
Abstract
A dual-component Mu-transposition system was modified for the integration/amplification of genes in Corynebacterium. The system consists of two types of plasmids: (i) a non-replicative integrative plasmid that contains the transposing mini-Mu(LR) unit bracketed by the L/R Mu ends or the mini-Mu(LER) unit, which additionally contains the enhancer element, E, and (ii) an integration helper plasmid that expresses the transposition factor genes for MuA and MuB. Efficient transposition in the C. glutamicum chromosome (≈ 2 × 10−4 per cell) occurred mainly through the replicative pathway via cointegrate formation followed by possible resolution. Optimizing the E location in the mini-Mu unit significantly increased the efficiency of Mu-driven intramolecular transposition–amplification in C. glutamicum as well as in gram-negative bacteria. The new C. glutamicum genome modification strategy that was developed allows the consequent independent integration/amplification/fixation of target genes at high copy numbers. After integration/amplification of the first mini-Mu(LER) unit in the C. glutamicum chromosome, the E-element, which is bracketed by lox-like sites, is excised by Cre-mediated fashion, thereby fixing the truncated mini-Mu(LR) unit in its position for the subsequent integration/amplification of new mini-Mu(LER) units. This strategy was demonstrated using the genes for the citrine and green fluorescent proteins, yECitrine and yEGFP, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since its discovery in 1957, as an l-glutamate-producing non-pathogenic Gram-positive soil bacterium from the Actinomyces branch and its classification as a “generally recognized as safe” (GRAS) organism, Corynebacterium glutamicum has become a workhorse for the large-scale industrial production of amino acids, chemicals, materials, fuels, and various proteins (Becker and Wittmann 2012). Recent progress in the characterization and targeted engineering of the metabolism of C. glutamicum is mainly based on high-throughput omics techniques such as genomics (Ikeda and Nakagawa 2003; Kalinowski et al. 2003), transcriptomics (Glanemann et al. 2003; Inui et al. 2007; Wendisch 2003), proteomics (Hermann et al. 2001; Li et al. 2007), metabolomics (Bartek et al. 2010; Woo et al. 2010), and fluxomics (Kjeldsen and Nielsen 2009; Marx et al. 1996; Shinfuku et al. 2009; Wittmann and Heinzle 2002), as well as the accelerated development of highly efficient genetic tools for this organism.
Currently, there are many approaches for chromosomal editing of C. glutamicum. Most of these techniques are based on the application of various types of plasmids (Nešvera and Pátek 2008; Tauch 2005), which allow the deletion, substitution, and overexpression of target genes (Kirchner and Tauch 2003; Nešvera and Pátek 2011). Unfortunately, to our knowledge, a perfect analogue of the λRed/RecET-based recombineering approach for the high-efficiency integration of double-stranded PCR products with rather short homologous arms into targeted loci of the bacterial chromosome (reviewed in (Court et al. 2002))—a method developed for Escherichia coli and several other gram-negative bacteria (Datsenko and Wanner 2000; Katashkina et al. 2009; Swingle et al. 2010)—has been recently described for C. glutamicum only in one publication (Huang et al. 2017), though the corresponding experiments have been earlier announced (Ma et al. 2015). Moreover, the already published RecT-dependent (Binder et al. 2013; Cho et al. 2017; Jiang et al. 2017) or annealing protein-independent (Krylov et al. 2014) recombination approaches between short single-stranded oligonucleotides and a targeted locus in the C. glutamicum chromosome are good starting points. Additionally, integrative plasmid vectors have also been constructed based on various corynephages, and these carry DNA elements that enable phage-governed site-specific recombinant DNA integration (Moreau et al. 1999; Oram et al. 2007).
In addition, several different mini-transposons that work according to the so-called cut-and-paste mechanism of transposition (miniTn31831, Tn5-based; Tn13655) have been successfully used for the integration of recombinant DNA at random locations in the C. glutamicum chromosome (Suzuki et al. 2006; Tsuge et al. 2007).
However, the integration and possible amplification of target genes in the chromosome of Escherichia coli and closely related gram-negative bacteria is known to be efficiently achieved using a system based on phage Mu-driven transposition that was initially characterized and practically exploited more than 30 years ago (Castilho et al. 1984; Chaconas et al. 1981a, b).
The Mu phage undergoes two alternative transposition pathways at different stages of its life cycle that differ in their donor substrate configuration and fate of the transposition products: (i) nick-join-reparative transposition, which results in the integration of linear Mu DNA bracketed by specific Mu L and R ends, into random sites spaced 5 bp apart in the bacterial chromosome during Mu phage infection; and (ii) nick-join-replicative transposition, which occurs through the formation of a cointegrate structure that is obligatory for replication during phage lytic growth (Au et al. 2006; Choi et al. 2014; Harshey 2012, 2014). The Mu-driven replicative transposition pathway provided by an artificial dual-component system was previously extensively used for genome editing of gram-negative bacteria (Akhverdyan et al. 2011). In this system, the first component is an “integrative” plasmid that contains transposing DNA in the form of either a mini-Mu(LR) unit bracketed by L and R ends or a mini-Mu(LER) unit in which an enhancer element, E, is properly arranged between L/R to positively influence the efficiency of transposition (Leung et al. 1989). The second component is an integration helper plasmid that contains inducible genes for the MuA and MuB transposition factors, thus enabling integration of the mini-Mu unit located in the first plasmid. Supplied in trans on an unlinked/non-transposed compatible helper plasmid, these MuAB genes can be eliminated from recipient cells after mini-Mu unit transposition into the bacterial genome (Akhverdyan et al. 2011). Among the alternative systems typically used for the Mu-driven integration of recombinant DNA into a heterologous host genome, electroporation of an in vitro-assembled linear mini-Mu unit in combination with the MuA transposase has successfully resulted in Mu-driven reparative transposition into the chromosome of different organisms, including not only gram-negative bacteria (Lamberg et al. 2002; Lanckriet et al. 2009) but also gram-positive bacterial species (Pajunen et al. 2005) and yeasts as well as mouse and human genomes (Paatero et al. 2008).
In the present study, an expressed dual-component Mu-driven system efficiently transposed target genes in Corynebacterium glutamicum mainly through the nick-join-replicative pathway. Moreover, this work confirmed that proper placement of the E element in the mini-Mu unit structure could increase the efficiency of Mu-driven integration, especially the efficiency of intrachromosomal amplification, in the C. glutamicum chromosome as well as in gram-negative strains (Akhverdyan et al. 2011). Using specially constructed Cre-excisable cassettes with an E element bracketed by lox-like sites as part of a mini-Mu(LER) unit, a new genome modification strategy was developed and adjusted, providing a new tool for the C. glutamicum genetic toolbox. This strategy consists of several repeated stages in which the ith stage includes three consecutive steps: (i) selective MuAB-dependent integration of the mini-Mu(LER)-i unit into a random location on the bacterial genome, followed by (ii) its Mu-driven intrachromosomal amplification and (iii) fixation of truncated mini-Mu(LR)-i units at their new positions, due to Cre-mediated excision of their E elements and selective markers artificially bracketed by lox-like elements. An analogous three-step genome modification strategy could be performed with a mini-Mu(LER)-(i + 1) unit, beginning with its MuAB-driven integration and amplification, provided that all previously transposed mini-Mu-k units (where k = 1, 2, …, i) lost the E element from their structures and are stably maintained. The application of this novel strategy was successfully demonstrated in C. glutamicum.
Materials and methods
Strains, plasmids, and growth conditions
All of the strains and plasmids used in this study are described in Table 1. The Corynebacterium glutamicum strains were grown at 32 °С on Brain Heart Infusion (BHI) medium (Difco, USA). When needed, the corresponding antibiotics were added at the following final concentrations: 1 μg/mL of gentamicin (Gm), 25 μg/mL of kanamycin (Km), 1 μg/mL of tetracycline (Tc), and 250 μg/mL (normal) or 750 μg/mL (high) of streptomycin (Sm).
E. coli strains were grown at 37 °C on Luria-Bertani (LB) medium (Sambrook and Russell 2001). When needed, the corresponding antibiotics were added at the following final concentration: 200 μg/mL of ampicillin (Ap), 20 μg/mL of Tc, 50 μg/mL of Sm, 10 μg/mL of Gm, and 40 μg/mL of Km.
Recombinant DNA experiments
All of the oligonucleotides used in this study are described in Table S1. The restriction, ligation, electrophoresis, and Ca2+-dependent transformation of E. coli cells were performed according to standard protocols (Sambrook and Russell 2001). Plasmid and genomic DNA were isolated using a QIAPREP spin kit (QIAGEN, Hilden, Germany) and Genomic DNA Purification Kit (Thermo Fisher Scientific, Waltham, Ma, USA), respectively. Restriction enzymes, T4 DNA ligase, Long PCR Enzyme Mix, and High Fidelity PCR Enzyme Mix were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and Taq DNA polymerase was purchased from Sileks-M (Moscow, Russia). These enzymes were used according to the manufacturers’ instructions. DNA sequencing was performed commercially by Genotekhnologiya (Moscow, Russia). The “Supplementary Material” contains detailed methods for the construction of the integration helper plasmid, pVK-lacIQ-P tac -MuAB (GenBank accession no. MG014199, Fig. S1); the excision helper plasmid, p06-P dapA -cre (GenBank accession no. MG014197, Fig. S2); and all of the integrative plasmids: pAH-mini-Mu(LR)-YK (Fig. S3), pAH-mini-Mu(LER)-YK (GenBank accession no. MG014198, Fig. S4), pAH-mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK (Fig. S4), and pAH-mini-Mu(LER)-GK (Fig. S5).
