
Abstract
Mechanisms whereby T lymphocytes contribute to synovial inflammation in rheumatoid arthritis are poorly understood. Here we review data that indicate an important role for cell contact between synovial T cells, adjacent macrophages and fibroblast-like synoviocytes (FLS). Thus, T cells activated by cytokines, endothelial transmigration, extracellular matrix or by auto-antigens can promote cytokine, particularly TNFα, metalloproteinase production by macrophages and FLS through cell-membrane interactions, mediated at least through β-integrins and membrane cytokines. Since soluble factors thus induced may in turn contribute directly to T cell activation, positive feedback loops are likely to be created. These novel pathways represent exciting potential therapeutic targets.
Similar content being viewed by others


Introduction
The elucidation of pathways by which T lymphocytes might contribute to synovial inflammation in rheumatoid arthritis (RA) has proved elusive. Thus, mechanisms by which synovial T cells are activated, and subsequently effect articular inflammation, remain incompletely understood. Moreover, the magnitude and nature of such effector pathways might not be constant over time or in different individuals. The predominance of macrophage-derived cytokines, particularly tumour necrosis factor (TNF)-α in the hierarchy of synovial inflammation has been clearly demonstrated in several recent clinical studies [1*]. Those factors that in turn promote such macrophage activation could represent effective therapeutic targets, but are currently ill-defined. This brief review will explore the hypothesis that a significant proportion of such pro-inflammatory activity might reside in synovial T cells through their capacity to modulate inflammation by cell-cell contact.
Potential T cell effector mechanisms in RA synovial membrane
Data from animal models, recognised HLA associations and the clinical efficacy of T cell targeted therapies implicate T cells in promoting RA synovitis [2,3]. Empirically, synovial T cells might drive inflammation through the release of inflammatory mediators, particularly cytokines, or through pathways dependent on cell contact. Several reports indicate that synovial CD4+, CD45RO+ T cells are predominantly of 'type 1' polarity [4,5,6,7,8,9,10]. Synovial T cell clones are usually of Th1 phenotype. Up to 80% of synovial T cells are CCR5+; when stimulated with mitogen they predominantly express interferon (IFN)-γ but not interleukin (IL)-4, as assessed by ELISPOT or intracellular fluorescence-activated cell sorting. T cell derived cytokines that can be detected in synovium, albeit at low levels, are of Th1 type and include IFN-γ and IL-17. Using double specific antibodies, we recently detected spontaneous IFN-γ production by less than 1% of RA synovial T cells ex vivo (JA Gracie and IB McInnes, unpublished data), commensurate with the foregoing immunohistochemical observations. A priori, therefore, it seems unlikely that such cytokine output by T cells could fully explain the extensive and perpetual synovial inflammation apparent in RA.
Evidence for interactions between T cells and macrophages in synovial membrane
T cells and macrophages lie in juxtaposition within and adjacent to cellular aggregates in the synovial membrane, providing for reciprocal cellular cross-talk. Such interactions can effectively activate T cells through the local release of cytokines (eg IL-12, IL-18), through co-stimulation (eg via pathways dependent on CD28, CD154 or CD47), and/or through the presentation of putative auto-antigens [11,12,13]. However, it has recently become clear that T cells in turn might be stimulatory to adjacent macrophages. Dayer and colleagues have provided elegant results demonstrating that T lymphocytes can modulate the activity of a variety of cell types through cell contact [14*,15*]. However, most such studies have employed mitogen-activated T cells. We recently demonstrated that freshly isolated, paraformaldehyde-fixed T-lymphocyte-enriched populations from synovial fluid might induce TNF-α production directly by blood or synovial macrophages through pathways dependent on cell contact [16*], without additional exogenous stimulation. Moreover, this property can be sustained or enhanced in vitro by the addition of cytokines that activate T cells, such as IL-15 [16*]. These observations indicate that a fundamental property of synovial T cells might be the activation of adjacent cells mediated by cell contact.
