs for the treatment of moderate to severe psoriasis [108]. These include therapeutic antibodies that neutralize IL-17 and IL-23 as well as TNF-α (Table 2). A broad range of further drugs is under development [138, 139].
Treg cells and melanoma
Clinical and histological picture of melanoma
The worldwide incidence of cutaneous melanoma has been increasing annually at a more rapid rate compared with any other type of cancer. In 2012, 232,000 new cases of melanoma and 55,000 related deaths were registered worldwide, ranking 15th among most common cancers [140]. About 90% of melanoma cases are diagnosed as primary tumors without evidence of metastasis, and their 10-year survival is between 75 and 80% [141]. Metastases, which can develop either via the lymphatic or the hematogenous route, are the main cause of death in melanoma patients [142]. Disease is subclassified to estimate prognosis and determine therapeutic interventions. This classification considers TNM criteria together with tumor thickness, ulceration, mitotic figures, and microscopic satellites of the primary tumor.
UV light radiation from sunlight, in particular the UV-B spectrum, is the main environmental risk factor for melanoma skin cancer development [143]. Melanoma in chronically sun-exposed skin usually manifests in older-aged individuals and has a high tumor mutational burden related to UV exposure. The main genetic drivers are mutations in the genes encoding B-Raf proto-oncogene (BRAF), neurofibromin 1 (NF1), NRAS, and others. Melanomas associated with intermittently sun-exposed skin cases arise in younger-aged individuals and are usually associated with the BRAFV600E mutation and a lower mutational load [144, 145]. Up to 90% of melanomas exhibit an aberrant MAPK pathway activation as central step in melanoma development [146]. Furthermore, SNPs in genetic loci that associate with the risk for developing malignant melanoma have also been reported in patients. Examples of such genes are CDKN2A, CDK4, and others. These genetic findings helped to establish small molecular inhibitors of signaling pathways that promote melanoma.
Immunopathophysiology of melanoma
Melanoma is deemed one of the most immunogenic types of cancer. In fact, several melanoma-specific antigens have been identified and large numbers of melanoma-specific antibodies and functional lymphocytes are present in patients with melanoma [147]. Moreover, spontaneous regression of melanoma with simultaneous onset of vitiligo has been reported [148] and metastatic melanoma responds to immune-stimulating agents, such as IFNs and IL-2 as well as the novel immune checkpoint inhibitors blocking cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and PD-1 [149,150,151]. The major base for the strong immunogenicity of melanoma is its often very high (UV-driven) tumor mutational burden, which allows for the creation of neoantigens recognizable as “non-self” by host immune cells [152, 153]. Accordingly, strong immune cell infiltration is an established positive prognostic parameter in advanced melanoma [154, 155]. Despite the immunogenicity of melanoma and induction of tumor-specific immune responses [156], current immunotherapies show limited efficacy and are restricted to subpopulations of patients with advanced melanoma. It has been suggested that several negative factors hinder antitumor immune activities. These include (i) immune-suppressive cells like Treg cells and myeloid-derived suppressive cells, (ii) anti-inflammatory cytokines like tumor growth factor (TGF)-β and IL-10, (iii) defective antigen presentation by tumor cells because of antigen expression loss and antigen processing defects, (iv) immune inhibitory molecules like CTLA-4 and PD-1, and (v) amino acid–catabolizing enzymes like arginase and indoleamine-2-3 dioxygenase (IDO) [157].
In both animal models and human beings, Treg cells infiltrate into the tumor microenvironment, dampening immune responses to tumor cells [158,159,160]. Cell-to-cell contact, production of immune-suppressive cytokines like IL-10 and TGF-ß, competing for growth factors with other effector cells, and modification of APCs are the four main strategies how Treg cells apply to exert their inhibitory effects [161, 162]. As most tumor antigens are normal self-antigens, such tumors could induce tumor-specific Treg cells, suppressing effective antitumor responses [163, 164]. In animal models of melanoma, transient Treg cell depletion induces immune responses against tumor and improves survival, indicating the importance of these cells [165]. Wang et al. were the first to isolate Treg cells that recognize epitopes from the tumor-associated antigen LAGE-1 from patients with melanoma, providing evidence for the relevance of this mechanism also in the melanoma setting [166]. Tumor-specific Treg cells that can recognize a broad range of melanoma-associated antigens and neoantigens can be detected in the tumors and in the blood of melanoma patients [167, 168]. Fourcade et al. showed that the same melanoma-associated antigens can stimulate both Th and Treg cells [169]. As a consequence, immunotherapeutic vaccinations with melanoma-associated antigens in patients with melanoma can result in expansion of both induced and naturally occurring melanoma-associated Treg cells [170].
Numerous researches have indicated increased numbers of Treg cells not only in the local tumor microenvironment including primary and metastatic lesions but also in peripheral blood of subjects with metastatic melanoma, as well as in affected draining lymph nodes [159, 160]. Treg cell accumulation in the tumor microenvironment was reported to be predictive of reduced survival of melanoma patients [171]. Subsequently, several other retrospective studies demonstrated the correlation between Treg cell infiltration and prognosis of melanoma patients [172]. Vice versa, the parameter that best correlates with favorable clinical outcome and survival of melanoma patients seems to be the ratio of CD8-positive effector T cells to Treg cells in the tumor microenvironment [173]. The chemokine CCL22 is known to mediate CCR4high Treg cell trafficking into tumors [174]. The CCR4-mediated Treg cell attraction into melanomas, however, seems to be caused by the alternative CCR4 ligand CCL2 [175].
