
Abstract
Background
Atopic dermatitis (AD) is a chronic inflammatory disease caused by the complex interaction of genetic, immune and environmental factors. There have many recent discoveries involving the genetic and epigenetic studies of AD.
Methods
A retrospective PubMed search was carried out from June 2009 to June 2016 using the terms “atopic dermatitis”, “association”, “eczema”, “gene”, “polymorphism”, “mutation”, “variant”, “genome wide association study”, “microarray” “gene profiling”, “RNA sequencing”, “epigenetics” and “microRNA”. A total of 132 publications in English were identified.
Results
To elucidate the genetic factors for AD pathogenesis, candidate gene association studies, genome-wide association studies (GWAS) and transcriptomic profiling assays have been performed in this period. Epigenetic mechanisms for AD development, including genomic DNA modification and microRNA posttranscriptional regulation, have been explored. To date, candidate gene association studies indicate that filaggrin (FLG) null gene mutations are the most significant known risk factor for AD, and genes in the type 2 T helper lymphocyte (Th2) signaling pathways are the second replicated genetic risk factor for AD. GWAS studies identified 34 risk loci for AD, these loci also suggest that genes in immune responses and epidermal skin barrier functions are associated with AD. Additionally, gene profiling assays demonstrated AD is associated with decreased gene expression of epidermal differentiation complex genes and elevated Th2 and Th17 genes. Hypomethylation of TSLP and FCER1G in AD were reported; and miR-155, which target the immune suppressor CTLA-4, was found to be significantly over-expressed in infiltrating T cells in AD skin lesions.
Conclusions
The results suggest that two major biologic pathways are responsible for AD etiology: skin epithelial function and innate/adaptive immune responses. The dysfunctional epidermal barrier and immune responses reciprocally affect each other, and thereby drive development of AD.
Similar content being viewed by others


Background
Atopic dermatitis (AD) is the most common skin disease worldwide, affecting up to 30 % of children and 3 % of adults [1]. Together with food allergy, allergic rhinitis and atopic asthma, AD belongs to the atopic group of disorders with common characteristics of allergen sensitization, epithelial barrier abnormalities and Type 2 immune responses [2]. A number of familial studies and twin studies have demonstrated that AD is a highly heritable disease [3–9]. The rapid increase in prevalence of AD has been attributed to changes in life style and the environment. A comprehensive review by Barnes summarized genetic studies on AD before June of 2009 [10]. In her review, outcomes of 5 linkage studies and 111 targeted gene association studies were analyzed. Looking retrospectively, the year of 2009 was a time point for transition to a new era in which genome-wide association studies (GWAS) emerged as a popular approach to elucidate the genetic susceptibility of human complex diseases such as AD. Concomitantly, epigenetic alterations in response to environmental exposures opened a novel area for researchers to explore AD etiology. After June 2009, targeted gene association studies were still actively used, however, traditional genetic linkage studies for AD had been substituted by GWAS studies with large scale sample sizes in different populations. During the last 5 years, next-generation sequencing technologies have been increasingly used in AD research: two whole exome analyses were reported for the purpose of finding rare genetic variants in coding sequences of the AD genome; and high-throughput gene expression profiling assays such as RNA-sequencing technology were performed on skin biopsy specimens from AD patients. Although several reviews on the topic of AD genetics and epigenetics have been published since 2009 [11–13], more than 30 new studies have been reported since January, 2015. In this article, we summarize genetics and epigenetics studies on AD between June of 2009 and June of 2016. Studies from both adults and children with different ethnicities are included in this review. Additionally, a few studies of genome wide comparative analyses of AD versus asthma and AD versus psoriasis are discussed in the current review.
Review
Methods
We performed the literature search in PubMed database using the terms “atopic dermatitis” or ‘eczema” and “association”, or “gene”, or “polymorphism”, or “mutation”, or “variant”, or “genome wide association study”, or “microarray”, or “gene profiling”, or “RNA-sequencing”, or “epigenetics”, or “microRNA”, from June 2009 to present. Abstracts of the PubMed results were reviewed to identify any target gene association case–control studies, GWAS studies, gene profiling studies and epigenetics studies including both DNA methylation studies and microRNA studies of AD. Articles published in non-English languages were excluded.
This review included all results obtained from children and adults in all ethnicities. Comparative studies on “AD” and “AD plus asthma”, “AD” and “atopic march”, “AD” and “psoriasis” were included.
Results
Candidate gene association studies
Candidate gene association studies have focused on the skin epidermal differentiation complex genes and Type 2 immune responses based on our understanding of the pathophysiology of AD. The review by Barnes, in 2010, had examined 81 genes that had been studied prior to June 2009 [10]. Using the same strategy of literature research as Barnes, we identified 92 published studies on candidate gene association studies in AD (Additional file 1: Table S1, Additional file 2: Reference). Among these 92 studies, 65 genes were investigated, more than half of which had at least 1 positive association. To date, FLG null mutation is the most replicated AD gene association. Genes in the Th2 signaling pathway are the second category that has been replicated by multiple independent studies. Beside IL-4, IL-13, IL-4RA, IL13RA1, IL-13RA2 and STAT6, newly tested genes in this category are thymic stromal lymphopoietin (TSLP), its receptors IL-7R and TSLPR [14], and IL-31 [15]. In the skin barrier gene category, LAMA3 [16], TMEM79, filaggrin-2(FLG2) [17] and Late Cornified Envelope-like Proline-rich 1 (LELP1) [18] were identified to be associated with AD. The vitamin D signaling pathway is a novel pathway that has been explored in AD. In this regard, vitamin D receptor (VDR) polymorphisms and CYD27A1 were found to be associated with AD severity [19–21]. Additionally, IL10, IL6, TNFA and IL-1 family members were studied in this time frame. A few candidates from GWAS were also tested. Genes studied are summarized in Fig. 1 which include the analyses from Barnes’ review.

Genes associated with AD in at least 1 publication. Genes are grouped based on the reported positive association studies (see Additional file 1: Table S1 in the supplemental materials for a complete summary of 91 published studies). The Y-axis indicates the number of genes. The X-axis indicates the corresponding number of positive association reported
Genome-wide association studies (GWAS)
Candidate gene association studies are extremely limited in scope because the selection of candidates is often from known genes with selection biases from the investigators. Thus, this approach usually does not identify novel genes or novel pathophysiological pathways. To date, of the estimated 30,000 human genes, only a very small portion of the transcriptome, have been carefully investigated. A hypothesis-free approach can significantly decrease biases and lead to identification of novel pathophysiology pathways for AD. Single nucleotide polymorphisms (SNP) are the most common class of genetic variations in humans. The haplotype structure of the human genome suggests that a set of 1 million SNP can capture approximately 90 % of genetic variation in the population. The data from the Hapmap project and development of dense genotyping chips allow GWAS assays to be effectively conducted on a large number of samples. Therefore, GWAS became a powerful method to comprehensively investigate associations between common SNP and complex diseases [22]. Using the key words “genome wide association study” and “atopic dermatitis” to search the Pubmed database, a total of 13 articles were published since 2009, 9 articles focused exclusively on AD, 4 other articles did genome-wide comparative analysis of AD with asthma/atopic march and psoriasis.
