
Abstract
Currently, contamination of indoor environment by fungi and molds is considered as a public health problem. The monitoring of indoor airborne fungal contamination is a common tool to help understanding the link between fungi in houses and respiratory problems. Classical analytical monitoring methods, based on cultivation and microscopic identification, depend on the growth of the fungi. Consequently, they are biased by difficulties to grow some species on certain culture media and under certain conditions or by noncultivable or dead fungi that can consequently not be identified. However, they could have an impact on human health as they might be allergenic. Since molecular methods do not require a culture step, they seem an excellent alternative for the monitoring of indoor fungal contaminations. As a case study, we developed a SYBR® green real-time PCR-based assay for the specific detection and identification of Aspergillus versicolor, which is frequently observed in indoor environment and known to be allergenic. The developed primers amplify a short region of the internal transcribed spacer 1 from the 18S ribosomal DNA complex. Subsequently, the performance of this quantitative polymerase chain reaction (qPCR) method was assessed using specific criteria, including an evaluation of the selectivity, PCR efficiency, dynamic range, and repeatability. The limit of detection was determined to be 1 or 2 copies of genomic DNA of A. versicolor. In order to demonstrate that this SYBR® green qPCR assay is a valuable alternative for monitoring indoor fungal contamination with A. versicolor, environmental samples collected in contaminated houses were analyzed and the results were compared to the ones obtained with the traditional methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The effects of allergenic molds on public health are well documented (Bellanger et al. 2009; Packeu et al. 2012; Reboux et al. 2010). Indeed, molds can produce mycotoxins, spores, hyphae, and wall fragments containing (1 → 3)-β-D-glucans and proteins which could induce allergic reactions of types I, III, and IV (Douwes et al. 2003; Seo et al. 2008). Some studies revealed an association between the fungal levels in the air and the occurrence of allergy (Horner et al. 1995; Meheust et al. 2014; Mendell et al. 2011; Reboux et al. 2010) and between water-damaged buildings and human exposure to fungal contamination present in indoor air (Jones et al. 2011; Vesper et al. 2013). Presently, with the climate change, the new energy conservation measures, the development of the urbanization, and the considerable amount of time spent inside buildings, people are increasingly exposed and might be more susceptible to developing a respiratory problem caused by a fungal contamination (de Ana et al. 2006; Mendell et al. 2011; Sharpe et al. 2014). More specifically, Aspergillus versicolor is one of the most important fungal contaminants of houses (Beguin and Nolard 1994; Packeu et al. 2012), and this species is known to produce allergenic compounds (Benndorf et al. 2008), which can have an implication in the development of asthma (de Ana et al. 2006).
Usually in routine analysis, the detection and identification of indoor airborne fungi are based on culture, microscopic visualization, and visual counts. However, this classical approach requires an important level of expertise and is time-consuming (Vesper 2011). Another drawback of this approach is that it depends on the growth of the culture, which is known to be affected by the growth media chosen, the culture conditions (especially temperature and humidity), the incubation time (some species grow faster than others), or competition between species. Furthermore, a part of the sampled biological fraction is dead and noncultivable and therefore is not always detected by traditional analysis methods, although it could have an impact on human health as it might be allergenic. Therefore, this could lead to an underestimation of the total fungal airborne community and level of the contamination classically determined by colony-forming units (CFU) on an agar plate (Pitkaranta et al. 2011; Vesper 2011).
Molecular techniques like the real-time quantitative polymerase chain reaction (qPCR), amplifying a specific DNA sequence in the fungal genome with a real-time detection of the amplification products, have been proposed as an alternative to these classical detection methods (Black et al. 2013; Haugland et al. 2004; Timothy et al. 2004). Besides being fast, sensitive, and specific, qPCR also gives the advantage of being independent from culture.
Most often qPCR methods designed for fungal detection are based on the ribosomal gene complex and particularly on its internal transcribed spacers (ITS) (Bellanger et al. 2009; Chemidlin Prevost-Boure et al. 2011; Costa et al. 2001; Johnson et al. 2012; Melkin et al. 2004; Michealsen et al. 2006; Roussel et al. 2013), which are noncoding regions of the fungal rDNA (ITS 1 and ITS 2) flanked by the small subunit (SSU) rRNA and by the large subunit (LSU) rRNA genes. The sequence variation of ITS regions has led to their use in phylogenetic studies of many different organisms (Nilsson et al. 2008). These ITS regions are selected for qPCR assays due to their good conservation and their weak level of polymorphism amongst DNA sequences of the same genus (Chemidlin Prevost-Boure et al. 2011; Costa et al. 2001; Iwen et al. 2002). Therefore, they offer the possibility to develop specific molecular methods for detection at the genus or even at the species level (Hinrikson et al. 2005; Nilsson et al. 2008; Schoch et al. 2012).
The qPCR methods are started to be more frequently used for environmental investigations and monitoring of the most common fungal contaminants present inside buildings (Bellanger et al. 2009; Roussel et al. 2013), such as performed by the US Environmental Protection Agency (EPA). Indeed, the EPA has developed a large set of qPCR methods based on the TaqMan® chemistry, as real-time detection system of the amplified products aimed for indoor fungi monitoring (A. versicolor included) (Haugland et al. 2004; United States Environmental Protection Agency 2014). These labeled probe-based qPCR assays are highly specific. An alternative to TaqMan® method is the SYBR® green chemistry. Here, the detection of amplification is based on an intercalating fluorescent dye, independent of the use of a labeled probe. The specificity of this method is determined by the primers’ specificity and by the melting temperature (T m) of the amplicon. This approach is also sensitive and fast but cheaper than the probe-based one. However, despite these advantages, not many SYBR® green assays exist yet for the detection and identification of airborne indoor molds. To the best of our knowledge, no SYBR® green based detection and identification method for A. versicolor has been reported yet.
Additionally, currently, even if many qPCR methods exist for the fungal detection, no specific guidelines are proposed concerning the assessment of their performance. This is in contrast to other domains where real-time PCR methods are already used during many years for the detection and identification of specific targets, such as for the detection of genetically modified organisms (GMO) in food and feed (Broeders et al. 2014; European Network of GMO Laboratories (ENGL) 2008; ENGL 2015). Recently, the guidelines established for the validation of real-time PCR methods for GMO detection have been used to evaluate qPCR assays for the detection and identification of bacterial pathogens (Barbau-Piednoir et al. 2013b). However, for fungal qPCR detection methods, this has not yet been done.
The present study reports on the development of a SYBR® green qPCR method for A. versicolor detection. The primers designed in this study are selected in the ITS region. The performance of the developed qPCR assay was subsequently assessed, using the guidelines for validation of qualitative real-time PCR methods based on criteria defined for GMO. The PCR efficiency, dynamic range, sensitivity, selectivity, and repeatability of the developed A. versicolor SYBR® green qPCR were evaluated and discussed. Finally, a proof of concept for the developed qPCR method was delivered using air samples collected in two contaminated houses, by comparing the qPCR results to the results obtained with traditional analysis.
Materials and methods
Fungal strains
All the fungal species (A. versicolor, Aspergillus creber, Aspergillus sydowii, Aspergillus fumigatus, Alternaria alternata, Cladosporium cladosporoïdes, Cladosporium herbarum, Cladosporium sphaerospermum, Penicillium chrysogenum, Stachybotrys charatum, Ulocladium botrytis) and strains used in this study are listed in Table 1. All of them were purchased from the BCCM/IHEM collection (Scientific Institute of Public Health in Brussels, Belgium).