Electroporation protocol for C. glutamicum
The protocol presented in this study is the result of electroporation (electrotransformation) optimization, which was performed to achieve the highest possible rate of C. glutamicum ATCC13869 cell transformation with native superhelical (SH) plasmid DNA (e.g., purified from E. coli cells). A 250-μL sample of an overnight culture of C. glutamicum was added to 5 mL of BHI liquid medium, and the cells were grown at 32 °С to an OD595 of 1.5–2 over approximately 1.5–2 h. Then, Ap was added (100 μg/mL), and the cells were incubated for one additional hour. Next, the cells were cooled to + 4 °C and 5 mL of cell culture was harvested by centrifugation. For electrotransformation with a MicroPulser™ (Bio-Rad, Hercules, CA, USA), the cells were washed three times in 10% glycerol at + 4 °C and concentrated to 50 μL in 10% glycerol. These electrocompetent cells were mixed with 100 ng of plasmid DNA immediately prior to transformation and transferred to a 0.1-cm sterile, cold electrode chamber for electroporation via a 2.0 kV, 25 μF и 200 Ω pulse. The cells were immediately diluted with 1 mL of BHI medium and incubated for approximately 2 h at 32 °C with shaking, followed by selection of the desirable transformants using solid selective media (1.5% agar) for 2–3 days at 32 °C. The typical transformation efficiency of the ATCC13869 strain was ≈ 1 × 10−3/100 ng DNA/surviving cells. To achieve high transformation efficiency, cells of the MB001 (DSM102070) and ATCC13032 strains were diluted with 1 mL of BHI medium and incubated at 46 °C for 6 min immediately after electrotransformation. This modification significantly increased the transformation efficiencies of these two latter strains, but the corresponding values still did not exceed 10−4/100 ng DNA/surviving cells for MB001 and 1.5 × 10−5/100 ng DNA/surviving cells for ATCC13032.
Integration of the mini-Mu unit in C. glutamicum chromosome
The electroporation protocol described above, with slight modification, was used for the MuAB-driven integration of a mini-Mu unit into the C. glutamicum chromosome. Briefly, C. glutamicum strain of interest that already possessed the integration helper plasmid, pVK-lacIQ-P tac -MuAB seeded densely (with start OD 0.4–0.8), were grown at 37 °С overnight. A 500-μL sample of an overnight culture of C. glutamicum was added to 5 mL of BHI liquid medium, and the cells were grown at 32 °С to an OD595 of 1.5–2 over approximately 1.5–2 h. Induction of the expression of the MuAB genes was achieved by adding 1.5 mМ IPTG into the BHI medium during the recovery step. The targeted integrants were selected on solid BHI medium supplemented with Km. Integration was confirmed by PCR using the primer pair P37/P38. For the pAH-mini-Mu(LER)-YK integrative plasmid in particular, the obtained KmR clones in which the replicative transposition pathway occurred through cointegrate formation were detected by their SmHR and TcR phenotypes. Conversely, the obtained KmR clones in which the reparative transposition pathway (or replicative transposition followed by fast cointegrate resolution) occurred had SmR and TcS phenotypes. The absence of the helper plasmid was determined by a GmS phenotype in the obtained clones.
In one specially indicated case, preparations of covalently closed but relaxed plasmid DNA were used for integration. For relaxation, the native integrative plasmid was hydrolyzed at a unique restriction site (SalI) located in the mini-Mu portion of the plasmid, followed by recircularization of the plasmid via treatment with T4 DNA ligase at a low DNA concentration (≈ 1 μg/mL).
Intrachromosomal amplification of the mini-Mu(LER) unit
The resolved cointegrate carrying the mini-Mu(LER) unit in the chromosome and the pVK-lacIQ-P tac -MuAB as a plasmid was grown overnight with aeration at 37 °C in liquid BHI medium supplemented with 1.5 mM IPTG. Then, cells were seeded in series of dilutions (from 10−2 to 10−5) onto solid BHI medium containing a high concentration of Sm. Single colonies that grew on these plates were considered SmHR cells and likely contained amplified mini-Mu units. Amplification efficiency was calculated as the number of SmHR colonies per total number of seeded SmR cells estimated by CFU. Finally, SmHR clones were cured of the integration helper plasmid by dual-reseeding and aerobic cultivation in liquid BHI medium at 37 °C for 48 h, followed by plating for the selection of GmS phenotypes.
Excision of the lox-bracketed DNA fragment from the mini-Mu units
Helper plasmid p06-P dapA -cre was transformed into the C. glutamicum integrants. Clones with this plasmid were selected on solid BHI medium supplemented with Cm at 30 °C after 48 h of growth. The selected CmR clones were seeded from single colonies onto solid BHI medium without antibiotics at 30 °C. The resulting clones were tested on solid BHI medium containing Km and Cm for the presence of the KmS, SmS, and CmR markers.
Southern blotting analysis
Southern hybridization was performed in accordance with conventional protocols (Sambrook and Russell 2001) using the following equipment: BrightStar™-Plus Positively Charged Nylon Membrane (Thermo Fisher Scientific, Waltham, MA, USA), VacuGene XL Vacuum Blotting System (GE Healthcare, Chicago, IL, USA), and a Hybridization Oven/Shaker (former Amersham Biosciences). DNA labeling with Biotin-11-dUTP (Thermo Fisher Scientific, Waltham, MA, USA) was performed in a standard 50 μL PCR reaction with the necessary pairs of primers and templates, and 0.2 mM Biotin labeling mix and Taq DNA polymerase. The Biotin labeling mix consists of 2 mM dGTP, 2 mM dATP, 2 mM dCTP, 1.3 mM dTTP, and 0.7 mM Biotin-11-dUTP aqueous solution. Biotin chromogenic detection kits (Thermo Fisher Scientific, Waltham, MA, USA) were used to detect the DNA probes after Southern hybridization. The primer pairs used for the PCR amplification of the probes were P37/P38 for kan and P22/P23 for yECitrine (Table S1).
Fluorescence intensity assay
Colonies of target cells contained the yECitrine and/or yEGFP genes; control cells without either of these genes were picked, and 200-μL cellular suspensions of these cells were prepared separately in 96-well plates (GBO, Kremsmünster, Austria). Optical density (OD595) and fluorescence intensity (F) were measured using the Safire™ plate reader (Tecan, Männedorf, Switzerland). The excitation/emission wavelengths for yECitrine and yEGFP were 522/560 nm and 490/522 nm, respectively. The fluorescence intensity of a blank sample with no cells was established as the background fluorescence (Fbackground). The average OD595 of the samples was between 0.2–0.3. Relative fluorescence intensity (RF) was calculated according to the equation RF = [(Ftarget – Fbackground)/ODtarget] and expressed in arbitrary units.
Determination of the mini-Mu unit integration points
Chromosomal DNA was purified from the mini-Mu unit integrants and hydrolyzed by the restriction enzymes RsaI or Sau3A, many cleavage sites of which are located in the Mu unit, and one of them rather close to the Mu-L or Mu-R ends, respectively (Fig. S6). After circularization of the obtained DNA fragments via treatment with T4 DNA ligase at a low DNA concentration, inverse PCR was used with the divergent primer sets P31/P32 and P29/P30, which correspond to the internal portion of the Mu-R or Mu-L ends, respectively, as described earlier (Zimenkov et al. 2004). The sequence of the host DNA at its border with the mini-Mu unit was established via DNA sequencing of the obtained PCR fragments using the same primers.
Shotgun cloning of the integrated mini-Mu unit
The chromosomal DNA of clone no. 10 was digested with the StuI restriction endonuclease, which does not have any recognition sites in the integrated mini-Mu(LER)-YK unit. The obtained DNA fragments were cloned into the wide-host-range plasmid vector pCM110-GmR at its SwaI site (detailed construction scheme in Fig. S7), followed by transformation into E. coli TG1 cells and KmR selection. Sequences adjacent to the Mu-L and Mu-R ends were determined by sequencing (Fig. S6).
Results
Designed elements of the Mu-driven transposition system for C. glutamicum
To investigate potential Mu-driven transposition in C. glutamicum cells, the previously developed dual-component system for gram-negative bacteria (Akhverdyan et al. 2011) was modified in the following fashion.