The first GWAS study of AD was published in May 2009 by Esparza-Gordillo et al. It was performed on a German cohort of 939 cases and 975 controls as well as 275 complete nuclear families with two affected siblings [23]. This study replicated FLG locus as an AD predisposing factor and identified a novel susceptibility region at chromosome 11q13.5 located 38 kb down-stream of C11orf30. Two year later in 2011, Sun et al. reported a GWAS study on Chinese Han population, FLG region was once more validated in Chinese population and two novel loci of 5q22.1 and 20q13.33 were identified. These two loci were validated using 1806 cases and 3256 controls from Germany [24]. Interestingly, the TSLP gene is located about 300 kb down-stream of the associated region of 5q22.1. In the same year of 2011, Paternoster et al. [25] published a meta-analysis of GWAS on European ancestry, in which they identified three more new risk loci for AD (11q31.1, 19p13.2, 5q31). In addition, this study reported a significant genome-wide association signal within the cytokine cluster on 5q31.1 due to two distinct signals, one centered on RAD50/IL13 and the other on IL4/KIF3A. In 2012, Hirota et al. reported GWAS study findings in the Japanese population, this study added eight other novel susceptibility loci, including the major histocompatibility complex (MHC) region on chromosome 6p21 and the IL1RL1–IL18R1–IL18RAP locus on chromosome 2q12 [26]. In 2013, Ellinghaus et al. reported the densely genotyped results of 2425 German cases and 5449 controls using an Immunochip array [27], followed by replication in 7196 cases and 15,480 controls from Germany, Ireland, Japan, and China. Four additional novel susceptibility loci for AD were identified (4q27 IL2/IL21, 11p13 PRR5L, 16p13.13 CLEC16A/DEXI, 17q21.32 ZNF652). Adjacent genes to these loci include TRAF6, RAG1, RAG2, SOCS1 and NGFR [27]. Additionally, Esparza-Gordillo et al. analyzed data in the public repository and validated selected markers in three different sets of cases and controls, and identified 4 SNPs, rs2040704 (RAD50), rs10903122 (RUNX3), rs2292239 (ERBB3) and rs2228145 (IL6R), as new susceptibility genes for AD. The authors further validated that the IL6R rs2228145 (C) genotype is associated with increased soluble IL-6R plasma levels in AD and persistent AD status using two independent population-based cohort [28].
In 2015, 3 more reports involving GWAS study and AD were published. Schaarschmidt et al. analyzed imputed SNP data from previous GWAS studies, followed by validation with additional case and control cohorts. This study validated 19 of previously established AD genetic risk loci and identified two new susceptibility loci (2q24.3 and 9p21.3) with genome-wide significance in German population [29]. Kim et al. conducted the first GWAS assay in Korean population which was aimed to identify genetic biomarkers for moderate-to-severe AD in children. Since the discovery cohort of this study only contained 246 AD cases, the results from this report are not discussed in this review [30]. To note, the AD GWAS study with the best statistic power so far is conducted by Paternoster et al. who led an international collaborated study. This study included 21,000 cases and 95,000 controls with multi-ancestry in the discovery cohort, the results were replicated in 32,059 cases and 228,628 controls, 15,539,996 variants with minor allele frequency (MAF) ≥ 1 % were analyzed [31]. This study had not only replicated 16 AD risk loci identified by previous GWAS studies, but also identified 11 more new risk loci for AD. The new loci include candidate genes of CD207 (langerin), PPP2R3C, IL-7R, STAT3 and ZBTB10, with known function in the regulation of innate host defenses and T cell function [32–36]. Taken together, these 8 GWAS assays and meta-analyses reported 34 AD risk loci as listed in Table 1.
Children with AD often develop asthma and other allergic conditions later in life. This phenomenon is termed “the atopic march” [37]. Identification of genetic risk factors for the atopic march is important for the development of asthma prevention strategies. Weidinger et al. conducted a GWAS study to specifically examine the genetic differences between the AD endophenotypes “AD plus asthma” and “AD no asthma” [38]. In this study, 1563 cases of children-onset AD with known asthma status and 4054 European controls were genotyped as discovery cohort. Association variants were further assessed for a replication cohort including 2286 European cases and 3160 European controls. The results found that FLG locus, 5q31 between RAD 50 and IL-13 locus, 6p21 MHC locus and 11q13.5 locus were associated with the co-morbidity of AD and asthma. Using more stringent criteria of inclusion for the atopic march cases (eczema up to the age of 3 years and asthma up to age of 16 years) as compared to the study of Weidinger et al., Marenholz et al. conducted a meta-analysis to search for genetic markers of the atopic march in European descents [39]. This study validated the results of the Weidinger et al. report. Additionally, two novel loci (6p12.3/EFHC1 and 12q21.3/SLC6A15) for the atopic march were reported. Importantly, both studies demonstrated a strong contribution of AD risk genes to subsequent asthma occurrence in the atopic march, supporting the epidemiology observation that infantile eczema is a predisposing factor for asthma (OR 4.33; 95 % confidence interval 3.72–5.01, p < 0.0001) [39].
The differences between AD and psoriasis (another common cutaneous inflammatory disease) were also investigated using meta-analysis of GWAS data [38, 40, 41]. AD is associated with Type 2—polarized immune responses, allergen sensitization and recurrent microbial skin infections, while psoriasis is associated with Type 1–polarized immune responses and is not associated with skin infections. Corresponding to their clinical phenotypes, opposing genetic effects were seen on Th2 locus and loci related to Th1 cytokines and host anti-viral genes between AD and psoriasis. Some genetic risk loci are in concordance for AD and psoriasis, suggesting that these two common dermatological disease share some genetic and inflammatory features.
Gene expression profiling assays
Disease related genetic variants usually either alter gene expression or change the function of gene products by changing protein amino acid structure. Epigenetic modification and microRNA are important mechanisms that can also alter gene expression. Investigation of the transcriptome in disease relevant tissues and cells is therefore an ideal strategy to identify molecular signatures of complex diseases. Guttman-Yassky et al. performed high-throughput expression profiling on skin biopsies from AD lesions compared with health control subjects [42]. This study observed that the expression of a large number of keratinocyte terminal differentiation genes were reduced in AD as compared to normal subjects. The affected genes included filaggrin, loricrin (LOR), involucrin, late cornified envelope protein LCE2B, S100 fusion gene TCHH and multiple S100 family members, etc. This study indicated AD was associated with broad defects of epidermal cornification in AD skin lesion. These results validated a previous microarray profiling study by Sugiura et al. which revealed down-regulation of LOR and FLG in AD skin lesions [43]. More recently, the Guttman-Yassky group used RNA-sequencing technology to compare the transcriptomes of nonlesional and lesional skin from patients with moderate-to-severe AD. This study identified increased expression of a novel TREM-1 signal pathway as well as IL-36 in AD [44]. Using laser capture microdissection to separate epidermis and dermis of AD lesional and nonlesional skin compared with the expression profiles of normal skin transcriptomes, Esaki et al. once again demonstrated that AD lesions had down-regulation of genes encoding skin barrier proteins including FLG, LOR, CLDN4 and CLDN8; and elevated gene expression of Th2 and Th17 cytokines such CCL22, CCL26, TSLP and IL-22 etc. [45].