Several results now indicate that cytokine-mediated 'bystander' activation can confer monocyte activatory capacity on memory T cells, representing a physiological 'surrogate' for mitogen. The cytokine-mediated activation (by IL-2, IL-1β and TNF-α) of resting human CD45RO+, CD4+ T cells can promote cytokine production and help from B cells in the absence of T cell receptor (TCR) ligation [17*]. Similarly, paraformaldehyde-fixed, IL-15-activated T cells derived from peripheral blood are capable of inducing TNF-α production by macrophages [16*]. Combinations of T cell activatory cytokines, including TNF-α, IL-6 and IL-15, which are present within inflamed synovial membrane, seem to be synergistic in this respect [18]. Soluble cytokines present in the local milieu, eg TNF-α, granulocyte-macrophage colony-stimulating factor, IL-10 and IL-11, will further modulate the magnitude of cognate interactions. Complex local autocrine regulatory loops have been proposed in which membrane-bound and secreted cytokines, together with adhesion molecule interactions, are implicated in defining the ratios of synthesis of pro-inflammatory and anti-inflammatory cytokines, eg TNF-α/IL-10 [19] (Fig. 1).
Studies investigating blood-derived lymphocyte-macrophage interactions clearly indicate that secretory activity arising from cell contact extends beyond TNF-α to include numerous cytokines such as IL-1β and IL-12, and cytokine inhibitors such as TNF receptor p75 and IL-1RA [14*,20, 21*,22]. We have also recently demonstrated that synovial macrophages consistently release low levels of IL-15 after contact with cytokine-activated synovial or blood-derived T cells (unpublished data). The production of matrix metalloproteinase (MMP), but not of tissue inhibitor of MMP (TIMP)1, by THP-1 cells and blood-derived macrophages might also be induced after contact with T cells [23]. The relative role of cell contact and soluble factors apparently vary for the production of different cytokines. During investigations of the pro-inflammatory activities of T cells and macrophages in glomerular inflammation, we recently defined discrete requirements for cell contact and for soluble factors during the induction of expression of cytokines, chemokines and adhesion molecules by mesangial or renal tubular epithelial cells [24,25]. For example, whereas either cell-contact or soluble factors induced monocyte chemotactic protein-1 (MCP-1) and IP-10, RANTES production by RTEC was dependent exclusively on contact. Similar studies are required to characterise such regulatory diversity within RA synovial membrane.
The precise membrane ligands implicated in interactions between synovial T cells and macrophages are currently unclear. Co-incubation with neutralising antibodies against LFA-1 (lymphocyte function associated antigen-1), intercellular cell-adhesion molecule-1 (ICAM-1) and CD69 effectively suppresses synovial T cell induced activation of macrophages [15*,16*]. By extension from studies with T cells derived from peripheral blood, CD11b, CD40L and CD45 might also be implicated [21*,22]. Membrane-bound cytokines, particularly IL-1α, TNF-α and IFN-γ, clearly represent logical additional candidate molecules [14*]. It is also of interest that membrane-bound IL-15 has recently been described in blood-derived monocytes [26]. However, such studies are not straightforward because steric hindrance by antibodies can confound, and the formal characterisation of critical pathways remains elusive. Similarly, the signalling consequences within macrophages after T cell contact have been little studied. These might not be easily predicted. For example, a recent comparison of TNF-α synthesis induced by LPS and CD45 in monocytes revealed the discrete use of phosphoinositide 3-kinase and the nuclear factor NF-κB, but common use of p38 mitogen-activated protein kinase (MAPK) pathways by these diverse stimuli [27]. Thus, the precise mechanisms by which T cells are activated, and the relative contribution of subsequent cell contact in comparison with macrophage activation mediated by soluble factors, might profoundly influence signalling events in synovial macrophages. Such detail will probably have important implications in the design of future targeted therapies.