Immunosuppressive factors that are locally secreted by melanomas, such as TGF-ß and IL-10, could promote both expansion of naturally occurring Treg cells and de novo generation of induced Treg cells [165]. Likewise, molecular mechanisms of tumor immunosuppression mediated by IDO have a direct anergic effect on effector T cells and enhance local Treg cell–mediated immunosuppression. Moreover, expression of IDO on tumor-infiltrating APCs stimulates the conversion of conventional T cells to Treg cells [176]. Upregulation of IDO expression in melanoma lymph-node metastases is associated with an increased number of tumor-infiltrating Treg cells and consequently shorter patient survival [177]. Interestingly, very recently, it has been reported in an inducible autochthonous model of melanoma that the expression of the oncogenic BRAFV600E mutation in melanocytes resulted in nevus formation, CCR4 induction, and Treg cell recruitment [178]. This suggests the BRAFV600E signaling is sufficient to recruit the Treg cells to melanomas and might add an additional mechanism for explaining the therapeutic activity of BRAF inhibition in patients with metastatic melanoma (see below). The pathophysiology of melanoma is depicted in Fig. 5.

Immunopathophysiology of melanoma
Immunopathology-based therapy of melanoma
As mentioned above, the majority of patients with newly diagnosed melanoma have early-stage disease, for which surgical excision represents the treatment of choice and is curative in the majority of cases [179]. However, approximately 10% of melanoma cases are diagnosed at an advanced stage and are unresectable or already metastatic. Due to the known immunogenicity of melanoma, experimental immunotherapy had a prominent position in the treatment of melanoma for decades.
Collected data from several clinical trials evaluating the efficacy of recombinant IL-2 therapy in the 1980s showed that a small fraction of melanoma patients experienced durable complete responses. Based on these results, in 1998, the FDA approved IL-2 for the treatment of unresectable melanoma [180] (Table 2). IL-2 is a key regulator in supporting proliferation and homeostasis of effector T cells but is also crucial for the development of Treg cells, therefore simultaneously leading to increased numbers of Treg cells in melanoma patients [167, 181]. However, IL-2 was also described to mask the suppressive function of Treg cells on effector T cell proliferation [182].
Since Treg cell–mediated immunosuppression is generally deemed one of the main hurdles for cancer immunotherapy, various approaches for depletion and/or modulation of Treg cells (cyclophosphamide, denileukin diftitox, anti-CD25 antibody (daclizumab), anti-CD25 immunotoxin) have been characterized and tested with different clinical outcome. New such experimental approaches include an anti-CCR4 antibody for Treg cell depletion as well as an agonistic antibody against the glucocorticoid-induced tumor necrosis factor receptor (GITR) for modulation of Treg cell activity [159, 160, 165].
The main treatment for melanoma patients in the early stages is surgical resection. For a long period, the only treatments for patients with metastatic melanoma included chemotherapy with dacarbazine and some other agents as well as immunotherapy with high doses of IL-2. In the last 10 years, MAP kinase pathway–targeted therapies (BRAF and MEK inhibitors) and immune checkpoint inhibitors blocking CTLA-4 and PD-1 have revolutionized the management of advanced melanoma and significantly prolonged patient survival [149, 151] (Table 2).
In BRAFV600E-mutated melanoma, the combination of BRAF and MEK inhibitors has led to high response rates (70%) and rapid response induction and symptom control, with a significant prolongation of progression-free survival [183, 184]. Interestingly, it was reported that BRAF inhibition could promote the immune response to melanoma [185].
CTLA-4 is an inhibitory receptor that is constitutively expressed by Treg cells. CTLA-4 binds to CD80 and CD86 on APCs and acts as a key negative regulator of peripheral T cell proliferation and function. In mice models, treatment with monoclonal anti-CTLA-4 antibodies increased the local infiltration of cytotoxic T cells while dramatically reducing Tregs at the tumor site [173, 186]. The human anti-CTLA-4 antibody ipilimumab was demonstrated to be effective in a phase III clinical trial and received FDA approval for treating metastatic melanoma in 2011 [187]. Remarkably, meta-analyses demonstrated the durability of long-term survival in a significant number of ipilimumab-treated patients [188]. Whether there is a depletion or reduction of Treg cells in melanoma microenvironment as mechanism of therapeutic anti-CTLA-4 antibodies in the patients is still under debate [189,190,191,192]. The PD-1 receptor is expressed on Treg cells and activated effector lymphocytes. It binds to PD-L1 and PD-L2, acts as a T cell co-inhibitory molecule, and suppresses T cell activation. Nivolumab is a high-affinity anti-PD-1 monoclonal antibody that inhibits the binding between the PD-1 receptor and its ligands PD-L1 and PD-L2. Nivolumab was approved (2014) by the FDA for the treatment of patients with metastatic melanoma [193]. Pembrolizumab, another anti-PD-1 antibody, was approved by the FDA in 2015 for the treatment of advanced melanomas [184]. Interestingly, a very recent paper suggests that PD-1 blockade can decrease the suppressive function of Tregs in vitro and that the therapeutic benefit of nivolumab in melanoma patients corresponds to a decreased suppressive function of blood Treg cells [194]. Moreover, a quite current paper described significantly increased responses to anti-PD1 in aged melanoma patients and correlated this with higher CD8+ effector /FoxP3+ Treg cell ratios in the tumor microenvironment in this population [195]. Interestingly, in some patients with successful anti-tumor response by checkpoint inhibitors, vitiligo-like depigmentation can be observed.