Loss-of-function mutations of the gene encoding FLG are the most significant genetic risk factor for AD. Cole et al. conducted a transcriptome profiling study using a RNA-sequencing approach to compare non-lesional skin from AD with site-matched samples from healthy controls [46]. This study found that differentially expressed genes between normal and AD subjects were enriched in pathways involved in the extracellular space, lipid metabolism and stress response. When the whole transcriptome data-set was stratified according to FLG genotype, FLG deficient skin expressed a type-I interferon-mediated stress response.
A small subset of AD are susceptible to disseminated herpes simplex virus skin infections, also called “eczema herpeticum” (EH). Our lab recently compared the transcriptomic changes of peripheral blood mononuclear cells (PBMCs) between AD with a history of EH (ADEH+) and AD without a history of AD (ADEH−). The results demonstrated that unstimulated ADEH+ and ADEH–PBMC had similar transcriptomes. However, following stimulation with herpes simplex virus 1, PBMCs from ADEH+ had distinct transcriptome profiles as compared to ADEH−, with striking down-regulation of anti-viral cytokines including both type I and type III interferons. These results are indicative of defective innate immune responses in ADEH+ subjects [47].
Epigenetic studies of AD: DNA modification and microRNAs (miRNAs)
The prevalence of AD has been increasing too rapidly to be accounted for by shifts in genetic variation. These changes are thought to be due to environmental factors such as industrialization or the western life style associated with increased stress, more sedentary life style, obesity, low vitamin D level, overuse of antibiotics, etc. [48]. Birth cohort studies suggest that both indoor and outdoor pollution are risk factors for the development of AD [49]. There is increasing evidence showing that environmental factors regulate gene expression through genomic DNA modifications and miRNA mechanisms [50]. A study by Liu et al. suggested that diesel exhaust resulted in hypermethylation of CpG sites in the IFNG gene promoter and hypomethylation of the IL4 gene promoter. This was positively correlated with increased induction of IgE in response to intranasal challenge with the allergen, Aspergillus fumigatus [51]. This observation is further supported by a recent study that poly-aromatic hydrocarbon such as benzopyrene, derived largely from incomplete combustion of organic material, such as fossil fuels, coal, wood and tobacco, is associated with increased serum IL-4 in children with asthma [52]. Several studies demonstrated that Benzo[a]pyrene can decrease global DNA methylation by inhibition of DNA methytransferase expression and interfere with assembling of the methylation machinery [53–55]. Another study showed that children with prenatal tobacco exposure had a global hypomethylation at AluYb8 repeat element in buccal cells collected from mouth swab [56].
Several epigenetic studies targeting AD have been conducted and summarized in Table 2. A Chinese group led by Lu et al. have reported that hypomethylation of the promoters of TSLP and FCER1G are responsible for gene over-expression in AD [57, 58]. A German group recently reported an integrated epigenetic and transcriptomic analyses using epidermal lesions from AD patients in comparison with healthy control epidermis. The results showed that methylation status in AD skin lesions was strikingly different from healthy control epidermis and was partly correlated with the transcript levels of genes involved in epidermal differentiation and immune response [59]. In addition, two preliminary studies on FLG gene methylation have been reported but gave conflicting results [60, 61].
Aside from modification of genomic DNA to transcriptionally regulate gene expression, miRNA mediated post-transcriptional regulation is another type of epigenetic gene expression regulation. miRNAs are a class of non-coding molecules that bind to the 3′-UTR of target mRNAs and regulate translation [62]. They are highly efficient in fine-tuning gene expression, exerting subtle yet significant effects throughout the genome, and their expression can be induced by environmental factors such as microbes and toxins. A few studies have explored whether miRNA are involved in AD pathogenesis. The first report by Sonkoly et al. used PCR-based miRNA arrays to compare healthy and AD lesional skin, and identified 44 miRNAs that were significantly different between AD and healthy controls with 34 down-regulated and 10 up-regulated miRNAs. The authors further validated miR-155 as significantly over-expressed in infiltrating T cells in AD skin lesions [63]. The authors found that environmental factors such as dust mite allergen and staphylococcal superantigens could induce miR-155 expression in atopic skin and identified the immune suppressor, CTLA-4, as its target gene [63]. Recently, a different group reported that miR-155 could be induced by LPS and IL-10 was its target gene [64]. In addition, a study conducted in a northern Chinese cohort found that miR-203 and miR-483-5p were significantly upregulated in serum of children with AD as compared with healthy children. The level of miR-483-5p in serum was significantly associated with AD and other atopic conditions including rhinitis and/or asthma [65].
Discussion
Limitations of current genetic and epigenetic studies on AD
Currently, all published studies of gene profiling assays with AD have involved relatively small sample sizes. Thus, replication and validation are significantly needed. Comparing the three different approaches, GWAS assays provide a relative high degree of replication, suggesting that the discovered genetic risk loci are robust. One caveat of the existing GWAS studies is that the SNPs identified in these studies often have a minor allele frequency greater than 1 %; therefore GWAS results cannot fully explain AD heritability due to their limited power to detect common variants with only a small effect [29]. The “missing heritability” of AD may require identification of rare genetic variants using deep sequencing technology such as the whole exome sequencing and whole genome sequencing. Two exome analyses of AD were reported: the study by Suzuki et al. did exome-sequencing on 37 AD with the extreme phenotype of serum IgE > 1000 units. The positive hits from this discovery cohort were then validated in replication cohort including 469 AD and 935 controls. This approach identified a rare genetic variant rs199691576(A/G) in CYP27A1 that is associated with AD of high serum total IgE [21]. Another study was performed on 60 Africa American patients with AD and found filaggrin-2 variation is associated with a more persistent phenotype [17]. The Atopic Dermatitis Research Network (ADRN) funded by National Institute of Health/National Institute of Allergy and Infectious Diseases have initiated whole genome sequencing analyses on AD genomes and the results are currently being analyzed [66].
Epigenetic studies of AD are currently at the stage of exploratory with small sample sizes investigated. Since the epigenetic modifications are tissue-specific and often result in gene expression changes, it is the best to investigate epigenetic alteration and gene profiling on the same tissue or cells taken from human subjects simultaneously. Currently, only one study did so [59].