The significance of the predominant Th1 polarity of synovial T cells in the context of cognate interactions is unclear. Recent studies indicate that cytokine-activated Th1 cells exhibit greater potential to enhance the expression of monocyte IL-1β through cell contact than do Th2 cells [21*]. This might in part be mediated through CD154-CD40 interactions. In contrast, Th1 and Th2 cells induce similar IL-10 production, whereas Th2 cells are more efficient in inducing IL-1RA synthesis [22]. We recently described the expression of IL-18 in RA synovial membrane, which together with IL-12 and IL-15 probably sustains a Th1 phenotype in synovial T cells [11]. In the absence of high levels of IFN-γ production in situ, cognate interactions involving synovial Th1 cells might be of critical importance. Studies addressing this important issue are under way in several laboratories.

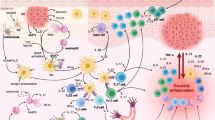
Pathways by which T cell interactions can contribute to synovial inflammation. T cells activated by cytokine combinations, by contact between extracellular matrix and endothelium and potentially by autoantigen, can activate the production of cytokines, MMPs and prostaglandins (PGs) by macrophages and FLS, creating potential positive feedback loops, and leading in turn to articular damage. Contact interactions might be variably mediated through adhesion molecules or membrane cytokines. The parallel secretion of pro-inflammatory and anti-inflammatory cytokines and cytokine receptors further modulate responses. The production by FLS of cytokines, such as IL-7, IL-15 and IL-18, that activate T cells is likely, but because few studies have yet directly addressed this issue, these pathways have been omitted. Interactions between synovial T and B lymphocytes are beyond the scope of this review.
Interactions of T cells with synovial fibroblasts
Cell contact between T lymphocytes and fibroblast-like synoviocytes (FLS) is a further important area of interest. Early studies demonstrated that cytokine-activated FLS bind T cells through pathways that are dependent on at least ICAM-1 [28]. The synthesis of FLS collagen types I and III is downregulated at the transcriptional level by cell contact with mitogen-activated T cells or membrane preparations derived from T cells [29]. These effects are not tissue-specific because similar effects can be mediated on dermal fibroblasts. Direct contact with blood-derived activated T cells, or synovial CD4+ and CD8+ T cell clones, induces the production of MMP and prostaglandin by FLS [30]. In contrast, TIMP1 is only briefly upregulated in this system. Membrane-bound cytokines, including TNF-α and IL-1α, but not CD69, CD154 or CD11b, seem crucial to such interactions between T cells and FLS [30]. The capacity of FLS to respond in co-cultures might be enhanced by TGF-β. Moreover, because FLS activated by IL-1β and TNF-α express in turn cytokines capable of T cell activation, including IL-7, IL-15 and IL-18 [11,16*,31], it is likely that local positive feedback loops, similar to those described above for T cells and monocytes, might be generated. Together these results suggest that interactions between T cells and FLS could contribute significantly to the extra-cellular matrix dysregulation characteristic of RA.
Which factors activate synovial T cells to promote cell-cell interactions?
The foregoing discussion has focused on 'bystander' cytokine-mediated T cell activation. This provides for TCR-independent activation, by which polyclonal memory T cell populations can contribute within an inflammatory lesion, irrespective of local antigen expression. This hypothesis does not preclude a role for T cell populations driven by autoantigens [32]. Several recent reports have identified oligoclonal T cell expansion in RA synovial membrane, particularly among CD4+, CD28- cells [33], although molecular targets remain unknown. However, whether such expanded antigen-specific T cell subsets can contribute in vivo to chronic inflammation in a quantitatively important manner through cell contact is as yet unclear.