Conclusion
Chronic T cell–mediated skin diseases represent a major health economy problem worldwide. Pathogenetically, these diseases are based on different mechanisms that are closely related to effector functions of respective T cell subtypes. Activity of T1 cells leads to destruction of cells expressing antigens recognized by these T cells. For example, when respective antigens are carried by melanocytes, the stimulation of the antigen-specific T1 cells results in pigment-free skin patches that characterize vitiligo. The activation of T2 cells leads to IL-4, IL-5, and IL-13 secretion, provoking skin barrier alteration, immune cell infiltration into skin, and itch as observed in atopic dermatitis. The stimulation of T17 and T22 cells is associated with highly increased production of IL-17, IL-22, and TNF-α that promote the proliferation of keratinocytes and alter their terminal differentiation. These cellular changes result in sharply demarcated, reddish-colored raised plaques with superficial silvery scaling as observed in psoriasis. In many other immune-mediated skin diseases, the exact phenotype of disease-responsible T cells is less clear. Examples are diseases such as lichen planus, hidradenitis suppurativa, or pemphigus. Apart from the dysregulated activity of T1, T2, T17, and T22 effector T cells, an increased activity of Treg cells can be involved in the development of skin diseases. An impressive example for this is skin melanoma, a malignant tumor of melanocytic origin, which presents as hyper-pigmented maculae or nodules. In that disease, local Treg cell–mediated immunosuppression is thought to be responsible for the dampened immune response (mainly T1) to the tumor cells.
The understanding of the immunopathogenetic mechanisms involved in skin diseases opens up great opportunities for the development of targeted therapeutic approaches for the respective patients. In fact, while, on the one hand, our knowledge of T cell biology has allowed the development of efficient strategies to control, e.g., psoriasis, the great success of these strategies in dermatology has, vice versa, decidedly contributed to the understanding of T cell biology and the pathways they are involved in. Further studies in immunodermatology are needed to improve the treatment options for many other inflammatory and neoplastic skin diseases beyond vitiligo, psoriasis, atopic dermatitis, and melanoma.
References
Fuchs E (2016) Epithelial skin biology: three decades of developmental biology, a hundred questions answered and a thousand new ones to address. Curr Top Dev Biol 116:357–374
Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD (2018) Atopic dermatitis. Nat Rev Dis Primers 4:1
Wolk K, Witte K, Sabat R (2010) Interleukin-28 and interleukin-29: novel regulators of skin biology. J Interf Cytokine Res 30:617–628
Fuchs E (2008) Skin stem cells: rising to the surface. J Cell Biol 180:273–284
Sabat R, Wolk K (2011) Research in practice: IL-22 and IL-20: significance for epithelial homeostasis and psoriasis pathogenesis. J Dtsch Dermatol Ges 9:518–523
Sabat R, Philipp S, Hoflich C, Kreutzer S, Wallace E, Asadullah K, Volk HD, Sterry W, Wolk K (2007) Immunopathogenesis of psoriasis. Exp Dermatol 16:779–798
Gebhardt T, Palendira U, Tscharke DC, Bedoui S (2018) Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev 283:54–76
Parisi R, Webb RT, Kleyn CE, Carr MJ, Kapur N, Griffiths CEM, Ashcroft DM (2019) Psychiatric morbidity and suicidal behaviour in psoriasis: a primary care cohort study. Br J Dermatol 180:108–115
Kurek A, Johanne Peters EM, Sabat R, Sterry W, Schneider-Burrus S (2013) Depression is a frequent co-morbidity in patients with acne inversa. J Dtsch Dermatol Ges 11:743–749 50
Weidinger S, Novak N (2016) Atopic dermatitis. Lancet 387:1109–1122
Richard MA, Corgibet F, Beylot-Barry M, Barbaud A, Bodemer C, Chaussade V, D'Incan M, Joly P, Leccia MT, Meurant JM, Petit A, Geffroy BR, Sei JF, Taieb C, Misery L, Ezzedine K (2018) Sex- and age-adjusted prevalence estimates of five chronic inflammatory skin diseases in France: results of the << OBJECTIFS PEAU >> study. J Eur Acad Dermatol Venereol 32:1967–1971
Krangel MS (2003) Gene segment selection in V(D)J recombination: accessibility and beyond. Nat Immunol 4:624–630
Taniuchi I (2016) Views on helper/cytotoxic lineage choice from a bottom-up approach. Immunol Rev 271:98–113
Klein L, Kyewski B, Allen PM, Hogquist KA (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 14:377–391
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712
Williams MA, Bevan MJ (2007) Effector and memory CTL differentiation. Annu Rev Immunol 25:171–192
Michie CA, McLean A, Alcock C, Beverley PC (1992) Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature 360:264–265
Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, Verschoor A, Schiemann M, Hofer T, Busch DH (2013) Disparate individual fates compose robust CD8+ T cell immunity. Science 340:630–635
Picker LJ, Singh MK, Zdraveski Z, Treer JR, Waldrop SL, Bergstresser PR, Maino VC (1995) Direct demonstration of cytokine synthesis heterogeneity among human memory/effector T cells by flow cytometry. Blood 86:1408–1419
Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R (2010) Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 107:14733–14738
Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellicci DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, Carbone FR, van Lier RA, Kallies A, van Gisbergen KP (2016) Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352:459–463
Kambayashi T, Laufer TM (2014) Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 14:719–730
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Pradeu T, Cooper EL (2012) The danger theory: 20 years later. Front Immunol 3:287
De Silva NS, Klein U (2015) Dynamics of B cells in germinal centres. Nat Rev Immunol 15:137–148
