
Abstract
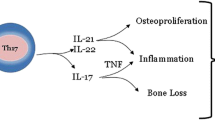
Th17 cells are implicated in human autoimmune diseases, such as rheumatoid arthritis (RA), although it has not been established whether this persistent destructive arthritis is driven by Th1 and/or Th17 cells. Interleukin-17A (IL-17A) contributes to the pathogenesis of arthritis as has been shown in several experimental arthritis models. Importantly, recent data from first clinical trials with anti-IL-17A antibody treatment in psoriatic arthritis patients and RA patients looks promising. This review summarizes the findings about the role of Th17 cells in arthritis and discusses the impact of the different Th17 cytokines in the pathogenesis of this disease. However, further studies are needed to unravel the interplay between IL-17A and other Th17 cytokines such as IL-17F, IL-22, and IL-21 in the pathoimmunological process of this crippling disease, in particular, whether regulating Th17 cell activity or specific combinations of Th17 cytokines will have additional value compared to neutralizing IL-17A activity alone. Moreover, tumor necrosis factor-positive Th17 cells are discussed as potential dangerous cells in driving persistent arthritis in human early RA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Is rheumatoid arthritis a Th1- and/or Th17-driven disease?
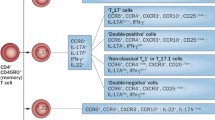
Rheumatoid arthritis (RA) is characterized by synovial inflammation and destruction of joint cartilage and bone mediated by persistent synthesis of proinflammatory cytokines and matrix metalloproteinases (MMP) [4]. T cell activation and migration occurs as an early consequence of RA, and these cells adopt a proinflammatory phenotype. Classically, autoimmune diseases such as RA were thought to be a Th1- and not a Th2-associated disorder [21]. At present, it is unclear whether RA is a Th1- and/or Th17-mediated disease [52] (Fig. 1). It has been shown that interleukin-17A (IL-17A) is produced by some proinflammatory Th1/Th0 cells isolated from synovial membranes and from synovial fluid of RA patients. In joints of patients with established RA, predominantly, Th1 but not Th17 cells were observed [1]. We detected a relatively high percentage of IL-17A-producing CCR6+ memory T cells in peripheral blood mononuclear cell (PBMC) from treatment-naïve early RA patients [14] (Van Hamburg et al., unpublished observations). Of note, the Th1 interferon (IFN)-gamma and the Th17 IL-17 cytokines are often coexpressed in human memory CD4+CD45+RO+ T cells from treatment-naïve early RA patients [14].

Schematic overview of the (potential) role of Th1 and Th17 cells in RA
Although the levels of IL-17A in sera of RA patients is hard to detect, elevated levels of this T cell cytokine have been demonstrated in synovial fluid of these patients [10, 41, 100]. In line with these enhance IL-17A levels in the joint, IL-17A-producing CCR6+ memory T cells have been identified in synovial fluid of RA patients [25].
Similar as for the human situation, in the autoimmune collagen-induced arthritis (CIA) model, the mouse model for RA, it is hard to detect IL-17A levels in the serum of arthritic mice. In contrast, elevated levels of IL-17A have been found in inflamed synovium [53]. IL-17A blocking experiments have shown the importance of this T cell cytokine as proinflammatory in the pathogenesis of CIA [8, 53, 54]. Furthermore, as in human RA, IL-17 plays an important role in the additive/synergistic effects induced together with tumor necrosis factor (TNF) and IL-1, two key cytokines in destructive arthritis [55, 62].
In CIA, it is clear that the IL-23/Th17 axis is critical for the development of autoimmune arthritis. Mice that were deficient for IL-23 were completely protected against the development of CIA. Interestingly, these IL-23-deficient mice had no IL-17+ CD4 T cells but normal antigen-specific IFN-gamma-producing Th1 cells [65]. These data show that IL-17-producing CD4+ T cells but not IFN-gamma-producing CD4+ T cells are critical for the initiation of CIA. In line with these results, we found in the T cell-dependent antigen-induced mono-arthritis (AIA) model that lack of IL-23 did not prevent the onset of AIA but protect against progression of joint inflammation into a destructive synovitis [15]. Lower proportion of IL-17-positive T cell receptor gamma delta and IL-17-positive CD4+ T cells was noted in the spleen, lymph nodes, and inflamed synovium in these IL-23-deficient arthritic mice. Interestingly, although mono-arthritis induction in IL-23-deficient mice did not result in a lower proportion of IFN-g-positive CD4+ T cells in the spleen and lymph node, a lower proportion of these cell was detected in the target organ, the joint [15]. Therefore, IL-23 could be required to promote Th1 and Th17 effector responses, especially at the target organ/tissue of inflammation, and further studies are needed to understand this phenomenon.
Are Th17 cells found in different arthropathies?
Although relatively high numbers of Th17 cells have been shown in PBMC from treatment-naïve early RA patients [14] (Van Hamburg JP et al., unpublished observations), the proportion of these cells in PBMC and synovial-fluid mononuclear cells in established RA varies between no Th17 to low Th17 cells (Table 1). Yamada et al. [96] showed that IL-17-positive cells were detected in CD45RO+CD4 T cells in RA, and most IL-17 positive cells produced neither IFN-gamma nor IL-4, but TNF [96]. The frequency of Th17 cells was neither increased in RA nor correlated with the DAS28 disease activity score. Unexpectedly, the frequency of Th17 cells was significantly decreased in the joints compared with PBMC of the same patients with RA, whereas Th1 cells were more abundant in the joints than in PBMC in these established RA patients [96]. Increased levels of IL-17A, IL-6, TGF-beta, and IFN-gamma concentrations in sera and synovial fluid of reactive arthritis (ReA) and undifferentiated spondyloarthropathy (uSpA) compared to RA have been shown, suggesting that Th1 and Th17 cells could be the major agents in inflammation in ReA/uSpA [81] (Table 1). Jandus et al. [34] found increased numbers of circulating Th17 cells in the peripheral blood of patients with seronegative spondylarthritides (psoriatic arthritis and ankylosing spondylitis), but not in patients with RA (Table 1). In addition, Th17 cells from the spondylarthritis patients showed advanced differentiation and were polyfunctional in terms of T cell receptor-driven cytokine production [34].
Subclinical gut inflammation is common in spondylarthritis. A strong and significant upregulation of IL-23p19 transcript was found in the terminal ileum in patients with AS and patients with Crohn's disease (CD). IL-23 was abundantly produced by infiltrating monocyte-like cells in inflamed mucosa from AS and CD patients [12]. Notably, Paneth cells were identified as a major source of IL-23 in patients with AS, patients with CD, and normal controls [12]. It has been shown that Paneth cells can also be a source of IL-17A in a TNF-induced experimental shock model in mice [84]. However, unlike CD, in AS patients, IL-23 was not associated with upregulation of IL-17 and the IL-17-inducing cytokines IL-6 and IL-1beta, indicating that overexpression of IL-23 but not IL-17 is a pivotal feature if subclinical gut inflammation in AS [12].
In patients with systemic sclerosis (SSc) increased IL-17A messenger RNA was expressed in unstimulated PBMC and lymphocytes from the skin and lungs of SSc patients, but not in similar samples from patients with systemic lupus erythematosus (SLE), polymyositis/dermatomyositis, or from healthy donors (Table 1). IL-17 levels were also increased in the serum of SSc patients, but not in that of SLE patients or healthy donors. IL-17 overexpression was significantly related to the early stage of SSc, but not to other clinical features of SSc [45] Furthermore, Radstake et al. showed that CD4, CD45RA, and CD45RO cells from SSc patients highly express the IL-23 receptor, which was associated with high IL-17 expression as well [75]. In contrast, IFN-gamma and TGF-beta were selectively upregulated in subsets of SSc patients. In addition, circulating levels of IL-17-inducing cytokines IL-6, IL-23, and IL-1alpha were increased in all or subsets of SSc patients. The combination of IL-17, IFN-gamma, and TGF-beta levels in CD45RO and CD45RA cells from SSc patients can be useful to distinguish between limited cutaneous SSc, early diffuse cutaneous SSc, or late diffuse cutaneous SSc [75].
Human data implicating IL-17 in lupus has become available [82, 94]. In contrast, some evidence argues against a role for IL-17 in human lupus [45, 61] (Table 1). In the mouse model for lupus-like autoimmunity, the BXD2 mouse model, a dramatic upregulation of serum IL-17 and numbers of Th17 cells have been demonstrated [27]. BXD2 mice form spontaneous germinal centers which was IL-17-dependent, and treatment with IL-17 promoted the secretion of both IgM and IgG autoantibodies which was reduced by crossing BXD2 mice to IL-17R KO mice [27]. A potential role for IL-17-producing T cells has been shown in Ets-1 knockout mice, another mouse model of lupus [63, 90] and in the spontaneous mouse model of lupus, the New Zealand Black (NZB) × SWR F1 cross (SNF1 mice) [36]. These studies reveal that there is increasing evidence in both humans and mouse models that IL-17-producing cells play a role in SLE progression.
Human Lyme arthritis is caused by Borrelia burgdorferi and is characterized by an inflammatory infiltrate that consists mainly of neutrophils and T cells. B. burgdorferi, Mycobacterium tuberculosis, and synthetic lipopeptides derived from B. burgdorferi outer surface lipoproteins induced IL-17 expression in both murine and human Th cells [30]. The IL-17-producing Th population is characterized by the coexpression of the proinflammatory cytokines IL-17, TNF, and granulocyte-macrophage colony-stimulating factor [30]. Anti-NapA (neutrophil-activating protein A of B. burgdorferi) antibodies were found in 48% of the patients with Lyme arthritis but were undetectable in the healthy controls [13]. T cells from the synovial fluid of patients with Lyme arthritis produced IL-17 in response to NapA. Moreover, NapA was able to induce the expression of IL-23 in neutrophils and monocytes, as well as the expression of IL-6, IL-1beta, and TGF-beta in monocytes via Toll-like receptor 2 [13]. Therefore, NapA of B. burgdorferi is able to elicit a synovial fluid Th17 cell response that might play a crucial role in the pathogenesis of Lyme arthritis [13] (Table 1). In addition, IL-23 is required for the development of arthritis in mice vaccinated and challenged with Borrelia species [42].
In patients with Wegener's granulomatosis (WG) in remission, the percentage of Th17 cells and Th2 cells within the activated CD69+, CD4+ T cell population were significantly increased, while no difference was found in Th1 cells compared with the percentage in healthy controls (Table 1). Increased percentages of Th17 cells in response to tetanus toxoid and staphylococcal enterotoxin B were found both in antineutrophil cytoplasmic antibody (ANCA)-positive and in ANCA-negative patients, while an increased frequency of PR3-specific Th17 cells was restricted to ANCA-positive patients. Therefore, a skewed Th17 response found in ANCA-positive WG patients following stimulation with the autoantigen PR3 suggests that IL-17 is involved in disease pathogenesis and could constitute a new therapeutic target for WG [3].