Culture conditions and DNA extraction
The fungal strains were grown in a S10 Sabouraud liquid medium (Bio-Rad, Temse, Belgium) at 25 °C with constant agitation between 3 and 10 days according to the species’ growth conditions.
After this incubation time, 300 mg of wet sample was transferred to cryotubes containing 0.25 ml of acid-washed glass beads (Sigma-Aldrich, Diegem, Belgium) put at −80 °C during 40 min and freeze-dried overnight with a freeze-dryer Epsilon 1-6D (Martin Christ, Osterode am Harz, Germany). Freeze-dried fungi were subsequently beat-beaten with a Mini bead beater (Biospec Products, OK, USA) during 1 min at maximal speed.
The total DNA was extracted with an adapted phenol chloroform (24:1) protocol (Ashktorab and Cohen 1992) and purified with the Qiagen CTAB genomic Tip-20 kit (Qiagen Benelux, B.V., KJ Venlo, the Netherlands) according to the manufacturer’s recommendation. DNA was eluted with 100 μl Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium). The DNA integrity was verified on a 2 % agarose gel. The DNA amount and purity were evaluated with a Nanodrop® 2000 (Thermo Scientific, Wilmington, USA).
Design of primers
All the at the time of primer design in NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) available 18S rDNA sequences from A. versicolor strains as well as from other closely related species (namely A. creber, A. fumigatus, A. sydowii, P. chrysogenum) were collected and aligned with the “MegAlign” software V10.0.1 (Lasergene, Madison, USA) to identify the sequence region of interest. The publicly available sequences of 18S rDNA used were for A. versicolor, AJ937751.1/AJ937753.1/AJ937754.1/AJ937755.1/AM883155.1/AM883156.1/AY728196.1/EF125026.1/EU042148.1/FJ878627.1/FJ878625.1/FJ461692.1/FJ904814.1/KJ466864.1/JN205048.1, for A. creber, KJ775474.1, for A. fumigatus, KC411924.1/KC237295.1/KC237291.1/KC237292.1/KC142152.1/HE864321.1/KC119199.1/KC119200.1/JX944178.1/JX944118.1, for A. sydowii, DQ114468.1/FJ807779.1/HQ625522.1/JN94914.1/KJ775568.1/KJ775569.1/KJ775570.1/KJ775571.1/KJ775574.1, and for P. chrysogenum, JN903544.1/JN798499.1/JX535315.1/AF033465.1/HQ336383.1/GU325676.1/EU709771.1/JX996985.1/JF834167.1/FJ004280.1.
Then, different primer pairs were designed in the regions of ITS 1 and ITS 2 with the “Primer 3 V.0.4” software (http://bioinfo.ut.ee/primer3-0.4.0/) (Untergasser et al. 2007). Primer dimers and secondary structure formation were evaluated during the design with Primer 3. An in silico specificity test was performed with the “wprimersearch” software (https://wemboss.uio.no/wEMBOSS/) (Sarachu and Colet 2005). This in silico PCR simulation allows selecting the primer pairs that only amplify the targeted sequences. The specificity of the primers was also verified using BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi). In total, seven primer pairs have been designed (Table 2).
Qualitative SYBR® green qPCR assay
The qPCR assay (Aversi_ITS qPCR method) was performed with the SYBR® green chemistry using a real-time PCR IQ5™ system from Bio-Rad (Temse, Belgium).
The standard reaction mix (25-μl final volume) contained 12.5 μl of 2× SYBR® green PCR Mastermix (Diagenode, Liège, Belgium), 0.25 μl of Aversi_ITS forward and reverse primers (0.2 μM), and 7 μl of Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium). To this mix, 5 μl of genomic DNA (gDNA) at 200 theoretical genomic copy numbers per microliter was added. The number of genomic DNA copies was calculated according to the formula presented below:
with C n as genomic copy number, m as the amount of gDNA (grams) and determined by Nanodrop® 2000 (Thermo Scientific, Wilmington, USA), Ac as the Avogadro’s constant (Mohr et al. 2008), M w as base pair mean molecular weight (649 Da), and G s as genome size (expressed in basepairs) of A. versicolor = 33,130,000 bp (Joint Genome Institute 2014). There is only publicly available information on the size of the genome of one specific A. versicolor strain. This size was taken to calculate the number of genomic DNA copies for all A. versicolor strains, although some strain-dependent deviations may exist.
All the runs were performed using following thermal cycling conditions: 1 cycle of 95 °C for 10 min (Taq activation), followed by 40 amplification cycles of 15 s at 95 °C (denaturing step) and the annealing and extension step at 60 °C for 1 min. Afterwards, a melting curve was performed with a gradual increase of temperature of 0.5 °C/6 s from 55 to 95 °C during 15 min. The threshold level for the reaction was automatically determined by the Bio-Rad IQ 5 software V. 2 (Bio-Rad, Temse, Belgium).
In each reaction, a “no template control” (NTC) was included for the analysis, i.e., the DNA template was replaced by ultrapure water in the reaction mix. This NTC allowed verifying that no contamination occurred and that no primer dimers were formed.
Strain confirmation: sequencing and theoretical T m calculation
In order to confirm their identity, the ITS 1 and 2 regions of A. versicolor (IHEM 18884, IHEM 1323, IHEM 1355, IHEM 2023, IHEM 2157, IHEM 2788, IHEM 6598, IHEM 9674, IHEM 10351, IHEM 19014, IHEM 19210, IHEM 19256, IHEM 29832, IHEM 22014, IHEM 2757, IHEM 24424), A. creber (IHEM 2646), and A. sydowii (IHEM 895, IHEM 1360, IHEM 20347) were verified by Sanger sequencing analysis on an ABI3130xl Genetic Analyzer apparatus (Applied Biosystems, Life Technologies, Gent, Belgium) with the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Life Technologies, Gent, Belgium) according to the manufacturer’s recommendations. The ITS 1 and 2 regions were firstly amplified with the primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) (Gardes and Bruns 1993) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (White et al. 1990) and then sequenced with the primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) (White et al. 1990). The consensus sequence of each of the targeted regions was made based on the forward and reverse sequence. These sequences were aligned with the MEGA v6.06 (http://www.megasoftware.net/) software and visualized with the CLC sequence viewer v7.0.2 (Qiagen Benelux, B.V., KJ Venlo, the Netherlands). The consensus sequences were also compared to the sequences available in the NCBI database by using BLASTn (http://blast.ncbi.nlm.nih.gov/) in order to confirm the amplification of the targeted DNA regions and their identity.
Based on these sequences, the theoretical T m of the amplicon obtained in the Aversi_ITS assay was calculated in silico with the online tool OligoAnalyzer 3.1 from IDT (http://eu.idtdna.com/calc/analyzer) (IDT, Leuven, Belgium) under the PCR conditions described above (White et al. 1990).
Aversi_ITS assay: performance assessment
Selectivity test
The selectivity test was composed of two steps as previously reported for the validation of qPCR methods for food pathogens (Barbau-Piednoir et al. 2013a, b). Firstly, a preliminary selectivity test was carried out on the target species (A. versicolor IHEM 18884) and two nontarget species (i.e., A. fumigatus IHEM 3562 and P. chrysogenum IHEM 20859), using 15,000 theoretical copies of gDNA.