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655–669
Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89:587–596
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527
Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646
Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H, Durham SR, Schmidt-Weber CB, Cavani A (2009) Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 119:3573–3585
Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B (2000) CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med 192:1553–1562
Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Forster R (2000) Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med 192:1545–1552
Josefowicz SZ, Lu LF, Rudensky AY (2012) Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30:531–564
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4:330–336
Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061
Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H (2009) Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol 10:864–871
Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F (1998) Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 187:129–134
Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, Querci V, Fambrini M, Liotta F, Levings MK, Maggi E, Cosmi L, Romagnani S, Annunziato F (2010) CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol 40:2174–2181
Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, Berrino L, Fambrini M, Caproni M, Tonelli F, Lazzeri E, Parronchi P, Liotta F, Maggi E, Romagnani S, Annunziato F (2008) Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med 205:1903–1916
Lee AY, Eri R, Lyons AB, Grimm MC, Korner H (2013) CC chemokine ligand 20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: odd couple or axis of evil? Front Immunol 4:194
Campbell DJ, Butcher EC (2002) Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med 195:135–141
Campbell JJ, Haraldsen G, Pan J, Rottman J, Qin S, Ponath P, Andrew DP, Warnke R, Ruffing N, Kassam N, Wu L, Butcher EC (1999) The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature 400:776–780
Soler D, Humphreys TL, Spinola SM, Campbell JJ (2003) CCR4 versus CCR10 in human cutaneous TH lymphocyte trafficking. Blood 101:1677–1682
Groom JR, Luster AD (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215
Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG (2000) A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406:309–314
Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, Fernandez B, Kelderman S, Schumacher TN, Corti D, Lanzavecchia A, Sallusto F (2015) T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 347:400–406
Halle S, Halle O, Forster R (2017) Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol 38:432–443
Cosmi L, Annunziato F, Galli MIG, Maggi RME, Nagata K, Romagnani S (2000) CRTH2 is the most reliable marker for the detection of circulating human type 2 Th and type 2 T cytotoxic cells in health and disease. Eur J Immunol 30:2972–2979
Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, Bruijnzeel-Koomen CA, Clark RA (2013) CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol 133:973–979
Frentsch M, Stark R, Matzmohr N, Meier S, Durlanik S, Schulz AR, Stervbo U, Jurchott K, Gebhardt F, Heine G, Reuter MA, Betts MR, Busch D, Thiel A (2013) CD40L expression permits CD8+ T cells to execute immunologic helper functions. Blood 122:405–412
Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, Hoog C, Tjernlund A, Michaelsson J, Folkersen L, Mjosberg J, Blomqvist L, Ehrstrom M, Stahle M, Bryceson YT, Eidsmo L (2017) CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity 46:287–300
Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, Collins N, Bouladoux N, Dzutsev A, Rosshart SP, Arbuckle JH, Wang CR, Kristie TM, Rehermann B, Trinchieri G, Brenchley JM, O'Shea JJ, Belkaid Y (2018) Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172:784–796 e18
Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, Sen SK, Byrd AL, Enamorado M, Yao C, Tamoutounour S, Van Laethem F, Hurabielle C, Collins N, Paun A, Salcedo R, O'Shea JJ, Belkaid Y (2019) Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science 363. https://doi.org/10.1126/science.aat6280
Wolk K, Warszawska K, Hoeflich C, Witte E, Schneider-Burrus S, Witte K, Kunz S, Buss A, Roewert HJ, Krause M, Lukowsky A, Volk HD, Sterry W, Sabat R (2011) Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol 186:1228–1239
Matusiak L, Szczech J, Bieniek A, Nowicka-Suszko D, Szepietowski JC (2017) Increased interleukin (IL)-17 serum levels in patients with hidradenitis suppurativa: implications for treatment with anti-IL-17 agents. J Am Acad Dermatol 76:670–675
Moran B, Sweeney CM, Hughes R, Malara A, Kirthi S, Tobin AM, Kirby B, Fletcher JM (2017) Hidradenitis suppurativa is characterized by dysregulation of the Th17:Treg cell axis, which is corrected by anti-TNF therapy. J Invest Dermatol 137:2389–2395
Witte-Handel E, Wolk K, Tsaousi A, Irmer ML, Mossner R, Shomroni O, Lingner T, Witte K, Kunkel D, Salinas G, Jodl S, Schmidt N, Sterry W, Volk HD, Giamarellos-Bourboulis EJ, Pokrywka A, Docke WD, Schneider-Burrus S, Sabat R (2018) The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J Invest Dermatol. https://doi.org/10.1016/j.jid.2018.11.018
Picardo M, Dell'Anna ML, Ezzedine K, Hamzavi I, Harris JE, Parsad D, Taieb A (2015) Vitiligo. Nat Rev Dis Primers 1:15011
Kumar S, Nayak CS, Padhi T, Rao G, Rao A, Sharma VK, Srinivas CR (2014) Epidemiological pattern of psoriasis, vitiligo and atopic dermatitis in India: hospital-based point prevalence. Indian Dermatol Online J 5:S6–S8
Boniface K, Seneschal J, Picardo M, Taieb A (2018) Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol 54:52–67
Alikhan A, Felsten LM, Daly M, Petronic-Rosic V (2011) Vitiligo: a comprehensive overview part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol 65:473–491