The fundamental importance of T cell activation after interactions with endothelial cells has long been recognised. CD45RO+ T cells from peripheral blood exhibit intrinsic migratory potential and are further activated during transmigration through endothelia [34,35]. This can be demonstrated directly by using the human synovium-SCID (severe combined immunodeficiency) mouse model in which human T cell migration is critically dependent on the expression of adhesion molecules and cytokines on endothelial cells [36]. This model provides further powerful evidence of a central role for polyclonal T cells in promoting synovial inflammation in vivo, because T cell depletion mediated by anti-CD2 within synovial grafts leads to the suppression of IL-1β, TNF-α and IL-15 levels and of MMP expression. The administration of T cells leads in turn to replenishment of these cytokine activities [37*].
Conclusion
The therapeutic success of TNF-α blockade illustrates the potential of selective immune pathway targeting in vivo. The foregoing results strongly suggest that T cells might promote the production of pro-inflammatory cytokines and MMP through cell-cell interactions within the synovial membrane (Fig. 1). It is intriguing that the response after anti-CD4 treatment is correlated with the proportion of synovial CD4+ T cells coated by antibody [38]. Moreover, the mechanism for action of anti-TNF-α antibodies, might potentially include the targeting of T cells through recognition of membrane TNF-α. Thus, therapeutic strategies that target both TNF-α and T cells, or critical pathways dependent on cell contact deserve consideration.
References
Maini RN, Taylor PC: Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000, 51: 207-229. 10.1146/annurev.med.51.1.207. This excellent review provides an account of the pathogenetic and clinicalrationale supporting the introduction of TNFa blockade in rheumatoid arthritis.
Fox DA: The role of T cells in the immunopathogenesis of rheumatoid arthritis: new perspectives. Arthritis Rheum. 1997, 40: 598-609.
Panayi G: T-cell-dependent pathways in rheumatoid arthritis. Curr Opin Rheum. 1997, 9: 236-240.
Ronnelid J, Berg L, Rogberg S, Nilsson A, Albertsson K, Klareskog L: Production of T-cell cytokines at the single-cell level in patients with inflammatory arthritides: enhanced activity in synovial fluid compared to blood. Br J Rheumatol. 1998, 37: 7-14. 10.1093/rheumatology/37.1.7.
Dolhain R, Van Der Heiden AN, Ter Haar NT, Breedveld FC, Miltenburg AMM: Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. 1996, 39: 1961-1969.
Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR: The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998, 101: 746-754.
Cohen SB, Katsikis PD, Chu CQ, Thomssen H, Webb LM, Maini RN, Londei M, Feldmann M: High level of interleukin-10 production by the activated T cell population within the rheumatoid synovial membrane. Arthritis Rheum. 1995, 38: 946-952.
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P: Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42: 963-970. 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E.
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W: High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000, 164: 2832-2838.
Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, Dayer JM: CCR5 is characteristic of Th1 lymphocytes. Nature. 1998, 391: 344-345. 10.1038/34814.
Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, Kennedy K, Carter R, Wei XQ, Xu D, Field M, Foulis A, Liew FY, McInnes IB: A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999, 104: 1393-1401.
Sfikakis PP, Zografou A, Viglis V, Iniotaki-Theodoraki A, Piskontaki I, Tsokos GC, Sfikakis P, Choremi-Papadopoulou H: CD28 expression on T cell subsets in vivo and CD28-mediated T cell response in vitro in patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38: 649-654.
Vallejo AN, Mugge LO, Klimiuk PA, Weyand CM, Goronzy JJ: Central role of thrombospondin-1 in the activation and clonal expansion of inflammatory T cells. J Immunol. 2000, 164: 2947-2954.
Vey E, Burger D, Dayer JM: Expression and cleavage of tumor necrosis factor-alpha and tumor necrosis factor receptors by human monocytic cell lines upon direct contact with stimulated T cells. Eur J Immunol. 1996, 26: 2404-2409.
Isler P, Vey E, Zhang JH, Dayer JM: Cell surface glycoproteins expressed on activated human T cells induce production of interleukin-1 beta by monocytic cells: a possible role of CD69. Eur Cytokine Netw. 1993, 4: 15-23. These two papers [14•,15•] are representative of a series of studies fromDayer’s group that have defined the role of T-cell-contact-mediated activationof macrophages and FLS.
McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY: Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nature Med. 1997, 3: 189-195. This paper documents the capacity of synovial T cells to induce TNFa production by macrophages. It clearly demonstrates that cytokines can sustain such activity in the synovial environment.
Unutmaz D, Pileri P, Abrignani S: Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J Exp Med. 1994, 180: 1159-1164. This paper was one of the first to document cytokine mediated T-cell activation.
Sebbag M, Parry SL, Brennan FM, Feldmann M: Cytokine stimulation of T lymphocytes regulates their capacity to induce monocyte production of tumor necrosis factor-alpha, but not interleukin-10: possible relevance to pathophysiology of rheumatoid arthritis. Eur J Immunol. 1997, 27: 624-632.
Parry SL, Sebbag M, Feldmann M, Brennan FM: Contact with T cells modulates monocyte IL-10 production: role of T cell membrane TNF-alpha. J Immunol. 1997, 158: 3673-3681. This paper demonstrates that the effector activities of Th1 and Th2 cells may be defined not only by their cytokine secretory actvity but also by their ability to drive macrophage activation through cell contact.
Avice MN, Demeure CE, Delespesse G, Rubio M, Armant M, Sarfati M: IL-15 promotes IL-12 production by human monocytes via T cell-dependent contact and may contribute to IL-12-mediated IFN-gamma secretion by CD4+ T cells in the absence of TCR ligation. J Immunol . 1998, 161: 3408-3415.
Chizzolini C, Chicheportiche R, Burger D, Dayer JM: Human Th1 cells preferentially induce interleukin (IL)-1beta while Th2 cells induce IL-1 receptor antagonist production upon cell/cell contact with monocytes. Eur J Immunol. 1997, 27: 171-177.
Ribbens C, Dayer JM, Chizzolini C: CD40-CD40 ligand (CD154) engagement is required but may not be sufficient for human T helper 1 cell induction of interleukin-2- or interleukin-15-driven, contact-dependent, interleukin-1beta production by monocytes. Immunology. 2000, 99: 279-286. 10.1046/j.1365-2567.2000.00948.x.
Miltenburg AM, Lacraz S, Welgus HG, Dayer JM: Immobilized anti-CD3 antibody activates T cell clones to induce the production of interstitial collagenase, but not tissue inhibitor of metalloproteinases, in monocytic THP-1 cells and dermal fibroblasts. J Immunol. 1995, 154: 2655-2667.
Kuroiwa T, Lee EG, Danning CL, Illei GG, McInnes IB, Boumpas DT: CD40 ligand-activated human monocytes amplify glomerular inflammatory responses through soluble and cell-to-cell contact-dependent mechanisms. J Immunol. 1999, 163: 2168-2175.
Kuroiwa T, Schlimgen R, Illei GG, McInnes IB, Boumpas DT: Distinct T cell/renal tubular epithelial cell interactions define differential chemokine production: implications for tubulointerstitial injury in chronic glomerulonephritides. J Immunol. 2000, 164: 3323-3329.
Musso T, Calosso L, Zucca M, Millesimo M, Ravarino D, Giovarelli M, Malavasi F, Ponzi AN, Paus R, Bulfone-Paus S: Human monocytes constitutively express membrane-bound, biologically active, and interferon-gamma-upregulated interleukin-15. Blood. 1999, 93: 3531-3539.
Hayes AL, Smith C, Foxwell BM, Brennan FM: CD45-induced tumor necrosis factor alpha production in monocytes is phosphatidylinositol 3-kinase-dependent and nuclear factor-kappaB-independent. J Biol Chem. 1999, 274: 33455-33461. 10.1074/jbc.274.47.33455.