First, the integration helper plasmid pVK9-lacIQ-P tac -MuAB (Fig. 1a) was constructed for the expression of the MuA and MuB transposition factor genes in C. glutamicum cells. This plasmid was designed on the basis of the pVK9-GmR vector for rather stable maintenance in C. glutamicum cells, but with the ability to be cured under non-selective conditions (without the addition of Gm in the medium). Expression of the transposition factor genes MuAB can be induced by IPTG addition via the introduction of P tac /O lac -promoter/operator region with the lacIQ unit as their control element, which has been repeatedly used in C. glutamicum (Eggeling and Bott 2005; Kirchner and Tauch 2003; Nešvera and Pátek 2011; Ravasi et al. 2012). The MuAB operon was cloned with the native ribosomal binding sites (RBSs) of its genes as no translation issues were anticipated in C. glutamicum according to calculations of RBS efficiency (https://salislab.net/software/) (Borujeni and Salis 2016). The lacIQ-P tac /O lac system is known to have an inherently high basal level of transcription in non-induced conditions (Billman-Jacobe et al. 1994; Xu et al. 2010), and some problems with cloning toxic genes may occur. However, we did not experience any problems with the MuAB genes.

Schematic map of the mini-Mu-based plasmids: integration helper plasmid pVK-lacIQ-P tac -MuAB (a); integrative plasmids pAH-mini-Mu(LER)-YK, pAH-mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK, pAH-mini-Mu(LR)-YK, and pAH-mini-Mu(LER)-GK (b); and excision helper plasmid p06-P dapA -cre (c)
As the second element of the Mu-driven transposition system, several integrative plasmids with mini-Mu units were constructed (Fig. 1b) using the conditionally replicated pir+-dependent (oriRγ) E. coli plasmid pAH162 (Haldimann and Wanner 2001; Posfai et al. 1994), which cannot autonomously replicate in C. glutamicum cells. The presence of Mu-L/R ends separates the integrative plasmids into two parts: non-Mu DNA and the mini-Mu unit. All integrative plasmids were named according to the specific features contained in their mini-Mu unit.
The non-Mu DNA of the plasmids carried the constitutively expressed gene tetA from Tn10 (Hillen and Berens 1994; Lawley et al. 2000). To our knowledge, no experimental data concerning the expression of this tetA gene in C. glutamicum exists. Putative expression of this gene in C. glutamicum was very important for the confirmation of cointegrate formation due to nick-join-replicative transposition followed by its possible resolution, as the cointegrates and resolvants were detected by their TcR and TcS phenotypes, respectively. The results presented below confirmed rather low TetA activity in C. glutamicum; even though the expression level of tetA resulted in TcR to only 1 μg/mL of Tc in the medium, this resistance was higher than the basal resistance of the TcS control C. glutamicum strain.
The mini-Mu units carried the strAB genes, which were expressed at relatively low levels and conferred resistance to Sm at approximately 250 μg/mL. We expected that strains with several copies of the mini-Mu cassettes in the C. glutamicum chromosome could be selected using increased concentrations of Sm, as previously shown for Methylophilus methylotrophus (Abalakina et al. 2008). Additionally, all of the cassettes contained either the yECitrine or yEGFP gene-encoded mutant citrine or green fluorescent protein, respectively (Sheff and Thorn 2004). The mini-Mu cassettes mainly differed by the presence of an E element and its orientation towards the L and R ends. In the mini-Mu(LR) unit, the E element is absent; in the mini-Mu(LER) unit, the E element is properly arranged in relation to the L/R ends, as in the native Mu genome; and in the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit, the E element is located in the opposite direction (Fig. 1b).
Lox66/lox71 sites (Albert et al. 1995) bracketed the DNA fragment containing the E element and KmR, SmR antibiotic-resistance markers, allowing irreversible excision of this fragment from the mini-Mu unit by the phage P1 Cre recombinase. For this purpose, an excision helper plasmid based on p06-CmR was constructed (Fig. 1c). Cre-mediated excision can occur due to the constitutive expression of the phage P1 gene encoding Cre, which was under the control of the C. glutamicum P dapA promoter. This promoter of medium strength (Pátek 2005; Pátek et al. 1996) was used to avoid the overexpression of Cre, which could result in intrachromosomal Cre-dependent recombination not only between lox-like sites that are closely located in the same copy of the mini-Mu(LER) unit, but as well as between those in different chromosomally integrated units that are separated by long distances.
When successful excision of lox-bracketed DNA fragments occur, all mini-Mu units retain a mini-Mu(LR)-like form, consisting of only their antibiotic-markerless parts with expressed fluorescent protein gene, yECitrine or yEGFP, bracketed by Mu-L/R ends, which can be detected in the bacterial genome.
Transposition of the mini-Mu(LER) unit from a superhelical integrative plasmid into the C. glutamicum chromosome
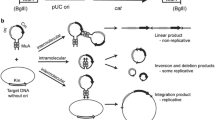
To test potential Mu-driven transposition into the C. glutamicum chromosome, the integration helper plasmid-carrying strain C. glutamicum ATCC 13869[pVK-lacIQ-P tac -MuAB] was initially obtained by electrotransformation (“Materials and methods”) of plasmid DNA and was then used as the recipient for electroporation with pAH-mini-Mu(LER)-YK, followed by the IPTG-induced expression of the MuAB genes during cell cultivation (“Materials and methods”). Since the pAH-based integrative plasmid cannot replicate in C. glutamicum cells, the appearance of KmR transformants putatively resulted from the integration of the mini-Mu unit into the host chromosome, through either the Mu-driven reparative or replicative transposition pathway (Fig. 2) (Craig 1996; Watson et al. 2004).

The two outcomes of Mu-driven DNA transposition from the mini-Mu unit-carrier integrative plasmid (IP) into bacterial chromosome (BC). On superhelical IP (supercoils not shown), in the presence of HU and divalent metal ions (Me2+), the transposase MuA generates endonucleolytic cleavages, producing 3′-OH nicks at Mu DNA L/R ends. Within the active site of MuA, in the subsequent strand-transfer step, the 3′-OH ends directly attack phosphodiester bonds in the target BC spaced 5 bp apart, Mu ends join to 5′-Ps in the BC, leaving 3′-OH nics on the target DNA, whose capture is promoted by MuB (a). The common θ intermediate can be resolved differently by the DNA repair/replication host-dependent machinery through reparative or replicative transposition pathways (b). The reparative transposition into the BC results in a “simple insertion” in which BC gains a copy of the mini-Mu unit. The replicative transposition, in turn, leads to a “cointegrate” formation in which I and BC fuse and two copies of the mini-Mu unit border this junction as direct repeats. The cointegrate can subsequently be resolved by homologous recombination between two mini-Mu units. Adapted from Akhverdyan et al. (2011) and Au et al. (2004)
Based on previous experience with M. methylotrophus (Abalakina et al. 2008), the SmR levels may be lower for the KmR clones obtained via the nick-join-reparative transposition pathway or for the rapidly resolved cointegrates that have only one copy of the integrated mini-Mu unit in the chromosome. For stable cointegrate formation, the entire integrative plasmid must be found in the bacterial chromosome of the KmR clones, with two copies of the mini-Mu unit bracketed as a direct repeat of the bacterial and non-Mu plasmid parts of the cointegrate DNA; SmHR and TcR phenotypes could be detected for these clones.
The selection of transformants on media containing Km resulted in a set of KmR clones that had a transformation frequency ≈ 1.6 × 10−4 (≈ 200 clones/100 ng DNA/1.2 × 106 cells that survived after electroporation). This Mu-driven transposition efficiency was only tenfold lower than the transformation efficiency of the SH plasmid DNA (≈ 10−3) under these conditions. These transformants additionally manifested SmHR (95–99%) or SmR (1–5%) phenotypes on media supplemented with 750 or 250 μg/mL Sm, respectively. Moreover, practically all of the SmHR clones were resistant to the 1 μg/mL Tc that was added to the medium. At this stage, one clone (clone no. 10) was determined to have stable SmHR and TcS phenotypes (see below). As expected, all of the SmR clones were TcS. The SmHR and TcR phenotypes of the obtained strains were rather stable: after five to eight generations, 97% of the single clones maintained this phenotype, and < 3% became SmR and TcS, likely due to resolution of the cointegrate by an intrachromosomal general recombination process that resulted in the deletion and loss of the non-replicative pAH-based plasmid. Finally, the obtained GmR and KmR strains of the integrants were cured of the helper plasmid by selecting for GmS and KmR clones, as described earlier. Analysis of yECitrine-mediated fluorescence in the obtained SmHR clones and their SmR derivates confirmed our suppositions (Fig. 3A).