Badri AM, Todd PM, Garioch JJ, Gudgeon JE, Stewart DG, Goudie RB (1993) An immunohistological study of cutaneous lymphocytes in vitiligo. J Pathol 170:149–155
Xie H, Zhou F, Liu L, Zhu G, Li Q, Li C, Gao T (2016) Vitiligo: how do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci 81:3–9
He Y, Li S, Zhang W, Dai W, Cui T, Wang G, Gao T, Li C (2017) Dysregulated autophagy increased melanocyte sensitivity to H2O2-induced oxidative stress in vitiligo. Sci Rep 7:42394
Mosenson JA, Zloza A, Nieland JD, Garrett-Mayer E, Eby JM, Huelsmann EJ, Kumar P, Denman CJ, Lacek AT, Kohlhapp FJ, Alamiri A, Hughes T, Bines SD, Kaufman HL, Overbeck A, Mehrotra S, Hernandez C, Nishimura MI, Guevara-Patino JA, Le Poole IC (2013) Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med 5:174ra28
Richmond JM, Bangari DS, Essien KI, Currimbhoy SD, Groom JR, Pandya AG, Youd ME, Luster AD, Harris JE (2017) Keratinocyte-derived chemokines orchestrate T-cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol 137:350–358
Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, Zhou Y, Deng A, Hunter CA, Luster AD, Harris JE (2014) CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med 6:223ra23
Wu J, Zhou M, Wan Y, Xu A (2013) CD8+ T cells from vitiligo perilesional margins induce autologous melanocyte apoptosis. Mol Med Rep 7:237–241
Witte E, Kokolakis G, Witte K, Warszawska K, Friedrich M, Christou D, Kirsch S, Sterry W, Volk HD, Sabat R, Wolk K (2016) Interleukin-29 induces epithelial production of CXCR3A ligands and T-cell infiltration. J Mol Med (Berl) 94:391–400
Dwivedi M, Kemp EH, Laddha NC, Mansuri MS, Weetman AP, Begum R (2015) Regulatory T cells in vitiligo: implications for pathogenesis and therapeutics. Autoimmun Rev 14:49–56
Lili Y, Yi W, Ji Y, Yue S, Weimin S, Ming L (2012) Global activation of CD8+ cytotoxic T lymphocytes correlates with an impairment in regulatory T cells in patients with generalized vitiligo. PLoS One 7:e37513
Elela MA, Hegazy RA, Fawzy MM, Rashed LA, Rasheed H (2013) Interleukin 17, interleukin 22 and FoxP3 expression in tissue and serum of non-segmental vitiligo: a case- controlled study on eighty-four patients. Eur J Dermatol 23:350–355
Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA (2012) A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol 132:1869–1876
Frisoli ML, Harris JE (2017) Vitiligo: mechanistic insights lead to novel treatments. J Allergy Clin Immunol 140:654–662
Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE (2017) CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol 137:982–985
Oyoshi MK, He R, Kumar L, Yoon J, Geha RS (2009) Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol 102:135–226
Tokura Y (2010) Extrinsic and intrinsic types of atopic dermatitis. J Dermatol Sci 58:1–7
Bohme M, Wickman M, Lennart Nordvall S, Svartengren M, Wahlgren CF (2003) Family history and risk of atopic dermatitis in children up to 4 years. Clin Exp Allergy 33:1226–1231
Uehara M, Kimura C (1993) Descendant family history of atopic dermatitis. Acta Derm Venereol 73:62–63
Otsuka A, Nomura T, Rerknimitr P, Seidel JA, Honda T, Kabashima K (2017) The interplay between genetic and environmental factors in the pathogenesis of atopic dermatitis. Immunol Rev 278:246–262
Addor FA, Takaoka R, Rivitti EA, Aoki V (2012) Atopic dermatitis: correlation between non-damaged skin barrier function and disease activity. Int J Dermatol 51:672–676
Bin L, Leung DY (2016) Genetic and epigenetic studies of atopic dermatitis. Allergy, Asthma Clin Immunol 12:52
Saeki H, Kuwata S, Nakagawa H, Etoh T, Yanagisawa M, Miyamoto M, Tokunaga K, Juji T, Shibata Y (1995) Analysis of disease-associated amino acid epitopes on HLA class II molecules in atopic dermatitis. J Allergy Clin Immunol 96:1061–1068
Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, Ramon M, Bergman R, Krueger JG, Guttman-Yassky E (2009) IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol 123:1244–1252 e2
Sabat R, Ouyang W, Wolk K (2014) Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov 13:21–38
Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Luscher-Firzlaff J, Luscher B, Baron JM (2012) IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol 129:426–433 33 e1-8
Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, Schneider L, Beck LA, Barnes KC, Leung DY (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120:150–155
Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, Vom Baur E, Witte K, Warszawska K, Philipp S, Johnson-Leger C, Volk HD, Sterry W, Sabat R (2009) IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med (Berl) 87:523–536
Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R (2006) IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36:1309–1323
Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, Khatcherian A, Novitskaya I, Carucci JA, Bergman R, Krueger JG (2008) Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol 181:7420–7427
Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, Darst MA, Gao B, Boguniewicz M, Travers JB, Leung DY (2003) Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol 171:3262–3269
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–1160
Wolk K, Mitsui H, Witte K, Gellrich S, Gulati N, Humme D, Witte E, Gonsior M, Beyer M, Kadin ME, Volk HD, Krueger JG, Sterry W, Sabat R (2014) Deficient cutaneous antibacterial competence in cutaneous T-cell lymphomas: role of Th2-mediated biased Th17 function. Clin Cancer Res 20:5507–5516
Wolk K, Witte K, Witte E, Raftery M, Kokolakis G, Philipp S, Schonrich G, Warszawska K, Kirsch S, Prosch S, Sterry W, Volk HD, Sabat R (2013) IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med 5:204ra129
Christophers E, Henseler T (1987) Contrasting disease patterns in psoriasis and atopic dermatitis. Arch Dermatol Res 279(Suppl):S48–S51
Klein PA, Greene WH, Fuhrer J, Clark RA (1997) Prevalence of methicillin-resistant Staphylococcus aureus in outpatients with psoriasis, atopic dermatitis, or HIV infection. Arch Dermatol 133:1463–1465
