
Abstract
The advantages of phage Mu transposition-based systems for the chromosomal editing of plasmid-less strains are reviewed. The cis and trans requirements for Mu phage-mediated transposition, which include the L/R ends of the Mu DNA, the transposition factors MuA and MuB, and the cis/trans functioning of the E element as an enhancer, are presented. Mini-Mu(LR)/(LER) units are Mu derivatives that lack most of the Mu genes but contain the L/R ends or a properly arranged E element in cis to the L/R ends. The dual-component system, which consists of an integrative plasmid with a mini-Mu and an easily eliminated helper plasmid encoding inducible transposition factors, is described in detail as a tool for the integration/amplification of recombinant DNAs. This chromosomal editing method is based on replicative transposition through the formation of a cointegrate that can be resolved in a recombination-dependent manner. (E-plus)- or (E-minus)-helpers that differ in the presence of the trans-acting E element are used to achieve the proper mini-Mu transposition intensity. The systems that have been developed for the construction of stably maintained mini-Mu multi-integrant strains of Escherichia coli and Methylophilus methylotrophus are described. A novel integration/amplification/fixation strategy is proposed for consecutive independent replicative transpositions of different mini-Mu(LER) units with “excisable” E elements in methylotrophic cells.
Similar content being viewed by others

Introduction
The relevance and practical significance of constructing plasmid-less recombinant bacterial strains for use in applied microbiology and biotechnology are increasing due to the potential for genetic instability to reduce the number of active recombinant alleles in plasmids (Friehs 2004) and restrictions on the application of plasmids in large-scale industries of the First World (European Council Directives 1990, 1998). In vivo chromosomal editing methods, which are primarily based on homologous and/or site-specific recombination of DNA as well as on transposition mechanisms, have been used to engineer plasmid-less bacteria (Balbás and Gosset 2001). The increase in genomic copy number of native or previously modified target genes is an important tool for chromosomal editing and the construction of stably maintained bacterial genomes. The development of new approaches for the integration (Minaeva et al. 2008; Rivero-Müller et al. 2007) and the increase in genomic copy number (Tyo et al. 2009) of recombinant DNA fragments remains relevant even for Escherichia coli and closely related Gram-negative bacteria for which recombineering-based technologies (Court et al. 2002; Sawitzke et al. 2007; Sharan et al. 2009) have already been developed to finely modulate the expression of chromosomal genes (De Mey et al. 2010; Katashkina et al. 2005; Meynial-Salles et al. 2005) and sometimes conditionally increase (Doroshenko et al. 2010b) or silence (Krylov et al. 2010) the level of their transcription. The methods of integration/amplification are especially important for bacteria for which the genetic tools for the chromosomal editing are not as comprehensive and diversified as those available for E. coli.
The integration of target genes in a bacterial chromosome followed by the increase in genomic copy number can be efficiently achieved using a phage Mu-driven transposition system originally developed for E. coli and initially characterized more than 20 years ago (Castilho et al. 1984; Chaconas et al. 1981a, b). Since the mid-1980s, several Mu derivatives, mini-Mu(s), have been constructed and extensively used for applications in classical in vivo transposition, including insertional mutagenesis, gene fusion, and mapping techniques, as well as for gene cloning and DNA sequencing strategies (Groisman 1991; Groisman and Casadaban 1986, 1987). The highly efficient Mu-based technology of the in vitro DNA transposition has been developed as a convenient tool for the functional analysis of genes, genomes, and proteins because of the accurate nature reaction and the low stringency for the target preference (Haapa et al. 1999; Haapa-Paananen et al. 2002; Savilahti et al. 1995; Savilahti and Mizuuchi 1996; Turakainen et al. 2009). The construction of recombinant plasmid-less l-threonine-overproducing E. coli strains may have been the first application of mini-Mu as a vector for the integration/amplification of target pathway genes for metabolic engineering (Kurahashi et al. 1990; Kurahashi and Takinami 1991).
Comprehensive investigations, primarily in vitro, of the Mu-driven system have provided extensive insights into the molecular aspects of the transposition mechanism. These works have been recently summarized in excellent research papers and overviews (Abdelhakim et al. 2008; Au et al. 2006; Chaconas and Harshey 2002; Choi and Harshey 2010; Gueguen et al. 2005; Harshey and Jayaram 2006; Nakai et al. 2001; Rice and Baker 2001). However, in vivo Mu-driven systems may be underutilized in applied microbiology and biotechnology. The main aim of this review is to highlight, especially for the new generation of investigators, the potential application of Mu-driven systems as a powerful genetic tool for the integration/amplification of target genes and the construction of plasmid-less engineered bacterial strains.
Cis and trans requirements of Mu phage-mediated transposition
Known as the most efficient transposon, Mu is a temperate bacteriophage that is capable of growth on many enteric bacteria, including E. coli K-12. The Mu phage undergoes two alternative transposition pathways (Fig. 1) at different stages of its life cycle (Howe 1997; Paolozzi and Ghelardini 2006; Symonds et al. 1987). The Mu-driven “integrative” (or “conservative” or “nonreplicative”) transposition, which is also designated as a “simple insertion” (Akroyd and Symonds 1983; Harshey 1984; Liebart et al. 1982), and the “replicative” transposition (Chaconas et al. 1981b, 1996) are temporally separated. During the Mu phage infection step, various flanking sequences from the previous host are lost during the integrative transposition of the linear Mu DNA into a random site of the bacterial chromosome (Au et al. 2006; Bukhari and Zipser 1972). Replicative transposition through the formation of a “cointegrate” structure is obligatory for replication and for the production of approximately 100 phages during lytic growth (Chaconas et al. 1981b; Chaconas and Harshey 2002; Craigie and Mizuuchi 1985). As a result of replicative transposition, new copies of the Mu DNA occur at many sites in the bacterial genome, frequently within several kilobase pairs of one another. The most obvious factor that affects the random integration of Mu DNA into bacterial genes is their efficient transcription, which has a clearly negative impact on transposition (Manna et al. 2004).

The Mu phage life cycle. As a temperate phage, Mu can undergo lysogenic or lytic growth. The life cycle begins with infection in which the phage adsorbs and transfers its linear double-stranded DNA [37 kb Mu genome flanked by short regions from the previous host (Daniell et al. 1973; Bukhari and Taylor 1975)] to a bacterial cell. The ends of the infecting DNA are bound by the MuN protein to form a noncovalent circular structure (Gloor and Chaconas 1986; Harshey and Bukhari 1983). In an integrative transposition that requires the MuA protein, the flanking sequences are lost and the Mu DNA is randomly inserted into the bacterial genome. After integration, approximately 1–10% of the phages become lysogens (Howe and Bade 1975). The majority of the phages immediately enter the lytic cycle (which is also the ultimate fate of the induced lysogens) during which MuA, assisted by the activities of several factors, catalyzes multiple replicative transpositions that eventually produce a burst size of approximately 100 new phages. Adapted from Choi and Harshey (2010) and Sokolsky and Baker (2003)
Both Mu-mediated transposition pathways are catalyzed by a high-order protein–DNA complex called a transpososome (reviewed in Gueguen et al. 2005; Harshey and Jayaram 2006). The Mu DNA sites and proteins involved in transpososome assembly and function are presented in Figs. 2 and 3, respectively (for detailed references, see Chaconas and Harshey 2002; Harshey and Jayaram 2006). The core of the Mu transpososome is composed of two Mu end DNA segments (L and R ends) that are synapsed by stably bound multiple subunits of the transposase MuA, which catalyzes the specific DNA cleavage and joining required for transposition (Craigie et al. 1984; Kuo et al. 1991; Lavoie et al. 1991). The L/R ends each contain three MuA-binding sites (L1–L3 and R1–R3, correspondingly) with different spacings (Fig. 2a) (Craigie et al. 1984). MuA binds as a monomer to each L/R site, introducing an 80° to 90° bend (Kuo et al. 1991). The function of the cis- or trans-encoded MuA protein is essential for the transposition in vivo of the mini-Mu that is presented on supercoiled DNA and consists of the L/R sites (Patterson et al. 1986). The six binding sites are not equally important for transposome assembly. Indeed, genetic experiments show the unimportance of the R3 site; a deletion of the L3 site results only in a 10-fold reduction in transposition in vivo (Groenen et al. 1985). It is shown in vitro that a MuA-mediated stable synaptic complex with only three (L1, R1, R2) sites could be formed in which the donor DNA strands are nicked and fully competent in the subsequent strand-transfer step of transposition (Kuo et al. 1991). The linear mini-Mu DNA flanked by the (R1R2) sites in an inverted orientation is efficiently used for in vitro MuA-mediated assemble the transpososome followed by integrative transposition in vitro (Haapa et al. 1999; Savilahti et al. 1995) or in vivo in genomes of different organisms (Lamberg et al. 2002; Paatero et al. 2008; Pajunen et al. 2005).