Secondly, a larger selectivity test was performed, evaluating the inclusivity (the selected primers should amplify DNA of each tested strain from the tested target species) and the exclusivity (DNA of nontarget species close to the target or described to frequently occur in the same environment with the target species should not be amplified by the selected primers) of the Aversi_ITS qPCR method.
The experimental design of the full selectivity test was adapted from Barbau-Piednoir et al. (2013a), i.e., 16 target strains (A. versicolor) and 14 nontarget strains were included, i.e., 4 from the Aspergillus section Versicolores (Jurjevic et al. 2012) (A. creber IHEM 2646, A. sydowii IHEM 895, IHEM 1360, IHEM 20347) and 10 of the most common indoor airborne fungi (Beguin and Nolard 1994) (Table 1).
Each qPCR was performed with the SYBR® green technology under the conditions described above using a total of 1000 theoretical copies of gDNA per reaction (evaluated for each target with its own corresponding genome size).
Dynamic range and efficiency estimation
The linearity of this SYBR® green qPCR assay was assessed based on the qPCR analysis of a serial dilution, in duplicate, of gDNA (1000, 500, 100, 50, 10, 5, 2, and 1 theoretical copy number of gDNA) obtained by two independent extractions of A. versicolor IHEM 18884. This analysis gives the possibility to assess two parameters, i.e., the coefficient of determination (R 2) and the PCR efficiency. R 2 is an indicator of the correlation of the data regarding the linear regression curve. The PCR efficiency (E) calculation was previously described in Rutledge and Cote (2003). According to the most recent guidelines developed for GMO detection with qPCR SYBR® green (ENGL 2015), the R 2 and amplification efficiency are not applicable to qualitative methods. However, a R 2 ≥ 0.98 and a PCR efficiency ranging between 80 and 120 % have previously been indicated as performance criteria for the validation of qualitative qPCR methods (Broeders et al. 2014).
Sensitivity test: limit of detection
To evaluate the sensitivity of the Aversi_ITS assay, a serial dilution of gDNA of A. versicolor IHEM 18884 was performed to determine the limit of detection (LOD). The LOD is defined as the lowest concentration of an analyte which is detected with a probability of 95 % (Barbau-Piednoir et al. 2013b).
To estimate this LOD, seven dilutions of gDNA of A. versicolor IHEM 18884 (i.e., 10, 5, 2, 1, 0.5, 0.2, and 0.1 theoretical copies of gDNA) were tested in six independent runs, each with six repetitions. The qPCR conditions applied here were the same as those described above. The LOD should be below 25 copies according to definition of minimum performance requirements for analytical methods of GMO testing (ENGL 2015).
Repeatability calculation
As previously described (Barbau-Piednoir et al. 2013b), the repeatability limit (r) of the Aversi_ITS qPCR method was evaluated with the same experimental design than that applied for the LOD evaluation (see section “Sensitivity test: limit of detection”). This r value is defined as the maximal difference of two results obtained under identical experimental conditions with a probability of 95 % (Barbau-Piednoir et al. 2013b).
The relative standard deviation of the repeatability (RSDr) was calculated as the absolute value of the coefficient variation and expressed in percentage. For these criteria, there is no limit fixed for qualitative qPCR methods (ENGL 2015). The RSDr, evaluated for the C q values, should be ≤25 % for all dilutions above the LOD for quantitative methods (ENGL 2015).
Environmental testing: inhibition test
Before qPCR analysis of the environmental samples, an inhibition test was performed in order to verify that no inhibition from the collection liquid, used for air sampling with the Coriolis µ air sampler (Bertin Technology, Montigny-le-Bretonneux, France), occurred during the qPCR. To this end, 300 mg of an A. versicolor IHEM 18884 culture was spiked, in duplicate, into 15 ml of collection liquid, i.e., ultrapure water containing Tween® 20 0.01 % (Sigma-Aldrich, St Louis, USA). Then, the spiked solutions were centrifuged at 5000g during 15 min. The supernatant was removed, the pellet was suspended in 1.5 ml of Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium), and the DNA was extracted according to the protocol described above. Afterward, each undiluted DNA extract and a 10-fold dilution thereof were analyzed with the Aversi_ITS qPCR assay. Theoretically, the C q difference (∆C q) between the C q value of the 10-fold diluted and the undiluted sample is 3.3 when the PCR efficiency is 100 %. The experimental ∆C q is calculated based on the obtained C q for both samples (difference between the obtained C q for the 10-fold diluted and the undiluted sample). Taking into account the PCR efficiency of each run, we considered that there are no PCR inhibitors in the DNA extract if the experimental ∆C q value for the 10-fold dilution equals 3.3 ± 0.5.
Environmental testing: proof of concept
To assess the performance of the Aversi_ITS assay to detect A. versicolor in real-life samples, two houses contaminated by fungi were studied in the framework of the activities of the CRIPI (Cellule Régionale d’Intervention en Pollution Intérieure from Brussels Environment, Brussels, Belgium). In each house, two sets of air samples were independently collected for classical and molecular analysis respectively, in minimum four different rooms, i.e., the bedroom, bathroom, living, and kitchen. In each procedure, air samples were taken at the height of a seated person. To avoid any contamination by air flow, each room was as much as possible isolated from outside by closing windows and doors.
A first set of samples of air (0.08 m3 for each sample) were collected and analyzed according to the procedure previously defined (Nolard et al. 2004) and used in routine by the CRIPI (Nolard et al. 2004). Briefly, air contaminants were directly sampled on Sabouraud chloramphenicol agar (Bio-Rad, Temse, Belgium) slipped on HYCON® Agar Strips (Merck, Darmstadt, Germany), using the RCS plus (Biotest, Rupperswil, Switzerland) air sampler following the manufacturer’s instructions and with a flow rate of 80 l/min during 1 min.
After an incubation of 5 days at 25 °C for mesophilic and 2 days at 45 °C for thermophilic fungi, the species determination was performed by microscopic visualization (Nolard et al. 2004). The level of contamination was evaluated by counting of the colony-forming units (CFU) on plate and expressed as CFU/m3 according to the guidelines defined by Nolard and coworkers (2004).
The second set of air samples (1.5 m3 for each sample) were taken in duplicate with the Coriolis µ air sampler (Bertin Technology, Montigny-le-Bretonneux, France) and collected in 15 ml of ultrapure water containing Tween® 20 (0.01 %) (Sigma-Aldrich, St Louis, USA) in an appropriate sterile cone (Bertin Technology, Montigny-le-Bretonneux, France). A flow rate of 300 l/min during 5 min was applied in each room. In each room, two cones were collected; the first one was analyzed by culturing and microscopic visualization, the second one by qPCR for the detection of A. versicolor. The first cone was put at −80 °C until analysis.
To analyze the Coriolis µ air samples with the culturing approach, the samples were centrifuged at 5000g during 15 min, and the supernatant was discarded. The pellet was suspended in 1.5 ml of Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium) and plated on Sabouraud chloramphenicol agar (Bio-Rad, Temse, Belgium), using an inclusion method. Hereto, under laminar flow, the 1.5-ml samples were put into empty petri dishes. Afterward, Sabouraud chloramphenicol medium was poured into the plates and kept at room temperature until the medium was solidified. The plates were incubated 5 days at 25 °C for mesophilic and 2 days at 45 °C for thermophilic fungi as it was done for the routine protocol described above. The species determination was performed by microscopic visualization (Nolard et al. 2004).