Ong PY (2014) Recurrent MRSA skin infections in atopic dermatitis. J Allergy Clin Immunol Pract 2:396–399
Ong PY, Leung DY (2016) Bacterial and viral infections in atopic dermatitis: a comprehensive review. Clin Rev Allergy Immunol 51:329–337
Totte JE, van der Feltz WT, Hennekam M, van Belkum A, van Zuuren EJ, Pasmans SG (2016) Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: a systematic review and meta-analysis. Br J Dermatol 175:687–695
Jinnestal CL, Belfrage E, Back O, Schmidtchen A, Sonesson A (2014) Skin barrier impairment correlates with cutaneous Staphylococcus aureus colonization and sensitization to skin-associated microbial antigens in adult patients with atopic dermatitis. Int J Dermatol 53:27–33
Wollenberg A, Wetzel S, Burgdorf WH, Haas J (2003) Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol 112:667–674
Paller AS, Tom WL, Lebwohl MG, Blumenthal RL, Boguniewicz M, Call RS, Eichenfield LF, Forsha DW, Rees WC, Simpson EL, Spellman MC, Stein Gold LF, Zaenglein AL, Hughes MH, Zane LT, Hebert AA (2016) Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol 75:494–503 e6
Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, Silverberg JI, Deleuran M, Kataoka Y, Lacour JP, Kingo K, Worm M, Poulin Y, Wollenberg A, Soo Y, Graham NM, Pirozzi G, Akinlade B, Staudinger H, Mastey V, Eckert L, Gadkari A, Stahl N, Yancopoulos GD, Ardeleanu M, Solo, Investigators S (2016) Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med 375:2335–2348
Parisi R, Symmons DP, Griffiths CE, Ashcroft DM, Identification, Management of P, Associated ComorbidiTy project t (2013) Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 133:377–385
Sterry W (2015) Plaque psoriasis. In: Sterry W, Sabat R, Philipp S (eds) Psoriasis, diagnosis and management. Wiley Blackwell, Chichester, pp 57–75
Wolk K, Röwert-Huber HJ, Sabat R (2015) Microscopic skin alterations. In: Sterry W, Sabat R, Philipp S (eds) Psoriasis, diagnosis and management. Wiley Blackwell, Chichester, pp 21–27
Heidenreich R, Rocken M, Ghoreschi K (2009) Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol 90:232–248
Eberle FC, Bruck J, Holstein J, Hirahara K, Ghoreschi K (2016) Recent advances in understanding psoriasis. F1000Res 5. https://doi.org/10.12688/f1000research.7927.1
Schon MP, Broekaert SM, Erpenbeck L (2017) Sexy again: the renaissance of neutrophils in psoriasis. Exp Dermatol 26:305–311
Henes JC, Ziupa E, Eisfelder M, Adamczyk A, Knaudt B, Jacobs F, Lux J, Schanz S, Fierlbeck G, Spira D, Horger M, Kanz L, Koetter I (2014) High prevalence of psoriatic arthritis in dermatological patients with psoriasis: a cross-sectional study. Rheumatol Int 34:227–234
Sterry W, Strober BE, Menter A, International Psoriasis C (2007) Obesity in psoriasis: the metabolic, clinical and therapeutic implications. Report of an interdisciplinary conference and review. Br J Dermatol 157:649–655
Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, Gelfand JM (2017) Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol 76:377–390
Henseler T, Christophers E (1985) Psoriasis of early and late onset: characterization of two types of psoriasis vulgaris. J Am Acad Dermatol 13:450–456
Tiilikainen A, Lassus A, Karvonen J, Vartiainen P, Julin M (1980) Psoriasis and HLA-Cw6. Br J Dermatol 102:179–184
Schakel K, Schon MP, Ghoreschi K (2016) Pathogenesis of psoriasis. Hautarzt 67:422–431
Witte E, Sabat R (2015) Genetics of psoriasis. In: Sterry W, Sabat R, Philipp S (eds) Psoriasis, diagnosis and management. Wiley Blackwell, Chichester, pp 49–54
Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, Eidsmo L (2014) Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 192:3111–3120
Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P, Nestle FO (2014) Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 134:984–991
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467:967–971
Sabat R, Wolk K (2015) Pathogenesis of psoriasis. In: Sterry W, Sabat R, Philipp S (eds) Psoriasis: diagnosis and management. Wiley-Blackwell, Hoboken, pp 28–48
Albanesi C, Cavani A, Girolomoni G (1999) IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol 162:494–502
Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG (2011) Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131:677–687
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S (1996) T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 183:2593–2603
Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, Catron D, Buchanan ME, Muller A, deWaal Malefyt R, Deng G, Orozco R, Ruzicka T, Lehmann P, Lebecque S, Caux C, Zlotnik A (2000) Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol 164:6621–6632
Katz Y, Nadiv O, Beer Y (2001) Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheum 44:2176–2184
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, Wittig BM, Warszawska K, Kurek A, Erdmann-Keding M, Kunz S, Asadullah K, Kadin ME, Volk HD, Sterry W, Wolk K, Sabat R (2014) IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol 134:2757–2767
Holstein J, Fehrenbacher B, Bruck J, Muller-Hermelink E, Schafer I, Carevic M, Schittek B, Schaller M, Ghoreschi K, Eberle FC (2017) Anthralin modulates the expression pattern of cytokeratins and antimicrobial peptides by psoriatic keratinocytes. J Dermatol Sci 87:236–245
Huang W, Na L, Fidel PL, Schwarzenberger P (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631
Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R (2004) IL-22 increases the innate immunity of tissues. Immunity 21:241–254
Wolk K, Witte E, Warszawska K, Schulze-Tanzil G, Witte K, Philipp S, Kunz S, Docke WD, Asadullah K, Volk HD, Sterry W, Sabat R (2009) The Th17 cytokine IL-22 induces IL-20 production in keratinocytes: a novel immunological cascade with potential relevance in psoriasis. Eur J Immunol 39:3570–3581