Cis and trans requirements for Mu transposition. a Relative arrangement of subsites within the L/R ends and the E element on the 37-kb Mu genome (Morgan et al. 2002). The genes regulating transposition (c, A, B) and the E. coli proteins HU and IHF, which bind to the L end and the E element, respectively, during transposition, are indicated. The Mu transposase can interact with the high-affinity L/R sites and weak-affinity E sites. b The structural organization of the transposase MuA with the assigned functions of the various domains and subdomains. Based on limited proteolysis, three domains (I–III) have been assigned to MuA. Amino acid numbers corresponding to the boundary of each major domain are shown beneath the structure. The function of each domain is indicated at the top. The N-terminal domain I contacts the L/R end and E element through separate regions. The central domain II of MuA is involved in catalysis (DDE in the catalytic triad). The C-terminal domain III is responsible for interaction with the MuB facilitating transposition factor and the host ClpX unfoldase protein. Isolated domain III displays, as well, the nonspecific DNA-binding and endonuclease activities designated as BAN (Wu and Chaconas 1995). The minimal region required for DNA binding, cleavage, and strand transfer in vitro, including subdomains Iβ, Iγ, the catalytic core (domain II), and the N-terminal portion of domain III (IIIα), is indicated according to Krementsova et al. (1998). Adapted from Baker and Luo (1994), Choi and Harshey (2010), and Haapa-Paananen (2002)

The simplified scheme of the Mu-driven transposition pathway. The assembly of the Mu transpososome is initiated by the interaction of the MuA subunits with the R end and the E element. HU promotes capture of the L end by MuA/ER complex to form the MuA/LER complex. MuA then acts in trans to first cleave and then join the cleaved ends of the Mu DNA to the target DNA. MuB captures and delivers the target DNA to the transpososome where the newly created 3′-OH groups now function in the joining or strand-transfer step catalyzed by the complex and ClpX-mediated exchanges MuA with a set of host proteins in preparation for repair or replication to finalize the integrative or replicative transposition pathways, respectively. For details, see Au et al. (2006), Choi and Harshey (2010), and Harshey and Jayaram (2006)
In E. coli, Mu transpososome assembly is facilitated by two host-encoded DNA bending proteins—HU and a sequence-specific integration host factor (IHF) (Swinger and Rice 2004). HU binds in vivo to the spacer between L1 and L2 (Lavoie et al. 1996), introducing a bend and presumably drawing the bound MuA protomers together (Gueguen et al. 2005). The binding site for IHF is in an E element that is located approximately 1 kb from the L end in the native Mu DNA.
The enhancer element, E [earlier named as the internal activation sequence or IAS (Mizuuchi et al. 1995; Mizuuchi and Mizuuchi 1989)], stimulates transposition more than 100-fold in vitro and in vivo (Castilho et al. 1984; Leung et al. 1989; Surette et al. 1989). The E element partially overlaps with the O1–O3 region (Krause and Higgins 1986), which was initially identified as the operator to which the Mu c-repressor binds to silence the transcription of early phage functions from the Pe promoter (Fig. 2a). The E element is composed of two clusters of MuA-binding sequences separated by a binding site for IHF (Mizuuchi and Mizuuchi 1989; Surette et al. 1989). The site-specific IHF-mediated bending of DNA at the E element is presumed to assist MuA-mediated end-enhancer interactions (Harshey and Jayaram 2006).
IHF is required in vitro for the efficient transposition of the mini-Mu carrying the E element when the superhelical density (σ) of the donor DNA is low (Surette and Chaconas 1989), i.e., σ decreases from −0.05 (a character value for naked bacterial superhelical DNA) to a level of σ ~ −0.025 [this density is typical for the “restrained” protein-bound DNA molecule in vivo (Dillon and Dorman 2010; Pettijohn 1996; Pettijohn and Pfenninger 1980)].
The in cis orientation of the E element in mini-Mu with respect to the L/R ends is critical to its function, while its distance from the ends does not seem to be significant. The E element can stimulate transposition if it is present in trans in an unlinked DNA molecule (Surette and Chaconas 1992). The presence of the E element is important for correct transpososome assembly (Fig. 3) (Allison and Chaconas 1992; Lavoie and Chaconas 1995; Watson and Chaconas 1996), and it remains associated with this complex throughout the transposition process (Pathania et al. 2002, 2003; Yin et al. 2007).
MuA is a 663-aa residue protein that can be divided into three domains by partial proteolysis; each domain can be functionally or structurally divided into subdomains (Fig. 2b, and reviewed in Rice and Baker 2001). The N-terminal domain of the MuA protein contacts the sequences of the L/R ends and the E element through separate regions (Mizuuchi et al. 1995). The central domain is involved in the catalysis of the transposition reactions. The C-terminal domain is responsible for the interaction with the host protein, ClpX (see below), and with the auxiliary transposition factor, the MuB protein. MuB not only modulates the activity of MuA (Faelen et al. 1978) but also delivers the target DNA to the transpososome (Chaconas and Harshey 2002; Roldan and Baker 2001) and protects the actively replicating/transposing Mu from self-integration (Ge et al. 2010; Han and Mizuuchi 2010). Both the DNA binding and activation of the MuA transposase functions of MuB are required for productive phage Mu replicative transposition, but only the activation of transposase is necessary for efficient integrative transposition (Roldan and Baker 2001). The MuB protein is not required for the integrative transposition (O’Day et al. 1978); however, it enhances the rate and extent of this pathway (Roldan and Baker 2001).
During the multistep reaction, the MuA present in the transpososome complex catalyzes the specific DNA single-stranded cleavages at the terminal CA dinucleotides of the L1 and R1 sites, using water as the nucleophile in such a way that 3′-OH groups of the transposable element are exposed (Mizuuchi 1984). These 3′ ends serve as nucleophiles for the subsequent joining or strand-transfer step, attacking phosphodiester bonds spaced 5 bp apart on target DNA in a one-step transesterification reaction (Mizuuchi and Adzuma 1991). The resulting θ-like DNA structure is a common intermediate for both transposition pathways (Chaconas and Harshey 2002; Harshey and Jayaram 2006). The host ClpX unfoldase ultimately destabilizes the transpososome, which facilitates the recruitment of the host-dependent DNA replication/repair machinery to finalize the “nick–join–replicative” or “nick–join–repair” transposition pathways (Fig. 4) (Abdelhakim et al. 2008; Nakai et al. 2001). In both cases, the strand transfer at staggered positions in target DNA strands will cause a duplication of 5 bp of the target DNA flanking the transposed Mu-based element. Probably, ClpX participates, as well, in stimulating the nuclease activity of the C-terminal domain of the transposase MuA (Wu and Chaconas 1995) in a highly regulated reaction that removes the attached host DNA after the incoming Mu genome has inserted into a bacterial chromosome according to the integrative transposition (Choi and Harshey 2010).

The two outcomes of Mu-driven transposition. The common θ intermediate can be resolved differently by the DNA repair/replication host-dependent machinery through integrative or replicative transposition. Transposition of the mini-Mu(LR) unit from the integrative plasmid (I) into the bacterial chromosome (BC) results in a “simple insertion” in which BC gains a copy of the unit or a “cointegrate” in which I and BC fuse and two copies of the mini-Mu unit border the junction of the fused replicons as direct repeats. The cointegrate can subsequently be resolved by homologous recombination between two mini-Mu units. Adapted from Abalakina et al. (2008a) and Choi and Harshey (2010)
Mini-Mu as a genetic tool for E. coli chromosomal editing
The structures of mini-Mus can differ significantly. Some mini-Mus carry all of the genetic elements essential for transposition, replication, and packaging. Other derivatives possess only the Mu ends (later designated as mini-Mu(LR) units) and can be complemented in trans to perform Mu-specific functions in E. coli (Chaconas et al. 1981a; Harshey 1983; Patterson et al. 1986). Mini-Mu(cts) chromosomal vectors, which lack the genes required for making phage particles, have been used for simultaneous thermo-induced replicative transposition and heterologous protein synthesis in E. coli (Weinberg et al. 1993).