To analyze the Coriolis µ air samples by qPCR for the presence of A. versicolor, samples were first centrifuged at 5000g during 15 min, and the supernatant was discarded. Next, the pellet was suspended in 1.5 ml of Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium), and the extraction was performed with the DNA extraction protocol used for pure cultures (see section “Culture conditions and DNA extraction”). The DNA amount and purity were evaluated with a Nanodrop® 2000 (Thermo Scientific, Wilmington, USA). The qPCR reactions were performed on 10 μl of eluted DNA, i.e., 10 % of the total DNA eluted from in 1.5 m3 of air sampled, corresponding to the amount of DNA mentioned in Table 5.
Results
Design and selection of A. versicolor qPCR primer pair
To select the A. versicolor primers, all the at the time of the primer design publicly available A. versicolor 18S rDNA sequences were searched. This collection of sequences was extended with those available for A. creber and A. sydowii. These species are both members of the Versicolores group as is also A. versicolor. These three species are difficult to be morphologically distinguished. Based on an in silico analysis of these sequences, seven forward primers and two reverse primers targeting a conserved region of the full ITS 1 region of A. versicolor were designed (Table 2, Fig. 1).

Alignment of selected forward and reverse Aversi_ITS primers on ITS1 region sequences of A. versicolor, A. creber, and A. sydowii. This alignment was made using publicly available ITS sequences of Aspergilus versicolor, A. creber, and A. sydowii, extended with ITS sequences from the strains from the BCCM/IHEM collection used during the validation of the qPCR assay (indicated with IHEM prefix) and with the primers designed in this study (Aversi_ITS_f and Aversi_ITS_r). Because no nucleotide variation was detected for each of the public sequences used, only one sequence was introduced for this alignment, as a representative for that species. The accession numbers of all the NCBI sequences used in this study are listed hereunder, i.e., for A. versicolor AJ937751.1/AJ937753.1/AJ937754.1/AJ937755.1/AM883155.1/AM883156.1/AY728196.1/EF125026.1/EU042148.1/FJ878627.1/FJ878625.1/FJ461692.1/FJ904814.1/KJ466864.1/JN205048.1, for A. creber KJ775474.1, for A. sydowii DQ114468.1/FJ807779.1/HQ625522.1/JN94914.1/KJ775568.1/KJ775569.1/KJ775570.1/KJ775571.1/KJ775574.1. The ITS1 region of the BCCM/IHEM strains of A. versicolor (16), A. creber (1) and A. sydowii (3) used during the performance assessment of the qPCR assay was sequenced and aligned to those publicly available. Consensus (last line of the alignment) corresponds to a consensus sequence defined by the software. The conservation level among each sequence (0 to 100 % of conservation) is represented by the pink rectangles at the bottom of the figure
Unfortunately, in the conserved ITS1 region for A. versicolor, it was not possible to design specific primers which are not conserved in A. creber and A. sydowii (Fig. 1). The amplicons obtained with the Aversi_ITS f/r primers present 100 % of identity when using the sequences for these three species available in NCBI (Fig. 1). Preliminary experimental specificity tests were performed on DNA of A. versicolor, A. fumigatus, and P. chrysogenum. The Aversi_ITS_1f/r primer pair (Fig. 1, Table 2) was selected for the detection of A. versicolor as it was the unique combination of forward and reverse primers allowing the amplification of A. versicolor DNA and not that of the closely genetically related A. fumigatus and P. chrysogenum (data not shown).
Aversi_ITS assay: performance assessment
The performance of this Aversi_ITS assay was assessed according to the guidelines defined for the validation of qPCR detection and identification methods in other fields (Barbau-Piednoir et al. 2013b; Broeders et al. 2014; ENGL 2015). The performance assessment includes the evaluation of the following criteria: the selectivity of the primers, the dynamic range, the PCR efficiency, the LOD, and the Aversi_ITS assay repeatability.
Selectivity of the qPCR SYBR® green Aversi_ITS assay
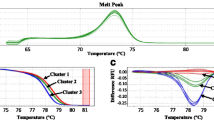
The assessment of the selectivity was performed in two steps, i.e., an inclusivity and an exclusivity test. To perform the inclusivity test, DNA extracted from 16 BCCM/IHEM strains of A. versicolor was amplified. DNA of all the strains was amplified (16/16) with a C q range of 24.26 ± 0.44 to 28.67 ± 0.23 for 1000 copies of gDNA. The T m values ranged between 76.25 ± 0.35 and 76.75 ± 0.29 °C (Table 1, Fig. 2). The sequence of these BCCM/IHEM strains matches perfectly the ones published on NCBI for the corresponding region. All of the amplicons showed 100 % identity (Fig. 1). The obtained T m for each amplicon corresponds to the theoretical T m (76.50 °C) (Fig. 2).

Melting curves obtained with the Avesi_ITS qPCR assay for the A. versicolor pure strains listed in Table 1. The melt curves were obtained with the Bio-Rad IQ 5 software V. 2 (Bio-Rad, Temse, Belgium). The x axis shows the temperature (°C). The y-axis presents the inverse of the first derivative of the best-fitted curve of the measured fluorescence decrease. The grey curves correspond to the A. versicolor listed in Table 1. The blue flat curves represent the nontemplate controls
Subsequently, the exclusivity test was performed using DNA of nontarget species (A. creber, A. sydowii, A. fumigatus, A. alternata, C. cladosporoïdes, C. herbarum, C. sphaerospermum, P. chrysogenum, S. charatum, U. botrytis), closely related to A. versicolor and/or occurring in the same environment, i.e., mold species from indoor air. As expected based on the in silico analysis, no DNA from nontargeted species considered as a background set of indoor air fungal species (i.e., A. fumigatus, A. alternata, C. cladosporoïdes, C. herbarum, C. sphaerospermum, P. chrysogenum, S. charatum, U. botrytis) was amplified during the test (Table 1), except for the A. sydowii (three strains tested IHEM 895, 1360, 20347) and A. creber (IHEM 2646) strain. Although the in silico analysis of the targeted ITS region of A. versicolor, A. creber, and A. sydowii with the available sequences in the NCBI database did not show any sequence dissimilarities (Fig. 1), for a same genome copy number (i.e., 1000 theoretical genomic copies), the C q values ranged between 30.18 ± 0.13 and 37.34 ± 1.84 for A. sydowii and 30.70 ± 0.70 for A. creber (Table 1). To clarify this issue, the obtained amplicons were sequenced. The sequence of the ITS1 amplicon obtained for the experimentally tested A. creber (IHEM 2646) differs from that of A. versicolor with 17 nucleotides, of which seven in the annealing site of the primers. Amplicons of A. sydowii IHEM 895, IHEM 1360, IHEM 20347 differ with eight nucleotides of which three in the annealing site of the forward primer (Fig. 1). But despite of the differences in nucleotide composition, the obtained T m values for the amplicon of A. creber and A. sydowii were in the same range as that of the target T m (76.25–76.75) with respectively 76.50 and 76.25 °C (Table 1). The BLAST analysis of the ITS1 and ITS2 region obtained by sequencing confirmed the IHEM 895, IHEM 1360, and IHEM 20347 as A. sydowii with 93 % of identity and the IHEM 2646 as A. creber with 81 % of identity. Despite this latter low identity, the strain was confirmed as a A. creber based on morphological analysis and sequencing of the regions 5.8 S and ITS2 by BCCM/IHEM (data not shown).