Also, in Sjogren's syndrome (SS), an upregulation has been shown of the Th17/IL-23 system at the time of disease [68] (Table 1). Salivary gland biopsy specimens from SS patients revealed strong positive staining for both IL-17 and IL-23 within lymphocytic foci and diffuse staining on epithelial tissues. In sera and saliva from SS patients, IL-17 and IL-6 were present at varying levels [68].
In addition, IL-18 and Th17 cells detected in the salivary glands in SS patients are associated with the pathogenesis of SS in the salivary glands [79].
IL-17+ T cells were also detected in the joints of children with juvenile idiopathic arthritis (JIA), and these cells were enriched in the joint compared to the blood of JIA patients [69] (Table 1). Of note, IL-17+ T cell numbers were higher in patients with extended oligoarthritis, the more severe subtype of JIA, as compared with patients with persistent oligoarthritis, the milder subtype [69]. Within the joint, there was an inverse relationship between IL-17+ T cells and FoxP3+ Treg cells [69].
These studies suggest that IL-17+ T cells contribute to the pathogenesis of different arthropathies and that the stage of the disease and the site of expression might be important for the role of these IL-17+ T cells in the pathogenesis of the disease. Furthermore, the balance between, on one hand, Th17 and Th1 and, the other hand, Th17 and Treg may be critical in disease outcome.
What is the functional role of Th17 and Th1 cells in arthritis?
Th17 cells have been detected in different arthropathies, but their functional role in the human disease has not been established. Both Th1 and Th17 cells have been recognized in RA; however, it remains unclear whether Th1 and/or Th17 cells drive disease chronicity [52]. In experimental arthritis models, Th17 cells have been recognized as important contributors to the inflammatory processes [52, 55], and preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in RA and its animal model has been shown [25]. However, in experimental autoimmune encephalitis (EAE) and experimental autoimmune uveoretinitis models, evidence also suggest the potential of Th1 cells as pathogenic mediators [44, 57]. In arthritis, both Th17- and Th1-dependent experimental models have been described [19, 25, 65, 67, 88]. Th17-dependent models can be induced by a specific antigen with complete Freund's adjuvant, but also Th17-dependent spontaneous arthritis models were reported [25, 65, 67, 88]. On the other hand, the development of proteoglycan-induced arthritis has been shown to be IL-17-independent, and the severity of arthritis is dependent on the production of IFN-gamma [19].
However, whether this is also the case in human arthritis needs further clarification including the functional role of these T cell subsets with focus on different stages of the disease.
Are IL-17+TNF+ memory T cells pathogenic Th17 cells in early rheumatoid arthritis?
T cells and the cytokines IL-17A and TNF-alpha have been shown to activate RA synovial fibroblast (RASF), resulting in the expression of proinflammatory cytokines such as IL-6 and IL-8, which are mediators of joint inflammation [6, 7, 10, 60, 62, 73, 85]. In addition, an IL-17A-triggered positive-feedback loop of IL-6 signaling in fibroblast has been described in experimental arthritis and discussed for human Th17-mediated autoimmune disorders [60, 72]. However, characterization of the T cell population that activates RASF and responsible for IL-6 and IL-8 cytokine induction has not been established. We identified IL-17+TNF+Th17 cells in PBMC from treatment-naïve early RA patients. These cells are identified as Th17 cells since they express CCR6, IL-17F, IL-22, IL-26, RORc, CCL20, low T-bet, low FoxP3, and low IFN-gamma. Their functional potential was examined using cocultures with early RASF. CCR6+ (Th17) but not CCR6− (Th1) memory T cells were potent inducers of IL-6, IL-8, and MMPs by the synovial fibroblast in these cocultures. Of interest, also IL-17A expression was increased in these cocultures with synovial fibroblast indicating the induction of a proinflammatory feedback loop. This Th17-RASF proinflammatory feedback loop might be an important mechanism driving persistent arthritis (Van Hamburg JP et al., unpublished observations; Fig. 1). Blocking experiments revealed that both TNF and IL-17A needed to be blocked for optimal suppressive effect, indicating additional value of blocking Th17 activity to anti-TNF neutralization in early RA (Van Hamburg JP et al., unpublished observations).
These are the first data indicating potential pathogenicity of Th17 cells in the human situation. Interestingly, data from first clinical trials with anti-IL-17A antibody treatment in psoriatic arthritis patients and RA patients looks promising. Since additional neutralization of the Th17 cytokine IL-17A to TNF-alpha was needed to suppress the mechanism that drives the proinflammatory feedback loop in cocultures of cells from treatment-naïve early RA patients, blocking Th17 activity in addition to anti-TNF in early RA and potentially other Th17-mediated disorders may be essential to reach the ultimate goals and that is (1) to prevent the development of persistent and destructive arthritis at the very early stage of the disease and (2) reach permanent remission of the disease.
Does plasticity exist between Th1, Th17, and Treg in arthritis?
Th17 cells appear to be associated with Treg cells, which are indicated by the use of a common inducer, TGF-beta, an overlapping chemokine receptor profile, and the expression of the Th17-associated transcription factor RORgammat [32, 87]. In mice, it has been shown that Treg cells can be converted to IL-17-producing T cells [74, 95, 98]. Additionally, also human Treg cells, defined as CD4+CD25highFoxP3+CD127-CD27+, were reported to differentiate to IL-17-producing T cells. This was accompanied by an upregulation of RORgammat and CCR6 expression [38].
Next to plasticity between Treg and Th17 cells new data emerged that Th17 cells are closely related to Th1 cells. In the mouse, differentiated Th17 cells responded rapidly in vitro to IL-12, by upregulating the expression of IFN-gamma and downregulating IL-17 expression. The development of IFN-gamma-producing effector T cells from IL-17-producing progenitor cells is inhibited in the presence of TGF-beta, which is important for the maintenance of IL-17 expression by Th17 polarized cells [49, 50, 58].
Recently, a role for epigenetic regulation of effector T cell plasticity was found. For example, the gene encoding for T-bet, the master regulator of Th1 differentiation, was found to be in an active state, according to histone methylation marks, in both Th17 and Treg cells. This implies that Th17 and Treg cells remain to have the potential to upregulate the expression of T-bet and to differentiate towards Th1 cells [92].
Associations between T cell subsets such as Th17, Th1, and Treg cells have been found in inflammation and progression of RA. In joint inflammation of RA, high levels of IL-17 can be found, and hardly any IFN-gamma expression can be detected in the joint synovium [10, 76]. This was in line with studies in mice, which indicate the importance for IL-17 in the induction of arthritis [55]. In contrast, IFN-gamma cytokine levels were detected in the joint synovium in later stages of RA, and IFN-gamma expression was detected in joint infiltrating CD4 T cells and in CD4 T cell clones, obtained from inflamed synovium [1, 18, 64]. In line with these findings, increased levels of IFN-gamma were detected in lymph nodes and synovium at later time points when adjuvant arthritis was induced in rats. In contrast, IL-17 was induced shortly after initiation and declined in time [9]. In a 2-year predictive study, it became even evident that IFN-gamma was playing a role in reducing joint progression. Inhibition of IFN-gamma in later disease was even exacerbating the disease [37].
Next to Th1 cells, an inverse correlation between IL-17-producing and FoxP3+ Treg cells has been identified. In children with JIA, the balance between IL-17+ cells and Treg cells may even be critical for the disease outcome [69]. In addition, Treg cells are present in higher numbers in inflamed joints in patients with a mild RA phenotype, compared to patients with a more severe disease [17, 78].
These observations indicated that plasticity between Treg, Th1, and Th17 subsets exist and that differentiation of these subsets was not completely restricted to separate lineages. All together, these recent advances provide a complete new concept of T cell plasticity that may be relevant to the arthritis process (Fig. 1).
Do Th17 driving cytokines have any influence on the pathogenicity of Th17 cells?
The role of IL-1, IL-6, IL-23, TGF-beta, and TNF-alpha in the differentiation of mouse and human Th17 cells has been reported [40]. However, the role of IL-23 in the effector function of Th17 cells is not clearly understood. The essential role for IL-23 in the pathogenic function of Th17 cells was shown in EAE [59]. In this experiment, Th17 cells, which were polarized under IL-23 polarizing conditions, were potent inducers of EAE, while the pathogenic function in TGF-beta/IL-6-polarized Th17 cells was completely abrogated. A possible explanation for this discrepancy could be the inhibited induction of IL-10 in the IL-23 polarized cells. Interestingly, TGF-beta/IL-6-polarized Th17 cells modulated the induction of EAE by IL-23-polarized Th17 cells [59]. These data indicate that IL-23 is essential for the pathogenic function of Th17 cells. In line with this, we have shown that IL-23 and TGF-beta/IL-6 differentially regulate T helper specific transcription factors during Th17 development in autoimmune experimental arthritis, which may explain the pathogenic potential of IL-23 [66]. In CD4+ T cells from naïve and collagen-type II immunized DBA/1 mice, IL-23 increased the expression of IL-17A, IL-17F, and RORgammat. In contrast to TGF-beta/IL-6, IL-23 inhibited the Th1 and regulatory T cell-specific transcription factors T-bet and FoxP3 respectively [66].
In addition to IL-23, IL-1beta signaling in T cells has been shown to induce a robust and durable primary and secondary CD4 responses [5]. Mice defective in IL-1R1 signaling were resistant to EAE and exhibit a severe defect in the generation of IL-17-producing T cells, suggesting that IL-1 is important for mouse Th17 cell regulation in vivo [83]. On the other hand, IL-1Ra-deficient mice spontaneously developed chronic inflammatory arthropathy which could be completely suppressed when crossing these mice with IL-17A-deficient mice or TNF-alpha-deficient mice [26, 67]. Moreover, IL-1 signaling regulated early Th17 cell differentiation [11]. IL-1 receptor 1 (IL-1R1) expression in T cells, which was induced by IL-6, was necessary for the induction of experimental autoimmune encephalomyelitis and for early Th17 cell differentiation in vivo [11]. Moreover, IL-1 signaling in T cells was required in dendritic cell-mediated Th17 cell differentiation from naïve or regulatory precursors and IL-1 synergized with IL-6 and IL-23 to regulate Th17 cell differentiation and maintain cytokine expression in effector Th17 cells [11]. IL-1 regulated the expression of the transcription factors IRF4 and RORgammat during Th17 cell differentiation [11].
Thus, similar to the human system, IL-1 plays a unique, nonredundant role during murine Th17 cell polarization [11], and its role in Th17 cell/Th17 cytokine regulation in arthritis may be important [2, 56].
What is the functional role of the Th17 cytokines IL-17F, IL-21, and IL-22 in arthritis?
IL-17F
Apart from the well-studied effects of IL-17A in arthritis [33, 52, 55, 62], little is known about the specific role of IL-17F in arthritis. It was shown that IL-17F is an important regulator of inflammatory responses that seems to function differently than IL-17A in immune responses and diseases [97]. Although IL-17F has many biologically overlapping effects with IL-17A, IL-17F is less potent, for example, in activating synovial fibroblast [101]. IL-17F has been shown to have cartilage destructive potential in vitro [28], IL-17F played only marginal roles, if at all, in CIA and arthritis in IL-1RA-deficient mice [31]. In contrast, both IL-17F and IL-17A were involved in host defense against mucoepithelial infection by Staphylococcus aureus and Citrobacter rodentium [31]. Of note, IL-17A was produced mainly in T cells, whereas IL-17F was produced in T cells, innate immune cells, and epithelial cells [31]. Further studies are needed to clarify the role of IL-17F in arthritis and the interaction of IL-17F with other Th17 cytokines.
IL-21
IL-21 was recently reported to play an important role in the generation of Th17 cells. IL-21 was shown to be potently induced by IL-6 [99]. IL-21 can be produced by Th17 cells [39, 71] and can also be involved in Th17 polarization since in the absence of IL-6, IL-21 in combination with TGF-beta could function as an alternative signal for the induction of Th17 cells [39].
In arthritis, IL-21R-deficient (IL-21R−/−)K/BxN mice were completely refractory to the development of spontaneous arthritis [35]. These mice contain fewer CD4+ T cells with a reduced proportion of homeostatically proliferating cells, fewer follicular Th cells and, surprisingly, more Th17 cells than the control mice [35]. Moreover, these mice also failed to develop IgG1+ memory B cells and autoantigen-specific IgG1 antibodies secreting cells [35]. These data suggest that IL-21 forms a positive-feedback autocrine loop involving homeostatically activated CD4+ cells and is essential in the development of autoimmune arthritis by mechanisms dependent on follicular Th cells development, autoreactive B cell maturation, and RANKL induction but independent of Th17 cell function [35]. In line with this, blocking IL-21 in CIA by in vivo administration of soluble IL-21R-Fc fusion protein delayed the onset and progression of arthritis [35]. In addition, in MLR/lpr mice, a spontaneous model for SLE, blocking IL-21 attenuated disease [24]. Genetic association of IL-21 polymorphisms with SLE has been suggested [80]. In addition, a polymorphism within IL-21R confers a risk for SLE [91]. Moreover, highly suggestive evidence has been provided for IL-2/IL-21 loci as a risk factor for RA [16]. These data suggest IL-21 to be an interesting therapeutic target in arthritis.
IL-22
IL-22 is a cytokine belonging to the IL-10 super family [77] and has been shown to act as an effector cytokine of the Th17 lineage [51]. IL-22 is primarily produced by activated T cells and natural killer cells [93]. High levels of IL-22 were expressed both in the lining and the sublining layers of RA synovial tissues [29]. The majority of IL-22-positive cells were synovial fibroblasts and macrophages. IL-22R1 expression was also expressed in both the lining and the sublining layers of RA synovial tissues [29]. IL-22 significantly increased proliferation of RASF and the production of MCP-1 in vitro [29].
In line with other inflammatory models such as EAE [43], we found that IL-23 is essential for IL-22 production in polarized CD4+ T cells from naïve and type II collagen-immunized DBA/1 mice [66]. IL-22-deficient mice were less susceptible to CIA than wild type mice, as evidenced by their decreased incidence of arthritis and decreased pannus formation [23]. Remarkably, less severe CIA in IL-22-deficient mice was associated with increased production of CII-specific and total IgG antibodies, whereas cellular CII responses were unchanged [23]. In vitro, IL-22 was found to promote osteoclastogenesis [23]. Although these data suggest a proinflammatory role of endogenous IL-22 in arthritis, promoting osteoclastogenesis and regulating antibody production, the uncoupling between low incidence and higher antibody production in IL-22-deficient mice during CIA is unclear and needs further investigation.
In addition to IL-22 produced by Th17 cells, new evidence exist that IL-22 can be produced by effector T cells without IL-17A by a subset of human skin-homing memory T cells [20, 86]. These so called “Th22” cells can also be found in RA (Van Hamburg JP and Lubberts E, unpublished observations); however, the function of these cells in the pathogenesis of RA is unknown.
Modulation of Th17/Th17 cytokine activity in arthritis
Since Th17 cells and Th17 cytokines can have a proinflammatory effect in arthritis, it will be of interest to understand how to regulate the activity of these cells/cytokines. It has been shown that overexpression of the Th2-specific transcription factor, GATA3, in T cells can modulate Th17 cell differentiation and protects against severe joint inflammation and bone erosion in experimental arthritis [88]. Moreover, GATA3 overexpression resulted in reduced gene expression of the Th17-associated transcription factor RORgammat [88].