However, the cis-encoded transposition factors in the mini-Mu(cts) constructs can cause instability in engineered strains even under noninduced conditions (Akhverdyan et al. 2007). Stabilization has been achieved through the use of dual-component systems in which the genes for the MuA and MuB transposition factors are encoded in trans on an unlinked/nontransposed DNA molecule and eliminated after mini-Mu transposition.
Different variants of this system have been developed (Akhverdyan et al. 2007; Castilho et al. 1984; Chaconas et al. 1981a; Groenen et al. 1985; Patterson et al. 1986). One of the most popular systems includes an “integrative” plasmid that consists of a mini-Mu(LR) unit as the first component and a compatible “helper” plasmid carrying the inducible MuA and MuB genes as the second component. Usually, the helper plasmid possesses an unstable replicon and can be easily eliminated from the cells. The transposed genes in a set of mini-Mu(LR) units are flanked by Rho-independent transcription terminators in a directly repeated orientation (Abalakina et al. 2008a, b; Gulevich et al. 2009). The upstream terminator prevents readthrough transcription of the randomly integrated mini-Mu unit from the chromosomal promoter. In turn, the downstream terminator interrupts internal readthrough transcription of the mini-Mu that could interfere with transpososome assembly in vivo (Patterson et al. 1986).
In the mini-Mu derivatives that could be assigned as mini-Mu(LER) units, an E element properly arranged between the L and R ends positively influences transposition (Leung et al. 1989). However, intense expression of the MuA and MuB genes, even when located in trans, leads to host cell lethality because of the overly efficient replicative transposition of the mini-Mu(LER) units (Akhverdyan et al. 2007; Lee 2002). Although artificial mini-Mu(LER) units are actively used in fundamental investigations of transpososome assembly and function in vitro and in vivo (Lee 2002), they have not been widely applied in stably engineered E. coli strains.
In contrast, in trans E element has been successfully used to significantly increase the transposition efficiency of the mini-Mu(LR) unit. A comparison of the E-carrier ((E-plus)-helperFootnote 1) and the helper that did not contain the E element ((E-minus)-helperFootnote 2) revealed a nearly two-order magnitude of increase in the transposition efficiency of the mini-Mu(LR) units when the (E-plus)-helper was used.
To facilitate the selection of the transposed mini-Mu, an antibiotic resistance marker (AntR) can be included.Footnote 3 Markers that are flanked by the sequences essential for site-specific recombination, λattL/R (Peredelchuk and Bennett 1997), loxP (Arakawa et al. 2001), or FRT (Datsenko and Wanner 2000), are usually used. These markers can be excised in vivo from the mini-Mu units by the corresponding recombinase to obtain the marker-less recombinant strain (Abalakina et al. 2008a; Gulevich et al. 2009; Wei et al. 2010).
Several protocols have been developed for mini-Mu(LR) unit transposition into the E. coli chromosome with the help of the dual-component Mu-driven system. The best results have been obtained when an (E-plus)-helper plasmid is first transferred into the recipient strain and is stably maintained under noninducing conditions. Then, an integrative plasmid is transferred into the helper–carrier strain; the efficacy of this step mainly determines the total efficiency of the detected mini-Mu(LR) unit transposition. Therefore, different strategies that are based on the presence of a selective marker in the mini-Mu(LR) unit are employed. For a marker–carrier mini-Mu, replication of the integrative plasmid is not necessary in the recipient cell. So, the transposase can be induced during plasmid entry followed by direct selection of the clones–integrants. Usually, transposition occurs in the majority of cells that receive the integrative plasmid (Zimenkov et al. 2004).
If the marker-less mini-Mu(LR) unit is used, the integrative plasmid has to be selectively transformed into the helper–carrier recipient. The transposase is then expressed in the cells that contain both autonomously replicating components of the Mu-driven system. Finally, the obtained clones–integrants are cured of the unstable plasmid(s). Usually, the transposition of the mini-Mu(LR) units can be detected in at least 10% of the induced cells (Savrasova et al. 2007).
Both strategies can lead to high-efficiency formation of clones possessing one or more (up to five to ten) copies of the mini-Mu(LR) unit in the chromosome (Akhverdyan et al. 2007). The copy number of the integrated mini-Mu can be increased further by reintroducing and expressing the (E-plus)-helper plasmid in the corresponding plasmid-less strain–integrant. The (E-minus)-helper plasmid has a significantly decreased but still detectable ability to facilitate intrachromosomal replicative transposition of mini-Mu(LR) units in E. coli (Gak et al., in preparation).
A dual-component Mu-driven system has been frequently used for E. coli chromosome editing and for constructing different bacterial strains for basic research and metabolically engineered amino acid production (Table 1). In most cases, the expression level of the Mu-integrated genes increased proportionally as the copy number in the chromosome increased. As a rule, the amplification of a biosynthetic operon leads to the increased accumulation of the corresponding amino acid. The amino acid production levels of the plasmid-less strains obtained were comparable to those of their plasmid-carrier recombinant analogs, and the former were significantly more stable during nonselective cultivation (Akhverdyan et al. 2007; Savrasova et al. 2007).
The transposition of the mini-Mu(LR) unit into the bacterial chromosome mainly occurs through a “nick–join–replicative” mechanism with the formation of a cointegrate in which the integrative plasmid and the chromosome fuse and two copies of the mini-Mu unit border the junction of the fused replicons as direct repeats (Fig. 4) (Abalakina et al. 2008a; Patterson et al. 1986; Tokmakova 2010). The cointegrate is subsequently resolved by a reciprocal recombination event between the two mini-Mu units. The chromosomal structure of the final strain that contains one copy of the mini-Mu unit is the same as if integrative transposition of this unit had occurred. The sites of mini-Mu insertion in the E. coli chromosome could be precisely determined (Wei et al. 2010; Zimenkov et al. 2004) by inverse polymerase chain reaction (Ochman et al. 1988).
Several copies of the mini-Mu(LR) units can be easily detected in the bacterial genome after using the dual-component Mu-driven system. It could not be excluded that these multiple copies are due to independent transpositions from several copies of the nonreplicated integrative plasmid that were initially transformed into one cell or from several copies of autonomously replicated integrative plasmid. However, especially when the nonreplicating integrative plasmid was used, it seems more likely that the amplification is achieved due to the mini-Mu intrachromosomal replicative transposition. Certainly, the genome of the final stable plasmid-less multi-integrant strain may contain transposition-mediated chromosomal rearrangements, including inversions and/or deletions of bacterial DNA fragments (Watson et al. 2004).
Adaptation of the Mu-driven system for Methylophilus methylotrophus AS1
Interest in the use of methylotrophic bacteria for applied microbiology and biotechnology has increased in recent years (Schrader et al. 2009). Several genomes of methylotrophs have been sequenced (Chistoserdova et al. 2007; Vuilleumier et al. 2009). Significant progress has been made in elucidating the metabolism of these bacteria (Chistoserdova et al. 2009), and the number of tools available for genetic and metabolic engineering has expanded greatly (Bélanger et al. 2004; Choi et al. 2006; Marx and Lidstrom 2001, 2004). Strategies to produce fine and bulk chemicals using methylotrophs have been described previously (Bourque et al. 1995; Fitzgerald and Lidstrom 2003; Motoyama et al. 1993, 2001). In particular, the obligate methylotroph Methylophilus methylotrophus AS1, which was extensively studied with respect to the industrial-scale production of single cell protein from methanol in the 1970s (Anthony 1982; Vasey and Powell 1984), was recently metabolically engineered for the biosynthesis of l-lysine (Gunji and Yasueda 2006; Tsujimoto et al. 2006) and l-phenylalanine (Tokmakova et al. 2010) and for the efficient secretion of recombinant proteins (Itaya et al. 2008).
The adaptation of mini-Mu transposition for chromosomal editing in M. methylotrophus AS1 (Abalakina et al. 2008a, b) was a significant development. Mu-driven integration of the E. coli transporter gene into the methylotrophic genome was the basis for the development of an efficient method of constructing auxotrophic mutants (Yomantas et al. 2010) with the following exploiting the developed system for metabolic engineering of l-phenylalanine overproduction (Iomantas and Abalakina 2002; Tokmakova et al. 2008, 2010).
The adaptation of the Mu-driven system to M. methylotrophus AS1 included several stages (Abalakina et al. 2008a). Initially, a DNA fragment containing the genes from E. coli plasmid, pMH10 (Akhverdyan et al. 2007), for the thermo-induced transposition factors was cloned into vectors containing the broad host range replicons of the IncQ (Chistoserdov and Tsygankov 1986) or IncPα groups (Ditta et al. 1985; Pansegrau et al. 1994). These new (E-plus)-helpers (Fig. 5a) were transformed into methylotrophic cells (by mobilization or electroporation) and maintained under strictly selective conditions without expressing the Mu transposition factors. All of the helpers could be easily eliminated from M. methylotrophus AS1 by aerobically culturing the cells in liquid medium without antibiotics (Abalakina et al. 2008a).

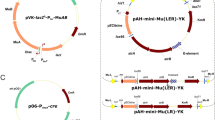
Schematic linear representation of the set of helper (a) and integrative (b, c) plasmids used in the dual-component Mu-driven system for integration/amplification of genes in the M. methylotrophus AS1 genome. These mini-Mu units carry excisable KmR marker bracketed by FRT sites, SmR marker used for selective amplification, and a gene of ZsGreen fluorescent protein (Matz et al. 1999) as a reporter. The internal parts of all mini-Mu units are bracketed by Rho-independent transcription terminators (ter). a The helpers are based on the mobilizable (oriT) replicon (oriV) of the IncPα group plasmid pRK310 (Ditta et al. 1985). The (E-plus)-helper, pTP310 (Abalakina et al. 2008b), carries the Mucts62 and MuA and MuB genes under the native Mu DNA early regulatory region, including the Pe promoter and (E; O1-O3) (Krause and Higgins 1986). In the (E-minus)-helper, p17TP310, MuA and MuB are under the transcriptional control of the M. methylotrophus constitutive P17 promoter (Abalakina et al. 2008a). b The integrative plasmids containing the mini-Mu(LR) or (LER) units are based on the conditionally replicated pir +-dependent (oriRγ) plasmid pAH162 (Haldimann and Wanner 2001). c One of the new integrative plasmids with a mini-Mu(L[exE]R) unit in which the E element is flanked by lox66/71 sites
The constructed integrative plasmids could not autonomously replicate in methylotrophic cells. Because it was unclear if it was possible for Mu-driven transposition to occur, the Mob +RP4 element (Simon et al. 1983, 1984) was included in the initial integrative plasmids to ensure their highly efficient transfer into M. methylotrophus by mobilization (Abalakina et al. 2008a). The next generation of integrative plasmids (Fig. 5b, see experimental details in Tokmakova 2010) did not carry the Mob+ element and could be autonomously maintained only in the pir +E. coli strain used to propagate γ-replicon originating from R6K (Bowers et al. 2007). These integrative plasmids were introduced into the methylotrophic recipient cells by electroporation. The KmR gene, flanked by FRT sites, was used as the “excisable” selective marker in the constructed mini-Mu(LR) units (Abalakina et al. 2008a).
The mini-Mu(LR) unit can be detected in the M. methylotrophus AS1 chromosome after the transfer of the integrative plasmid into the helper–carrier cells in the presence of the partially induced (37°C) transposition factors. Curing the selected integrants of the helper plasmid and Flp-mediated elimination of the marker results in a plasmid-less marker-less recombinant strain that is ready for the next round of mini-Mu transposition (Abalakina et al. 2008a).
The efficiencies (10−2–10−3) of plasmid mobilization and Mu-driven integration are correlated. This means that, under the developed conditions, a mini-Mu(LR) unit is transposed into the bacterial chromosome of each cell of the recipient strain that received the integrative plasmid. As in E. coli, the transposition of the mini-Mu(LR) unit into the M. methylotrophus AS1 chromosome occurs through “nick–join–replicative” formation of the cointegrate, followed by its recombination-mediated resolution (Abalakina et al. 2008a). These factors suggested that the mini-Mu(LR) units might be amplified in M. methylotrophus with the assistance of the same set of host proteins (Au et al. 2006; Gueguen et al. 2005; North and Nakai 2005). However, the MuA- and MuB-mediated intrachromosomal duplication of the mini-Mu(LR) unit was detected only with a low (10−4) frequency (Abalakina et al. 2008a).
The efficiency of the Mu-driven increase in genomic copy number in M. methylotrophus AS1 was increased 100-fold by exploiting the mini-Mu(LER) unit (Fig. 5b). When transferred as part of the integrative plasmid into M. methylotrophus, these units, like earlier used mini-Mu(LR), could be integrated into the bacterial chromosome with the formation of a cointegrate; the transposition efficiency of this process was equally high whether the helper was (E-plus) or (E-minus). On the contrary, the increase in genomic copy number efficacy of the mini-Mu(LER) or mini-Mu(LR) units was significantly dependent on the type of helper used (Table 2). The detected capacities for increase in genomic copy number for the mini-Mu(LER) and mini-Mu(LR) units differed by approximately four orders of magnitude in the presence of the expressed (E-minus)-helper.
These results served as the basis for a novel integration/amplification/fixation strategy that was developed for the M. methylotrophus AS1 Mu-driven system. The E element in the mini-Mu(LER) unit was substituted with its excisable analog, which was bracketed by loxP-like sites [lox66 and lox71 (Albert et al. 1995)] that could serve as the target for irreversible excision by phage P1 Cre recombinase (Abremski and Hoess 1984). Thus, the mini-Mu(L[exE]R) unit (Fig. 5c) could be integrated and amplified in the M. methylotrophus AS1 genome according to the standard procedure (using (E-plus)- or (E-minus)-helpers), followed by Cre-mediated excision of the E element. The “residual” copies of the mini-Mu(LR) units in the chromosome could not be further efficiently amplified in the presence of the (E-minus)-helper plasmid. This allows the integration and independent amplification of another mini-Mu(L[exE]R) unit into the M. methylotrophus AS1 genome without changing the number and location of the previously integrated genes (Fig. 6) (Tokmakova 2010).

Application of the integration/amplification/fixation strategy. Initially, a mini-Mu(L[exE]R) unit is integrated and amplified in the chromosome of M. methylotrophus AS1 according to the replicative transposition in the presence of the expressed (E-minus)-helper plasmid. Then, the E element is excised by Cre-mediated site-specific recombination. The “truncated” mini-Mu(LR) units could be further amplified with very low frequency by (E-minus)-helper, i.e., their positions in the chromosome could be considered as fixated. So, the obtained multi-integrant could be served as the recipient for integration and independent amplification of new Mu(L[exE]R) units
There are still many fundamental questions concerning the differences in the efficiencies of the Mu-driven increase in genomic copy number in E. coli and M. methylotrophus. The answers likely depend on the precise nature of the respective transpososome assembly processes. These processes could depend, in particular, on the intracellular concentrations of the expressed transposition factors and the DNA-bending host proteins that facilitate transposition at the “restrained” host DNAs.
Concluding remarks
We have reviewed the adaptation of the dual-component Mu-driven system for chromosomal editing and the construction of plasmid-less marker-less strains of several Gram-negative bacteria due to the integration/amplification of target genes by the “nick–join–replicative” pathway in vivo. Certainly, other methods could be used for the chromosomal amplification of the target genes as well. The same purposes could be achieved through the single insertion of a DNA cassette with multiple copies of the target gene (Choi et al. 2006). The marker-containing single insertions in different specific (Haldimann and Wanner 2001; Minaeva et al. 2008) or random (De Lorenzo and Timmis 1994; Peredelchuk and Bennett 1997; Wei et al. 2010) sites could be combined in one strain by general P1-mediated transduction, followed by marker curing. These alternative methods, each of which possesses its own specific advantages, are significantly inferior to the Mu-driven amplification approach with respect to ease-of-use and the speed with which a target can be achieved, especially if selection for the best variants of the multi-integrant is possible.
Up today, in vitro-assembled Mu transpososome complexes have been efficiently applied for the in vivo random insertion of recombinant DNAs into different bacterial genomes by the “nick–join–repair” pathway (Haapa et al. 1999; Laasik et al. 2005; Lamberg et al. 2002; Lanckriet et al. 2009; Pajunen et al. 2005; Savilahti et al. 1995; Savilahti and Mizuuchi 1996; Tu Quoc et al. 2007; Wei et al. 2010; Wu et al. 2009). Moreover, efficient Mu transpososome-based integration has been verified even in yeast, mouse, and human genomes (Paatero et al. 2008; Turakainen et al. 2009). Many more host proteins likely participate in the process of Mu-driven replicative transposition than in the simple insertion of mini-Mu into the host chromosome (Au et al. 2006; North and Nakai 2005). It is possible that only integrative, but not replicative, transposition occurs in this broad range of host organisms, even when efficient intracellular expression of Mu transposition factors is provided. However, to broaden the range of hosts in which the Mu-driven system can be used for the integration/amplification of target genes, its adaptation should be attempted, especially by using mini-Mu(LR)/(LER) units in combination with different (E-plus)/(E-minus)-helpers expressed in different genetic backgrounds.
The combined Mu-driven system could be proposed when the in vitro-assembled MuA-mediated transpososome results single-copy integrants due to the integrative transposition, followed by the increase in genomic copy number of the integrated mini-Mu in vivo by the replicative transposition in the presence of the helper plasmid expressing MuA and MuB factors.
Notes
The p15A-based plasmid pMH10 was used as a helper in this dual-component system (Akhverdyan et al. 2007). pMH10 carried the fragment from pMud4041 (Symonds et al. 1987), which consisted of the Mucts62 repressor gene and MuA, MuB with the native cts-controlled regulatory region including Pe promoter, and (E; O1–O3).
MuA and MuB genes controlled by the λPL/OL regulatory region were located in the nontransposing part of the single plasmid that carried the transposed mini-Mu(LR) unit as well (Patterson et al. 1986).
References
Abalakina EG, Tokmakova IL, Gorshkova NV, Gak ER, Akhverdyan VZ, Mashko SV, Yomantas YAV (2008a) Phage Mu-driven two-plasmid system for integration of recombinant DNA in the Methylophilus methylotrphus genome. Appl Microbiol Biotechnol 81:191–200
Abalakina EG, Tokmakova IL, Gorshkova NV, Smirnov SV, Akhverdyan VZ, Mashko SV, Yomantas YAV (2008b) Exploiting of the Mu-phage transposition system for recombinant DNA integration in the Methylophilus methylotrophus chromosome. Biotechnologiya 3:13–26 (in Russian)
Abdelhakim AH, Oakes EC, Sauer RT, Baker TA (2008) Unique contacts direct high-priority recognition of the tetrameric Mu transposase-DNA complex by the AAA+ unfoldase ClpX. Moll Cell 30:39–50
Abremski K, Hoess R (1984) Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem 259:1509–1514
Akhverdyan VZ, Savrasova EA, Kaplan AM, Lobanov AO, Vavilova EY, Kozlov YI (2007) Development of the mini-Mu system providing an effective integration and amplification of the genetic material in the Esherichia coli chromosome. Biotechnologiya 3:3–20 (in Russian)
Akroyd JE, Symonds N (1983) Evidence for a conservative pathway of transposition of bacteriophage Mu. Nature 303:84–86
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7:649–659
Allison RG, Chaconas G (1992) Role of the A protein-binding sites in the in vitro transposition of Mu DNA. II. A complex circuit of interactions involving the Mu-ends and the transpositional enhancer. J Biol Chem 267:19963–19970
Anthony C (1982) The commercial exploitation of methylotrophs. In: Anthony C (ed) The biochemistry of methylotrophs. Academic, London, pp 328–378
Arakawa H, Lodygin D, Buerstedde JM (2001) Mutant loxP vectors for selective marker recycle and conditional knock-outs. BMC Biotechnol 1:7
Au TK, Agrawal P, Harshey RM (2006) Chromosomal integration mechanism of infecting Mu virion DNA. J Bacteriol 188:1829–1834
Baker TA, Luo L (1994) Identification of residues in the Mu transposase essential for catalysis. Proc Natl Acad Sci USA 91:6654–6658
Balbás P, Gosset G (2001) Chromosomal editing in Escherichia coli. Vectors for DNA integration and excision. Mol Biotechnol 19:1–12
Bélanger L, Figueira MM, Bourque D, Morel L, Béland M, Laramée L, Groleau D, Míguez CB (2004) Production of heterologous protein by Methylobacterium extorquens in high cell density fermentation. FEMS Microbiol Lett 231:197–204
Bourque D, Pomerleau Y, Groleau D (1995) High cell density production of poly-beta-hydrobutyrate (PHB) from methanol by Methylobacterium extorquens: production of high-molecular-mass PHB. Appl Microbiol Biotechnol 44:367–376
Bowers LM, Krüger R, Filutowicz M (2007) Mechanism of origin activation by monomers of R6K-encoded π protein. J Mol Biol 368:928–938
Bukhari AI, Taylor AL (1975) Influence of insertions on packaging of host sequences covalently linked to bacteriophage Mu DNA. Proc Natl Acad Sci USA 72:4399–4403
Bukhari AI, Zipser D (1972) Random insertion of Mu-1 DNA within a single gene. Nature New Biol 236:240–243
Castilho BA, Olfson P, Casadaban MJ (1984) Plasmid insertion mutagenesis and lac gene fusion with mini-Mu bacteriophage transposons. J Bacteriol 158:488–495
Chaconas G, Harshey RM (2002) Transposition of phage Mu DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. American Society for Microbiology, Washington, pp 384–402
Chaconas G, deBruijn FG, Casadaban M, Lupski JR, Kwok TJ, Harshey RM, DuBow MS, Bukhari AI (1981a) In vitro and in vivo manipulations of bacteriophage Mu DNA: cloning of Mu ends and construction of mini-Mu’s carrying selectable markers. Gene 13:37–46
Chaconas G, Harshey RM, Sarvetnick N, Bukhari AI (1981b) Predominant end-products of prophage Mu DNA transposition during the lytic cycle are replicon fusions. J Mol Biol 150:341–359
Chaconas G, Lavoie BD, Watson MA (1996) DNA transposition: jumping gene machine, some assembly required. Curr Biol 6:817–820
Chistoserdov AY, Tsygankov YD (1986) Broad host range vectors derived from an RSF1010::Tn1 plasmid. Plasmid 16:161–167
Chistoserdova L, Lapidus A, Han C, Goodwin L, Saunders L, Brettin T, Tapia R, Gilna P, Lucas S, Richardson PM, Lidstrom ME (2007) Genome of Methylobacillus flagellatus, molecular basis for obligate methylotrophy, and polyphyletic origin of methylotrophy. J Bacteriol 189:4020–4027
Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME (2009) The expanding world of methylotrophic metabolism. Annu Rev Microbiol 63:477–499
Choi W, Harshey RM (2010) DNA repair by the cryptic endonuclease activity of Mu transposase. Proc Natl Acad Sci USA 107:10014–10019
Choi YJ, Bourque D, Morel L, Groleau D, Míguez CB (2006) Multicopy integration and expression of heterologous genes in Methylobacterium extorquens ATCC 55366. Appl Environ Microbiol 72:753–759
Court DL, Sawitzke JA, Thomason LC (2002) Genetic engineering using homologous recombination. Annu Rev Genet 36:361–388
Craigie R, Mizuuchi K (1985) Mechanism of transposition of bacteriophage Mu: structure of a transposition intermediate. Cell 41:867–876
Craigie R, Mizuuchi M, Mizuuchi K (1984) Site-specific recognition of the bacteriophage Mu ends by the Mu A protein. Cell 39:387–394
Daniell E, Abelson J, Kim JS, Davidson N (1973) Heteroduplex structures of bacteriophage Mu DNA. Virology 51:237–239
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
De Lorenzo V, Timmis KN (1994) Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405
De Mey M, Maertens J, Boogmans S, Soetaert WK, Vandamme EJ, Cunin R, Foulquié-Moreno MR (2010) Promoter knock-in: a novel rational method for the fine tuning of genes. BMC Biotechnol 10:26
Dillon SC, Dorman CJ (2010) Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nature Rev Microbiol 8:185–195
Ditta G, Schmidhauser T, Yakobson E, Lu P, Liang XW, Finlay DR, Guiney D, Helinski DR (1985) Plasmid related to the broad host range vector, pRK290, useful for gene cloning and for monitoring gene expression. Plasmid 13:149–153
Doroshenko V, Airich L, Vitushkina M, Kolokolova A, Livshits V, Mashko S (2007) YddG from Escherichia coli promotes export of aromatic amino acids. FEMS Microbiol Let 275:312–318
Doroshenko VG, Shakulov RS, Kazakova SM, Kivero AD, Yampolskaya TA, Mashko SV (2010a) Construction of an l-phenylalanine-producing tyrosine-prototrophic Escherichia coli strain using tyrA ssrA-like tagged alleles. Biotechnol Lett 32:1117–1121
Doroshenko VG, Tsyrenzhapova IS, Kiseleva EM, Ermishev VY, Kazakova SM, Biryukova IV, Mashko SV (2010b) Pho regulon promoter-mediated transcription of the key pathway gene aroG Fbr improves the performance of an l-phenylalanine-producing Escherichia coli strain. Appl Microbiol Biotechnol 88:1287–1295
Eremina NS, Yampolskaya TA, Altman IB, Mashko SV, Stoynova NV (2010) Overexpression of ydbK-encoding putative pyruvate synthase improves l-valine production and aerobic growth on ethanol media by an Escherichia coli strain carrying an oxygen-resistant alcohol dehydrogenase. J Microbial Biochem Technol 2:77–83. doi:https://doi.org/10.4172/1948-5948.1000028
European Council Directive—90/220/EEC of 23 April 1990 on the deliberate release into the environment of genetically modified organisms
European Council Directive—98/81/EEC of 26 October 1998 amending Directive 90/219/EEC on the contained use of genetically modified microorganisms
Faelen M, Huisman O, Toussaint A (1978) Involvement of phage Mu-1 early functions in Mu-mediated chromosomal rearrangements. Nature 271:580–582
Fitzgerald KA, Lidstrom ME (2003) Overexpression of a heterologous protein, haloalkane dehalogenase, in a poly-β-hydroxybutyrate-deficient strain of the facultative methylotroph Methylobacterium extorquens AM1. Biotechnol Bioeng 81:263–268
Friehs K (2004) Plasmid copy number and plasmid stability. In: Scheper TH (ed) New trends and developments in biochemical engineering, vol 86. Springer, Berlin, pp 47–82
Ge J, Lou Z, Harshey RM (2010) Immunity of replicating Mu to self-integration: a novel mechanism employing MuB protein. Mob DNA 1:8
Gloor G, Chaconas G (1986) The bacteriophage Mu N gene encodes the 64-kDa virion protein which is injected with, and circularizes, infecting Mu DNA. J Biol Chem 261:16682–16688
Groenen MAM, Timmers E, van de Putte P (1985) DNA sequences at the ends of the genome of bacteriophage Mu essential for transposition. Proc Natl Acad Sci USA 82:2087–2091
Groisman EA (1991) In vivo genetic engineering with bacteriophage Mu. Methods Enzymol 204:180–212
Groisman EA, Casadaban MJ (1986) Mini-Mu bacteriophage with plasmid relicons for in vivo cloning and lac gene fusing. J Bacteriol 168:357–364
Groisman EA, Casadaban MJ (1987) Cloning of genes from members of the family Enterobacteriaceae with mini-Mu bacteriophage containing plasmid replicons. J Bacterial 169:687–693
Gueguen E, Rousseau P, Duval-Valentin G, Chandler M (2005) The transpososome: control of transposition at the level of catalysis. Trends Microbiol 13:543–549
Gulevich AI, Skorokhodova AI, Ermishev VI, Krylov AA, Minaeva NI, Polonskaia ZM, Zimenkov DV, Biriukova IV, Mashko SV (2009) New method of construction of artificial translational-coupled operons in bacterial chromosome. Mol Biol (Mosk) 43:547–557
Gunji Y, Yasueda H (2006) Enhancement of l-lysine production in methylotroph Methylophilus methylotrophus by introducing a mutant LysE exporter. J Biotechnol 127:1–13
Haapa S, Taira S, Heikkinen E, Savilahti H (1999) An efficient and accurate integration of mini-Mu transposons in vitro: a general methodology for functional genetic analysis and molecular biology applications. Nucl Acids Res 27:2777–2784
Haapa-Paananen S (2002) The mechanisms, applications, and target site selection of bacteriophage Mu minimal in vitro DNA transposition reaction. Academic dissertation (Savilahti H—supervisor), Institute of Biotechnology and Department of Biosciences, University of Helsinki
Haapa-Paananen S, Rita H, Savilahti H (2002) DNA transposition of bacteriophage Mu. A quantitative analysis of target site selection in vitro. J Biol Chem 277:2843–2851
Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision, and retrieval plasmid–host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393
Han YW, Mizuuchi K (2010) Phage Mu transposition immunity: protein pattern formation along DNA by a diffusion-ratchet mechanism. Mol Cell 39:48–58
Harshey RM (1983) Switch in the transposition products of Mu DNA mediated by proteins: cointegrates versus simple insertions. Proc Natl Acad Sci USA 80:2012–2016
Harshey RM (1984) Transposition without duplication of infecting bacteriophage Mu DNA. Nature 311:580–581
Harshey RM, Bukhari AI (1983) Infecting bacteriophage Mu DNA forms a circular DNA–protein complex. J Mol Biol 167:427–441
Harshey RM, Jayaram M (2006) The Mu transpososome through a topological lens. Crit Rev Biochem Mol Biol 41:387–405
Howe MM (1997) Bacteriophage Mu. In: Busby SJW, Thomas CM, Brown NL (eds) Molecular microbiology. Springer, Heidelberg, pp 65–80
Howe MM, Bade EG (1975) Molecular biology of bacteriophage Mu. Science 190:624–632
Iomantas YV, Abalakina EG (2002) Method for producing l-phenylalanine. Patent US6350596
Itaya H, Kikuchi Y, Date M (2008) Method for producing protein. Patent EP1882743
Katashkina JI, Skorokhodova AY, Zimenkov DV, Gulevich AY, Minaeva NI, Doroshenko VG, Biryukova IV, Mashko SV (2005) Tuning of expression level of the genes of interest located in the bacterial chromosome. Mol Biol (Mosk) 39:823–831
Klyachko EV, Shakulov RS, Kozlov YI (2008) Mutant phosphoribosylpyrophosphate synthetase and method for producing l-histidine. Patent EP1529839
Krause HM, Higgins NP (1986) Positive and negative regulation of the Mu operator by Mu repressor and Escherichia coli integration host factor. J Biol Chem 261:3744–3752
Krementsova E, Giffin MJ, Pincus D, Baker TA (1998) Mutational analysis of the Mu transposase: contributions of two distinct regions of domain II to recombination. J Biol Chem 273:31358–31365
Krylov AA, Airich LG, Kiseleva EM, Minaeva NI, Biryukova IV, Mashko SV (2010) Conditional silencing of the Escherichia coli pykF gene results from artificial convergent transcription protected from Rho-dependent termination. J Mol Microbiol Biotechnol 18:1–13
Kuo CF, Zou AH, Jayaram M, Getzoff E, Harshey R (1991) DNA–protein complexes during attachment-site synapsis in Mu DNA transposition. EMBO J 10:1585–1591
Kurahashi O, Takinami K (1991) Transposon Mu as a genetic tool for stabilized amplification of genetic information. Proceedings of the 10th International Symposium on Life Sciences, Nara-shi, Japan, pp 109–111
Kurahashi O, Beyou A, Takinami K, Jarry B, Richaud F (1990) Stabilized amplification in Gram negative bacteria with Mu phage and its application in l-threonine production. Proceedings of the International Symposium on Industrial Microorganisms, Societé Française de Microbiologie, Paris, p 178
Kutukova E, Livshits V, Altman I, Ptitsyn L, Zyiatdinov M, Tokmakova I, Zakataeva N (2005) The yeaS (leuE) gene of Escherichia coli encodes an exporter of leucine, and the Lrp protein regulates its expression. FEBS Lett 579:4629–4634
Laasik E, Ojarand M, Pajunen M, Savilahti H, Mäe A (2005) Novel mutants of Erwinia carotovora subsp. carotovora defective in the production of plant cell wall degrading enzymes generated by Mu transpososome-mediated insertion mutagenesis. FEMS Microbiol Lett 243:93–99
Lamberg A, Nieminen S, Qiao M, Savilahti H (2002) Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl Environ Microbiol 68:705–712
Lanckriet A, Timbermont L, Happonen LJ, Pajunen MI, Pasmans F, Haesebrouck F, Ducatelle R, Savilahti H, Van Immerseel F (2009) Generation of single-copy transposon insertions in Clostridium perfringens by electroporation of phage Mu DNA transposition complexes. Appl Environ Microbiol 75:2638–2642
Lavoie BD, Chaconas G (1995) Transposition of phage Mu DNA. Curr Top Microbiol Immunol 204:83–102
Lavoie BD, Chan BC, Allison RG, Chaconas G (1991) Structural aspects of a higher order nucleoprotein complex: induction of an altered DNA structure at the Mu–host junction of the Mu type 1 transpososome. EMBO J 10:3051–3059
Lavoie BD, Shaw GS, Millner A, Chaconas G (1996) Anatomy of a flexer–DNA complex inside a higher-order transposition intermediate. Cell 85:761–771
Lee I (2002) Importance of the conserved TG/CA dinucleotide termini in phage Mu transposition: similarities to transposable elements in the human genome. Ph.D. dissertation (Harshey RM—superviser), University of Texas at Austin
Leung PC, Teplow DB, Harshey R (1989) Interaction of distinct domains in Mu transposase with Mu DNA ends and an internal transpositional enhancer. Nature 338:656–658
Liebart JC, Ghelardini P, Paolozzi L (1982) Conservative integration of bacteriophage Mu DNA into pBR322 plasmid. Proc Natl Acad Sci USA 79:4362–4366
Livshits VA, Doroshenko VG, Mashko SV, Akhverdian VZ, Kozlov YI (2005) Methods of making amino acids using E. coli transformed with csc genes. Patent US6960455
Manna D, Breier AM, Higgins NP (2004) Microarray analysis of transposition targets in Escherichia coli: the impact of transcription. Proc Natl Acad Sci USA 101:9780–9785
Marx CJ, Lidstrom ME (2001) Development of improved versatile broad-host-range vectors for use in methylotrophs and other gram-negative bacteria. Microbiology 147:2065–2075
Marx CJ, Lidstrom ME (2004) Development of an insertional expression vector system for Methylobacterium extorquens AM1 and generation of null mutants lacking mtdA and/or fch. Microbiology 150:9–19
Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA (1999) Fluorescent proteins from nonbioluminescent Anthozoa species. Nature Biotechnol 17:969–973
Meynial-Salles I, Cervin MA, Soucaille P (2005) New tool for metabolic pathway engineering in Escherichia coli: one step method to modulate expression of chromosomal genes. Appl Environ Microbiol 71:2140–2144
Minaeva NI, Gak ER, Zimenkov DV, Skorokhodova AY, Biryukova IV, Mashko SV (2008) Dual in/out strategy for genes integration into bacterial chromosome: a novel approach to step-by-step construction of plasmid-less marker-less recombinant E. coli strains with predesigned genome structure. BMC Biotechnol 8:63
Mizuuchi K (1984) Mechanism of transposition of bacteriophage Mu: polarity of the strand transfer reaction at the initiation of transposition. Cell 39:395–404
Mizuuchi K, Adzuma K (1991) Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: evidence for a one-step transesterification mechanism. Cell 66:129–140
Mizuuchi M, Mizuuchi K (1989) Efficient Mu transposition requires interaction of transposase with a DNA sequence at the Mu operator: implication for regulation. Cell 58:399–408
Mizuuchi M, Baker TA, Mizuuchi K (1995) Assembly of phage Mu transpososomes: cooperative transitions assisted by protein and DNA scaffolds. Cell 83:375–385
Morgan GJ, Hatfull GF, Casjens S, Hendrix RW (2002) Bacteriophage Mu genome sequence: analysis and comparison with Mu-like prophage in Haemophilus, Neisseria and Deinococcus. J Mol Biol 317:337–359
Motoyama H, Anazawa H, Katsumata R, Araki K, Teshiba S (1993) Amino acid production from methanol by Methylobacillus glycogens mutants: isolation of l-glutamic acid hyper-producing mutants from M. glucogenes strains, and derivation of l-threonine- and l-lysine-producing mutants from them. Biosci Biotechnol Biochem 57:82–87
Motoyama H, Yano H, Terasaki Y, Anazawa H (2001) Overproduction of l-lysine from methanol by Methylobacillus glycogens derivatives carrying a plasmid with a mutated dapA gene. Appl Environ Microbiol 67:3064–3070
Nakai H, Doseeva V, Jones JM (2001) Handoff from recombinase to replisome: insights from transposition. Proc Natl Acad Sci USA 98:8247–8254
North SH, Nakai H (2005) Host factors that promote transpososome disassembly and the PriA-PriC pathway for restart primosome assembly. Mol Microbiol 56:1601–1616
O’Day KJ, Schultz DW, Howe MM (1978) Search for integration-deficient mutants of bacteriophage Mu. In: Schlessinger D (ed) Microbiology. American Society for Microbiology, Washington, pp 48–51
Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120:621–623
Paatero AO, Turakainen H, Happonen LJ, Olsson C, Palomaki T, Pajunen MI, Meng X, Otonkoski T, Tuuri T, Berry C, Malani N, Frilander MJ, Bushman FD, Savilahti H (2008) Bacteriophage Mu integration in yeast and mammalian genomes. Nucl Acids Res 36:e148
Pajunen MI, Pulliainen AT, Finne J, Savilahti H (2005) Generation of transposon insertion mutant libraries for Gram-positive bacteria by electroporation of phage Mu DNA transposition complexes. Microbiology 151:1209–1218
Pansegrau W, Lanka E, Barth PT, Figurski DH, Guiney DG, Haas D, Helinski DR, Schwab H, Stanisich VA, Thomas CM (1994) Complete nucleotide sequence of Birmingham IncP alpha plasmids. Compilation and comparative analysis. J Mol Biol 239:623–663
Paolozzi L, Ghelardini P (2006) The bacteriophage Mu. In: Calendar R (ed) The bacteriophages. Oxford University Press, New York, pp 469–496
Pathania S, Jayaram M, Harshey RM (2002) Path of DNA within the Mu transpososome: transposase interacts bridging two Mu ends and the enhancer trap five DNA supercoils. Cell 109:425–436
Pathania S, Jayaram M, Harshey RM (2003) A unique right end-enhancer complex precedes synapsis of Mu ends: the enhancer is sequestered within the transpososome throughout transposition. EMBO J 22:3725–3736
Patterson TA, Court DL, Dubuc G, Michniewicz JJ, Goodchild J, Bukhari AI, Narang SA (1986) Transposition studies of mini-Mu plasmids constructed from the chemically synthesized ends of bacteriophage Mu. Gene 50:101–109
Peredelchuk MY, Bennett GN (1997) A method for construction of E. coli strains with multiple DNA insertions in the chromosome. Gene 187:231–238
Pettijohn DE (1996) The nucleoid. In: Neidhardt FC (ed) Escherichia coli and Salmonella: cellular and molecular biology, 2nd edn. ASM, Washington, pp 158–166
Pettijohn D, Pfenninger O (1980) Supercoils in prokaryotic DNA restrained in vivo. Proc Natl Acad Sci USA 77:1331–1335
Rice PA, Baker TA (2001) Comparative architecture of transposase and integrase complexes. Nature Struct Biol 8:302–307
Rivero-Müller A, Lajic S, Huhtaniemi I (2007) Assisted large fragment insertion by red/ET-recombination (ALFIRE)—an alternative and enhanced method for large fragment recombineering. Nucl Acids Res 35:e78
Roldan LAS, Baker TA (2001) Differential role of the MuB protein in phage Mu integration vs. replication: mechanistic insights into two transposition pathways. Mol Microbiol 40:141–155
Savilahti H, Mizuuchi K (1996) Mu transpositional recombination: donor DNA cleavage and strand transfer in trans by the Mu transposase. Cell 85:271–280
Savilahti H, Rice PA, Mizuuchi K (1995) The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J 14:4893–4903
Savrasova EA, Sycheva EV, Michurina TA, Kozlov JI (2006) Method for preparing l-amino acids by fermentation of mixture of glucose and pentoses. Patent RU2273666
Savrasova EA, Akhverdyan VZ, Lobanov AO, Kaplan AM, Kozlov YI (2007) Creation of mini-Mu system without selective markers for integration of genes into Escherichia coli chromosome. Biotechnologiya 4:3–17 (in Russian)
Savrasova EA, Kivero AD, Shakulov RS, Stoynova NV (2011) Use of the valine biosynthetic pathway to convert glucose into isobutanol. J Ind Microbiol Biotechnol. doi:https://doi.org/10.1007/s10295-010-0907-2
Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL (2007) Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Meth Enzymol 421:171–199
Schrader J, Schilling M, Holtmann D, Sell D, Filho MV, Marx A, Vorholt JA (2009) Methanol-based industrial biotechnology: current status and future perspectives of methylotrophic bacteria. Trends Biotechnol 27:107–115
Sharan SK, Thomason LC, Kuznetsov SG, Court DL (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4:206–223
Simon R, Priefer U, Puhler A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nature Biotechnol 1:784–791
Simon R, Priefer U, Puhler A (1984) High frequency mobilization of gram-negative bacterial replicons by the in vitro constructed Tn5-mob transposon. Mol Gen Genet 196:413–420
Skorokhodova AY, Katashkina JI, Zimenkov DV, Smirnov SV, Gulevich AY, Biryukova IV, Mashko SV (2004) Design and study on characteristics of auto- and smoothly regulated genetic element O3/PlacUV5/Olac→lacI. Biotechnologiya 5:3–21 (in Russian)
Sokolsky TD, Baker TA (2003) DNA gyrase requirements distinguish the alternate pathways of Mu transposition. Mol Microbiol 47:397–409
Surette MG, Chaconas G (1989) A protein factor which reduces the negative supercoiling requirement in the Mu DNA strand transfer reaction is Escherichia coli integration host factor. J Biol Chem 264:3028–3034
Surette MG, Chaconas G (1992) The Mu transpositional enhancer can function in trans: requirement of the enhancer for synapsis but not strand cleavage. Cell 68:1101–1108
Surette MG, Lavoie BD, Chaconas G (1989) Action at a distance in Mu DNA transposition: an enhancer-like element is the site of action of supercoiling relief activity by integration host factor (IHF). EMBO J 8:3483–3489
Swinger KK, Rice PA (2004) IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol 14:28–35
Sycheva EV, Stoynova NV, Savrasova EA, Kozlov YI, Bocharov EV, Sobol AG, Arseniev AS (2003) Aerobic catabolism of threonine in Escherichia coli strain with feedback resistant biosynthetic threonine deaminase. Biotechnologiya 4:22–34 (in Russian)
Symonds NA, Toussaint A, van de Putte P, Howe MM (1987) Phage Mu. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Tokmakova IL (2010) Development of chromosomal editing methods for metabolic engineering of an obligate methylotrophic bacteria Methylophilus methylotrophus AS1. Ph.D. thesis (Gak ER and Yomantas YAV—supervisors), Ajinomoto-Genetika Research Institute, Moscow, Russia
Tokmakova IL, Gorshkova NV, Abalakina EG, Yomantas YAV (2008) Method for production of aromatic l-amino acids using bacterium belonging to genus Methylophilus. Patent RU2333950
Tokmakova IL, Abalakina EG, Gorshkova NV, Yomantas YAV (2010) Method for imparting auxotrophicity on l-amino acid to Methylophilus genus bacteria, and method of producing l-amino acid. Patent RU2395569
Tsujimoto N, Gunji Y, Ogawa-Miyata Y, Shimaoka M, Yasueda H (2006) l-lysine biosynthetic pathway of Methylophilus methylotrophus and construction of an l-lysine producer. J Biotechnol 124:327–337
Tu Quoc PH, Genevaux P, Pajunen M, Savilahti H, Georgopoulos C, Schrenzel J, Kelley WL (2007) Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect Immun 75:1079–1088
Turakainen H, Saarimäki-Vire J, Sinjushina N, Partanen J, Savilahti H (2009) Transposition-based method for the rapid generation of gene-targeting vectors to produce Cre/Flp-modifiable conditional knock-out mice. PLoS ONE 4:e4341. doi:https://doi.org/10.1371/journal.pone.0004341
Tyo KEJ, Ajikumar PK, Stephanopoulos G (2009) Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nature Biotechnol 27:760–765
Vasey RB, Powell KA (1984) Single-cell protein. Biotechnol Genet Eng Rev 2:285–310
Vuilleumier S, Chistoserdova L, Lee MC, Bringel F, Lajus A, Zhou Y, Gourion B, Barbe V, Chang J, Cruveiller S, Dossat C, Gillett W, Gruffaz C, Haugen E, Hourcade E, Levy R, Mangenot S, Muller E, Nadalig T, Pagni M, Penny C, Peyraud R, Robinson DG, Roche D, Rouy Z, Saenampechek C, Salvignol G, Vallenet D, Wu Z, Marx CJ, Vorholt JA, Olson MV, Kaul R, Weissenbach J, Médigue C, Lidstrom ME (2009) Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PloS One 4:e5584
Watson MA, Chaconas G (1996) Three-site synapsis during Mu DNA transposition: a critical intermediate preceding engagement of the active site. Cell 85:435–445
Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004) Molecular biology of the gene, 5th edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 316–334
Wei XX, Shi ZY, Li ZJ, Cai L, Wu Q, Chen GQ (2010) A mini-Mu transposon-based method for multiple DNA fragment integration into bacterial genomes. Appl Microbiol Biotechnol 87:1533–1541
Weinberg RA, De Ciechi PA, Obukowicz M (1993) A chromosomal expression vector for Escherichia coli based on the bacteriophage Mu. Gene 126:25–33
Wu Z, Chaconas G (1995) A novel DNA binding and nuclease activity in domain III of Mu transposase: evidence for a catalytic region involved in donor cleavage. EMBO J 14:3835–3843
Wu Z, Xuanyuan Z, Li R, Jiang D, Li C, Xu H, Bai Y, Zhang X, Turakainen H, Saris PE, Savilahti H, Qiao M (2009) Mu transposition complex mutagenesis in Lactococcus lactis—identification of genes affecting nisin production. J Appl Microbiol 106:41–48
Yin Z, Suzuki A, Lou Z, Jayaram M, Harshey RM (2007) Interactions of phage Mu enhancer and termini that specify the assembly of a topologically unique interwrapped transpososome. J Mol Biol 372:382–396
Yomantas YAV, Tokmakova IL, Gorshkova NV, Abalakina EG, Kazakova SM, Gak ER, Mashko SV (2010) Aromatic amino acid auxotrophs constructed by recombinant marker exchange in Methylophilus methylotrophus AS1 cells expressing the aroP encoded transporter of Escherichia coli. Appl Environ Microbiol 76:75–83
Zimenkov DV, Skorokhodova AY, Katashkina JI, Minaeva NI, Savrasova EA, Biryukova IV, Doroshenko VG, Akhverdyan VZ, Mashko SV (2004) The regions in E. coli chromosome preferable for phage Mu-driven system of gene integration. Biotechnologiya 6:3–18 (in Russian)
Acknowledgements
This paper is devoted to the memory of Prof. Yurii I. Kozlov (1944–2007) who initiated the investigation and application of the Mu-driven integration/amplification systems at AGRI. The authors are extremely thankful to all of the AGRI members who participated in these investigations. In particular, we thank Drs. E.G. Abalakina, I.V. Biryukova, V.G. Doroshenko, A.M. Kaplan, A.O. Lobanov, V.V. Samsonov, E.A. Savrasova, Mrs. N.V. Gorshkova, and E.Y. Vavilova for their fruitful experimental cooperation and helpful discussions of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Akhverdyan, V.Z., Gak, E.R., Tokmakova, I.L. et al. Application of the bacteriophage Mu-driven system for the integration/amplification of target genes in the chromosomes of engineered Gram-negative bacteria—mini review. Appl Microbiol Biotechnol 91, 857–871 (2011). https://doi.org/10.1007/s00253-011-3416-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3416-y