In each assay, an NTC was included to verify that no contamination occurred during the preparation of the qPCR mixes and filling of the 96-well plates. In none of the assays, a signal was observed for the NTC. This absence of signal also demonstrates that no dimerization of primers occurred during the analysis, as predicted during the in silico test (Fig. 2).
Limit of detection and PCR repeatability
Based on 6 independent runs with a total of 18 repetitions (Table 3), the LOD for this SYBR® green assay was determined as being one or two copies of A. versicolor genomes (C q 35.32 ± 0.78 and 34.65 ± 0.91) (Table 4). The r and RSDr were respectively 2.4 and 6.889 % for the C q value at LOD.
Dynamic range and PCR efficiency
Based on a dilution series of 8 levels, corresponding to 1000 to 1 theoretical genomic copies of A. versicolor, the dynamic range and efficiency of the Aversi_ITS assay were determined. Between the tested range, a linear model with a R 2 of 0.9919 and an efficiency of 88.34 % were obtained for this qPCR method (Fig. 3).

R 2 and PCR efficiency of the Aversi_ITS qPCR assay. The PCR efficiency (E) was evaluated in duplicate on a serial dilution of gDNA (1000 to 1 theoretical copy number of gDNA) obtained by two independent extractions of A. versicolor IHEM 18884. The coefficient of determination (R 2) regarding a linear correlation curve. Log copy number/logarithm of the theoretical copy number of gDNA. C q/C q values obtained by qPCR for each repetition of each gDNA dilution
Proof of concept: environmental testing
At first, an inhibition test was performed to verify that no inhibition occurred during the amplification of the DNA extracted from the environmental samples. Theoretically, with a 100 % efficient amplification, a 10-fold dilution corresponds to a C q difference of 3.3. The obtained C q value was 25.06 ± 0.34 for the undiluted DNA extract of the collection fluid spiked with pure A. versicolor and 28.28 ± 0.64 for the 10-fold dilution. This test showed that no inhibition of the amplification occurs.
The Aversi_ITS assay was subsequently tested on environmental samples collected with the Coriolis µ-air sampler and compared to the results obtained with classical detection and identification method used by CRIPI in routine. Additionally, environmental samples collected with the Coriolis µ were also analyzed using culturing and microscopic visualization, in order to be able to evaluate whether possible observed differences are due to the differences in the sampling method or in the detection method used (Table 5).
With the classical routine method (culturing, counting and microscopic visualization), A. versicolor was detected in the two sampled houses with a quantity ranging between 13 and 50 CFU/m3. P. chrysogenum was found as the most important contaminant in each of the sampled houses with a range of 75 to 300 CFU/m3. Other taxa, i.e., Aspergillus glaucus, Cladosporium spp., and an undetermined species, were also observed, though in the same range of quantities as A. versicolor. These taxa occur in the list of most frequently found fungal contaminants of indoor environments. Infertile mycelia were also present in the sampled houses. The range of counts for A. versicolor correlated with the results obtained with the Aversi_ITS qPCR assay. For each sample where A. versicolor was identified by microscopic analysis, a positive qPCR signal ranging between C q 30.26 ± 0.49 and C q 35.85 ± 0.07 was obtained (Table 5). The samples where A. versicolor was not detected on plate gave a negative signal in qPCR.
Similar observations were made for the results obtained with culturing of the Coriolis µ air samples. For the different sampling places, the same species as identified for the samples collected with the RCS plus sampler were found, within two rooms (room of house 1 and the bathroom of house 2) some additional undetermined species detected for the Coriolis µ sampler. The number of colonies on plate for these species was for both air samplers in the same range, including for A. versicolor. As for the RCS plus-based sampling, also for the air samples collected with the Coriolis µ, for each sample where A. versicolor was identified by microscopic analysis, a positive qPCR signal was obtained (Table 5). A negative signal in qPCR was obtained for the samples where A. versicolor was not detected on plate.
Discussion
Currently, indoor fungal contamination is considered as an important public health problem (World Health Organization 2009). Among the different species described, A. versicolor is considered as one of the most important (Beguin and Nolard 1994; Benndorf et al. 2008; de Ana et al. 2006; Melkin et al. 2004) and is suspected to have a link with asthma (Mendell et al. 2011; Sharpe et al. 2014; Verhoeff and Burge 1997). However, to provide more scientific-based, causal evidence on this, efficient screening and monitoring of indoor airborne fungal communities are crucial and necessary.
In this study, we developed a SYBR® green qPCR method for the detection of A. versicolor. This Aversi_ITS assay targets the ITS1 region, which is the same sublocus than the one targeted by the TaqMan®-based EPA assays (including the one for A. versicolor) (Haugland et al. 2004). This region from the 18S rDNA is considered as the most variable locus of this complex, and therefore, it is the most efficient species marker for fungi (Nilsson et al. 2008), making it the most appropriate region of the fungal DNA for the development of a molecular screening tool. This some intraspecies variability of ITS sequence might impact TaqMan®-based methods and the probe’s hybridization when different strains of a specific species need to be detected with the same probe. An alternative to the hydrolysis probe can be found in the SYBR® green chemistry. Less expensive than TaqMan-based assays, SYBR® green assays theoretically allow for species specificity and species discrimination based on a melting curve and the T m value of the amplicon.
Most published PCR and qPCR assays to detect molds are not uniformly assessed for their performance with clear guidelines or norms (Costa et al. 2001; Haugland et al. 2004; Johnson et al. 2012; Melkin et al. 2004; Roussel et al. 2013). In the present study, we propose a strategy to evaluate the performance of qPCR assays applied to mold detection. Therefore, using the same strategy than the one published for foodborne pathogens (Barbau-Piednoir et al. 2013b), the guidance existing for GMO was used to select a set of performance criteria for the qPCR assay (Broeders et al. 2014; ENGL 2015); namely, the selectivity, PCR efficiency, dynamic range, sensitivity, and repeatability parameters were assessed to evaluate the performance of our developed Aversi_ITS assay.
First, the results of the specificity test showed that all of the tested A. versicolor strains were detected with the Aversi_ITS assay. However, for an identical number of genomic copies (i.e., 1000 gDNA copy numbers), a variation of approximately 4 C q between these strains was observed. Because no differences in the ITS sequences were observed between the tested BCCM/IHEM stains (Fig. 1), including the sites of primer annealing, this difference in C q might be explained by variation in the copy number of the 18S rDNA operon to which the targeted ITS region belongs. This ITS region is known to vary not only between species but also within the species (Black et al. 2013; Corradi et al. 2007; Iwen et al. 2002; Schoch et al. 2012). For example, for A. fumigatus, the intraspecies variation factor was estimated at 2.5, implying that the 18S rDNA gene complex could vary between 38 and 91 copies per genome (Herrera et al. 2009). A similar variation rate for A. versicolor might explain the observed difference in C q values between different strains of this species. However, this hypothesis needs more investigation of the copy number of 18S rDNA in A. versicolor strains, similar to what was described by Herrera et al. for A. fumigatus (Herrera et al. 2009). Consequently, qPCR assays developed on the ITS region, such as this Aversi_ITS assay, are qualitative detection methods. To develop a quantitative method based on ITS, the copy number of the 18S rDNA should be determined for each target strain of the species.
The Aversi_ITS assay did not amplify DNA extracted from 10 nontarget strains selected among the most common airborne fungal species. This indicates a good selectivity of the Aversi_ITS assay (Beguin and Nolard 1994). However, the assay did amplify DNA of two other members of the Aspergillus section Versicolores, i.e., A. creber and A. sydowii. This was expected, based on the primer sequences and the alignments of already publicly available ITS sequences, which were identical to the one of A. versicolor in the selected region. These species are very close and their ITS sequences in general differ only in a few nucleotides (Hinrikson et al. 2005; Jurjevic et al. 2012). However, the C q obtained for the amplicons of A. sydowii and A. creber were not in the expected range based on the identity of the publicly available sequences, although they resulted in the expected T m. Therefore, additional tests for strain identification were performed. These additional tests confirmed the identity of the strains used for the exclusivity test. They also confirmed the obtained experimental T m, coinciding with the one calculated based on the obtained sequence which however shows mismatches as compared to the ITS1 target region of A. versicolor or of the publicly available sequences of A. creber and A. sydowii used for the primer design. Although normally the T m allows for species discrimination, in the selected amplicon region this is not the case for A. versicolor, A. creber, and A. sydowii. The obtained C q is however higher for A. creber and A. sydowii for the same amount of template DNA, which might be due to the mismatches in the primer annealing sites, in addition to the difference in ITS copy number. These observations demonstrate the need for more publicly available sequences for these closely related species to be used for qPCR assay development.
These nontarget amplifications of A. sydowii were also previously reported for the TaqMan® assays of EPA with probes Avers2 and Asydo3 developed respectively for the detection of A. versicolor and A. sydowii (Haugland et al. 2004; United States Environmental Protection Agency 2014). These probes amplified each time both species. A. creber is a recently described species, isolated from indoor air samples and identified through multilocus DNA sequencing (Jurjevic et al. 2012), and has not yet been commonly used for the testing of TaqMan® A. versicolor specific assays. However, these aspecific detections should have a limited impact on a possible use of the Aversi_ITS assay in routine analysis in Belgium. Indeed, A. creber and A. sydowii are not frequently detected in indoor environment, as compared to A. versicolor. Further investigations are needed to evaluate the difference between these three species concerning the impact of their presence in indoor air on public health.
To improve the discrimination between these three closely related species based on merely molecular methods, an alternative approach should be developed. One possibility could be the use of a combination of different markers similar to what has already been done for the detection of GMO (Van den Bulcke et al. 2010) and foodborne pathogens (Barbau-Piednoir et al. 2015). The presence or the absence of an amplification signal for each of the markers and their combination defines a decision-taking tree. For fungi, this approach to improve the species discrimination could be possible if we would combine our primers of the Aversi_ITS assay with primers targeting another gene marker or a discriminatory SNP. In case that the sample is composed of a mixture of DNA from different species (e.g., from an air sample), the results will hint to a “candidate mold” which should be further confirmed by downstream analysis, such as sequencing. This idea is supported by Schoch and coworkers in 2012 who suggested that the use of a combination of different DNA regions, like ITS 1 and β-tubulin or another region, i.e., the DNA barcode principle, should improve the phylogenetic analysis of fungi (Schoch et al. 2012). Hereto, more genomic sequences of both species (A. creber and A. sydowii) should become publicly available in order to design primers for such discriminatory regions.
Subsequently, the PCR linearity and efficiency were evaluated as quality criteria for the developed assay. The BCCM/IHEM 18884 strain was collected and purified from a contaminated house by CRIPI and is used as a reference strain for allergy studies by the CRIPI. Therefore, this strain was selected as a reference to determine the parameters of our qPCR assay during the performance assessment.
The efficiency of this assay (E) was estimated to be 88.34 %, which complies with the guidelines for qualitative qPCR methods (Broeders et al. 2014). Our method is also characterized by a high R 2 value (0.9919) which demonstrated the linearity of our assay and of the accuracy of our experimental setup.
Moreover, the Aversi_ITS assay is sensitive with a LOD defined as one or two genomic copies per reaction, and therefore, it complies with the acceptance criteria, i.e., LOD below 25 copies (ENGL 2015). This LOD is similar to the results reported by Johnson et al. (2012) for their qPCR detection method of A. fumigatus which has an LOD ranging between 6 and 0.6 genomes (Johnson et al. 2012). Our Aversi_ITS showed also to be repeatable, with a RSD r, for all dilutions above the LOD, lower than 25 %, thereby fulfilling the requirements of the validation guidelines used in this study.
As discussed above, we mainly used the guidelines for GMO detection for the evaluation of our qPCR assay. As it was recently shown (Barbau-Piednoir et al. 2013a), these guidelines were successfully used to evaluate with high standard the performance of the SYBR® green qPCR methods for the detection of bacterial pathogens. However, requirements defined for food and feed analysis are not necessarily adapted for indoor airborne fungi monitoring, as the laws and the control measures are more numerous for food than for microbiological air pollution and the impact of food contamination on public health is more documented and better understood than for fungal air contamination. The increasing development and use of molecular tools for the identification and the monitoring of indoor airborne microbiological contamination imposes the establishment of guidelines for harmonization of performance requirements and threshold values for the parameters of the qPCR assays. Ultimately, this may contribute to the establishment of standardized and reproducible microbiological methods, highly needed to determine a causal link between indoor fungal contamination and health effects.
To deliver a proof-of-concept of our developed qPCR assay aimed to be used for indoor fungal contamination monitoring, the Aversi_ITS assay was tested on environmental samples and results were compared to those obtained with traditional methods of screening (i.e., culture, CFU counting and microscopic identification). This last part of our study confirmed that the Aversi_ITS assay is a valuable alternative for the currently used classical methods. The results of the qPCR assay were comparable to the ones obtained with the classical routine protocol as to indicate whether the targeted species A. versicolor was present or not, and this using a shorter time period to obtain these results with the qPCR method, i.e., 2 days maximum for the qPCR analysis (including DNA extraction with an overnight lyophilization step and qPCR analysis) compared to 5 to 10 days for the classical methods. Based on the obtained C q and an extrapolation from the A. versicolor data obtained for the LOD estimation, a theoretical copy number of A. versicolor gDNA per cubic meters of air could be estimated in order to attempt a more detailed comparison of the results from molecular and the classical methods (Table 5). The theoretical number of genomic copies for A. versicolor ranged between 7 and 67 for 1 m3 of sampled air for a number of CFU/m3 ranging between 13 and 50 for those collected with the RCS plus air sampler in the routine classical method. A similar range of colonies of A. versicolor was obtained for the samples collected with the Coriolis µ. The culturing of Coriolis µ samples was included as a control to verify that different samplers do not lead to different species detected on plate, especially for A. versicolor.
The A. versicolor amounts obtained are in a similar range when comparing the culturing results to the qPCR results, despite the following considerations. The small difference in amounts could be attributed to the sampling method, the performance of the sampler used, and the conversion factor used to express the results in m3, which are different for the RCS plus and Coriolis µ sampling methods. Also, the possibility that one colony would grow from an aggregate of fungal cells should be taken into account, explaining why the results are not exactly coinciding. Additionally, qPCR will also include the nonculturable A. versicolor fraction, while this fraction remains undetected using the classical methods. It is also important to note that the estimation of gDNA copies was based on the strain IHEM 18884, which is not necessarily the strain that was present in the contaminated houses. As observed during the evaluation of the assay, the real copy number of the ITS regions of the strains detected here could differ from that of the strain IHEM 18884. This would lead to another amount of gDNA copies. Nevertheless, the trend for the contamination by A. versicolor was similar as detected by the different methods, i.e., the house 1 was the most contaminated and bathrooms presented more contamination than other rooms.