In addition, treatment of mice with CIA with anti-IL-6R mAb on day 0 markedly suppressed the induction of Th17 cells and arthritis development, but treatment with this antibody on day 14 failed to suppress both Th17 differentiation and arthritis [22]. Of note, treatment of mice with TNFR-Fc from day 0 to day 14 suppressed neither Th17 differentiation nor arthritis, but treatment from day 21 to day 35 successfully ameliorated arthritis without inhibiting Th17 induction [22]. This study indicate that the protective effect of IL-6 blockade, but not TNF blockade in CIA, correlated with the inhibition of Th17 differentiation and suggest that IL-6 blockade in RA in human likely involve a therapeutic mechanism distinct from that of TNF blockade [22]. Moreover, Notley et al. [70] showed that TNF blockade using TNFR-Fc fusion protein or anti-TNF monoclonal antibody reduced collagen arthritis severity but, unexpectedly, expanded populations of Th1 and Th17 cells, which were shown by adoptive transfer to be pathogenic [70]). Th1 and Th17 cell populations were also expanded in CII-immunized TNFR p55KO but not p75KO. The expansion of Th1/Th17 cells was abrogated by blockade of p40 [70]. However, although TNF blockade increased numbers of Th1 an Th17 cells in lymph nodes, it inhibited their accumulation in the joint, thereby providing an explanation for the paradox that anti-TNF therapy ameliorates arthritis despite increasing numbers of pathogenic T cells [70].
Lai Kwan Lam et al. showed that local B cell-activating factor (BAFF) gene silencing suppresses Th7 cell generation and ameliorates autoimmune arthritis [46]. TNF superfamily member BAFF plays an important role in humoral immunity and in autoimmune diseases, including RA. Local BAFF gene targeting inhibited proinflammatory cytokine expression, suppressed generation of plasma cells and Th17 cells, and markedly ameliorated joint pathology [46]. This study revealed a previously unrecognized role for BAFF in promoting the expansion of Th17 cells and demonstrated IL-17 as a crucial effector cytokine for BAFF-mediated proinflammatory effects during CIA development [46].
Apart from regulation of Th17 cells in experimental mouse models for arthritis, Colin et al. showed modulatory effects of 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3) on the memory Th17 activity in untreated early RA patients [14]. 1,25-(OH)2D3 reduced the levels of IL-17A and IFN-gamma and increased IL-4 in stimulated PBMC from treatment-naïve early RA patients [14]. Interestingly, 1,25-(OH)2D3, in contrast to dexamethasone, directly modulated human Th17 polarization accompanied with suppression of IL-17A, IL-17F, TNF-alpha, and IL-22 production by FACS sorted memory T cells from these early RA patients [14].
Conclusions
Ample evidence exist that Th17 cells are important in the initiation of experimental autoimmune arthritis. The occurrence of this T cell subset seems to be dependent on the stage of the disease and localization (systemically and/or at site of inflammation). The functional role of Th17 cells and their cytokines in human arthritis is still not known. However, the first data from clinical trials using anti-IL-17A antibody treatment in psoriatic arthritis and RA are promising. Whether regulating Th17 cell activity or specific combinations of Th17 cytokines will have additional value compared to neutralizing IL-17A activity alone or TNF alone needs to be elucidated.
The identification and diagnosis of RA early in the disease course is becoming increasingly important because early and intensive treatment has been demonstrated to prevent joint damage, to preserve joint function, and to improve work participation of patients [47, 48, 89]. Interestingly, the recognition of Th17 cells and Th17 cytokines in the very early stage of RA fits in the concept that the IL-23/Th17 immune pathway may be important in the development of this autoimmune disease [14, 52] (Van Hamburg et al., unpublished observations). However, the exact biological role of this immune pathway in the development of arthritis needs further examination. In addition, further understanding of the plasticity of T cell subsets in the pathogenesis of chronic destructive arthritis especially at different stages of the disease will be essential to understand the T cell biology in RA. Moreover, understanding of the effects of T cell plasticity on the efficacy of different therapies may improve treatment. Modulation of the Th17 pathway seems to be an interesting approach alone or in addition to anti-TNF to reach the ultimate goal of permanent remission or even to prevent the development of this crippling disease.
References
Aarvak T, Chabaud M, Kallberg E, Miossec P, Natvig JB (1999) Change in the Th1/Th2 phenotype of memory T-cell clones from rheumatoid arthritis synovium. Scand J Immunol 50:1–9
Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, van den Berg WB (2008) Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest 118:205–216
Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG (2008) Skewed distribution of Th17 lymphocytes in patients with Wegener's granulomatosis in remission. Arthritis Rheum 58:2196–2205
Arend WP, Dayer JM (1995) Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum 38:151–160
Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE (2009) IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA 106:7119–7124
Bombara MP, Webb DL, Conrad P, Marlor CW, Sarr T, Ranges GE, Aune TM, Greve JM, Blue ML (1993) Cell contact between T cells and synovial fibroblasts causes induction of adhesion molecules and cytokines. J Leukoc Biol 54:399–406
Brennan FM, McInnes IB (2008) Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest 118:3537–3545
Bush KA, Farmer KM, Walker JS, Kirkham BW (2002) Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum 46:802–805
Bush KA, Walker JS, Lee CS, Kirkham BW (2001) Cytokine expression and synovial pathology in the initiation and spontaneous resolution phases of adjuvant arthritis: interleukin-17 expression is upregulated in early disease. Clin Exp Immunol 123:487–495
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P (1999) Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 42:963–970
Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30:576–587
Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, Peralta S, Franco V, Giardina E, Craxi A, Pitzalis C, Triolo G (2009) Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum 60:955–965
Codolo G, Amedei A, Steere AC, Papinutto E, Cappon A, Polenghi A, Benagiano M, Paccani SR, Sambri V, Del Prete G, Baldari CT, Zanotti G, Montecucco C, D'Elios MM, de Bernard M (2008) Borrelia burgdorferi NapA-driven Th17 cell inflammation in Lyme arthritis. Arthritis Rheum 58:3609–3617