yECitrine relative fluorescence intensity (A) and Southern blot analysis (B) of the parental strain (1) independent co-integrants (SmHR and TcR) and their resolvants (SmR and TcS) (2, 3, 4, and 5). For the Southern blot analysis, genomic DNA from the individual clones was digested with SmaI and hybridized with a kan-carrying DNA fragment amplified by PCR. (10) Results for clone no. 10, which had an unusual phenotype (SmHR and TcS) after the standard Mu-driven integration procedure. Averages of two experiments are shown and in all cases standard deviation (SD) does not exceed 15%
Southern hybridization experiments confirmed the nature of the obtained integrants. Chromosomal DNA from several SmHR and TcR clones and their SmR and TcS progenies was purified and hydrolyzed using the SmaI restriction endonuclease, which has a unique recognition site in the mini-Mu unit of the pAH-mini-Mu(LER)-YK plasmid that is located outside of the KmR gene (see Fig. S8). After electrophoresis of the obtained DNA fragments in an agarose gel, Southern hybridization (“Material and methods”) was performed using the structural part of the KmR gene, which was amplified by PCR in the presence of fluorescent oligonucleotide precursors, as a marker for the mini-Mu unit. All tested SmHR and TcR clones had two copies of the KmR carrier mini-Mu unit in the bacterial chromosome, fully in accordance with their proposed cointegrate structure (Fig. 3B and its detailed explanation in Fig. S8). Moreover, all of the SmR and TcS derivatives retained only one copy of the mini-Mu unit at its initial point of integration: their hybridized DNA fragments consisted of the KmR carrier part of the mini-Mu unit from the SmaI site in the mini-Mu to the nearest SmaI site in the bacterial chromosome, which is the same for the parental DNA and these derivatives. Furthermore, for the SmHR and TcR clones, the second hybridized DNA fragment was identical for all of the probes and corresponded to the SmaI-hydrolyzed, full-size pAH-mini-Mu(LER)-YK plasmid (Fig. S8).
On the basis of these experiments, the mini-Mu unit can be confidently concluded to transpose from the integrative SH plasmid into the C. glutamicum chromosome, mainly through the nick-join-replicative transposition pathway with the formation of a cointegrate. For a minor fraction (< 5%) of initially obtained integrants, we could not determine which of the two transposition pathways led to clone formation, reparative, which results in simple insertion, or replicative, which is accompanied by fast cointegrate resolution via general recombination.
According to the literature (Harshey 1983), MuA has no resolvase activity that could facilitate cointegrate resolution prior to finalizing the replication of the mini-Mu unit during the nick-join-replicative transposition process. So, cointegrate resolution is dependent only on host general recombinogenic activity, mainly on the activity of the recA gene product (Fitzpatrick et al. 1994). Thus, a RecA− mutant of the C. glutamicum ATCC13869 strain was used as the recipient for the Mu-driven transposition of pAH-mini-Mu(LER)-YK, according to the standard protocol. The total efficiency of KmR transformant formation in this experiment was (0.5 ± 0.2) × 10−4 (KmR clones/100 ng plasmid DNA/surviving cells). The same with RecA+ isogenic recipient strain, approximately 97–98% of the obtained KmR transformants manifested SmHR and TсR phenotypes, and the residual 2–3% were SmR and TсS for RecA− strain. In contraposition to the SmHR and TсR cointegrates obtained in the Rec+ background, the phenotype of their Rec− analogs was significantly more stable; at a minimum, SmR and TcS resolvants could not be detected under standard conditions (after 5–8 generations grown in non-selective conditions). Thus, the initial integrants that possessed SmR and TcS phenotypes in both the RecA+ and Rec− strains were obtained mainly through nick-join-reparative transposition of the mini-Mu unit from the SH integrative plasmid into the C. glutamicum chromosome.
Amplification of the mini-Mu(LER)-YK unit in the C. glutamicum chromosome
The data presented in Fig. 3 helped identify the nature of clone no. 10, which had stable SmHR and TcS phenotypes. The chromosome of clone no. 10 contained two copies of the KmR gene that were likely obtained due to either (i) two independent mini-Mu(LER)-YK unit simple insertions resulting from reparative transposition from two integrative plasmids transformed into one recipient cell; (ii) two cointegrate resolutions obtained after consequent replicative transposition of mini-Mu units from two integrative plasmids into the chromosome; or, most likely, (iii) amplification due to intramolecular replicative transposition of the initially integrated mini-Mu(LER)-YK unit during growth of the bacterial cell with induced MuAB expression. In the latter case, the origin of the first integrated mini-Mu unit (through either reparative or replicative transposition coupled to cointegrate resolution) is not essential for the final conclusion of intrachromosomal mini-Mu unit amplification.
Each occurrence of intramolecular nick-join-replicative Mu-driven transposition is known (Craig 1996; Watson et al. 2004) to lead to (i) mini-Mu unit amplification, causing chromosomal inversion separated by inversely repeated mini-Mu units or (ii) deletion of non-replicative chromosomal DNA fragments due to the formation of two circular products, each carrying one copy of the mini-Mu unit: the first is capable of autonomous replication, while the second involves non-replicated parts of the bacterial DNA. Furthermore, if the circular DNA formed in the second instance consists of any essential gene(s) in a non-replicated part of bacterial DNA, only the fused circular DNA that results from the general recombination process between mini-Mu units in these two transposition products can be detected in the surviving clones possessing two directly repeated copies of mini-Mu units in their circular bacterial chromosomes (Fig. S9). Thus, the nature of this two-copied integrant (clone no. 10) could possibly be determined by investigating mini-Mu unit integration points. If two copies of the inversely repeated Mu units are located in random (independent) points of the chromosome, then two independent acts of mini-Mu unit integration must have occurred. However, inverse fragments of the C. glutamicum chromosome between two copies of inversely repeated mini-Mu units would unambiguously correspond to intrachromosomal Mu unit amplification. Directly repeated mini-Mu units in the chromosome could result either from two independent acts of unit integration or from intrachromosomal amplification of an initially integrated mini-Mu unit followed by fusion of two circular transposition products due to intermolecular general recombination between mini-Mu units.
Molecular cloning of chromosomal DNA fragment carrying the integrated mini-Mu unit was performed for clone no. 10 (“Materials and methods”). Among the plasmid DNA purified from three independently obtained KmR E. coli transformants, we detected a plasmid with only one of two mini-Mu unit copies in a StuI-hydrolyzed DNA fragment of approximately 11.2 kb that was bracketed by C. glutamicum DNA. Sequence analysis indicated that the host bordering DNA of the cloned mini-Mu unit corresponded to an inverted C. glutamicum genome structure. Additional confirmation of the estimated locations of both of the mini-Mu units was obtained via the application of a previously developed strategy (Zimenkov et al. 2004) based on inverse PCR with divergently oriented primers that correspond to the internal part of the Mu-R (or Mu-L) ends (data not shown). In addition, the locations of the host DNA that were linked by the two mini-Mu units were determined to be 484,726 and 2,370,010 bp according to the sequence of the C. glutamicum ATCC 13869 genome (GenBank AN AP017557.2). The detected structure of the cloned StuI-fragment (Fig. S10) could only be obtained by the intramolecular nick-join-replicative Mu-driven amplification of an initially integrated mini-Mu unit via cointegrate formation, with inversion of the C. glutamicum ATCC13869 genome.
To provide artificial amplification, a C. glutamicum ATCC13869 strain carrying a single copy of the mini-Mu(LER)-YK unit in its chromosome was electrotransformed with the integration helper plasmid, followed by aerobic cultivation of a single transformant at 37 °C overnight in liquid BHI medium in the presence of Gm and IPTG with the final selection of SmHR variants. SmHR clones were detected at a frequency ≈ 5.0 × 10−4/cells, with several tens to hundreds of clones obtained in total (~ 105 SmR and GmR cells). Notably, the detected efficiency of intrachromosomal amplification was three orders of magnitude lower than the efficiency of intracellular cointegrate formation of the already penetrated SH integrative mini-Mu(LER) unit-carrier plasmid with the bacterial chromosome of the C. glutamicum ATCC13869 strain (note the total frequency of cointegrant formation was ≈ 1.6 × 10−4, and the SH plasmid had to penetrate the cell (frequency with ≈ 1 × 10−3) before initiation of transpososome formation).
In the final stage, the helper plasmid was cured from the obtained SmHR cells. Sets of SmHR clones were subjected to yECitrine-originated fluorescence analysis (“Materials and methods”). The fluorescence data and Southern hybridization results of the corresponding chromosomal DNA with the structural part of the KmR gene from the mini-Mu(LER)-YK unit labeled are presented in Fig. 4A and B, respectively. The results confirmed that all of the tested clones were obtained via intramolecular Mu-driven replicative amplification of an initially integrated mini-Mu unit and ultimately contained up to two or three copies per chromosome.