Our environmental test also showed, as expected, that other species than A. versicolor are present inside buildings. Therefore, it will be interesting to develop and assess a more exhaustive multiplex tool targeting different indoor airborne fungal species. Different types of multiplex technologies can be envisaged, including the SYBR® green technology, where the discrimination between species could be based on the T m values of the amplicons obtained with universal primers for the ITS region. However, this might imply the use of more accurate technologies such as high-resolution melting (HRM) qPCR to perform the discrimination of some closely related taxa. Indeed, some species differ by a few nucleotides and are hardly discriminated with traditional qPCR methods. Based on a very fine temperature gradient, HRM could be an interesting tool to perform the discrimination and the identification of the airborne fungal community (McCarthy et al. 2013). It would also be interesting to investigate whether HRM in combination with the primers of the Aversi_ITS assay might offer a solution to discriminate the three species of the Aspergillus section Versicolores.
In conclusion, the developed Aversi_ITS method based on the SYBR® green chemistry is a convenient qualitative assay for the detection of A. versicolor. With the increased risk for public health linked to the augmentation of indoor fungal contamination, such a molecular assay is a first step in offering a valuable alternative to the currently used classical methods in the framework of routine monitoring of indoor air fungal contamination by a public health laboratory. A reduced time of detection of both the culturable and nonculturable fungal fraction allows to reduce the time of reaction and taking of measurements aiming at reducing the impact on human health. With an extension toward other important indoor airborne fungal contaminants, tools as the one developed in this study, based on harmonized guidelines to be urgently established, will contribute to an improved indoor air quality monitoring and ultimately to an improved insight into the causal effect between indoor airborne fungal contaminants and health effects. Once this causal link is established, meaningful regulatory individual exposure standards for well-defined airborne mold allergens can be established (AIHA 2011; Verhoeff and Burge 1997).
References
AIHA (2011) Facts about mold. American Industrial Hygiene Association https://www.aiha.org/getinvolved/VolunteerGroups/Documents/BiosafetyVGFactsAbout%20MoldDecember2011.pdf. Accessed 15 Dec 2014
Ashktorab H, Cohen RJ (1992) Facile isolation of genomic DNA from filamentous fungi. Biotechniques 13:198–200
Barbau-Piednoir E, Bertrand S, Mahillon J, Roosens NH, Botteldoorn N (2013a) SYBR(R)Green qPCR Salmonella detection system allowing discrimination at the genus, species and subspecies levels. Appl Microbiol Biotechnol 97(22):9811–9824. doi:10.1007/s00253-013-5234-x
Barbau-Piednoir E, Botteldoorn N, Yde M, Mahillon J, Roosens NH (2013b) Development and validation of qualitative SYBR(R)Green real-time PCR for detection and discrimination of Listeria spp. and Listeria monocytogenes. Appl Microbiol Biotechnol 97(9):4021–4037. doi:10.1007/s00253-012-4477-2
Barbau-Piednoir E, Botteldoorn N, Mahillon J, Dierick K, Roosens NH (2015) Fast and discriminative CoSYPS detection system of viable Salmonella spp. and Listeria spp. in carcass swab samples. Int J Food Microbiol 192:103–110. doi:10.1016/j.ijfoodmicro.2014.09.018
Beguin H, Nolard N (1994) Mould biodiversity in homes. I. Air and surface analysis of 130 dwellings. Aerobiologia 10:157–166. doi:10.1007/BF02459231
Bellanger AP, Reboux G, Roussel S, Grenouillet F, Didier-Scherer E, Dalphin JC, Millon L (2009) Indoor fungal contamination of moisture-damaged and allergic patient housing analysed using real-time PCR. Lett Appl Microbiol 49:260–266. doi:10.1111/j.1472-765X.2009.02653.x
Benndorf D, Müller A, Bock K, Manuwald O, Herbarth O, von Bergen M (2008) Indentification of spore allergens from the indoor mould Aspergillus versicolor. Allergy 63:454–460. doi:10.1111/j.1398-9995.2007.01603.x
Black J, Dean T, Byfield G, Foarde K, Menetrez M (2013) Determining fungi rRNA copy number by PCR. JBT 24:1–7. doi:10.7171/jbt.13-2401-004
Broeders S, Huber I, Grohmann L, Berben G, Taverniers I, Mazzara M, Roosens NH, Morisset D (2014) Guidelines for validation of qualitative real-time PCR methods. Trends Food SciTech 37:115–126. doi:10.1016/j.tifs.2014.03.008
Chemidlin Prevost-Boure N, Christen R, Dequiedt S, Mougel C, Lelievre M, Jolivet C, Shahbazkia HR, Guillou L, Arrouays D, Ranjard L (2011) Validation and application of a PCR primer set to quantify fungal communities in the soil environment by real-time quantitative PCR. PLoS ONE 6, e24166. doi:10.1371/journal.pone.0024166
Corradi N, Croll D, Colard A, Kuhn G, Ehinger M, Sanders IR (2007) Gene copy number polymorphisms in an arbuscular mycorrhizal fungal population. Appl Environ Microbiol 73:366–369. doi:10.1128/AEM.01574-06
Costa C, Vidaud D, Olivi M, Bart-Delabesse E, Vidaud M, Bretagne S (2001) Development of two real-time quantitative TaqMan PCR assays to detect circulating Aspergillus fumigatus DNA in serum. J Microbiol Methods 44:263–269. doi:10.1016/S0167-7012(01)00212-3
de Ana SG, Torres-Rodriguez JM, Ramirez EA, Garcia SM, Belmonte-Soler J (2006) Seasonal distribution of Alternaria, Aspergillus, Cladosporium and Penicillium species isolated in homes of fungal allergic patients. J Investig Allergol Clin Immunol 16:357–363
Douwes J, Thorne P, Pearce N, Heederik D (2003) Bioaerosol health effects and exposure assessment: progress and prospects. Ann Occup Hyg 47:187–200. doi:10.1093/annhyg/meg032
European Network of GMO Laboratories (ENGL) (2008) Definition of Minimum Performance requirements for analyticals Methods of GMO testing. EUR - Scientific and Technical Research Reports, Luxembourg, Luxembourg. doi: 10.2788/65827
European network of GMO Laboratories (ENGL) (2015) Definition of minimum preformance requirements for analytical methods of GMO testing. EUR - Scientific and Technical Research Reports., Luxembourg, Luxembourg
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. doi:10.1111/j.1365-294X.1993.tb00005.x
Haugland RA, Varma M, Wymer LJ, Vesper SJ (2004) Quantitative PCR analysis of selected Aspergillus, Penicillium and Paecilomyces species. Syst Appl Microbiol 27:198–210. doi:10.1078/072320204322881826
Herrera ML, Vallor AC, Gelfond JA, Patterson TF, Wickes BL (2009) Strain-dependent variation in 18S ribosomal DNA Copy numbers in Aspergillus fumigatus. JCM. doi:10.1128/JCM.02073-08