Colin EM, Asmawidjaja PS, Van Hamburg JP, Mus AMC, van Driel M, Hazes JMW, van Leeuwen JPTM, Lubberts E (2010) 1, 25-Dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheum 62:132–142
Cornelissen F, Mus AMC, Asmawidjaja PS, van Hamburg JP, Tocker J, Luberts E (2009) Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORγt in γδ T cells. Arthritis Res Ther 11(6):R194
Daha NA, Kurreeman FA, Marques RB, Stoeken-Rijsbergen G, Verduijn W, Huizinga TW, Toes RE (2009) Confirmation of STAT4, IL2/IL21, and CTLA4 polymorphisms in rheumatoid arthritis. Arthritis Rheum 60:1255–1260
de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, de Jager W, Pugayung G, Giannoni F, Rijkers G, Albani S, Kuis W, Prakken B (2004) CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol 172:6435–6443
Dolhain RJ, van der Heiden AN, ter Haar NT, Breedveld FC, Miltenburg AM (1996) Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum 39:1961–1969
Doodes PD, Cao Y, Hamel KM, Wang Y, Farkas B, Iwakura Y, Finnegan A (2008) Development of proteoglycan-induced arthritis is independent of IL-17. J Immunol 181:329–337
Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F (2009) Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol 10:857–863
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature 423:356–361
Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, Ohsugi Y, Nishikawa T, Ripley B, Kimura A, Kishimoto T, Naka T (2008) Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 58:3710–3719
Geboes L, Dumoutier L, Kelchtermans H, Schurgers E, Mitera T, Renauld JC, Matthys P (2009) Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum 60:390–395
Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K (2007) IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol 178:3822–3830
Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S (2007) Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 204:2803–2812
Horai R, Nakajima A, Habiro K, Kotani M, Nakae S, Matsuki T, Nambu A, Saijo S, Kotaki H, Sudo K, Okahara A, Tanioka H, Ikuse T, Ishii N, Schwartzberg PL, Abe R, Iwakura Y (2004) TNF-alpha is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist-deficient mice. J Clin Invest 114:1603–1611
Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD (2008) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9:166–175
Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, Pan G, Gurney AL, de Vos AM, Starovasnik MA (2001) IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J 20:5332–5341
Ikeuchi H, Kuroiwa T, Hiramatsu N, Kaneko Y, Hiromura K, Ueki K, Nojima Y (2005) Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum 52:1037–1046
Infarte-Durante C, Horton HF, Byrne MC, Kamradt T (2000) Microbial lipopeptides induce the production of IL-17 in T cells. J Immunol 165:6107–6115
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y (2009) Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Iwakura Y, Nakae S, Saijo S, Ishigame H (2008) The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev 226:57–79
Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P (2008) Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum 58:2307–2317
Jang E, Cho SH, Park H, Paik DJ, Kim JM, Youn J (2009) A positive feedback loop of IL-21 signaling provoked by homeostatic CD4 + CD25- T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J Immunol 182:4649–4656
Kang HK, Liu M, Datta SK (2007) Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. J Immunol 178:7849–7858
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ (2006) Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum 54:1122–1131
Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112:2340–2352
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448:484–487
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103:1345–1352
Kotloski NJ, Nardelli DT, Peterson SH, Torrealba JR, Warner TF, Callister SM, Schell RF (2008) Interleukin-23 is required for development of arthritis in mice vaccinated and challenged with Borrelia species. Clin Vaccine Immunol 15:1199–1207
Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B (2007) IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol 179:8098–8104
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM (2008) IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med 205:1535–1541
Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, Takabayashi K, Iwamoto I (2000) Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum 43:2455–2463
Lai Kwan Lam Q, King Hung Ko O, Zheng BJ, Lu L (2008) Local BAFF gene silencing suppresses Th17-cell generation and ameliorates autoimmune arthritis. Proc Natl Acad Sci USA 105:14993–14998
Landewe RB, Boers M, Verhoeven AC, Westhovens R, van de Laar MA, Markusse HM, van Denderen JC, Westedt ML, Peeters AJ, Dijkmans BA, Jacobs P, Boonen A, van der Heijde DM, van der Linden S (2002) COBRA combination therapy in patients with early rheumatoid arthritis: long-term structural benefits of a brief intervention. Arthritis Rheum 46:347–356
Lard LR, Visser H, Speyer I, vander Horst-Bruinsma IE, Zwinderman AH, Breedveld FC, Hazes JM (2001) Early versus delayed treatment in patients with recent-onset rheumatoid arthritis: comparison of two cohorts who received different treatment strategies. Am J Med 111:446–451
Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT (2008) Late developmental plasticity in the T helper 17 lineage. Immunity 30:92–107
Lexberg MH, Taubner A, Forster A, Albrecht I, Richter A, Kamradt T, Radbruch A, Chang HD (2008) Th memory for interleukin-17 expression is stable in vivo. Eur J Immunol 38:2654–2664
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Lubberts E (2008) IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine 41:84–91
Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van de Loo FA, van den Berg WB (2001) IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol 167:1004–1013
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50:650–659
Lubberts E, Koenders MI, van den Berg WB (2005) The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther 7:29–37
Lubberts E, Schwarzenberger P, Huang W, Schurr JR, Peschon JJ, van den Berg WB, Kolls JK (2005) Requirement of IL-17 receptor signaling in radiation-resistant cells in the joint for full progression of destructive synovitis. J Immunol 175:3360–3368
Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR (2008) Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med 205:799–810
Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C (2009) Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol 39:216–224
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8:1390–1397
McInnes IB, Leung BP, Liew FY (2000) Cell-cell interactions in synovitis. Interactions between T lymphocytes and synovial cells. Arthritis Res 2:374–378
Meyers JA, Mangini AJ, Nagai T, Roff CF, Sehy D, van Seventer GA, van Seventer JM (2006) Blockade of TLR9 agonist-induced type I interferons promotes inflammatory cytokine IFN-gamma and IL-17 secretion by activated human PBMC. Cytokine 35:235–246
Miossec P (2003) Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum 48:594–601
Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC (2007) Ets-1 is a negative regulator of Th17 differentiation. J Exp Med 204:2825–2835
Morita Y, Yamamura M, Kawashima M, Harada S, Tsuji K, Shibuya K, Maruyama K, Makino H (1998) Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum 41:1669–1676
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198:1951–1957
Mus AMC et al (2010) Arthritis Rheum (in press)
Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y (2003) IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci USA 100:5986–5990
Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB (2008) Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren's syndrome: findings in humans and mice. Arthritis Rheum 58:734–743
Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR (2008) Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum 58:875–887
Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO (2008) Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med 205:2491–2497
Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C (2007) Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448:480–483
Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T (2008) Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29:628–636
Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, Church LD, Raza K, Wong SH, Trebilcock E, Scheel-Toellner D, Salmon M, Lord JM, Buckley CD (2008) Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis Res Ther 10:R47
Radhakrishnan S, Cabrera R, Schenk EL, Nava-Parada P, Bell MP, Van Keulen VP, Marler RJ, Felts SJ, Pease LR (2008) Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. J Immunol 181:3137–3147
Radstake TR, van Bon L, Broen J, Hussiani A, Hesselstrand R, Wuttge DM, Deng Y, Simms R, Lubberts E, Lafyatis R (2009) The pronounced Th17 profile in systemic sclerosis (SSc) together with intracellular expression of TGFbeta and IFNgamma distinguishes SSc phenotypes. PloS one 4:e5903
Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M (2005) Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther 7:R784–R795
Renauld JC (2003) Class II cytokine receptors and their ligands: key antiviral and inflammatory modulators. Nat Rev 3:667–676
Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F (2005) Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med 201:1793–1803
Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S (2008) Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren's syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol 181:2898–2906
Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, Wakeland EK, Li QZ, Wandstrat AE, Karp DR, James JA, Merrill JT, Lipsky P, Harley JB (2008) Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis 67:458–461
Singh R, Aggarwal A, Misra R (2007) Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J Rheumatol 34:2285–2290
Sullivan KE, Piliero LM, Dharia T, Goldman D, Petri MA (2000) 3′ polymorphisms of ETS1 are associated with different clinical phenotypes in SLE. Hum Mutat 16:49–53
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203:1685–1691
Takahashi N, Vanlaere I, de Rycke R, Cauwels A, Joosten LA, Lubberts E, van den Berg WB, Libert C (2008) IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med 205:1755–1761
Tran CN, Lundy SK, White PT, Endres JL, Motyl CD, Gupta R, Wilke CM, Shelden EA, Chung KC, Urquhart AG, Fox DA (2007) Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am J Pathol 171:1588–1598
Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H (2009) Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nature Immunol 10:864–871
van Hamburg JP, de Bruijn MJ, de Almeida CR, van Zwam M, van Meurs M, de Haas E, Boon L, Samsom JN, Hendriks RW (2008) Enforced expression of GATA3 allows differentiation of IL-17-producing cells, but constrains Th17-mediated pathology. Eur J Immunol 38:2573–2586
van Hamburg JP, Mus AM, de Bruijn MJ, de Vogel L, Boon L, Cornelissen F, Asmawidjaja P, Hendriks RW, Lubberts E (2009) GATA-3 protects against severe joint inflammation and bone erosion and reduces differentiation of Th17 cells during experimental arthritis. Arthritis Rheum 60:750–759
Visser H, le Cessie S, Vos K, Breedveld FC, Hazes JM (2002) How to diagnose rheumatoid arthritis early: a prediction model for persistent (erosive) arthritis. Arthritis Rheum 46:357–365
Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA (2005) Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol 17:1179–1191
Webb R, Merrill JT, Kelly JA, Sestak A, Kaufman KM, Langefeld CD, Ziegler J, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Alarcon GS, Vila LM, Alarcon-Riquelme ME, James JA, Gilkeson GS, Jacob CO, Moser KL, Gaffney PM, Vyse TJ, Nath SK, Lipsky P, Harley JB, Sawalha AH (2009) A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum 60:2402–2407
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O'Shea JJ, Zhao K (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30:155–167
Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R (2006) IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36:1309–1323
Wong CK, Ho CY, Li EK, Lam CW (2000) Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 9:589–593
Xu L, Kitani A, Fuss I, Strober W (2007) Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol 178:6725–6729
Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y (2008) Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis 67:1299–1304
Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C (2008) Regulation of inflammatory responses by IL-17F. J Exp Med 205:1063–1075
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28:29–39
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature Immunol 8:967–974
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W (2000) High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol 164:2832–2838
Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P (2009) Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol 182:3112–3120
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lubberts, E. Th17 cytokines and arthritis. Semin Immunopathol 32, 43–53 (2010). https://doi.org/10.1007/s00281-009-0189-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-009-0189-9