yECitrine relative fluorescence intensity (A) and Southern blot analysis (B) of clones selected as SmHR after mini-Mu(LER)-YK amplification (1–10) and parental clones with an initial single copy of the mini-Mu unit (11). For the Southern blot analysis, genomic DNA was isolated, digested with SmaI (which has a recognition site located in the non-kan part of the mini-Mu unit DNA) and hybridized with a kan-carrying DNA fragment amplified by PCR. Averages of two experiments are shown and in all cases standard deviation (SD) does not exceed 15%
DNA superhelicity and the E element influence the efficiency of mini-Mu unit transposition from the integrative plasmid into the C. glutamicum chromosome
Three integrative plasmids, containing mini-Mu(LER)-YK, mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK, and mini-Mu(LR)-YK, were used to investigate the influences of plasmid superhelicity and the presence/location of the E element on the efficiency of Mu-driven transposition in the C. glutamicum chromosome. All plasmids were examined in both SH and covalently closed but relaxed forms (“Materials and methods”). The best integration efficiency was detected with SH plasmid DNA carrying the mini-Mu(LER) unit (Table 2). Relaxation of this plasmid decreased the efficiency of its transposition 1000-fold; nevertheless, this plasmid was still the best donor for transposition among the other relaxed plasmids. The plasmids with the mini-Mu(LR) unit demonstrated the lowest integration efficiency: for the SH plasmid, the transposition level was approximately 20-fold lower than that of the mini-Mu(LER) unit-carrying SH plasmid. Again, the relaxed form of the plasmid with the mini-Mu(LR) unit showed a 1000-fold decrease compared to its SH form. The plasmids with the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit manifested intermediate levels of integration efficiency. In its relaxed form, this plasmid had threefold lower integration efficiency than the relaxed plasmid with the mini-Mu(LER) unit, and it had approximately tenfold higher efficiency than the relaxed plasmid without the E element. At the same time, the plasmid with the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit in its SH form had an integration efficiency closer to its analog with mini-Mu(LR) than to others with mini-Mu(LER) units.
Mu-driven intrachromosomal amplification of different mini-Mu units in C. glutamicum
Evaluating Mu-driven intrachromosomal amplification efficiency for mini-Mu units with converse orientations and/or the absence of an E element was interesting. For this, each of three different integrative plasmids in their SH forms was initially used to obtain corresponding resolved cоintegrates that had not lost the integration helper plasmid, pVK-lacIQ-P tac -MuAB. For each strain, a single clone was grown in liquid medium with induced expression of MuAB, and derivatives were selected at a high concentration of Sm. The efficiency of mini-Mu(LER) unit amplification was 4.0 × 10−3/cell (≈ 400 SmHR clones per 105 SmR clones plated in total), which fully coincides with previous results. Approximately fivefold fewer SmHR clones were detected in a strain with mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK, and only a few SmHR clones were detected in a strain with mini-Mu(LR)-YK unit. Additionally, Southern hybridization confirmed the amplification of up to two to three copies of the KmR gene in the chromosomes of SmHR clones with the mini-Mu(LER)-YK unit as well as several copies in SmHR clones with the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK unit, but only one copy of the tested marker was maintained in the few SmHR clones with mini-Mu(LR)-YK (Fig. 5). Thus, an inverse orientation of the E element, \( \overleftarrow{\mathbf{E}} \), can be concluded to result in a decreased amplification frequency of the corresponding mini-Mu units of approximately 6.0 × 10−4/cell. The absence of the E element in the mini-Mu(LR) unit structure decreased the frequency of the Mu-driven intramolecular replicative transposition to a level that was below 10−5, and amplification was not detected in the experiments with this type of unit.

Southern blot analysis of the parental strain (1) and clones with a single integrated copy of a mini-Mu(LER)-YK unit (2), a mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \))-YK unit (10), and a mini-Mu(LR)-YK unit (19) as well as their independent derivatives obtained after growth with MuAB expression (3–9), (11–18), and (20–22), respectively. For the Southern blot analysis, genomic DNA was digested with SmaI and hybridized with a kan-carrying DNA fragment amplified by PCR
Consecutive independent integration/amplification/fixation of different mini-Mu(LER) units in the C. glutamicum chromosome
Earlier, an integration/amplification/fixation strategy for Mu-driven transposition of different mini-Mu(LER) units with excisable E elements was proposed on the basis of differences in the intrachromosomal transposition efficiencies of the mini-Mu(LER) and mini-Mu(LR) units detected in gram-negative bacteria (Akhverdyan et al. 2011). Data presented in the previous section serves as a background for testing the same strategy in C. glutamicum. One single-copy mini-Mu(LER)-YK-integrant of the C. glutamicum ATCC13869 strain (named 1YK) and two of its progeny obtained via Mu-driven amplification of this unit to two and three copies per chromosome (named 2YK and 3YK, respectively) were cured of the integration helper plasmid pVK-lacIQ-P tac -MuAB. The internal parts of the intrachromosomal mini-Mu unit(s) bracketed by lox66/lox71 sites and the included E element as well as the KmR and SmR markers (Fig. 1b) were excised by the Cre recombinase (“Materials and methods”), followed by the elimination of the excision helper plasmid p06-P dapA -cre at the final stage of the experiment.
Targeted excision of the internal parts of the mini-Mu units was confirmed via the KmS phenotype of the obtained strains. yECitrine-originated fluorescence was quantitatively evaluated for three parental mini-Mu(LER)-YK integrants, 1YK, 2YK, and 3YK, and for their KmS derivatives with mini-Mu(LR)-Y type units (named 1Y, 2Y, and 3Y, respectively). The fluorescence data and subsequent Southern hybridization results probed with the structural part of the yECitrine gene confirmed the maintenance of the expected copy number of the truncated mini-Mu(LR)-like unit(s) after Cre-mediated chromosomal editing for all of the obtained strains (data not shown).
These three strains (iY, where i = 1, 2, 3, indicating the number of the mini-Mu(LR)-Y units in the chromosome) were used for new Mu-dependent integration followed by amplification using the mini-Mu-(LER)-GK unit, which differs from the previously used mini-Mu-(LER)-YK in that it contains the yEGFP gene rather than the yECitrine gene. All of the procedures used for Mu-driven nick-join-replicative transposition were performed according to the designed and described protocols above. At both transposition stages, after integration and amplification, the obtained strains were evaluated using both yECitrine and yEGFP-originated fluorescence as well as Southern hybridization with the structural parts of the yECitrine and yEGFP genes as markers (Fig. 6).

yECitrine and yEGFP relative fluorescence intensity (A) and Southern blot analysis (B) (SphI restricted genomic DNA) using yECitrine or yEGFP as probes of a parental strain with a single mini-Mu(LR)-Y unit, 1Y (1); a derivative of the 1Y clone with an introduced single mini-Mu(LER)-GK unit (2) and its derivatives with amplified mini-Mu(LER)-GK units (3–9); a parental strain with two mini-Mu(LR)-Y units, 2Y (10); a derivative of the 2Y clone with an introduced single mini-Mu(LER)-GK unit (11) and its derivative clones with amplified mini-Mu(LER)-GK units (12–21); a parental strain with three mini-Mu(LR)-Y units, 3Y (22, no Southern blot data); and a derivative of the 3Y clone with an introduced single mini-Mu(LER)-GK unit (30) and its derivative clones with amplified mini-Mu(LER)-GK units (23–29). SphI has a unique recognition site in the mini-Mu unit structure that is beyond the yECitrine or yEGFP genes; the resulting step-by-step intramolecular amplification position of the hybridized bands in the Southern blots is retained in subsequent steps. Averages of three experiments are shown on graphs and in all cases SD do not exceed 15%
All of the data confirmed that integration followed by amplification leading to one to three copies of the mini-Mu(LER)-GK unit in the chromosomes of all three recipients used was successfully realized, and a set of double C. glutamicum integrants iY-jGK (i, j = 1, 2, 3, indicating the number of yECitrine- and yEGFP-gene carrier units in the chromosome, respectively) was obtained. Moreover, the chromosomal positions of the mini-Mu(LR)-Y units during Mu-driven intrachromosomal amplification of the mini-Mu(LER)-GK units were maintained (fixed).
Discussion
Mu-driven replicative transposition is a highly efficient, convenient method for recombinant DNA integration and amplification in plasmid-less industrial strains that are based on gram-negative bacteria (Akhverdyan et al. 2011). This method is especially relevant and useful for organisms without developed powerful and comprehensive genetic tools for chromosomal editing. Considering the possible inclusion of a Mu-based integration/amplification strategy in a set of genetic tools for gram-positive microorganisms of industrial interest, in particular, we decided to modify a previously developed dual-component plasmid system (Akhverdyan et al. 2011) for expression in different strains of Corynebacterium glutamicum.
In this study, the C. glutamicum ATCC13869 strain was tested for Mu-driven transposition. Replicative transposition of the mini-Mu(LER) unit through cointegrate formation was confirmed as the main pathway of mini-Mu unit integration (> 95%) into the bacterial chromosome from the SH integrative plasmid. It was interesting to note that efficiency of initial transposition and intrachromosomal mini-Mu(LER) unit amplification were significantly increased if C. glutamicum cells were grown at 37 °C before inducing MuAB transposition factors as described in “Materials and methods.” Probably, this effect could be based on the possible synthesis of some “heat-shock proteins” and chaperones facilitated the transposition.
As shown from the results obtained for the isogenic C. glutamicum ATCC13869-(recA−) strain, a minor fraction of the recA+ strain integrants (< 5%) was likely obtained via nick-join-reparative transposition. Whether host proteins participate in this pathway of transposition in C. glutamicum is unknown; however, direct analogs of the E. coli RecBCD nuclease that collaborates with the transpososome in the repair of simple Mu insertions (Choi et al. 2014) are absent in this bacterium (Nakamura et al. 2003).
Both E. coli Mu-mediated reparative and replicative transposition pathways are known to be catalyzed by a higher order DNA protein complex called the transpososome, which is organized by bridging interactions among three DNA sites, the L/R ends of Mu and the E element. This complex is mediated by six subunits of the MuA transposase and assisted by the host proteins HU and IHF to form an LER synapse (Harshey 2014). The presence of the E element in the native transpososome LER structure increases the efficiency of transposition over two orders of magnitude in vivo (Leung et al. 1989) and accelerates the initial rate of transposition in vitro by a similar amount (Surette et al. 1989). In E. coli, the site-specific IHF-mediated bending of DNA at the E element located in the cis orientation with respect to the L/R ends facilitates efficient transposition of the mini-Mu(LER) unit, especially when the SH density (σ) of the transposed DNA becomes low (Surette and Chaconas 1989). To our knowledge, the presence of functionally active DNA-binding analogs of the E. coli IHF and HU proteins have not been reliably identified among corynebacterial proteins; however, a gene encoding the putative integration host protein cIHF (GenBank accession number: CG1811 (CorglutaCyc)) was annotated in the C. glutamicum ATCC 13032 genome (Kalinowski et al. 2003).
In the present study, the dependence of Mu-driven transposition efficiency in C. glutamicum on the presence of the E element in mini-Mu units and on the superhelicity of integrative plasmids was tested. A 20-fold difference in transposition efficiency was observed for the SH integrative plasmids, and the mini-Mu(LER) unit with properly located E element had the highest yield of transformants (Table 3) compared to the mini-Mu(LR) unit-carrying SH plasmid without E, which demonstrated the lowest yield. The plasmid with the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit had a transposition efficiency tenfold lower than that of the mini-Mu(LER) unit and only twofold higher than that of the mini-Mu(LR) unit. For rather small integrative plasmids, due to the close spatial location of the Mu-L/R ends in the SH DNA structure, the process of minimal transpososome formation occurred rather efficiently even without E element facilitation. However, inversely located enhancer, \( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \), in the corresponding SH plasmid had insufficient structural freedom to significantly increase the efficiency of full-sized transpososome assembly.
Based on the data obtained in the present study, Mu-driven transposition from the relaxed form of the integrative plasmid could be a closed experimental model of intrachromosomal replicative amplification under conditions where DNA-binding proteins constrain supercoils in bacterial DNA and significantly decrease chromosomal SH density (Dillon and Dorman 2010). Indeed, the estimated electroporation/penetration efficiency of the C. glutamicum ATCC 13869 strain with SH plasmid DNA was ≈ 1 × 10−3/100 ng DNA/surviving cells. At the same time, the efficiency of transposition (penetration + cointegrate formation) of the mini-Mu(LER) unit-carrying relaxed plasmid into the same strain was ≈ 9 × 10−7/100 ng DNA/surviving cells. Thus, the efficiency of intracellular cointegrate formation between the relaxed integrative plasmid and the C. glutamicum chromosome was estimated to be approx. 9 × 10−4/cell. This estimation is very close to the experimentally detected efficiency of mini-Mu(LER) unit intrachromosomal amplification, which was ≈ 5 × 10−4/cell. This assumption was additionally confirmed by the detection of threefold decreases in the transposition efficiency of relaxed integrative plasmids with the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit compared to the analogous mini-Mu(LER) unit-carrying plasmid, which correlated well with the twofold difference in the intramolecular amplification level detected for the corresponding mini-Mu units.
The results obtained for the mini-Mu(LR) unit were rather interesting and not easily predicted. Cointegrate formation between the relaxed integrative plasmid with the mini-Mu(LR) unit and the C. glutamicum chromosome occurred with a tenfold lower transposition efficiency than the mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) unit-carrying relaxed plasmid. At the same time, the difference in the frequencies of intramolecular amplification of the mini-Mu(LR) and mini-Mu(\( \mathbf{L}\overleftarrow{\mathbf{E}}\mathbf{R} \)) units was significantly higher, and this amplification could not be detected for the mini-Mu(LR) unit under the experimental conditions. This formation of the transpososome structure resulted in Mu end pairing without participation of the E element, which presented no significant difficulties for the relaxed integrative plasmid substrate, but was a serious problem when the constrained bacterial chromosome served as the substrate. Notably, the centrally located strong gyrase binding site (SGS) is required for efficient synapsis and formation of the transpososome, which is the obligatory first step for the initiation of Mu DNA replication in the whole Mu prophage genome, even though it carries the native E enhancer element. DNA gyrase bound at the SGS site allows the rapid, efficient synapsis of Mu prophage L/R ends within the constraints imposed by the structure of the bacterial nucleoid, doing so by promoting the formation of a supercoiled loop, with the apex site and prophage ends synapsed at the base of the loop (Pato 2004; Pato and Banerjee 2000).
The dramatic decrease in the intramolecular transposition efficiency of mini-Mu(LR) units compared to the mini-Mu(LER) units located in the restrained protein-bound DNA of the C. glutamicum chromosome allowed the application of a genome modification strategy previously developed for the Methylophilus methylotrophus AS-1 (Akhverdyan et al. 2011) strain. A consecutive independent integration/amplification/fixation process via excision of the E element in different mini-Mu(LER) units in the C. glutamicum chromosome was successfully demonstrated in the present study.
Note that application of the pVK9-lacIQ-P tac -MuAB helper plasmid resulted in amplification of the initially transposed mini-Mu(LER) unit up to two to three copies. At the same time, using the analogous helper plasmid with decreased expression level of MuAB due to their transcription with “weaker” promoter led to only one additional copy of mini-Mu (data not shown). So, amplification events could be controlled by the expression level of MuAB.
Additionally, Mu-driven replicative integration with the formation of cointegrate structure, followed by its resolution, was demonstrated using the same two-plasmid system in the widely used, wild-type lab strain of C. glutamicum, ATCC13032, and its prophage-free derivative MB001, which has also applications in biotechnology (Baumgart et al. 2013). The efficiency of initial Mu-driven integration (KmR clones/100 ng SH plasmid DNA/surviving cells) provided by optimized electrotransformation conditions for each strain was ≈ 10−7 for ATCC13032 (≈ 200 KmR integrants/100 ng plasmid DNA/2.5 × 109 surviving cells) and 10−6 for MB001.
To widely use this confirmed Mu-mediated transposition system as a useful tool for chromosomal editing in C. glutamicum, the more convenient mini-Mu(LER)-type plasmid, pAH-mini-Mu(LER)-YS (Fig. 7), was designed as a potential vector for cloning genes of interest. Application of the dependable KmR marker, located in the non-Mu DNA part of this plasmid, is more convenient for the selection of cointegrates than the TcR marker. However, the expressed Bacillus subtilis sacB gene can be used as a counter-selective marker and facilitates the detection of C. glutamicum resolvants in sucrose-containing media (Jäger et al. 1992). Application selection on 20% sucrose-containing medium resulted in 15–30% of colonies with resolved structure among all colonies against 2–3% in case of pAH-miniMu(LER)-YK. The SmR marker located in the mini-Mu unit of this new plasmid can be used for the direct selection of integrants, followed by the possible selective intrachromosomal amplification of the integrated (LER)-like unit. Moreover, the presence of the yECitrine gene can help to semi-quantitatively estimate the obtained mini-Mu unit copy number in the selected SmHR clones by fluorescence. All of the technical facilitating genes (E element, SmR, and yECitrine) can be excised from the integrated mini-Mu units in a Cre-dependent fashion due to the proper location of lox66/71 sites, and only the small markerless part of the vector plasmid containing the gene(s) of interest cloned into its multiple cloning site (MCS) is retained. This plasmid could be used as an integrative vector for cloning, followed by Mu-driven transposition in the chromosomes of different organisms and in C. glutamicum, in particular. Up to date, the maximal 8-kb target DNA fragments were successfully inserted and amplified in our lab via pAH-mini-Mu(LER)-YS–like vector.

Scheme of the new integrative plasmid vector pAH-mini-Mu(LER)-YS (GenBank accession no. MG014200)
References
Abalakina EG, Tokmakova IL, Gorshkova NV, Gak ER, Akhverdyan VZ, Mashko SV, Yomantas YAV (2008) Phage Mu-driven two-plasmid system for integration of recombinant DNA in the Methylophilus methylotrphus genome. Appl Microbiol Biotechnol 81(1):191–200. https://doi.org/10.1007/s00253-008-1696-7
Akhverdyan VZ, Gak ER, Tokmakova IL, Stoynova NV, Yomantas YAV, Mashko SV (2011) Application of the bacteriophage Mu-driven system for the integration/amplification of target genes in the chromosomes of engineered Gram-negative bacteria. Appl Microbiol Biotechnol 91(4):857–871. https://doi.org/10.1007/s00253-011-3416-y
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7(4):649–659. https://doi.org/10.1046/j.1365-313X.1995.7040649.x
Au TK, Pathania S, Harshey RM (2004) True reversal of Mu integration. EMBO J 23(16):3408–3420. https://doi.org/10.1038/sj.emboj.7600344
Au TK, Agrawal P, Harshey RM (2006) Chromosomal integration mechanism of infecting Mu virion DNA. J Bacteriol 188(5):1829–1834. https://doi.org/10.1128/JB.188.5.1829-1834.2006
Backman K, Betlach M, Boyer HW, Yanofsky S (1979) Genetic and physical studies on the replication of ColE1-type plasmids. Cold Spring Harb Symp Quant Biol 43(0):69–76. https://doi.org/10.1101/SQB.1979.043.01.012
Bartek T, Zönnchen E, Klein B, Gerstmeir R, Makus P, Lang S, Oldiges M (2010) Analysing overexpression of l-valine biosynthesis genes in pyruvate-dehydrogenase deficient Corynebacterium glutamicum. J Ind Microbiol Biotechnol 37(3):263–270. https://doi.org/10.1007/s10295-009-0669-x
Baumgart M, Unthan S, Rückert C, Sivalingam J, Grünberger A, Kalinowski J, Bott M, Noack S, Frunzke J (2013) Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol 79(19):6006–6015. https://doi.org/10.1128/AEM.01634-13
Becker J, Wittmann C (2012) Bio-based production of chemicals, materials and fuels – Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23(4):631–640. https://doi.org/10.1016/j.copbio.2011.11.012
Billman-Jacobe H, Hodgfon ALM, Lightowlers AIM, Wood PR, Radford AJ (1994) Expression of ovine gamma interferon in Escherichia coli and Corynebacterium glutamicum. Appl Environ Microbiol 60:1641–1645
Binder S, Siedler S, Marienhagen J, Bott M, Eggeling L (2013) Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res 41(12):6360–6369. https://doi.org/10.1093/nar/gkt312.
Borujeni AE, Salis HM (2016) Translation initiation is controlled by RNA folding kinetics via a ribosome drafting mechanism. J Am Chem Soc 138(22):7016–7023. https://doi.org/10.1021/jacs.6b01453
Castilho BA, Olfson P, Casadaban MJ (1984) Plasmid insertion mutagenesis and lac gene fusion with mini-Mu bacteriophage transposons. J Bacteriol 158:488–495
Chaconas G, deBruijn FG, Casadaban M, Lupski JR, Kwok TJ, Harshey RM, DuBow MS, Bukhari AI (1981a) In vitro and in vivo manipulations of bacteriophage Mu DNA: cloning of Mu ends and construction of mini-Mu’s carrying selectable markers. Gene 13(1):37–46. https://doi.org/10.1016/0378-1119(81)90041-X
Chaconas G, Harshey RM, Sarvetnick N, Bukhari AI (1981b) Predominant end-products of prophage Mu DNA transposition during the lytic cycle are replicon fusions. J Mol Biol 150(3):341–359. https://doi.org/10.1016/0022-2836(81)90551-9
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167. https://doi.org/10.1016/j.ymben.2017.06.010
Choi W, Jang S, Harshey RM (2014) Mu transpososome and RecBCD nuclease collaborate in the repair of simple Mu insertions. Proc Natl Acad Sci U S A 111(39):14112–14117. https://doi.org/10.1073/pnas.1407562111
Court DL, Sawitzke JA, Thomason LC (2002) Genetic engineering using homologous recombination. Annu Rev Genet 36(1):361–388. https://doi.org/10.1146/annurev.genet.36.061102.093104
Craig NL (1996) Transposition. In: Neidhardt FC (ed) Escherichia coli and Salmonella: cellular and molecular biology, 2nd edn. ASM, Washington, DC pp
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K12 using PCR products. Proc Natl Acad Sci U S A 97(12):6640–6645. https://doi.org/10.1073/pnas.120163297
Dillon SC, Dorman CJ (2010) Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol 8(3):185–195. https://doi.org/10.1038/nrmicro2261
Eggeling L, Bott M (2005) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton. https://doi.org/10.1201/9781420039696
Fitzpatrick R, O’Donohue M, Joy J, Heery DM, Dunican LK (1994) Construction and characterization of recA mutant strains of Corynebacterium glutamicum and Brevibacterium lactofermentum. Appl Microbiol Biotechnol 42(4):575–580. https://doi.org/10.1007/BF00173923
Glanemann C, Loos A, Gorret N, Willis L, O’Brien XM, Lesard P, Sinskey AJ (2003) Disparity between changes in mRNA abundance and enzyme activity in Corynebacterium glutamicum: implications for DNA microarray analysis. Appl Microbiol Biotechnol 61(1):61–68. https://doi.org/10.1007/s00253-002-1191-5
Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183(21):6384–6393. https://doi.org/10.1128/JB.183.21.6384-6393.2001
Harshey RM (1983) Switch in the transposition products of Mu DNA mediated by proteins: cointegrates versus simple insertions. Proc Natl Acad Sci U S A 80(7):2012–2016. https://doi.org/10.1073/pnas.80.7.2012
Harshey RM (2012) The Mu story: how a maverick phage moved the field forward. Mob DNA 3(1):21. https://doi.org/10.1186/1759-8753-3-21
Harshey RM (2014) Transposable phage Mu. Microbiol Spectr 2(5). https://doi.org/10.1128/microbiolspec.MDNA3-0007-2014
Hermann T, Pfefferle W, Baumann C, Busker E, Schaffer S, Bott M, Sahm H, Dusch N, Kalinowski J, Pühler A, Bendt AK, Krämer R, Burkovski A (2001) Proteome analysis of Corynebacterium glutamicum. Electrophoresis 22(9):1712–1723. https://doi.org/10.1002/15222683(200105)22:9<1712::AID-ELPS1712>3.0.CO;2-G
Hillen W, Berens C (1994) Mechanisms underlying expression of Tn10 encoded tetracycline resistance. Annu Rev Microbiol 48(1):345–369. https://doi.org/10.1146/annurev.mi.48.100194.002021
Huang Y, Li L, Zhao N, Han S, Lin Y, Zheng S (2017) Recombineering using ResET in Corynebacterium glutamicum ATCC14067 via a self-excisable cassette. Sci Rep 7(1):7916. https://doi.org/10.1038/s41598-017-08352-9.
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62(2-3):99–109. https://doi.org/10.1007/s00253-003-1328-1
Inui M, Suda M, Okino S, Nonaka H, Puskas LG, Vertes AA, Yukawa H (2007) Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 153(8):2491–2504. https://doi.org/10.1099/mic.0.2006/005587-0
Ishikawa K, Toda-Murakoshi Y, Ohnishi F, Kondo K, Osumi T, Asano K (2008) Medium composition suitable for L-lysine production by Methylophilus methylotrophus in fed-batch cultivation. J Biosci Bioeng 106(6):574–579 https://doi.org/10.1263/jbb.106.574
Jäger W, Schäfer A, Pühler A, Labes G, Wohlleben W (1992) Expression of the Bacillus subtilis sacB gene leads to sucrose sensitivity in the gram-positive bacterium Corynebacterium glutamicum but not in Streptomyces lividans. J Bacteriol 174(16):5462–5465. https://doi.org/10.1128/jb.174.16.5462-5465.1992
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, Wang R, Yang S (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179. https://doi.org/10.1038/ncomms15179
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J Biotechnol 104(1-3):5–25. https://doi.org/10.1016/S01681656(03)00154-8
Katashkina JI, Hara Y, Golubeva LI, Andreeva IG, Kuvaeva TM, Mashko SV (2009) Use of the lambda Red-recombineering method for genetic engineering of Pantoea ananatis. BMC Mol Biol 10(1):34. https://doi.org/10.1186/1471-2199-10-34
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104(1-3):287–299. https://doi.org/10.1016/S01681656(03)00148-2
Kjeldsen KR, Nielsen J (2009) In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol Bioeng 102(2):583–597. https://doi.org/10.1002/bit.22067
Krylov AA, Kolontaevsky EE, Mashko SV (2014) Oligonucleotide recombination in corynebacteria without the expression of exogeneous recombinases. J Microbiol Methods 105:109–115. https://doi.org/10.1016/j.mimet.2014.07.028
Lamberg A, Nieminen S, Qiao M, Savilahti H (2002) Efficient insertion mutagenesis strategy for bacterial genomes involving of in vitro-assempled DNA transposition complexes of bacteriophage Mu. Appl Environ Microbiol 68(2):705–712. https://doi.org/10.1128/AEM.68.2.705712.2002
Lanckriet A, Timbermont L, Happonen LJ, Pajunen MI, Pasmans F, Haesebrouck F, Ducatelle R, Savilahti H, Van Immerseel F (2009) Generation of single-copy transposon insertions in Clostridium perfringens by electroporation of phage Mu DNA transposition complexes. Appl Environ Microbiol 75(9):2638–2642. https://doi.org/10.1128/AEM.02214-08
Lawley TD, Burland V, Taylor DE (2000) Analysis of the complete nucleotide sequence of the tetracylcline-resistance transposon Tn10. Plasmid 43(3):235–239. https://doi.org/10.1006/plas.1999.1458