Weiss B, Wolk K, Grunberg BH, Volk HD, Sterry W, Asadullah K, Sabat R (2004) Cloning of murine IL-22 receptor alpha 2 and comparison with its human counterpart. Genes Immun 5:330–336
Martin JC, Wolk K, Beriou G, Abidi A, Witte-Handel E, Louvet C, Kokolakis G, Drujont L, Dumoutier L, Renauld JC, Sabat R, Josien R (2017) Limited presence of IL-22 binding protein, a natural IL-22 inhibitor, strengthens psoriatic skin inflammation. J Immunol 198:3671–3678
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189
Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S (2015) CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev 26:311–327
Beutler B, Cerami A (1989) The biology of cachectin/TNF--a primary mediator of the host response. Annu Rev Immunol 7:625–655
Witte K, Witte E, Sabat R, Wolk K (2010) IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev 21:237–251
Hawkes JE, Chan TC, Krueger JG (2017) Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 140:645–653
Welsch K, Holstein J, Laurence A, Ghoreschi K (2017) Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol 47:1096–1107
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386
Garbe C, Peris K, Hauschild A, Saiag P, Middleton M, Bastholt L, Grob JJ, Malvehy J, Newton-Bishop J, Stratigos AJ, Pehamberger H, Eggermont AM, European Dermatology F, European Association of D-O, European Organisation for R, Treatment of C (2016) Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline - update 2016. Eur J Cancer 63:201–217
Zbytek B, Carlson JA, Granese J, Ross J, Mihm MC Jr, Slominski A (2008) Current concepts of metastasis in melanoma. Expert Rev Dermatol 3:569–585
Gilchrest BA, Eller MS, Geller AC, Yaar M (1999) The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med 340:1341–1348
Bastian BC (2014) The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 9:239–271
Candido S, Rapisarda V, Marconi A, Malaponte G, Bevelacqua V, Gangemi P, Scalisi A, McCubrey JA, Maestro R, Spandidos DA, Fenga C, Libra M (2014) Analysis of the B-RafV600E mutation in cutaneous melanoma patients with occupational sun exposure. Oncol Rep 31:1079–1082
Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N, Arbiser JL (2002) Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin Cancer Res 8:3728–3733
Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P (2006) Human T cell responses against melanoma. Annu Rev Immunol 24:175–208
Speeckaert R, van Geel N, Vermaelen KV, Lambert J, Van Gele M, Speeckaert MM, Brochez L (2011) Immune reactions in benign and malignant melanocytic lesions: lessons for immunotherapy. Pigment Cell Melanoma Res 24:334–344
Azijli K, Stelloo E, Peters GJ, AJ VDE (2014) New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res 34:1493–1505
Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM (2011) Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist 16:5–24
Silva IP, Long GV (2017) Systemic therapy in advanced melanoma: integrating targeted therapy and immunotherapy into clinical practice. Curr Opin Oncol 29:484–492
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, Saw RP, Thompson JF (2012) Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol 30:2678–2683
Tuthill RJ, Unger JM, Liu PY, Flaherty LE, Sondak VK, Southwest Oncology G (2002) Risk assessment in localized primary cutaneous melanoma: a Southwest Oncology Group study evaluating nine factors and a test of the Clark logistic regression prediction model. Am J Clin Pathol 118:504–511
Jandus C, Speiser D, Romero P (2009) Recent advances and hurdles in melanoma immunotherapy. Pigment Cell Melanoma Res 22:711–723
Fourcade J, Zarour HM (2013) Strategies to reverse melanoma-induced T-cell dysfunction. Clin Dermatol 31:251–256
Antony PA, Restifo NP (2005) CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother 28:120–128
Jacobs JF, Nierkens S, Figdor CG, de Vries IJ, Adema GJ (2012) Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? Lancet Oncol 13:e32–e42
Ouyang Z, Wu H, Li L, Luo Y, Li X, Huang G (2016) Regulatory T cells in the immunotherapy of melanoma. Tumour Biol 37:77–85
Baumgartner J, Wilson C, Palmer B, Richter D, Banerjee A, McCarter M (2007) Melanoma induces immunosuppression by up-regulating FOXP3(+) regulatory T cells. J Surg Res 141:72–77
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500
Bluestone JA, Abbas AK (2003) Natural versus adaptive regulatory T cells. Nat Rev Immunol 3:253–257
von Herrath MG, Harrison LC (2003) Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol 3:223–232
Nizar S, Meyer B, Galustian C, Kumar D, Dalgleish A (2010) T regulatory cells, the evolution of targeted immunotherapy. Biochim Biophys Acta 1806:7–17
Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, Wang RF (2004) Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity 20:107–118
Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF, Rosenberg SA (2019) Tumor-infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol 4. https://doi.org/10.1126/sciimmunol.aao4310
Vence L, Palucka AK, Fay JW, Ito T, Liu YJ, Banchereau J, Ueno H (2007) Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci U S A 104:20884–20889
Fourcade J, Sun Z, Kudela P, Janjic B, Kirkwood JM, El-Hafnawy T, Zarour HM (2010) Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J Immunol 184:6709–6718
Francois V, Ottaviani S, Renkvist N, Stockis J, Schuler G, Thielemans K, Colau D, Marchand M, Boon T, Lucas S, van der Bruggen P (2009) The CD4(+) T-cell response of melanoma patients to a MAGE-A3 peptide vaccine involves potential regulatory T cells. Cancer Res 69:4335–4345
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Knol AC, Nguyen JM, Quereux G, Brocard A, Khammari A, Dreno B (2011) Prognostic value of tumor-infiltrating Foxp3+ T-cell subpopulations in metastatic melanoma. Exp Dermatol 20:430–434
Quezada SA, Peggs KS, Curran MA, Allison JP (2006) CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest 116:1935–1945
Wei S, Kryczek I, Zou W (2006) Regulatory T-cell compartmentalization and trafficking. Blood 108:426–431
Kimpfler S, Sevko A, Ring S, Falk C, Osen W, Frank K, Kato M, Mahnke K, Schadendorf D, Umansky V (2009) Skin melanoma development in ret transgenic mice despite the depletion of CD25+Foxp3+ regulatory T cells in lymphoid organs. J Immunol 183:6330–6337
Munn DH, Mellor AL (2007) Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest 117:1147–1154
Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, Emmons RV, Witkiewicz AK (2009) Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle 8:1930–1934
Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, Boni A, Curiel TJ, Angeles CV, Turk MJ (2018) Oncogenic BRAF(V600E) governs regulatory T-cell recruitment during melanoma tumorigenesis. Cancer Res 78:5038–5049
Ross MI, Gershenwald JE (2011) Evidence-based treatment of early-stage melanoma. J Surg Oncol 104:341–353
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17:2105–2116
Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL (2006) Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol 24:1169–1177
Thornton AM, Donovan EE, Piccirillo CA, Shevach EM (2004) Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol 172:6519–6523
Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Yan Y, Wongchenko M, Chang I, Hsu JJ, Koralek DO, Rooney I, Ribas A, Larkin J (2016) Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17:1248–1260
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30–39
Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA (2012) Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 18:1386–1394
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ (2013) Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 1:32–42
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33:1889–1894
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE (2015) Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A 112:6140–6145
Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, Sharma P (2019) Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin Cancer Res 25:1233–1238
Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caraco C, Curvietto M, Esposito A, Paone M, Palla M, Cavalcanti E, Sandomenico F, Petrillo A, Botti G, Fulciniti F, Palmieri G, Queirolo P, Marchetti P, Ferraresi V, Rinaldi G, Pistillo MP, Ciliberto G, Mozzillo N, Ascierto PA (2014) Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother 63:675–683
Tang F, Du X, Liu M, Zheng P, Liu Y (2018) Anti-CTLA-4 antibodies in cancer immunotherapy: selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci 8:30
Specenier P (2016) Nivolumab in melanoma. Expert Rev Anticancer Ther 16:1247–1261
Woods DM, Ramakrishnan R, Laino AS, Berglund A, Walton K, Betts BC, Weber JS (2018) Decreased suppression and increased phosphorylated STAT3 in regulatory T cells are associated with benefit from adjuvant PD-1 blockade in resected metastatic melanoma. Clin Cancer Res 24:6236–6247
Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, Weiss SA, Darvishian F, Al-Rohil RN, Ndoye A, Behera R, Alicea GM, Ecker BL, Fane M, Allegrezza MJ, Svoronos N, Kumar V, Wang DY, Somasundaram R, Hu-Lieskovan S, Ozgun A, Herlyn M, Conejo-Garcia JR, Gabrilovich D, Stone EL, Nowicki TS, Sosman J, Rai R, Carlino MS, Long GV, Marais R, Ribas A, Eroglu Z, Davies MA, Schilling B, Schadendorf D, Xu W, Amaravadi RK, Menzies AM, McQuade JL, Johnson DB, Osman I, Weeraratna AT (2018) Age correlates with response to anti-PD1, reflecting age-related differences in Intratumoral effector and regulatory T-cell populations. Clin Cancer Res 24:5347–5356
Acknowledgments
The authors would like to acknowledge Julia Triebus for her assistance in manuscript editing and picture production as well as Katrin Witte and Georgios Kokolakis for their help with the tables and manuscript proofreading.
Funding
The authors acknowledge the support by the Deutsche Forschungsgemeinschaft (DFG) FOR 2497/TP02 (GH133/2-1 to Kamran Ghoreschi), the German Federal Ministry of Education and Research (http://www.bmbf.de/; grant 01ZX1312A to Kerstin Wolk and RobertSabat), and Novartis Pharma GmbH (to Charité – Universitätsmedizin Berlin/Robert Sabat).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
KG has been a consultant, lecturer, or investigator for AbbVie Deutschland GmbH & Co. KG, Almirall, Biogen IDEC GmbH, Boehringer Ingelheim Pharma GmbH & Co. KG, Celgene GmbH, Eli Lilly and Company, Janssen-Cilag GmbH, MSD Sharp & Dohme, Novartis Pharmaceuticals, Pfizer Deutschland GmbH, and Roche.
RS has received research grants or honoraria for participation in advisory boards, clinical trials or as speaker for one or more of the following: AbbVie Inc., AbbVie Deutschland GmbH & Co. KG, Bayer Schering Pharma AG, Biogen IDEC GmbH, Boehringer Ingelheim Pharma GmbH & Co. KG, Celgene GmbH, Celgene International II Sàrl, Charité Research Organisation GmbH, Dr. Willmar Schwabe GmbH & Co. KG, Flexopharm GmbH & Co. KG, Generon Corporation Ltd., Janssen-Cilag GmbH, Novartis Pharma GmbH, Parexel International GmbH, Pfizer Deutschland GmbH, Sanofi–Aventis Deutschland GmbH, TFS GmbH, UCB Biopharma SPRL.
KW has been a consultant, lecturer, or investigator for AbbVie Deutschland GmbH & Co. KG, Bayer Schering Pharma AG, Celgene GmbH, Celgene International II Sàrl, Generon Corporation Ltd., Janssen-Cilag GmbH, Novartis Pharma GmbH, Sanofi–Aventis Deutschland GmbH, and UCB Biopharma SPRL.
Additional information
This article is a contribution to the special issue on The Pathogenicity of Acquired Immunity in Human Diseases - Guest Editor: Kiyoshi Hirahara
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sabat, R., Wolk, K., Loyal, L. et al. T cell pathology in skin inflammation. Semin Immunopathol 41, 359–377 (2019). https://doi.org/10.1007/s00281-019-00742-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00742-7