Based on results of target gene association assays, GWAS and transcriptome profiling analyses as well as epigenetic studies for AD accomplished so far, genes involved in disease pathogenesis mainly fall into two pathophysiologic groups: skin barrier genes and immune response genes. In most patients, both of these two major pathways cross talk with each other to form complex pathways leading to the development of AD.
Filaggrin and other skin barrier genes
It is now commonly accepted that skin barrier dysfunction is an essential feature for the pathogenesis of AD [67]. A disrupted skin barrier allows penetration of microbes, allergens, toxins and pollutants, leading to skin inflammation, allergen sensitization and bacterial colonization. Normal epidermal skin barrier function requires an intact stratum corneum and tight junctions in the stratum granulosum. An earlier linkage study for AD had implicated chromosome 1q21 where a very large cluster of genes involved in the epidermal differentiation process is located. This group of genes is also referred to as “the epidermal differentiation complex (EDC)” and includes FLG, loricrin, involucrin, small proline-rich proteins (SPRRs), S100A family, S100-fusion protein family and late cornified envelope proteins. To date, FLG from the EDC cluster is the most significant risk factor for AD pathogenesis, and two null mutations R501X and 2282del4 of FLG in Caucasians have demonstrated the strongest association for AD (18 and 48 % of moderate to severe AD, respectively) [68]. Furthermore, the frequency of R501X in AD with a history of EH is three times higher (24 vs 8 %, respectively, OR 11.8 vs 6.2; P = 0.0008) [69] than ADEH− subjects. The association of FLG null mutation with AD and EH have demonstrated ethnic differences: In Asians, FLG P478 S and C3321delA are associated with increased risk to AD, the atopic march and recurrent skin infection [70–76]; In African populations, FLG mutations are not common [77–80]. In contrast to European and Asian AD, loss-of–function mutations in FLG2, but not FLG, are associated with increased risk in African American children [17].
The FLG gene comprises three exons. The third exon is the largest and consists of nearly identical tandem repeats of about 972 base pair in length and has allelic variants of 10, 11 and 12 repeats [81]. A study of an Irish case–control cohort found that the number of repeats was significantly lower in AD cases than controls, suggesting that common copy number variations contributes to AD risk [82].
Because AD is a complex disease caused by the combination of genetic variation and environmental factors, it is important to evaluate the impact of gene-environment interactions. There are two recent articles reported interesting results on the interaction of environmental factors and FLG gene mutations [83, 84]. Phthalates are chemicals commonly used in a variety of cosmetic and personal care products. Wang et al. recently reported that phthalate metabolite levels are significantly associated with increased AD risk in Chinese children, and children with FLG P478S mutations have increased skin absorption of phthalate. These data suggest that FLG mutations may increase skin permeability leading to higher skin absorption of phthalate and thus confer an increased risk for AD [84, 85]. Another recent study conducted in Europe reported that maternal FLG mutations increase the risk of AD in children and this increased risk is independent of mutation inheritance, indicating that maternal FLG mutations can act as strong environmental risk factors for the offspring [83].
There is evidence that additional EDC genes may be associated with AD [86]. Recently, a case–control study found that a 24-bp deletion in SPRR3 was associated with AD in European cohorts [87]. However, deletion of LCE3B and LCE3C genes is not associated with AD in Caucasians [88]. Although a case–control study evaluating polymorphisms across 21 EDC genes in a German cohort did not find evidence for associations apart from FLG, several transcriptomic profiling studies have reported that EDC genes of IVL, LOR and LCE2B, and the cell–cell adhesion protein, CDSN, were significantly down-regulated in AD skin lesions, suggesting that elevated inflammatory cytokines in the disease loci also play an important role for dysregulation of epidermal barrier genes [42, 43].
Tight junctions in the granular layer of the epidermis play an important role in maintaining skin barrier integrity for regulation of transepidermal water loss. The claudin family represents one type of tight junction transmembrane proteins [89, 90]. The gene expression of claudin-1 (CLDN1) has been found to be reduced in the AD skin. Both Th2 cytokines and genetic variants were responsible for CLDN1 reduction in AD [91, 92]. Desmosomes are also important structures to maintain skin barrier functions that connect the cell surface to the intermediate filament cytoskeleton [93]. Samuelov et al. reported that loss-of-function mutations in gene DSG1, which encodes an important desmosome protein desmoglein 1, cause severe dermatitis, multiple allergies and metabolic wasting in human [94]. Additionally, two groups demonstrated that homozygous mutation of TMEM79 is responsible for the spontaneous dermatitis phenotype in flaky tail mice which was originally thought caused solely by FLG mutations [95–97]. Sasaki et al. demonstrated that TMEM79 has a function in the lamellar granule secretory system [95], indicating that this skin barrier deficiency can lead to AD skin inflammation. Moreover, a missense SNP (rs6694514) of human TMEM79 was identified to significantly associate with AD using a meta-analysis of 4245 AD cases and 10,558 population-matched control subjects [96]. Several other genes including LAMA3 encoding the alpha chain of laminin 5, OVOL1 and ACTL9 have been reported to be associated with AD [16, 25]. ACTL9 variants, however, were not replicated in two independent studies [98, 99].
The serine protease inhibitor of Kazal type 5 (SPINK5) gene encoding lympho-epithelial kazal type-related inhibitor type 5 (LEKTI) is a crucial protease inhibitor for epidermis homeostasis. This gene was found to have loss-of-function mutations in Netherton syndrome, a severe autosomal recessive skin disease including AD and sensitization [100]. Gene variants of SPINK5 was found to associate with AD in a Japanese population [101]. Functional studies demonstrated that enhanced protease activity due to defective function of SPINK5 led to increased proteolytic activity within the epidermis, subsequently resulting in reduced DSG1 and FLG, as well as enhanced TSLP expression. These changes all contribute to AD pathogenesis [102, 103]. Additionally, there is evidence shown that deficient FLG in keratinocytes leads to increased IL-1 and TSLP expression [104, 105]. These studies demonstrate that functional lack of epidermal enzyme inhibitors and structural proteins not only compromise the skin barrier integrity, but also mediate immunologic responses of allergic inflammation.
Adaptive/innate immune response genes
Since AD is associated with allergen sensitization, elevated serum IgE and increased expression of Type 2 cytokines (IL-4, IL5, and IL-13) in both unaffected skin and skin lesions of AD, candidate gene studies for AD have also focused on the Th2 pathway. Indeed, the GWAS assays have repeatedly identified AD genetic risk loci around Th2 genes regions at 5q31. Genetic variants of genes in the Th2 signaling pathway including IL4, IL13, and the IL4 receptor are positively associated with AD [10, 106–112]. IL-4 receptor down-stream genes such as STAT6 have also been reported to have positive correlations to AD as well [113–115]. Additionally, gene variants in the alpha and beta chains of the high-affinity IgE receptor (FCER1A/B) have been implicated in AD pathogenesis [116].