Shingu M, Hashimoto M, Ezaki I, Nobunaga M: Effect of cytokine-induced soluble ICAM-1 from human synovial cells on synovial cell-lymphocyte adhesion. Clin Exp Immunol. 1994, 97: 46-51.
Rezzonico R, Burger D, Dayer JM: Direct contact between T lymphocytes and human dermal fibroblasts or synoviocytes down-regulates types I and III collagen production via cell-associated cytokines. J Biol Chem. 1998, 273: 18720-18728. 10.1074/jbc.273.30.18720.
Burger D, Rezzonico R, Li JM, Modoux C, Pierce RA, Welgus HG, Dayer JM: Imbalance between interstitial collagenase and tissue inhibitor of metalloproteinases 1 in synoviocytes and fibroblasts upon direct contact with stimulated T lymphocytes: involvement of membrane-associated cytokines. Arthritis Rheum. 1998, 41: 1748-1759. 10.1002/1529-0131(199810)41:10<1748::AID-ART7>3.3.CO;2-V.
Harada S, Yamamura M, Okamoto H, Morita Y, Kawashima M, Aita T, Makino H: Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42: 1508-1516. 10.1002/1529-0131(199907)42:7<1508::AID-ANR26>3.0.CO;2-L.
Cope AP, Patel SD, Hall F, Congia M, Hubers HA, Verheijden GF, Boots AM, Menon R, Trucco M, Rijnders AW, Sonderstrup G: T cell responses to a human cartilage autoantigen in the context of rheumatoid arthritis-associated and nonassociated HLA-DR4 alleles. Arthritis Rheum. 1999, 42: 1497-1507. 10.1002/1529-0131(199907)42:7<1497::AID-ANR25>3.0.CO;2-#.
Goronzy JJ, Zettl A, Weyand CM: T cell receptor repertoire in rheumatoid arthritis. Int Rev Immunol. 1998, 17: 339-363.
Brezinschek RI, Lipsky PE, Galea P, Vita R, Oppenheimer-Marks N: Phenotypic characterization of CD4+ T cells that exhibit a transendothelial migratory capacity. J Immunol. 1995, 154: 3062-3077.
Iannone F, Corrigall VM, Kingsley GH, Panayi GS: Evidence for the continuous recruitment and activation of T cells into the joints of patients with rheumatoid arthritis. Eur J Immunol. 1994, 24: 2706-2713.
Oppenheimer-Marks N, Brezinschek RI, Mohamadzadeh M, Vita R, Lipsky PE: Interleukin 15 is produced by endothelial cells and increases the transendothelial migration of T cells in vitro and in the SCID mouse- human rheumatoid arthritis model in vivo. J Clin Invest. 1998, 101: 1261-1272.
Klimiuk PA, Yang H, Goronzy JJ, Weyand CM: Production of cytokines and metalloproteinases in rheumatoid synovitis is T cell dependent. Clin Immunol. 1999, 90: 65-78. 10.1006/clim.1998.4618. This important paper demonstrates in the murine SCID-human synovium model that T cells can maintain the monokine rich environment in synovial membrane.
Choy EH, Pitzalis C, Cauli A: Percentage of anti-CD4 monoclonal antibody-coated lymphocytes in the rheumatoid joint is associated with clinical improvement. Implications for the development of immunotherapeutic dosing regimens. Arthritis Rheum. 1996, 39: 52-56.
Acknowledgement
We thank the Arthritis Research Campaign (UK), The Wellcome Trust, The Scottish Chief Scientist Office, and the Medical Research Council (UK) for their invaluable support.
Author information
Authors and Affiliations
Additional information
Articles of particular interest have been highlighted as:
• of special interest
•• of outstanding interest
Rights and permissions
About this article
Cite this article
McInnes, I.B., Leung, B.P. & Liew, F.Y. Cell-cell interactions in synovitis Interactions between T lymphocytes and synovial cells. Arthritis Res Ther 2, 374 (2000). https://doi.org/10.1186/ar115
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar115