
Abstract
Atopic dermatitis (AD) is the most common allergic skin disease in the general population. It is a chronic inflammatory skin disease complicated by recurrent bacterial and viral infections that, when left untreated, can lead to significant complications. The current article will review immunologic and molecular mechanisms underlying the propensity of AD patients to microbial infections. These infections include Staphylococcus aureus (S. aureus) skin infections, eczema herpeticum, eczema vaccinatum, and eczema coxsackium. Previous studies have shown that skin barrier defects, a decrease in antimicrobial peptides, increased skin pH, or Th2 cytokines such as IL-4 and IL-13 are potential contributing factors for the increased risk of skin infections in AD. In addition, bacterial virulence such as methicillin-resistant S. aureus (MRSA) produces significantly higher number of superantigens that increase their potential in causing infection and more severe cutaneous inflammation in AD patients. More recent studies suggest that skin microbiome including Staphylococcus epidermidis or other coagulase-negative staphylococci may play an important role in controlling S. aureus skin infections in AD. Other studies also suggest that genetic variants in the innate immune response may predispose AD patients to increased risk of viral skin infections. These genetic variants include thymic stromal lymphopoietin (TSLP), type I interferon (α, ß, ω), type II interferon (γ), and molecular pathways that lead to the production of interferons (interferon regulatory factor 2). A common staphylococcal toxin, α-toxin, may also play a role in enhancing herpes simplex virus skin infections in AD. Further understanding of these disease processes may have important clinical implications for the prevention and treatment of skin infections in this common skin disease.
Similar content being viewed by others
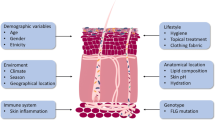

Introduction
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease in the general population. It is associated with chronic itching; sleep loss; and disruption in daily, school, and work activities [1]. Patients with AD are also at increased risk for bacterial and viral skin infections that, left untreated, can lead to invasive infections. In this review, we will summarize recent advances in the pathogenesis of AD and the mechanisms of infections in AD.
Pathogenesis of AD
The pathogenesis of AD consists of complex interactions between skin barrier defects, the immune response, and environmental exposures including allergens and microbes. Barrier defects in AD facilitate penetration of allergens and microbes into the skin. Loss-of-function filaggrin gene mutations and chronic skin inflammation lead to reduced skin hydration as measured by trans-epidermal water loss (TEWL). AD skin lesions and normal-looking AD skin are known to have significantly greater TEWL as compared to healthy skin [2]. The interplay between the cutaneous immune response and environmental triggers, enhanced by skin barrier defects, results in a vicious cycle of cutaneous inflammation in AD. The basis of barrier defects in AD is due to a deficiency of vital components of the stratum corneum and lack of keratinocyte differentiation in the epidermis. This leads to reduced protein levels of involcrin, filaggrin, loricrin, claudins, and lipid molecules including ceramide, cholesterol, and fatty acids. Various genetic causes of skin barrier defects have recently been elucidated. The loss-of-function mutations in the filaggrin genes (FLG) was initially associated with ichthyosis vulgaris, a dry skin condition that is characterized by scaly lesions on the lower extremities and hyperlinear palms, and was subsequently associated with AD [3]. De Benedetto et al. showed a deficiency of tight junction proteins in AD, including single-nucleotide polymorphisms in claudin-1, an important component of tight junctions [4]. In patients with AD, barrier defects may also be acquired. Th2 cytokines, IL-4 and IL-13, are present in both lesional and non-lesional AD skin. These cytokines have been shown to suppress the expression of filaggrin [3]. More recently, Omori-Miyake et al. showed that IL-4 and IL-13 are also capable of suppressing the expression of filaggrin and occludin in the late stage of keratinocyte differentiation (stratum granulosum), as well as keratins, desmogleins, and desmocollins at the early stage of keratinocyte differentiation (stratum spinosum)[5]. Keratins are the structural proteins derived from keratinocytes. They form the cytoskeleton of the epidermis. Desmogleins and desmocollins link the intracellular network of keratins and connect neighboring keratinocytes. Defects in these proteins may contribute to barrier dysfunction in AD. In addition to IL-4 and IL-13, IL-22, which is highly expressed in AD lesions, is also capable of suppressing filaggrin expression [6] and contributes to an immune-mediated suppression of barrier function in AD.
FLG has been associated with AD patients who have a history of recurrent skin infections [7]. Compared to AD subjects without FLG, patients with FLG mutations had a seven times higher risk of having more than four episodes of skin infections that required antibiotics in the past year. The role of filaggrin in the prevention of skin infections may be twofold: as a physical barrier, as well as a modulator of Staphylococcus aureus (S. aureus) growth. In the skin, filaggrin is broken down into hygroscopic amino acids including urocanic acid (UCA) and pyrrolidone carboxylic acid (PCA). UCA and PCA are components of the natural moisturizing factor (NMF) that hydrate and maintain an acidic pH of the stratum corneum. Acidic pH reduced the expression of two staphylococcal surface proteins, clumping factor B and fibronectin binding protein, which bind to host protein cytokeratin 10 and fibronectin, respectively. In addition, UCA and PCA may directly inhibit the proliferation of S. aureus [8]. The decreased expression of filaggrin, and hence, decreased UCA and PCA levels, may lead to the growth of S. aureus in AD. Multiple studies have shown that FLG mutations predispose to allergic sensitization [9], which is associated with increased skin infections in AD (discussed below). This concept was initially demonstrated by skin barrier defects and an increased allergic sensitization in the flaky tail mouse model of AD [10]. However, it was subsequently shown that mutation in the transmembrane protein 79 (Tmem79)/matt gene, rather than filaggrin deficiency, was responsible for the spontaneous development of allergic sensitization and dermatitis in the flaky tail AD mouse model [11, 12]. Tmem79/matt encodes for lamella granular proteins which are required for the processing of filaggrin, lipid molecules, proteases, and antimicrobial peptides (AMPs) [13]. A Tmem79/matt single-nucleotide polymorphism has been associated with AD in human subjects [12]. Further studies of Tmem79/matt gene are needed regarding its role in the pathogenesis of AD and skin infections.
Barrier defects are a universal feature of all AD subjects. In contrast, only a subset of AD subjects is known to be at increased risk for skin infections. A case in point is eczema herpeticum (EH), which is caused by herpes simplex virus (HSV). HSV is a ubiquitous pathogen in the general population (20 % of children and over 60 % of adults are seropositive to HSV); however, only about 3 % of AD subjects develop EH, suggesting that this skin infection in AD is a function of host immune response [14]. Both ichthyosis vulgaris and psoriasis are chronic skin conditions with barrier defects; however, they are not known to have more skin infections than the general population. These clinical observations further support the host immune response as playing a primary role in determining the subsets of AD patients who are at increased risk of skin infections. Recent whole genome sequencing from the Atopic Dermatitis Research Network (ADRN), the largest registry of AD subjects for the study of S. aureus colonization/infection in AD, showed that S. aureus colonization in AD is not associated with mutations in skin barrier genes, which include FLG and other crucial skin barrier genes in the epidermal differentiation complex (EDC) of chromosome 1 [15]. This was confirmed by only a modest association between S. aureus colonization and the five most common FLG mutations (2282del4, R501X, R2447X, 3702delG, S3247X) among Caucasian Americans [16].
Keratinocytes are key skin cells that are in direct contact with the outside environment. They play an important role as a sentinel and regulator of the cutaneous innate immunity. They are the key producers of AMPs, which are crucial in the cutaneous immune response against bacterial and viral infections. It has been shown that stimulated keratinocytes from AD patients produced significantly less AMPs than those from healthy and psoriasis subjects [17]. The mechanisms of decreased AMP production in AD are not fully understood. Interactions between microbial pathogens and pattern recognition receptors (PRR) on keratinocytes may play a role in the decreased immune response of these cells in AD patients [18]. This is supported by the presence of genetic polymorphisms of TLR-2, a key PRR for S. aureus, in a subset of severe AD patients who have increased skin infections [19, 20]. TLR-2 expression may also be downregulated in AD [21], leading to an attenuated innate immune response against microbial pathogens. In contrast, it has recently been shown that the activation of TLR-2 and TLR-6 heterodimers leads to increased keratinocyte expression of IL-6, which in turn induces an influx of myeloid-derived suppressor cells (MDSC) in the skin [22]. MDSC suppress T cell-mediated immunity, leading to increased risk of skin infections. Further studies of TLR functions and MDSC are needed in AD. Stimulation of TLR-3 by double-stranded RNA released from damaged epithelial cells leads to an increased production of thymic stromal lymphopoietin (TSLP) from keratinocytes [23]. TSLP is an IL-7-like cytokine that is highly expressed in AD lesions [24]. TSLP activates dermal dendritic cells (DC) to produce the chemokines, thymus, and activation-regulated chemokine (TARC/CCL17) and macrophage-derived chemokine (MDC/CCL22), which are chemokines that attract Th2 cells to AD lesions [25]. This leads to an increased expression of IL-4 and IL-13, which are known to suppress AMPs [26]. TSLP also activates group two innate lymphoid cells (ILC-2), which produce IL-13, IL-5, and/or IL-4 [27, 28], which may further suppress AMPs. ILC-2 cells are also activated by IL-33 and IL-25, both of which are highly expressed in AD lesions [29, 30]. IL-25 has been shown to enhance HSV and vaccinia virus (VV) replication by inhibiting filaggrin expression [31]. On the other hand, IL-33 has been shown to downregulate the expression of AMPs in keratinocytes [32].
Bacterial Infections
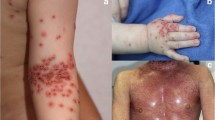
The most common skin infections in AD are caused by S. aureus (Fig. 1). Streptococcus pyogenes (S. pyogenes) skin infection is also relatively common in AD [33]. Both types of bacterial skin infections may lead to invasive infections in AD. The risk factors for severe or invasive bacterial infections in AD are not fully understood. It likely involves a complex interaction between host immune response and bacterial virulence. Benson et al. compiled a case series of AD patients who had bacteremia, infectious endocarditis, septic arthritis, and osteomyelitis. In that study, it was shown that invasive bacterial infections were associated with moderate to severe AD [34]. Such patients are generally known to have more allergic tendency with higher skin expression of Th2 cytokines, serum total IgE, and specific IgE. Multiple studies have confirmed that decreased expression of AMPs in AD lesions, as compared to psoriasis, and that the decrease in AMPs may be partly explained by the suppressive effect of Th2 cytokines. In addition, a relative decrease in IL-17, which is a known inducer of AMPs in keratinocytes, may also contribute to the decreased expression of AMPs in AD [35]. The relatively low expression of IL-17 in AD lesions may be partly explained by the low expression of IL-17 in CX3CR1+ T cells in AD patients [36]. CX3CR1 is the chemokine receptor for CX3CL1 (fractalkine), which has been shown to be highly expressed in AD lesions [37]. IFN-γ and IL-13 are present at a higher level than IL-17 in CD4 + CX3CR1+ T cells of AD patients. An infiltration of these cells into AD lesions leads to a more dominant Th1 and Th2 profile, which are characteristics of chronic and acute AD lesions, respectively.

S. aureus impetigo in a child with atopic dermatitis
The best-studied bacterial virulence factors in AD are staphylococcal enterotoxins (superantigens). Classic staphylococcal enterotoxins include staphylococcal enterotoxin (SE) A, SEB, SEC, SED, and toxic shock syndrome toxin-1 (TSST-1). Non-classical staphylococcal superantigens include SEE and SEG to SEQ. More than 80 % of S. aureus isolated from AD patients are superantigen-producing [38]. Staphylococcal superantigens are presented by antigen-presenting cells to activate polyclonal T cells via the variable beta chain of the T cell receptor, resulting in robust T cell-mediated inflammation in AD lesions. Staphylococcal superantigens are also capable of inducing host production of specific IgE directed to superantigens, leading to basophil release of histamines. Methicillin-resistant S. aureus (MRSA) produces more superantigens than methicillin-sensitive S. aureus (MSSA) [39]. The prevalence of MRSA colonization in AD patients is as high as 12 %. This is tenfold higher than the rate of MRSA colonization in the general population (1–3 %) [40]. The presence of MRSA is significantly higher in moderate to severe AD patients, as compared to mild AD patients (90 vs 10 %)[41]. It has been shown that AD patients who are colonized with MRSA are significantly more likely to develop skin and soft tissue infection than AD patients who are colonized with MSSA [42]. MRSA skin and soft tissue infections often lead to a vicious cycle of AD flare and recurrent infections [43]. AD flare associated with MRSA skin infections often has inadequate response to topical anti-inflammatory agents, including corticosteroids. This inadequate response may be due to corticosteroid resistance induced by staphylococcal superantigens [44]. S. aureus strains isolated from more severe AD patients produce significantly more superantigen types, as compared to those isolated from a general population of AD patients [39].
SEB has been shown to increase IL-31 expression in AD [45]. This cytokine is best known for causing pruritus in AD. In addition, IL-31 has also been shown to suppress filaggrin expression [46]. IL-31 may also suppress the S. aureus-induced expression of the AMPs [47], human β-defensin-2 and 3, and RNAse7, contributing to an increase in S. aureus colonization in AD. The suppression of these AMPs by IL-31 seems to be specific, as this cytokine did not suppress the expression of psoriasin, another AMP produced by keratinocytes. In addition to staphylococcal superantigens, S. aureus also produces α-toxin, which causes keratinocyte cytotoxicity and lymphocyte apoptosis [48, 49]. Filaggrin-deficient keratinocytes are particularly susceptible to the toxicity of α-toxin, as these cells lack sphingomyelinase, which is required to cleave the α-toxin receptor, sphingomyelin [50]. IL-4 and IL-13 also further increase the susceptibility of keratinocytes to α-toxin toxicity [51]. Using lactate dehydrogenase release as a marker of α-toxin-induced cell death, it was shown that undifferentiated keratinocytes are particularly susceptible to α-toxin-induced cell death. The toxicity of α-toxin on undifferentiated keratinocytes is further enhanced by IL-4/IL-13. IFN-γ, on the other hand, protects undifferentiated keratinocytes from α-toxin-induced cell death. Calcium-induced differentiation of keratinocytes also protects these cells from the toxicity of α-toxin. However, calcium-induced differentiation of keratinocytes did not protect these cells from α-toxin-induced cytotoxicity when incubated with IL-4/IL-13. The potentiation of IL-4/IL-13 on α-toxin-induced keratinocyte cytotoxicity was abolished using siRNA knockdown of signal transducer and activator of transcription 6 (STAT6), providing further evidence for the specificity of these cytokines in enhancing the cytotoxicity of α-toxin in keratinocytes. Interestingly, exogenous supplementation of sphingomyelinase protects IL-4/IL-13-treated keratinocytes from α-toxin-induced cytotoxicity. Endogenous expression of sphingomyelinase was shown to be significantly inhibited by IL-4/IL-13. Therefore, the mechanisms for the potentiation IL-4/IL-13 on α-toxin-induced cytotoxicity of keratinocytes are twofold: by suppressing both filaggrin and sphingomyelinase expression.
Other staphylococcal toxins/virulent factors include delta toxin, which increases mast cell degranulation; staphylococcal protein A, which acts via TNFR1; and lipoteichoic acid (LTA), which signals through TLR2 and platelet-activating factor receptor [52]. LTA and its lipoproteins are TLR2 agonists that are capable of exacerbating acute AD and convert acute AD into chronic AD [53]. The effect of TLR2 agonists may be mediated via TLR2 on cutaneous DC. The combined effect of TLR2 agonists and IL-4 leads to an increased expression of IL-12 and a decreased expression of IL-10 in skin DC. The decreased expression of IL-10, a well-known suppressive cytokine of inflammation, allows the development and perpetuation of cutaneous inflammation in AD. Diacylated lipoproteins of S. aureus also trigger TLR2, in association with TLR6, on keratinocytes to produce TSLP [54]. TSLP activates skin DC and ILC-2, which leads to further production of Th2 cytokines.
Skin microbiome may play a role in the pathogenesis of bacterial infections in AD. Grice et al. used 16S rRNA gene to study the microbial diversity on the flexural elbows of five healthy individuals [55]. They found a predominance of Pseudomonas and Janthinobacterium species, rather than Staphylococcus species. These results did not differ whether the analyses were obtained by swabs, scrapings, or skin biopsies. They postulated that a change in the integrity of the skin barrier may change the microbial diversity and lead to disease like AD. In a follow-up study, they analyzed the microbiome from the flexural elbows of 12 children with moderate to severe AD [56]. They found that increased AD severity inversely correlated with decreased microbial diversity, as measured by the Shannon diversity index. The decrease in microbial diversity was also observed in AD patients who did not use any treatment prior to an eczema flare. On the other hand, AD patients who used topical corticosteroids (TCS) had a significantly higher microbial diversity, even during an eczema flare. Staphylococcus was the predominant species during an AD flare. S. aureus was found to be the most abundant species during AD flare in patients who were not on any treatment. They did not find any association between AD flare and any specific strains of S. aureus. Interestingly, they also observed a parallel increase in the proportion of S. epidermidis during AD flare in patients who did not receive treatment. Given the potential competitive role of S. epidermidis or other coagulase-negative staphylococci via the production of their own AMPs [57], this increase in S. epidermidis during AD flare may represent an attempt to control S. aureus.
The Role of Viral Skin Infections in AD
Like bacterial skin infections, AD patients are also susceptible to viral skin infection [23]. However, as compared to bacterial skin infections, viral skin infections are relatively less common in AD patients. EH is one of the most common viral infections in AD patients. This skin infection may progress to systemic infection that manifests as fever, malaise, keratoconjunctivitis, encephalitis, and septic shock. The availability of anti-virals such as acyclovir has decreased the mortality of EH significantly. Identifying the risk factors among AD patients and the mechanisms of disease is also crucial in the prevention of EH. Severe disease, early-onset AD, high total serum IgE/peripheral eosinophils, and the presence of other allergic diseases such as food allergy and asthma are among the most important risk factors of AD patients in the development of EH [58, 59]. The clinical observation that AD patients with polarized Th2 response are more susceptible to EH is consistent with laboratory findings that IL-4 and IL-13 suppress keratinocyte expression of the AMP, LL-37, which has potent anti-viral activity against HSV [60, 61]. AD patients with a history of EH have been found to have the lowest levels of the cathelicidin, LL-37, in their skin lesions [14]. Two genetic variants of TSLP, rs1898671 and rs2416259, are significantly associated with AD patients with history of EH [62]. STAT6 is an important transcription factor for the expression of IL-4. Four genetic variants of STAT6, rs167769, rs841718, rs3024975, and rs703817, were found to be significantly associated with AD patients with a history of EH [63]. Interferons (IFN) are crucial cytokines in the immunity against HSV via cell-mediated response and direct anti-viral activities. Leung et al. compared the gene transcription profiles of peripheral blood mononuclear cells (PBMC) of AD patients with and without history of EH [64]. The receptors for type I IFN (α, β, and ω) and type II IFN (γ) were significantly downregulated in patients with history of EH, as compared to those without EH. In addition, the cytokine, IFN-γ, was also significantly downregulated in AD patients with history of EH. HSV-stimulated PBMC from patients with history of EH also produced significantly less IFN-γ protein than that of patients without history of EH. Genetic analysis showed five variants of IFN-γ receptor 1 (IFNGR1), rs7749390, rs10457655, rs9376269, rs1327475, and rs3799488, were significantly associated with AD patients with history of EH, as compared to those without a history of EH. Specifically, rs10457655 showed the largest difference with more than tenfold difference in significance between the two groups. Haplotype analysis of IFN-γ cytokine gene showed a significant difference in a four-marker haplotype (rs2069727, rs2069718, rs2430561, rs2069705) between the two groups. IFN-γ production from the PBMC of two of these variants (rs2069727 and rs2430561) was found to be significantly lower than that of wild types. On the other hand, PBMC from the genetic variant in IFNGR1, rs7749390, was found to have a significant increase in IFN-γ production, as compared to that of wild types. Another gene critical in the IFN-γ response pathway, interferon regulatory factor 2 (IRF2), has more recently been found to be associated with AD patients with history of EH [65]. IRF2 is an antagonist to IRF1 to block the IFN-γ-mediated pathway; five genetic variants of IRF2, rs17488073, rs809909, rs11132242, rs1342852, and rs1124191, were significantly associated with Caucasian American AD patients with history of EH. Of these variants, rs809909 showed the strongest association. Two different genetic variants of IRF2 were also found to be associated with African American AD patients with history of EH. This study suggests a potential defect in the IFN-γ pathway of AD patients with history of EH, regardless of ethnicity. However, the sample size for the African American patients was small in that study; a larger cohort will therefore be needed for replication of these results. More recent global transcriptomic analysis using the next-generation RNA sequencing (RNA-seq) technology showed that type I IFN-α4 and IFN-α5 gene transcripts were significantly downregulated in AD patients with a history of EH [66]. These findings were confirmed by quantitative PCR as well as at the protein level. In addition, the type III IFN, IL-29, was found to be significantly downregulated in AD patients with history of EH. Consistent with these findings, IRF3 and IRF7, which are transcription factors for the upregulation of type I and type III IFN, were found to be significantly downregulated in AD patients with history of EH.
A history of skin infections with S. aureus is also a major risk factor for the development of EH among AD patients [58]. This is consistent with the clinical observation that EH frequently occur concurrently with secondary S. aureus skin infection in AD patients. It has been shown that α-toxin increases the number of HSV in keratinocytes [67]. In contrast, SEB or TSST-1 did not increase the replication of HSV in keratinocytes. In addition to the lipid receptor sphingomyelin for α-toxin, a disintegrin, and metalloprotease 10 (ADAM 10), a host cell protein has been found to mediate the binding of α-toxin to host cells [68]. α-Toxin-enhanced replication of HSV was blocked in ADAM-10-deficient keratinocytes, suggesting that α-toxin-mediated viral enhancement is a receptor-dependent event and requires its binding to keratinocytes. Low expression of the AMP and LL-37 has been shown to predispose AD patients to EH. Incubation of keratinocytes with α-toxin did not decrease the gene expression of LL-37 and β defensins. However, α-toxin was shown to increase the number of HSV DNA copies in keratinocytes, in the presence of acyclovir, an HSV polymerase inhibitor that blocks HSV replication. These findings suggest that α-toxin enhances the entry of HSV into keratinocytes.
Another life-threatening viral infection in AD is eczema vaccinatum (EV), which is caused by VV in smallpox vaccines. Since the eradication of smallpox virus in the early 1970s, routine vaccination with VV had been discontinued in the general population in 1971 and in military personnel in 1990. However, due to the concern that unknown sources of smallpox virus may still exist and that it could be used as an agent of bioterrorism, the US government has implemented programs to vaccinate military personnel and public health workers. However, smallpox vaccines are contraindicated in AD patients due to the potential development of EV. This is one of the reasons that led CDC to revise its current recommendations on conventional smallpox vaccinations for the public and AD patients due to the risk of EV in case of a postevent emergency [69]. AD patients who are at risk for smallpox infection without known exposure to smallpox virus will receive the new attenuated, modified vaccinia Ankara virus vaccine, Imvamune, due to its safety profile in AD patients. On the other hand, AD patients who have been exposed to smallpox virus will receive the live VV vaccine (smallpox vaccine), ACAM2000. Due to the higher risk of the latter vaccine in causing EV in AD patients, it is therefore essential to understand the risk factors among AD patients in the development of EV in order to prevent those who are at high risk for EV from getting the live vaccines.
This was the main objective of Atopic Dermatitis Vaccinia Network (ADVN) in identifying biomarkers and genetic variants that lead to early identification of AD patients who are prone to develop EV. Similar to EH, EV only affects a small subset of AD patients. Data on AD patients with history of EH in ADVN has been utilized as a model for EV. In vitro and animal studies in ADVN have generated much needed information on the mechanisms of VV infection in AD models. IFN-γ receptor knockout mice that received VV vaccine by scarification developed disseminated VV skin lesions, significant weight loss, and reduced survival, as compared to control mice [64]. Ex vivo studies using human skin explants for microarray analyses of AD, psoriasis, and healthy subjects suggest the importance of innate immunity genes in VV infection [70]. These genes include leukotriene B4 receptor (LTB4R), orosomucoid 1 (ORM1), coagulation factor II (thrombin) receptor (F2R), complement component 9 (C9), and lipopolysaccharide binding protein (LBP), all of which were found to be significantly downregulated in VV-treated AD explants, as compared to that of healthy individuals or psoriasis patients. In addition, downregulation of innate immune response genes ORM1, TLR4, and NACHT leucine-rich repeat protein 1 (NLRP1) correlated with the severity of AD. Patera et al. first noted that IL-17 increased the virulence of VV [71]. In AD mouse model, increased IL-17 expression was found to be associated with filaggrin-deficient flaky tail mouse model [72] and was responsible for the dissemination of VV in AD lesions [73]. The same mouse model was recently backcrossed onto Balb/c mice to generate flaky tail mice without matt gene mutation for further studies of dissemination of VV in AD [74]. The study showed that flaky tail mice without matt mutation developed similar disseminated VV infections as flaky tail mice with matt mutation, suggesting that matt gene does not play a role in VV infection. Topical application of ovalbumin on flaky tail mice induced a Th2 response and led to a more severe disseminated VV infection with larger lesions, higher number of satellite lesions, and higher viral load. These mice also had higher expression of IL-17a, IL-4, IL-13, and IFN-γ mRNA. Flaky tail mice with knockout IL-17a, on the other hand, developed less severe skin lesions with VV infection. This study shows the importance of filaggrin deficiency, allergic inflammation, and IL-17a in the dissemination of VV infection.
A more recently described viral skin infection of AD is eczema coxsackium (EC) [75], which is caused by coxsackie viruses in the enterovirus group. AD patients with EC may present with oral sores and papules on palms and soles like hand, foot, and mouth disease. Superinfected AD lesions on the flexural or extensor areas may appear eroded and vesicular. In addition, these lesions may also appear on the buttock areas (Fig. 2), which is not a typical distribution of AD. The treatment for EC is not different from that for AD, which includes skin hydration, moisturizer, and TCS [76]. The clinical significance of EC is that it may be confused with EH. A lesional PCR for enterovirus may be considered if clinical signs are not apparent. EC has also been described in adults recently [77].

Erosions in the buttock of a child with eczema coxsackium
Conclusion
Skin infections cause significant morbidity in AD patients. In addition, infections such as EH and EV may be life-threatening. Therefore, there is a need to understand the mechanisms and risk factors of infections in AD. Of interest, recent studies suggest the lack of association between skin barrier genes and S. aureus colonization in AD. These studies implicate a primary role of the host immune functions in the infections of AD (summarized in Table 1). It is now well-established that enhanced allergic responses (increased serum total IgE, specific IgE, and skin Th2 cytokine expression) are associated with both bacterial and viral infections in AD [40, 58]. Understanding these mechanisms may lead to potential therapeutic interventions by suppressing Th2 response or by upregulating immunity that has been suppressed by Th2 inflammation. These advances also emphasize the need to focus on delineating the genetic basis of immune response against infections in AD. Recent genetic data from ADRN indicate that defects in IFN type I, II, and III and their receptors may be involved in the pathogenesis of EH. Of note, IFN type I and III defects likely reside in the innate immune cells or antigen-presenting cells [66]. These studies may provide a rationale for the replacement of these cytokines to protect AD patients who are at a higher risk of developing EH. Finally, further understanding on the virulence of microbial pathogens is needed in AD. Targeting MRSA or toxin-producing S. aureus and microbiome may lead to the prevention of invasive bacterial infections in AD.
References
Leung DYM, Guttman-Yassky E (2014) Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol 134:769–779
Grice K, Sattar H, Baker H, Sharratt M (1975) The relationship of transepidermal water loss to skin temperature in psoriasis and eczema. J Invest Dermatol 64(5):313–315
Irvine AD, McLean WH, Leung DY (2011) Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 365:1315–1327
De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, Berger A, Zhang K, Vidyasagar S, Yoshida T, Boguniewicz M, Hata T, Schneider LC, Hanifin JM, Gallo RL, Novak N, Weidinger S, Beaty TH, Leung DY, Barnes KC, Beck LA (2011) Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 127:773–786, e1-7
Omori-Miyake M, Yamashita M, Tsunemi Y, Kawashima M, Yagi J (2014) In vitro assessment of IL-4- or IL-13-mediated changes in the structural components of keratinocytes in mice and humans. J Invest Dermatol 134:1342–1350
Gutowska-Owsiak D, Schaupp AL, Salimi M, Taylor S, Ogg GS (2011) Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br J Dermatol 165:492–498
Cai SC, Chen H, Koh WP, Common JE, van Bever HP, McLean WH, Lane EB, Giam YC, Tang MB (2012) Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Br J Dermatol 166:200–203
Miajlovic H, Fallon PG, Irvine AD, Foster TJ (2010) Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J Allergy Clin Immunol 126:1184–1190
Brown SJ, McLean WH (2012) One remarkable molecule: filaggrin. J Invest Dermatol 132:751–762
Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, Callanan JJ, Kawasaki H, Shiohama A, Kubo A, Sundberg JP, Presland RB, Fleckman P, Shimizu N, Kudoh J, Irvine AD, Amagai M, McLean WH (2009) A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet 41:602–608
Sasaki T, Shiohama A, Kubo A, Kawasaki H, Ishida-Yamamoto A, Yamada T, Hachiya T, Shimizu A, Okano H, Kudoh J, Amagai M (2013) A homozygous nonsense mutation in the gene for Tmem79, a component for the lamellar granule secretory system, produces spontaneous eczema in an experimental model of atopic dermatitis. J Allergy Clin Immunol 132:1111–1120
Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, Campbell LE, Gierlinski M, Barton GJ, Schneider G, Balmain A, Prescott AR, Weidinger S, Baurecht H, Kabesch M, Gieger C, Lee YA, Tavendale R, Mukhopadhyay S, Turner SW, Madhok VB, Sullivan FM, Relton C, Burn J, Meggitt S, Smith CH, Allen MA, Barker JN, Reynolds NJ, Cordell HJ, Irvine AD, McLean WH, Sandilands A, Fallon PG (2013) Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol 132:1121–1129
Elias PM, Wakefield JS (2014) Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol 134:781–791
Leung DY (2013) Why is eczema herpeticum unexpectedly rare? Antiviral Res 98:153–157
Mathias RA, Chavan S, Iyer K, Rafaels N, Boorgula M, Potee J, Hanifin JM, Paller A, and others. (2015) Identifying genetic determinants of atopic dermatitis and bacterial colonization using whole genome sequencing. J Allergy Clin Immunol 135(2):AB391 (abstract)
Yoshida T, Rafaels N, Babineau D, Artis K, Lockhart A, David G, Boguniewicz M, Ong P, DeBenedetto A, Hanifin J, Simpson EL, Paller AS, Guttman-Yassky E, Schneider L, Mathias R, Barnes K, Leung D, Beck LA. (2015) Filaggrin mutations do not associate with skin colonization in European American atopic dermatitis subjects. J Invest Dermatol 135:S58–S69 (abstract)
De Koning HD, Kamsteeg M, Rodijk-Olthuis D, Van Vlijmen-Willems IM, Van Erp PE, Schalkwijk J, Zeeuwen PL (2011) Epidermal expression of host response genes upon skin barrier disruption in normal skin and uninvolved skin of psoriasis and atopic dermatitis patients. J Invest Dermatol 131:263–266
Niebuhr M, Heratizadeh A, Wichmann K, Satzger I, Werfel T (2011) Intrinsic alterations of pro-inflammatory mediators in unstimulated and TLR-2 stimulated keratinocytes from atopic dermatitis patients. Exp Dermatol 20:468–472
Ahmad-Nejad M-DS, Breuer K, Klotz M, Werfel T, Herz U, Heeg K, Neumaier M, Renz H (2004) The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J Allergy Clin Immunol 113:565–567
Oh DY, Schumann RR, Hamann L, Neumann K, Worm M, Heine G (2009) Association of the toll-like receptor 2 A-16934T promoter polymorphism with severe atopic dermatitis. Allergy 64:1608–1615
Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, Gallo RL, Borkowski AW, Yamasaki K, Leung DY, Georas SN, De Benedetto A, Beck LA (2013) Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair. J Invest Dermatol 133:988–998
Skabytska Y, Wölbing F, Günther C, Köberle M, Kaesler S, Chen KM, Guenova E, Demircioglu D, Kempf WE, Volz T, Rammensee HG, Schaller M, Röcken M, Götz F, Biedermann T (2014) Cutaneous innate immune sensing of Toll-like receptor 2–6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity 41:762–775
Vu AT, Chen X, Xie Y, Kamijo S, Ushio H, Kawasaki J, Hara M, Ikeda S, Okumura K, Ogawa H, Takai T (2011) Extracellular double-stranded RNA induces TSLP via an endosomal acidification- and NF-kB-dependent pathway in human keratinocytes. J Invest Dermatol 131:2205–2212
Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, De Waal-Malefyt RR, Bazan F, Kastelein RA, Liu YJ (2002) Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol 3:673–680
Nakajima S, Igyártó BZ, Honda T, Egawa G, Otsuka A, Hara-Chikuma M, Watanabe N, Ziegler SF, Tomura M, Inaba K, Miyachi Y, Kaplan DH, Kabashima K (2012) Langerhans cells are critical in epicutaneous sensitization with protein antigen via thymic stromal lymphopoietin receptor signaling. J Allergy Clin Immunol 129:1048–1055, e6
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–1160
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D. (2013) TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 5:170ra16
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149
Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimäki S, Karisola P, Reunala T, Wolff H, Lauerma A, Alenius H (2012) IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol 132:1392–1400
Hvid M, Vestergaard C, Kemp K, Christensen GB, Deleuran B, Deleuran M (2011) IL-25 in atopic dermatitis: a possible link between inflammation and skin barrier dysfunction? J Invest Dermatol 131:150–157
Kim BE, Bin L, Ye YM, Ramamoorthy P, Leung DY (2013) IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J Invest Dermatol 133:2678–2685
Alase A, Seltmann J, Werfel T, Wittmann M (2012) Interleukin-33 modulates the expression of human β-defensin 2 in human primary keratinocytes and may influence the susceptibility to bacterial superinfection in acute atopic dermatitis. Br J Dermatol 167:1386–1389
Hayakawa K, Hirahara K, Fukuda T, Okazaki M, Shiohara T (2009) Risk factors for severe impetiginized atopic dermatitis in Japan and assessment of its microbiological features. Clin Exp Dermatol 34:e63–e65
Benenson S, Zimhony O, Dahan D, Solomon M, Raveh D, Schlesinger Y, Yinnon AM (2005) Atopic dermatitis—a risk factor for invasive Staphylococcus aureus infections: two cases and review. Am J Med 118:1048–1051
Wolk K, Mitsui H, Witte K, Gellrich S, Gulati N, Humme D, Witte E, Gonsior M, Beyer M, Kadin ME, Volk HD, Krueger JG, Sterry W, Sabat R (2014) Deficient cutaneous antibacterial competence in cutaneous T-cell lymphomas: role of Th2-mediated biased Th17 function. Clin Cancer Res 20:5507–5516
Staumont-Sallé D, Fleury S, Lazzari A, Molendi-Coste O, Hornez N, Lavogiez C, Kanda A, Wartelle J, Fries A, Pennino D, Mionnet C, Prawitt J, Bouchaert E, Delaporte E, Glaichenhaus N, Staels B, Julia V, Dombrowicz D (2014) CX3CL1 (fractalkine) and its receptor CX3CR1 regulate atopic dermatitis by controlling effector T cell retention in inflamed skin. J Exp Med 211:1185–1196
Echigo T, Hasegawa M, Shimada Y, Takehara K, Sato S (2004) Expression of fractalkine and its receptor, CX3CR1, in atopic dermatitis: possible contribution to skin inflammation. J Allergy Clin Immunol 113:940–948
Leung DY, Hanifin JM, Pariser DM, Barber KA, Langley RG, Schlievert PM, Abrams B, Hultsch T (2009) Effects of pimecrolimus cream 1% in the treatment of patients with atopic dermatitis who demonstrate a clinical insensitivity to topical corticosteroids: a randomized, multicentre vehicle-controlled trial. Br J Dermatol 161(2):435–443
Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DYM (2010) Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol 125:39–49
Warner JA, McGirt LY, Beck LA (2009) Biomarkers of Th2 polarity are predictive of staphylococcal colonization in subjects with atopic dermatitis. Br J Dermatol 160:183–185
Jagadeesan S, Kurien G, Divakaran MV, Sadanandan SM, Sobhanakumari K, Sarin A (2014) Methicillin-resistant Staphylococcus aureus colonization and disease severity in atopic dermatitis: a cross-sectional study from South India. Indian J Dermatol Venereol Leprol 80:229–234
Lo WT, Wang SR, Tseng MH, Huang CF, Chen SJ, Wang CC (2010) Comparative molecular analysis of meticillin-resistant Staphylococcus aureus isolates from children with atopic dermatitis and healthy subjects in Taiwan. Br J Dermatol 162:1110–1116
Ong PY (2014) Recurrent MRSA infections in atopic dermatitis. J Allergy Clin Immunol Pract 2:396–399
Leung DYM (2005) Superantigens, steroid insensitivity and innate immunity in atopic eczema. Acta Derm Venereol Suppl (Stockh) 215:11–15
Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, Alenius H, Dieu-Nosjean MC, Meller S, Rieker J, Steinhoff M, Hoffmann TK, Ruzicka T, Zlotnik A, Homey B (2006) IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol 117:411–417
Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Lüscher-Firzlaff J, Lüscher B, Baron JM (2012) IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol 129:426–433, e1-8
van Drongelen V, Haisma EM, Out-Luiting JJ, Nibbering PH, El Ghalbzouri A (2014) Reduced filaggrin expression is accompanied by increased Staphylococcus aureus colonization of epidermal skin models. Clin Exp Allergy 44:1515–1524
Breuer K, Wittmann M, Kempe K, Kapp A, Mai U, Dittrich-Breiholz O, Kracht M, Mrabet-Dahbi S, Werfel T (2005) Alpha-toxin is produced by skin colonizing Staphylococcus aureus and induces a T helper type 1 response in atopic dermatitis. Clin Exp Allergy 35:1088–1095
Ezepchuk YV, Leung DYM, Middleton MH, Bina P, Reiser R, Norris DA (1996) Staphylococcal toxins and protein A differentially induce cytotoxicity and release of tumor necrosis factor-alpha from human keratinocytes. J Invest Dermatol 107:603–609
Brauweiler AM, Bin L, Kim BE, Oyoshi MK, Geha RS, Goleva E, Leung DY (2013) Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin-induced keratinocyte death. J Allergy Clin Immunol 131:421–427
Brauweiler AM, Goleva E, Leung DY (2014) Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J Invest Dermatol 134:2114–2121
Travers JB (2014) Toxic interaction between Th2 cytokines and Staphylococcus aureus in atopic dermatitis. J Invest Dermatol 134:2069–2071
Kaesler S, Volz T, Skabytska Y, Köberle M, Hein U, Chen KM, Guenova E, Wölbing F, Röcken M, Biedermann T (2014) Toll-like receptor 2 ligands promote chronic atopic dermatitis through IL-4-mediated suppression of IL-10. J Allergy Clin Immunol 134:92–99
Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, Kamijo S, Hiramatsu K, Ikeda S, Ogawa H, Okumura K, Takai T (2010) Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol 126:985–993, 993.e1-3
Grice EA, Kong HH, Renaud G, Young AC, Comparative Sequencing Program NISC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA (2008) A diversity profile of the human skin microbiota. Genome Res 18:1043–1050
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Program NSCCS, Murray PR, Turner ML, Segre JA (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22:850–859
Gallo RL, Hooper LV (2012) Epithelial antimicrobial defense of the skin and intestine. Nat Rev Immunol 12:503–516
Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, Paller AS, Lieff S, Reese J, Zaccaro D, Milgrom H, Barnes KC, Leung DY (2009) Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol 124(2):260–269
Wollenberg A, Zoch C, Wetzel S, Plewig G, Przybilla B (2003) Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol 49:198–205
Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY (2006) Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol 117(4):836–841
Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY (2006) Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 24(3):341–348
Gao PS, Rafaels NM, Mu D, Hand T, Murray T, Boguniewicz M, Hata T, Schneider L, Hanifin JM, Gallo RL, Gao L, Beaty TH, Beck LA, Leung DY, Barnes KC (2010) Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol 25:1403–1407, e4
Howell MD, Gao P, Kim BE, Lesley LJ, Streib JE, Taylor PA, Zaccaro DJ, Boguniewicz M, Beck LA, Hanifin JM, Schneider LC, Hata TR, Gallo RL, Kaplan MH, Barnes KC, Leung DY (2011) The signal transducer and activator of transcription 6 gene (STAT6) increases the propensity of patients with atopic dermatitis toward disseminated viral skin infections. J Allergy Clin Immunol 128:1006–1014
Leung DY, Gao PS, Grigoryev DN, Rafaels NM, Streib JE, Howell MD, Taylor PA, Boguniewicz M, Canniff J, Armstrong B, Zaccaro DJ, Schneider LC, Hata TR, Hanifin JM, Beck LA, Weinberg A, Barnes KC (2011) Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J Allergy Clin Immuno 127:965–973, e1-5
Gao PS, Leung DY, Rafaels NM, Boguniewicz M, Hand T, Gao L, Hata TR, Schneider LC, Hanifin JM, Beaty TH, Beck LA, Weinberg A, Barnes KC (2012) Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol 132:650–657
Bin L, Edwards MG, Heiser R, Streib JE, Richers B, Hall CF, Leung DY (2014) Identification of novel gene signatures in patients with atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol 134:848–855
Bin L, Kim BE, Brauweiler A, Goleva E, Streib J, Ji Y, Schlievert PM, Leung DY (2012) Staphylococcus aureus α-toxin modulates skin host response to viral infection. J Allergy Clin Immunol 130:683–691, e2
Wilke GA, Bubeck WJ (2010) Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A 107:13473–13478
Petersen BW, Damon IK, Pertowski CA, Meaney-Delman D, Guarnizo JT, Beigi RH, Edwards KM, Fisher MC, Frey SE, Lynfield R, Willoughby RE (2015) Clinical guidance for smallpox vaccine use in a postevent vaccination program. MMWR Recomm Rep 64:1–26
Grigoryev DN, Howell MD, Watkins T, Chen YC, Cheadle C, Boguniewicz M, Barnes KC, Leung DY (2010) Vaccinia virus-specific molecular signature in atopic dermatitis skin. J Allergy Clin Immunol 125:153–159, e28
Patera AC, Pesnicak L, Bertin J, Cohen JI (2002) Interleukin 17 modulates the immune response to vaccinia virus infection. Virology 299(1):56–63
Oyoshi MK, Murphy GF, Geha RS (2009) Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol 124(3):485–493
Oyoshi MK, Elkhal A, Kumar L, Scott JE, Koduru S, He R, Leung DY, Howell MD, Oettgen HC, Murphy GF, Geha RS (2009) Vaccinia virus inoculation in sites of allergic skin inflammation elicits a vigorous cutaneous IL-17 response. Proc Natl Acad Sci U S A 106(35):14954–14959
Oyoshi MK, Beaupré J, Venturelli N, Lewis CN, Iwakura Y, Geha RS. (2015) Filaggrin deficiency promotes the dissemination of cutaneously inoculated vaccinia virus. J Allergy Clin Immunol 135(6):1511–1518.e6
Mathes EF, Oza V, Frieden IJ, Cordoro KM, Yagi S, Howard R, Kristal L, Ginocchio CC, Schaffer J, Maguiness S, Bayliss S, Lara-Corrales I, Garcia-Romero MT, Kelly D, Salas M, Oberste MS, Nix WA, Glaser C, Antaya R (2013) “Eczema coxsackium" and unusual cutaneous findings in an enterovirus outbreak. Pediatrics 132:e149–e157
Johnson VK, Hayman JL, McCarthy CA, Cardona ID (2014) Successful treatment of eczema coxsackium with wet wrap therapy and low-dose topical corticosteroid. J Allergy Clin Immunol Pract 2:803–804
Harris PN, Wang AD, Yin M, Lee CK, Archuleta S (2014) Atypical hand, foot, and mouth disease: eczema coxsackium can also occur in adults. Lancet Infect Dis 14:1043
Acknowledgments
The authors wish to thank JoAnn Ferguson for her assistance in preparing this manuscript. We thank the Edelstein Family Chair for partially funding this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ong, P.Y., Leung, D.Y.M. Bacterial and Viral Infections in Atopic Dermatitis: a Comprehensive Review. Clinic Rev Allerg Immunol 51, 329–337 (2016). https://doi.org/10.1007/s12016-016-8548-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8548-5