The Th2 cytokine, IL-31, is also increased in AD lesions and serum [117–121]. It is not only involved in AD inflammatory responses and dysregulation of skin barrier [122–124], but also causes severe itching [125–127]. Recently, antibodies against IL-31 receptor A has begun to be tested in clinical trials to treat AD and reduced itching [128]. IL31 polymorphisms have been reported in AD by several groups [15, 129, 130].
Both GWAS studies and targeted gene association studies have provided evidence for the association of AD risk with TSLP gene variants [14, 24]. TSLP can be induced in epidermal epithelial cells by a variety of stimuli including scratching, viral infections, inflammatory cytokines, protease allergens, bacteria and bacterial products [131]. The expression of TSLP is significantly increased in AD skin lesions [45]. The major function of TSLP is to promote Th2 immune response, thus it has been considered to play an important role in AD pathogenesis. A targeted gene association study reported that genetic variants in TSLP are associated with AD, and the association is stronger in patients with the ADEH+ phenotype [14]. Additionally, polymorphisms of IL-7R (T244I at exon 6 and T46I at exon 2), a TSLP receptor, are associated with AD [132]. Two other epithelium-derived Th2-promoting cytokines IL-33 and IL-25 also play an important role in the pathophysiology of AD [133, 134]. No genetic polymorphism, however, of these two genes has been reported to be associated with AD.
The elevated Type 2 response in AD could be the secondary effect of impaired Th1 responses or reduced inhibitory feedback. Based on this notion, targeted gene association studies have also been performed on genes in Th1 responses and immune suppression genes. IL12B, IL12 receptor beta 1(IL12RB1) and IL-18 promote Th1 development [135–137]. There are studies reported that both IL12B A1188C and IL12RB1 A-111T were associated with the risk of AD in a Japanese population [138, 139], and IL18 variants were associated with AD in both German and Korean populations [140, 141]. Both suppressor of cytokine signaling 3 (SOCS3) and IL-10 can suppress adaptive immune responses [142, 143]. SOCS3 polymorphism was associated with elevated expression in European AD patients [144]; and IL10 (-819 and -592 promoter polymorphisms) was found to be associated with AD in a Korean population [145].
Aside from AD patients prone to EH, AD is often complicated by recurrent bacterial infection and other types of virus infection. The most common bacterial pathogen for AD is Staphylococcus aureus. Molluscum contagiosum virus can cause eczema molluscatum, and exposure to vaccinia virus can cause eczema vaccinatum after smallpox vaccine inoculation [146]. The increased prevalence of skin infections in AD suggest that the innate defense system is impaired in AD skin. Indeed, genetic variants in multiple genes functioning in pattern recognition receptors (PRR) have been implicated in the pathogenesis of AD. A number of genes in PRR signaling pathways, including TLR2, TLR9, CD14, TOLLIP, MYD88, MAL, NOD1, NOD2 and NALP12, has been reported to be associated with AD [147–149]. Polymorphisms in anti-microbial peptides of S100 protein, human defensins α and β and sphingosine have also been implicated in AD pathogenesis [147, 148]. In addition to the direct effect of genetic modifications in innate immune response genes, the attenuation of the normal antimicrobial response caused by overexpression of Th2 cytokines in skin is especially relevant in AD. For example, it has been demonstrated that Th2 cytokines can inhibit gene expression of human β-defensin 3 and LL-37 in epidermal keratinocytes [150, 151]. Taken together, both genetic variation/mutation and acquired impairment of innate immune responses may contribute to Th2 polarization of AD. However, the genetic loci identified by GWAS need further fine mapping in order to identify the genes exactly involved; most of the associations mentioned by candidate gene assays here involved relatively small cohorts, and replication in independent large populations are needed. It is critical to note that functional validation for these candidate genes represents a significant unmet need in the field, and that should be part of the future direction in AD research.
Conclusions
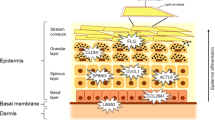
In summary, candidate genes for AD suggest that epidermal barrier dysfunction, enhanced Th2 immune signaling, weakened innate immune responses, IL-1 signaling and the vitamin D pathway etc. have roles in the pathogenesis of AD. Epigenetic studies also indicate the modifications of genes involved in these pathways as well. The dysfunctional epidermal barrier and immune responses reciprocally affect each other, and thereby drive the development of AD (Fig. 2). Interventions targeting either of these pathways can lead to remission of this disease.

The schematic illustration of AD etiology. Genetic and epigenetic reasons lead to the alteration of gene expression and function of AD associated genes. AD associated genes majorly belong to two pathways: skin barrier and innate/adaptive immunity. Dysregulation of innate/adaptive immune responses and impaired skin barrier reciprocally affect each other to drive AD development
In the future, it will be important to identify biomarkers with prognostic and predictive value for AD. Such biomarkers will lead to opportunities for precision medicine in AD. Nevertheless, research accomplishment to date further confirmed that FLG mutations and the Type 2 pathway are major risk factors for AD. Therapeutic reagents improving FLG function and biologics blocking Th2 cytokines such as anti-IL-4 receptor alpha, anti-IL-4/IL-13 or TSLP [152, 153] are evolving as treatment for severe AD patients. Based on GWAS studies of the atopic march, arrest of AD in infancy may be beneficial for prevention of asthma in this subset of AD patients.
Abbreviations
- AD:
-
atopic dermatitis
- ADEH+:
-
atopic dermatitis with a history of eczema herpeticum
- ADEH-:
-
atopic dermatitis without a history of eczema herpeticum
- EDC:
-
epidermal differentiation complex
- FLG:
-
filaggrin
- GWAS:
-
genome-wide association study
- PRR:
-
pattern recognition receptor
- SNP:
-
single nucleotide polymorphism
- Th:
-
T helper lymphocyte
- TLR:
-
toll-like receptor
- TSLP:
-
thymic stromal lymphopoietin
References
Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109–22.
Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol. 2010;105(2):99–106.
Sneddon IB. The management of infantile eczema. Med Press. 1951;226(14):329–33.
Schaffer N. Atopic dermatitis in the older child. J Asthma Res. 1966;3(3):189–91.
Edfors-Lubs ML. Allergy in 7000 twin pairs. Acta Allergol. 1971;26(4):249–85.
Schultz Larsen FV, Holm NV. Atopic dermatitis in a population based twin series. Concordance rates and heritability estimation. Acta dermato-venereologica. Suppl. 1985;114:159.
Larsen FS, Holm NV, Henningsen K. Atopic dermatitis. A genetic-epidemiologic study in a population-based twin sample. J Am Acad Dermatol. 1986;15(3):487–94.
Strachan DP, Wong HJ, Spector TD. Concordance and interrelationship of atopic diseases and markers of allergic sensitization among adult female twins. J Allergy Clin Immunol. 2001;108(6):901–7.
van Beijsterveldt CE, Boomsma DI. Genetics of parentally reported asthma, eczema and rhinitis in 5-year-old twins. Euro Respir J. 2007;29(3):516–21.
Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. J Allergy Clin Immunol. 2010;125(1):16–29.
Tamari M, Hirota T. Genome-wide association studies of atopic dermatitis. J Dermatol. 2014;41(3):213–20.
Hoffjan S, Stemmler S. Unravelling the complex genetic background of atopic dermatitis: from genetic association results towards novel therapeutic strategies. Arch Dermatol Res. 2015;307(8):659–70.
Peng W, Novak N. Recent developments in atopic dermatitis. Curr Opin Allergy Clin Immunol. 2014;14(5):417–22.
Gao PS, Rafaels NM, Mu D, Hand T, Murray T, Boguniewicz M, Hata T, Schneider L, Hanifin JM, Gallo RL, Gao L. Genetic Variants in TSLP are Associated with Atopic Dermatitis and Eczema Herpeticum. J Allergy Clin Immunol. 2010;125(6):1403.
Sokolowska-Wojdylo M, et al. The frequencies of haplotypes defined by three polymorphisms of the IL-31 gene: -1066, -2057, and IVS2 + 12 in Polish patients with atopic dermatitis. Int J Dermatol. 2015;54(1):62–7.
Stemmler S, Parwez Q, Petrasch-Parwez E, Epplen JT, Hoffjan S. Association of variation in the LAMA3 gene, encoding the alpha-chain of laminin 5, with atopic dermatitis in a German case-control cohort. BMC Dermatol. 2014;14:17.
Margolis DJ, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol. 2014;133(3):784–9.
Trzeciak M, et al. Association of a Single Nucleotide Polymorphism in a Late Cornified Envelope-like Proline-rich 1 Gene (LELP1) with Atopic Dermatitis. Acta dermato-venereologica. 2016;96(4):459–63.
Heine G, et al. Association of vitamin D receptor gene polymorphisms with severe atopic dermatitis in adults. Br J Dermatol. 2013;168(4):855–8.
Kilic S, et al. Vitamin D receptor gene BSMI, FOKI, APAI, and TAQI polymorphisms and the risk of atopic dermatitis. J Investig Allergol Clin Immunol. 2016;26(2):106–10.
Suzuki H, et al. A rare variant in CYP27A1 and its association with atopic dermatitis with high serum total IgE. Allergy. 2016;71(10):1486–9.
Manolio TA. Genomewide association studies and assessment of the risk of disease. N Eng J Med. 2010;363(2):166–76.
Esparza-Gordillo J, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet. 2009;41(5):596–601.
Sun LD, et al. Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nat Genet. 2011;43(7):690–4.
Paternoster L, et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat Genet. 2012;44(2):187–92.
Hirota T, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet. 2012;44(11):1222–6.
Ellinghaus D, et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat Genet. 2013;45(7):808–12.
Esparza-Gordillo J, et al. A functional IL-6 receptor (IL6R) variant is a risk factor for persistent atopic dermatitis. J Allergy Clin Immunol. 2013;132(2):371–7.
Schaarschmidt H, et al. A genome-wide association study reveals 2 new susceptibility loci for atopic dermatitis. J Allergy Clin Immunol. 2015;136(3):802–6.
Kim KW, Myers RA, et al. Genome-wide association study of recalcitrant atopic dermatitis in Korean children. J Allergy Clin Immunol. 2015;136(3):678–84.
Genetics EA, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–56.
Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol. 2014;14(6):417–28.
de Jong MA, Geijtenbeek TB. Langerhans cells in innate defense against pathogens. Trends Immunol. 2010;31(12):452–9.
Uehira M, Matsuda H, Nakamura A, Nishimoto H. Immunologic abnormalities exhibited in IL-7 transgenic mice with dermatitis. J Investig Dermatol. 1998;110(5):740–5.
Steward-Tharp SM, et al. A mouse model of HIES reveals pro- and anti-inflammatory functions of STAT3. Blood. 2014;123(19):2978–87.
Hinds DA, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. 2013;45(8):907–11.
Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112(6 Suppl):S118–27.
Weidinger S, et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum Mol Genet. 2013;22(23):4841–56.
Marenholz I, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6:8804.
Tamari M, et al. An association study of 36 psoriasis susceptibility loci for psoriasis vulgaris and atopic dermatitis in a Japanese population. J Dermatol Sci. 2014;76(2):156–7.
Baurecht H, et al. Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am J Human Gen. 2015;96(1):104–20.
Guttman YE, Suárez FM, Chiricozzi A, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009;124(6):1235–44.
Sugiura H, et al. Large-scale DNA microarray analysis of atopic skin lesions shows overexpression of an epidermal differentiation gene cluster in the alternative pathway and lack of protective gene expression in the cornified envelope. Br J Dermatol. 2005;152(1):146–9.
Suarez-Farinas M, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol. 2015;135(5):1218–27.
Esaki H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Aller Clin Immunol. 2015;135(1):153–63.
Cole C, et al. Filaggrin-stratified transcriptomic analysis of pediatric skin identifies mechanistic pathways in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134(1):82–91.
Bin L, et al. Identification of novel gene signatures in patients with atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol. 2014;134(4):848–55.
Jurakic Toncic R, Marinovic B. What is new and hot in genetics of human atopic dermatitis: shifting paradigms in the landscape of allergic skin diseases. Acta dermatovenerologica Croatica: ADC. 2014;22(4):313–5.
Ahn K. The role of air pollutants in atopic dermatitis. J Allergy Clin Immunol. 2014;134(5):993–9.
Amarasekera M, Prescott SL, Palmer DJ. Nutrition in early life, immune-programming and allergies: the role of epigenetics. Asian Pac J Allergy Immunol. 2013;31(3):175–82.
Liu J, et al. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxic Sci. 2008;102(1):76–81.
Al-Daghri NM, et al. Increased IL-4 mRNA expression and poly-aromatic hydrocarbon concentrations from children with asthma. BMC Pediatr. 2014;14:17.
Wilson VL, Jones PA. Inhibition of DNA methylation by chemical carcinogens in vitro. Cell. 1983;32(1):239–46.
Zhang N, et al. Methylation of cytosine at C5 in a CpG sequence context causes a conformational switch of a benzo[a]pyrene diol epoxide-N2-guanine adduct in DNA from a minor groove alignment to intercalation with base displacement. J Mol Biol. 2005;346(4):951–65.
Weisenberger DJ, Romano LJ. Cytosine methylation in a CpG sequence leads to enhanced reactivity with Benzo[a]pyrene diol epoxide that correlates with a conformational change. J Biol Chem. 1999;274(34):23948–55.
Breton CV, et al. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180(5):462–7.
Luo Y, Zhou B, Zhao M, Tang J, Lu Q. Promoter demethylation contributes to TSLP overexpression in skin lesions of patients with atopic dermatitis. Clin Exp Dermatol. 2014;39(1):48–53.