Leung PC, Teplow DB, Harshey R (1989) Interaction of distinct domains in Mu transposase with Mu DNA ends and an internal transpositional enhancer. Nature 338(6217):656–658. https://doi.org/10.1038/338656a0
Li L, Wada M, Yokota A (2007) Cytoplasmic proteome reference map for a glutamic acid-producing Corynebacterium glutamicum ATCC 14067. Proteomics 7(23):4317–4322. https://doi.org/10.1002/pmic.200700269
Ma W, Wang X, Mao Y, Wang Z, Chen T, Zhao X (2015) Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter-selection marker. Biotechnol Lett 37(3):609–617. https://doi.org/10.1007/s10529-014-1718-8
Marx A, de Graaf AA, Wiechert W, Eggeling L, Sahm H (1996) Determination of the fluxes in central metabolism of Corynebacterium glutamicum by NMR spectroscopy combined with metabolite balancing. Biotechnol Bioeng 49(2):111–129. https://doi.org/10.1002/(SICI)10970290(19960120)49:2<111::AID-BIT1>3.0.CO;2-T
Marx CJ, Lidstrom ME (2001) Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology 147(8):2065–2075 https://doi.org/10.1099/00221287-147-8-2065
Moreau S, Blanco C, Trautwetter A (1999) Site-specific integration of corynephage phi16: construction of an integration vector. Microbiology 145(3):539–548. https://doi.org/10.1099/13500872145-3-539
Nakamura Y, Nishio Y, Ikeo K, Gojobori T (2003) The genome stability in Corynebacterium species due to lack of the recombinational repair system. Gene 317(1-2):149–155. https://doi.org/10.1016/S0378-1119(03)00653-X
Nakamura J, Hirano S, Ito H (2006) l-glutamic acid producing microorganism and a method for producing l -glutamic acid. US patent US20060141588A1
Nešvera J, Pátek M (2008) Plasmids and promoters in corynebacteria and their applications. In: Burkovski A (ed) Corynebacteria. Genomics and molecular biology. Caister, Academic Press, Norfolk, pp 113−154
Nešvera J, Pátek M (2011) Tools for genetic manipulations in Corynebacterium glutamicum and their applications. Appl Microbiol Biotechnol 90(5):1641–1654. https://doi.org/10.1007/s00253-011-3272-9
Oram M, Woolston JE, Jacobson AD, Holmes RK, Oram DM (2007) Bacteriophage-based vectors for site-specific insertion of DNA in the chromosome of corynebacteria. Gene 391(1-2):53–62. https://doi.org/10.1016/j.gene.2006.12.003
Ozaki A, Katsumata R, Oka T, Furuya A (1984) Functional expression of the genes of Escherichia coli in gram-positive Corynebacterium glutamicum. Mol Gen Genet 196(1):175–178. https://doi.org/10.1007/BF00334113
Paatero AO, Turakainen H, Happonen LJ, Olsson C, Palomaki T, Pajunen MI, Meng X, Otonkoski T, Tuuri T, Berry C, Malani N, Frilander MJ, Bushman FD, Savilahti H (2008) Bacteriophage Mu integration in yeast and mammalian genomes. Nucleic Acids Res 36(22):e148. https://doi.org/10.1093/nar/gkn801
Pajunen MI, Pulliainen AT, Finne J, Savilahti H (2005) Generation of transposon insertion mutant libraries for Gram-positive bacteria by electroporation of phage Mu DNA transposition complexes. Microbiology 151(4):1209–1218. https://doi.org/10.1099/mic.0.27807-0
Pátek M (2005) Regulation of gene expression. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Rato, pp 81–98. https://doi.org/10.1201/9781420039696.ch5
Pátek M, Eikmanns BJ, Pátek J, Sahm H (1996) Promoters from Corynebacterium glutamicum: cloning, molecular analysis and search for a consensus motif. Microbiology 142(5):1297–1309. https://doi.org/10.1099/13500872-142-5-1297
Pato ML (2004) Replication of Mu phage lacking the central strong gyrase site. Res Microbiol 155(7):553–558. https://doi.org/10.1016/j.resmic.2004.03.006
Pato ML, Banerjee M (2000) Genetic analysis of the strong gyrase site (SGS) of bacteriophage Mu: localization of determinants required for promoting Mu replication. Mol Microbiol 37(4):800–810. https://doi.org/10.1046/j.1365-2958.2000.02042.x
Posfai G, Koob M, Hradecna Z, Hasan N, Filutowicz M, Szybalski W (1994) In vivo excision and amplification of large segments of the Escherichia coli genome. Nucl Acids Res 22(12):2392–2398. https://doi.org/10.1093/nar/22.12.2392
Ravasi P, Peiru S, Gramajo H, Menzella HG (2012) Design and testing of a synthetic biology framework for genetic engineering of Corynebacterium glutamicum. Microb Cell Factories 11(1):147. https://doi.org/10.1186/1475-2859-11-147
Sambrook J, Russell DW (2001) Molecular cloning: laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sheff MA, Thorn KS (2004) Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21(8):661–670. https://doi.org/10.1002/yea.1130
Shinfuku Y, Sorpitiporn N, Sono M, Furusawa C, Hirasawa T, Shimizu H (2009) Development and experimental verification of a genome-scale metabolic model for Corynebacterium glutamicum. Microb Cell Factories 8(1):43. https://doi.org/10.1186/1475-2859-8-43
Surette MG, Chaconas G (1989) A protein factor which reduces the negative supercoiling requirement in the Mu DNA strand transfer reaction is Escherichia coli integration host factor. J Biol Chem 264(5):3028–3034
Surette MG, Lavoie BD, Chaconas G (1989) Action at a distance in Mu DNA transposition: an enhancer-like element is the site of action of supercoiling relief activity by integration host factor (IHF). EMBO J 8(11):3483–3489
Suzuki N, Okai N, Nonaka H, Tsuge Y, Inui M, Yukawa H (2006) High-throughput transposon mutagenesis of Corynebacterium glutamicum and construction of a single-gene disruptant mutant library. Appl Environ Microbiol 72(5):3750–3755. https://doi.org/10.1128/AEM.72.5.3750-3755.2006
Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S (2010) Recombineering using RecTE from Pseudomonas syringae. Appl Environ Microbiol 76(15):4960–4968. https://doi.org/10.1128/AEM.00911-10
Tauch A (2005) Native plasmids of amino acid-producing corynebacteria. In: Eggling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 57–80. https://doi.org/10.1201/9781420039696.ch4
Tsuge Y, Suzuki N, Inui M, Yukawa H (2007) Random segment deletion based on IS31831 and Cre/loxP excision system in Corynebacterium glutamicum. Appl Microbiol Biotechnol 74(6):1333–1341. https://doi.org/10.1007/s00253-006-0788-5
Vieira J, Messing J (1991) New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene 100:189–194. https://doi.org/10.1016/0378-1119(91)90365-I
Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004) Molecular biology of the gene, 5th edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 316–334
Wendisch VF (2003) Genome-wide expression analysis in Corynebacterium glutamicum using DNA microarray. J Biotechnol 104(1-3):273–285. https://doi.org/10.1016/S0168-1656(03)00147-0
Wittmann C, Heinzle E (2002) Genealogy profiling through strain improvement by using metabolic network analysis: metabolic flux genealogy of several generations of lysine-producing corynebacteria. Appl Environ Microbiol 68(12):5843–5859. https://doi.org/10.1128/AEM.68.12.5843-5859.2002
Woo HM, Noack S, Seibold GM, Willbold S, Eikmanns BJ, Bott M (2010) Link between phosphate starvation and glycogen metabolism in Corynebacterium glutamicum, revealed by metabolomics. Appl Environ Microbiol 76(20):6910–6919. https://doi.org/10.1128/AEM.01375-10
Xu D, Tan Y, Huan X, Hu X, Wang X (2010) Construction of a novel shuttle vector for use in Brevibacterium flavum, an industrial amino acid producer. J Microb Methods 80(1):86–92. https://doi.org/10.1016/j.mimet.2009.11.003
Zimenkov DV, Skorokhodova AY, Katashkina ZI, Minaeva NI, Savrasova EA, Biriukova IV, Doroshenko VG, Akhverdyan VZ, Mashko SV (2004) The regions in E. coli chromosome preferable for phage M-driven system of gene integration. Biotekhnologiya (Russian) No 6:3–18
Acknowledgements
The authors are extremely thankful to Drs. Y.A.V. Yomantas and E.R. Gak, who actively participated in broadening the host range of the dual-component Mu-driven transposition system and performed the first experiments with C. glutamicum. We are also grateful to Dr. E.G. Abalakina for her helpful advice and for the construction of the RecA− variant of the C. glutamicum ATCC13869 strain used in the present study. The authors also thank many other AGRI and Ajinomoto members, who, in addition to the above-named scientists, not only participated in helpful discussions related to the experiments and text of this manuscript but also exploited the developed integration/amplification system in their own investigations.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 1254 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gorshkova, N.V., Lobanova, J.S., Tokmakova, I.L. et al. Mu-driven transposition of recombinant mini-Mu unit DNA in the Corynebacterium glutamicum chromosome. Appl Microbiol Biotechnol 102, 2867–2884 (2018). https://doi.org/10.1007/s00253-018-8767-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8767-1