Hinrikson HP, Hurst SF, Lott TJ, Warnock DW (2005) Assessment of ribosomial large-subunit D1-D2 Internal transcribed spacer 1, and internal transcribed spacer 2 regions as targets for molecular identification of medically important Aspergillus species. JCM 43:2092–2103. doi:10.1128/JCM.43.5.2092-2103.2005
Horner WE, Helbling A, Salvaggio JE, Lehrer SB (1995) Fungal allergens. Clin Microbiol Rev 8:161–179
Iwen PC, Hinrichs SH, Rupp ME (2002) Utilization of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med Mycol 40:87–109. doi:10.1080/mmy.40.1.87.109
Johnson GL, Bibby DF, Wong S, Agrawal SG, Bustin SA (2012) A MIQE-compliant real-time PCR assay for Aspergillus detection. PLoS ONE 7, e40022. doi:10.1371/journal.pone.0040022
Jones R, Recer GM, Hwang SA, Lin S (2011) Association between indoor mold and asthma among children in Buffalo, New York. Indoor Air 21:156–164. doi:10.1111/j.1600-0668.2010.00692.x
Jurjevic Z, Peterson SW, Horn BW (2012) Aspergillus section Versicolores: nine new species and multilocus DNA sequence based phylogeny. IMA Fungus 3:59–79. doi:10.5598/imafungus.2012.03.01.07
McCarthy JK, Didham RK, Brockerhoff EG, van Bysterveldt KA, Varsani A (2013) High-resolution DNA melt-curve analysis for cost-effective mass screening of pairwise species interactions. Mol Ecol Resour 13:908–917. doi:10.1111/1755-0998.12143
Meheust D, Le Cann CP, Reboux G, Millon L, Gangneux JP (2014) Indoor fungal contamination: health risks and measurement methods in hospitals, homes and workplaces. Crit Rev Microbiol 40:248–260. doi:10.3109/1040841X.2013.777687
Melkin T, Haugland RA, Reponen T, Varma M, Lummus Z, Bernstein D, Wymer LJ, Vesper SJ (2004) Quantitative PCR analysis of houses dust can reveal abnormal mold conditions. J Environ Monit 6:615–620. doi:10.1039/b400250d
Mendell MJ, Mirer AG, Cheung K, Tong M, Douwes J (2011) Respiratory and allergic health effects of dampness, mold, and dampness-related agents: a review of the epidemiologic evidence. Environ Health Perspect 119:748–756. doi:10.1289/ehp.1002410
Michealsen A, Pinzari F, Ripka K, Lubitz W, Piñar G (2006) Application of molecular techniques fors identification of fungal communities colonising paper materials. Int Biodeterior Biodegradation 58:133–141. doi:10.1016/j.ibiod.2006.06.019
Mohr PJ, Taylor BN, Newell DB (2008) CODATA recommended values of the fundamental physical constants: 2006. Rev Mod Phys 80:633. doi:10.1103/RevModPhys.84.1527
Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H (2008) Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform Online 4:193–201. doi:10.4137/EBO.S653
Nolard N, Chasseur C, Marlier M, and Lognay G (2004) Validation des méthodes microbiologiques et chimiques de contrôle des lieux de travail. http://www.belspo.be/belspo/organisation/publ/pub_ostc/PS/rPS19_fr.pdf. Accessed 15 Dec 2014.
Packeu A, Chasseur C, Bladt S, Detandt M (2012) The role of indoor pollution in the development and maintenance of chronic airway inflammation in children. B-ENT 8(Suppl 19):73–79
Pitkaranta M, Meklin T, Hyvarinen A, Nevalainen A, Paulin L, Auvinen P, Lignell U, Rintala H (2011) Molecular profiling of fungal communities in moisture damaged buildings before and after remediation - a comparison of culture-dependent and culture-independent methods. BMC Microbiol 11:235. doi:10.1186/1471-2180-11-235
Reboux G, Bellanger AP, Roussel S, Grenouillet F, Millon L (2010) Moulds in dwellings: health risks and involved species. Rev Mal Respir 27:169–179. doi:10.1016/j.rmr.2009.09.003
Roussel S, Reboux G, Naegele A, Martinez J, Vacheyrou M, Scherer E, Millon L (2013) Detecting and quantifying mites in domestic dust: a novel application for real-time PCR. Environ Int 55:20–24. doi:10.1016/j.envint.2013.02.002
Rutledge RG, Cote C (2003) Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res 31:e93
Sarachu M, Colet M (2005) wEMBOSS: a web interface for EMBOSS. Bioinformatics 21:540–541. doi:10.1093/bioinformatics/bti031
Schoch CL, Seifert KA, Huhndorf S, Robert V, Pouge JL, Levesque A, Chen W, Fungal Barcoding Consortium (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. PNAS 109:6241–6246. doi:10.1073/pnas.1117018109
Seo S-C, Reponen T, Levin L, Brochelt T, Grinshpun SA (2008) Aerosolization of particulate (1-3)-β-d-glucan from moldy materials. Appl Environ Microbiol 74:585–593. doi:10.1128/AEM.01791-07
Sharpe R, Thornton CR, Osborne NJ (2014) Modifiable factors governing indoor fungal diversity and risk of asthma. Clin Exp Allergy. doi:10.1111/cea.12281
Timothy D, Betancourt D, Menetrez M (2004) A rapid DNA extraction method for PCR identification of fungal indoor air contaminants. J Microbiol Methods 56:431–434. doi:10.1016/j.mimet.2003.11.015
United States Environmental Protection Agency EPA (2014) Technology for mold identification and enumeration. http://www.epa.gov/nerlcwww/moldtech.htm#references. Accessed 15 Dec 2014.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. doi:10.1093/nar/gkm306
Van den Bulcke M, Lievens A, Barbau-Piednoir E, Mgongolo Mbella G, Roosens NH, Sneyers M, Leunda Casi A (2010) A theoretical introduction to "Combinatory SYBR®Green qPCR Screening", a matrix-based approach for the detection of materials derived from genetically modified plants. Anal Bioanal Chem 396:2113–2123. doi:10.1007/s00216-009-3286-7
Verhoeff AP, Burge HA (1997) Health risk assessment of fungi in home environments. Ann Allergy Asthma Immunol 78:544–556. doi:10.1016/S1081-1206(10)63214-0
Vesper SJ (2011) Traditional mould analysis compared to a DNA-based method of mould analysis. Crit Rev Microbiol 37:15–24. doi:10.3109/1040841X.2010.506177
Vesper SJ, Wymer L, Kennedy S, Grimsley LF (2013) Decreased pulmonary function measured in children exposed to high environmental relative moldiness index homes. Open Respir Med J 7:83–86. doi:10.2174/1874306401307010083;TORMJ-7-83
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand GH, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
World Health Organization (2009) Building dampness and its effect on indoor exposure to biological and non-biological pollutants. In WHO guidelines for indoor air quality: dampness and mould. World Health Organization, Copenhagen, pp 8–30
Acknowledgments
We would like to thank the CRIPI team for their help during sampling and the excellent technicians of Mycology team for the classical analysis. Sequencing was performed at the Platform Biotechnology and Molecular Biology at the Scientific Institute of Public Health (WIV-ISP). This work was supported by RP/PJ WIV-ISP (mycoMOLAIR), project number W4044.0105.1.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Libert, X., Chasseur, C., Bladt, S. et al. Development and performance assessment of a qualitative SYBR® green real-time PCR assay for the detection of Aspergillus versicolor in indoor air. Appl Microbiol Biotechnol 99, 7267–7282 (2015). https://doi.org/10.1007/s00253-015-6785-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6785-9