Liang Y, et al. Demethylation of the FCER1G promoter leads to FcepsilonRI overexpression on monocytes of patients with atopic dermatitis. Allergy. 2012;67(3):424–30.
Rodriguez E, et al. An integrated epigenetic and transcriptomic analysis reveals distinct tissue-specific patterns of DNA methylation associated with atopic dermatitis. J Investig Dermatol. 2014;134(7):1873–83.
Tan HT, et al. Methylation of the filaggrin gene promoter does not affect gene expression and allergy. Pediatr Allergy Immunol. 2014;25(6):608–10.
Ziyab AH, et al. DNA methylation of the filaggrin gene adds to the risk of eczema associated with loss-of-function variants. J Eur Acad Dermatol Venereol. 2013;27(3):e420–3.
Dai R, Ahmed SA. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res. 2011;157(4):163–79.
Sonkoly E, Janson P, Majuri ML, Savinko T, Fyhrquist N, Eidsmo L, Xu N, Meisgen F, Wei T, Bradley M, Stenvang J. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte–associated antigen 4. J Allergy Clin Immunol. 2010;126(3):581–9.
Quinn SR, et al. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J Biol Chem. 2014;289(7):4316–25.
Lv Y, et al. Profiling of serum and urinary microRNAs in children with atopic dermatitis. PLoS ONE. 2014;9(12):e115448.
Mathias RA, Chavan S, Iyer KR, Rafaels NM, Boorgula M, Potee J, et al. Identifying genetic determinants of atopic dermatitis and bacterial colonization using whole genome sequencing. J Allergy Clin Immunol. 2015;135(2):AB391.
Czarnowicki T, Krueger JG, Guttman YE. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol. 2014;2(4):371–9.
O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2008;122(4):689–93.
Gao PS, Rafaels NM, Hand T, Murray T, Boguniewicz M, Hata T, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol. 2009;124(3):507–13.
Wang IJ, Lin TJ. FLG P478S polymorphisms and environmental risk factors for the atopic march in Taiwanese children: a prospective cohort study. Ann Allergy Asthma Immunol. 2015;114(1):52–7.
Meng L, et al. Filaggrin gene mutation c.3321delA is associated with various clinical features of atopic dermatitis in the Chinese Han population. PLoS ONE. 2014;9(5):e98235.
Kim SY, et al. Association between P478S polymorphism of the filaggrin gene & atopic dermatitis. Indian J Med Res. 2013;138(6):922–7.
Yu HS, et al. Mutations in the filaggrin are predisposing factor in korean children with atopic dermatitis. Allergy Asthma Immunol Res. 2013;5(4):211–5.
Li M, et al. Interactions between FLG mutations and allergens in atopic dermatitis. Arch Dermatol Res. 2012;304(10):787–93.
Cai SC, et al. Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Br J Dermatol. 2012;166(1):200–3.
Wang IJ, et al. Filaggrin polymorphism P478S, IgE level, and atopic phenotypes. Br J Dermatol. 2011;164(4):791–6.
Brown SJ, McLean WH. One remarkable molecule: filaggrin. J Investig Dermatol. 2012;132(3 Pt 2):751–62.
Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol. 2010;162(3):472–7.
Margolis DJ, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol. 2012;130(4):912–7.
Winge MC, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br J Dermatol. 2011;165(5):1074–80.
Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Eng J Med. 2011;365(14):1315–27.
Brown SJ, et al. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Investig Dermatol. 2012;132(1):98–104.
Esparza-Gordillo J, et al. Maternal filaggrin mutations increase the risk of atopic dermatitis in children: an effect independent of mutation inheritance. PLoS Genet. 2015;11(3):e1005076.
Wang IJ, Karmaus WJ. The effect of phthalate exposure and filaggrin gene variants on atopic dermatitis. Environ Res. 2015;136:213–8.
Wang IJ, Lin CC, Lin YJ, Hsieh WS, Chen PC. Early life phthalate exposure and atopic disorders in children: a prospective birth cohort study. Environ Int. 2014;62:48–54.
Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Investig Dermatol. 2007;127(7):1667–72.
Marenholz I, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Investig Dermatol. 2011;131(8):1644–9.
Bergboer JG, et al. Deletion of late cornified envelope 3B and 3C genes is not associated with atopic dermatitis. J Investig Dermatol. 2010;130(8):2057–61.
Sugawara T, et al. Tight junction dysfunction in the stratum granulosum leads to aberrant stratum corneum barrier function in claudin-1-deficient mice. J Dermatol Sci. 2013;70(1):12–8.
Kirschner N, et al. Contribution of tight junction proteins to ion, macromolecule, and water barrier in keratinocytes. J Investig Dermatol. 2013;133(5):1161–9.
De Benedetto A, et al. Tight junction defects in patients with atopic dermatitis. J Aller Clin Immunol. 2011;127(3):773–86.
De Benedetto A, et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol. 2011;128(1):242–6.
Desai BV, Harmon RM, Green KJ. Desmosomes at a glance. J Cell Sci. 2009;122(Pt 24):4401–7.
Samuelov L, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45(10):1244–8.
Sasaki T, et al. A homozygous nonsense mutation in the gene for Tmem79, a component for the lamellar granule secretory system, produces spontaneous eczema in an experimental model of atopic dermatitis. J Allergy Clin Immunol. 2013;132(5):1111–20.
Saunders SP, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol. 2013;132(5):1121–9.
Sprecher E, Leung DY. Atopic dermatitis: scratching through the complexity of barrier dysfunction. J Allergy Clin Immunol. 2013;132(5):1130–1.
Lepre T, et al. Association of KIF3A, but not OVOL1 and ACTL9, with atopic eczema in Italian patients. Br J Dermatol. 2013;168(5):1106–8.
Kang Z, et al. Correlation of KIF3A and OVOL1, but not ACTL9, with atopic dermatitis in Chinese pediatric patients. Gene. 2015;571(2):249–51.
Chavanas S, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–2.
Nishio Y, et al. Association between polymorphisms in the SPINK5 gene and atopic dermatitis in the Japanese. Genes Immun. 2003;4(7):515–7.
Briot A, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135–47.
Fortugno P, et al. The 420 K LEKTI variant alters LEKTI proteolytic activation and results in protease deregulation: implications for atopic dermatitis. Human Mol Genet. 2012;21(19):4187–200.
Kezic S, O’Regan GM, Lutter R, Jakasa I, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129(4):1031–9.
Lee KH, et al. Filaggrin knockdown and Toll-like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp Dermatol. 2011;20(2):149–51.
Hummelshoj T, et al. Association between an interleukin-13 promoter polymorphism and atopy. Euro J Immunogenet. 2003;30(5):355–9.
Liu X, et al. An IL13 coding region variant is associated with a high total serum IgE level and atopic dermatitis in the German multicenter atopy study (MAS-90). J Allergy Clin Immunol. 2000;106(1 Pt 1):167–70.
Namkung JH, et al. Association of polymorphisms in genes encoding IL-4, IL-13 and their receptors with atopic dermatitis in a Korean population. Exp Dermatol. 2011;20(11):915–9.
de Guia RM, Ramos JD. The -590C/TIL4 single-nucleotide polymorphism as a genetic factor of atopic allergy. Int J Mol Epidemiol Genet. 2010;1(1):67–73.
He JQ, et al. Genetic variants of the IL13 and IL4 genes and atopic diseases in at-risk children. Genes Immun. 2003;4(5):385–9.
Kawashima T, et al. Linkage and association of an interleukin 4 gene polymorphism with atopic dermatitis in Japanese families. J Med Genet. 1998;35(6):502–4.
Oiso N, Fukai K, Ishii M. Interleukin 4 receptor alpha chain polymorphism Gln551Arg is associated with adult atopic dermatitis in Japan. Br J Dermatol. 2000;142(5):1003–6.
Casaca VI, et al. STAT6 polymorphisms are associated with neonatal regulatory T cells and cytokines and atopic diseases at 3 years. Allergy. 2013;68(10):1249–58.
Tamura K, et al. Novel dinucleotide repeat polymorphism in the first exon of the STAT-6 gene is associated with allergic diseases. Clin Exp Allergy. 2001;31(10):1509–14.
Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol. 2003;131(1):33–8.
Niwa Y, et al. FcepsilonRIalpha gene (FCER1A) promoter polymorphisms and total serum IgE levels in Japanese atopic dermatitis patients. Int J Immunogenet. 2010;37(2):139–41.
Takaoka A, et al. Involvement of IL-31 on scratching behavior in NC/Nga mice with atopic-like dermatitis. Exp Dermatol. 2006;15(3):161–7.
Sonkoly E, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117(2):411–7.
Neis MM, et al. Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J Allergy Clin Immunol. 2006;118(4):930–7.
Raap U, et al. Correlation of IL-31 serum levels with severity of atopic dermatitis. J Allergy Clin Immunol. 2008;122(2):421–3.
Szegedi K, et al. Increased frequencies of IL-31-producing T cells are found in chronic atopic dermatitis skin. Exp Dermatol. 2012;21(6):431–6.
Danso MO, et al. TNF-alpha and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Investig Dermatol. 2014;134(7):1941–50.
van Drongelen V, Haisma EM, Out-Luiting JJ, Nibbering PH, El Ghalbzouri A. Reduced filaggrin expression is accompanied by increased Staphylococcus aureus colonization of epidermal skin models. Clin Exp Allergy. 2014;44(12):1515–24.
Gruber R, et al. Diverse Regulation of Claudin-1 and Claudin-4 in Atopic Dermatitis. Am J Pathol. 2015;185(10):2777–89.
Arai I, Tsuji M, Takeda H, Akiyama N, Saito S. A single dose of interleukin-31 (IL-31) causes continuous itch-associated scratching behaviour in mice. Exp Dermatol. 2013;22(10):669–71.
Kato A, et al. Distribution of IL-31 and its receptor expressing cells in skin of atopic dermatitis. J Dermatol Sci. 2014;74(3):229–35.
Grimstad O, et al. Anti-interleukin-31-antibodies ameliorate scratching behaviour in NC/Nga mice: a model of atopic dermatitis. Exp Dermatol. 2009;18(1):35–43.
Nemoto O, Furue M, Nakagawa H, Shiramoto M, Hanada R, et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br J Dermatol. 2016;174(2):296–304.
Lan CC, et al. Distinct SPINK5 and IL-31 polymorphisms are associated with atopic eczema and non-atopic hand dermatitis in Taiwanese nursing population. Exp Dermatol. 2011;20(12):975–9.
Sokolowska-Wojdylo M, et al. Association of distinct IL-31 polymorphisms with pruritus and severity of atopic dermatitis. J Euro Acad Dermatol Venereol. 2013;27(5):662–4.
Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int. 2012;61(1):3–17.
Hoffjan S, et al. Analysis of variation in the IL7RA and IL2RA genes in atopic dermatitis. J Dermatol Sci. 2009;55(2):138–40.
Salimi M, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50.
Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol. 2015;15(1):98–103.
Manetti R, et al. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177(4):1199–204.
Trinchieri G. Interleukin-12 and its role in the generation of TH1 cells. Immunol Today. 1993;14(7):335–8.
Xu D, et al. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J Exp Med. 1998;188(8):1485–92.
Tsunemi Y, et al. Interleukin-12 p40 gene (IL12B) 3′-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J Dermatol Sci. 2002;30(2):161–6.
Takahashi N, et al. Association of the IL12RB1 promoter polymorphisms with increased risk of atopic dermatitis and other allergic phenotypes. Human Mol Genet. 2005;14(21):3149–59.
Novak N, et al. Single nucleotide polymorphisms of the IL18 gene are associated with atopic eczema. J Allergy Clin Immunol. 2005;115(4):828–33.
Kim E, et al. Association of the single-nucleotide polymorphism and haplotype of the interleukin 18 gene with atopic dermatitis in Koreans. Clin Exp Allergy. 2007;37(6):865–71.
Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. 2014;5:58.
Mosmann TR, Moore KW. The role of IL-10 in crossregulation of TH1 and TH2 responses. Immunol Today. 1991;12(3):A49–53.
Ekelund E, et al. Elevated expression and genetic association links the SOCS3 gene to atopic dermatitis. Am J Human Genet. 2006;78(6):1060–5.
Sohn MH, et al. Association of interleukin-10 gene promoter polymorphism in children with atopic dermatitis. J Pediatr. 2007;150(1):106–8.
Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124(2):260–9.
McGirt LY, Beck LA. Innate immune defects in atopic dermatitis. J Allergy Clin Immunol. 2006;118(1):202–8.
De Benedetto A, Agnihothri R, McGirt LY, Bankova LG, Beck LA. Atopic dermatitis: a disease caused by innate immune defects? J Investig Dermatol. 2009;129(1):14–30.
An Y, et al. Genetic variations in MyD88 adaptor-like are associated with atopic dermatitis. Int J Mol Med. 2011;27(6):795–801.
Howell MD, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006;121(3):332–8.
Howell MD, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24(3):341–8.
Beck LA, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Eng J Med. 2014;371(2):130–9.
Nemoto O, et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br J Dermatol. 2016;174(2):296–304.
Authors’ contributions
LB acquired the data and drafted the manuscript; DL conceived the review, participated in the acquisition of data, and revised the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
L.B. is supported by Nature Science Foundation of China (No. 81371716). L.B. is an advisory board member for Astellas Pharma Inc. D.L. is supported by the Edelstein Family Foundation, NIH Grants R01 AR41256 and The Atopic Dermatitis Research Network (NIH/NIAID contract NIH/NIAID HHSN272201000020C).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bin, L., Leung, D.Y.M. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin Immunol 12, 52 (2016). https://doi.org/10.1186/s13223-016-0158-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13223-016-0158-5