
Abstract
Background
The development of modern producer strains with metabolically engineered pathways poses special problems that often require manipulating many genes and expressing them individually at different levels or under separate regulatory controls. The construction of plasmid-less marker-less strains has many advantages for the further practical exploitation of these bacteria in industry. Such producer strains are usually constructed by sequential chromosome modifications including deletions and integration of genetic material. For these purposes complex methods based on in vitro and in vivo recombination processes have been developed.
Results
Here, we describe the new scheme of insertion of the foreign DNA for step-by-step construction of plasmid-less marker-less recombinant E. coli strains with chromosome structure designed in advance. This strategy, entitled as Dual-In/Out, based on the initial Red-driven insertion of artificial φ80-attB sites into desired points of the chromosome followed by two site-specific recombination processes: first, the φ80 system is used for integration of the recombinant DNA based on selective marker-carrier conditionally-replicated plasmid with φ80-attP-site, and second, the λ system is used for excision of inserted vector part, including the plasmid ori-replication and the marker, flanked by λ-attL/R-sites.
Conclusion
The developed Dual-In/Out strategy is a rather straightforward, but convenient combination of previously developed recombination methods: phages site-specific and general Red/ET-mediated. This new approach allows us to detail the design of future recombinant marker-less strains, carrying, in particular, rather large artificial insertions that could be difficult to introduce by usually used PCR-based Recombineering procedure. The developed strategy is simple and could be particularly useful for construction of strains for the biotechnological industry.
Similar content being viewed by others

Background
Escherichia coli is widely used in fundamental investigations and in modern biotechnology for production of biologically active compounds such as recombinant proteins, amino acids, vitamins etc. Construction of plasmid-less marker-less strains has advantages for extending the practical exploitation of these bacteria in industry [1–5]. Such producer strains are usually constructed by sequential chromosome modifications, mainly including deletions and integration of genetic material. For gene deletions, the Red/RecET recombination method developed by several groups [6–11] is considered as the most useful now. This method has been named Recombinogenic Engineering or Recombineering and reviewed in several papers [12–15]. It usually (but not always) based on λRed- or RecET-mediated recombination between bacterial chromosome and amplified DNA fragment carrying the removable selective marker, in which PCR primers provide the rather short homology to the targeted sequence. The integrated marker could be excised out of the chromosome by site-specific recombination. The Recombineering approach based on generated PCR products might be used not only for target genes disruption, but for integration of relatively short DNA fragments, for example, for substitution of recombinant regulatory regions (i.e. promoter, RBS) of a particular gene for its native regulatory region [16–19]. Although the special modifications of Recombineering procedure have been already developed for integration of large DNA fragments carrying several genes/operons (see, [20], for instance), more often special tools based on modified transposons [1, 21–23], non-replicative [3] or conditionally-replicative plasmids [4, 24–28] are using for the same purposes. Different transposons and phage Mu systems are exploited for introduction of the DNA cassettes into random points of the bacterial chromosome [1, 21, 29]. Integration into the native coliphages attB sites [30] or into artificially inserted recombinogenic sequences [3] is based on exploitation of corresponding site-specific recombination systems. By using cloned fragments of chromosomes as so-called "guides" [4] it is possible to integrate the cassette by general homologous recombination. In addition, combinations of different systems in one integration strategy are also used. For example, site-specific insertion of cassettes can be carried out in preliminary randomly integrated recombinogenic sites [3], or the "marked" cassettes can be randomly integrated, followed by excision of the marker by a site-specific recombination system [1].
For expansion of this group of methods, we propose a new strategy of DNA fragments integration in the process of plasmid-less marker-less recombinant E. coli strain construction. It initiates from Red-driven insertion of the antibiotic resistance marker flanked by φ80-attL/R sites, into the desired point of bacterial chromosome (this part of the total procedure is named as – the first "In") followed by φ80-Int/Xis-mediated excision of the marker (the first "Out) with retaining of φ80-attB-site. After that two sequential site-specific recombination processes are provided: first, the φ80-Int is used for integration (the second – "In") of the recombinant DNA constructed on the basis of conditionally-replicated plasmid with φ80-attP-site, and second, the λ-Int/Xis system [1, 10, 19] is used for excision of inserted vector part, including the plasmid ori-replication and the selective marker, that flanked by λ-attL/R-sites (the second – "Out"). So, the new strategy schematically presented in Fig. 1, could be named Dual-In/Out.

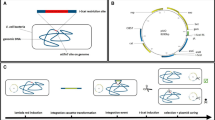
Dual In/Out method for plasmid-less marker-less strain construction. A: deletion of the native φ80-attB by Red recombination. B: Red integration of the φ80-removable marker to the desired loci of MG-Δ(φ80-attB) chromosome. C: curing of the marker by φ80-Int/Xis-system. D: φ80-driven integration of the CRIM plasmid with target cassette into the different of φ80-attB sites. E: construction of "marker-less" cassette-carrier-strains by λ-Int/Xis excision of the vector part of integrative plasmids.
Results
Construction of strains with different locations of the φ80-attBsite
The native φ80-attB-site is located at ~28 min of E. coli MG1655 chromosome (http://www.genetics.wisc.edu/ and http://mol.genes.nig.ac.jp/ecoli/). Insertion of the artificial φ80-attB into the chromosome of strain with deleted φ80-attB native site will allow us to introduce the DNA cassette into the new loci by φ80-Int-dependent system. If cassettes, integrated in the set of strains in different points, possess excisable selective markers, it will be possible to bring them in one strain by P1vir-mediated generalized transduction (P1-duction) due to removing the marker from the recipient genome before the next step of transduction.
The desired locations of φ80-attB sites can be spread in non-essential parts of bacterial genome at different distances from oriC. In this case, the level of expression of the same integrated cassette will depend on its position due to the θ-like structure of replicating chromosome and gene-dosage effect: the cassettes closer to oriC will be expressed at higher level than those located near the terminus of the chromosome replication [31].
We consider as good candidates for the further integration points the positions of the "native" IS elements (in our case IS5.7–IS5.11) since insertions into the genes already disrupted by IS elements will not be detrimental to cell viability and IS elements are almost randomly distributed along the chromosome of MG1655 [32]. Accordingly, the set of genes disrupted by IS5-elements insertion was chosen as the future locations of the artificial φ80-attB sites. These points are rather far from each other, so the future integrated cassettes can be combined in one strain by independent acts of P1-duction.
Realization of this strategy includes several steps. At first, the deletion of the native φ80-attB was carried out by Recombineering between E. coli MG1655 chromosome and the constructed "λ-excisable" CmR-marker amplified by PCR from pMW118-(λattL- CmR-λattR) [19]. After Red recombination, the DNA locus modification was verified by PCR, and followed by λ-Int/Xis-mediated excision of the marker from the chromosome. The marker-less strain, MG-Δ(φ80-attB), was used as the recipient for the insertion of the artificial φ80-attB. The latter includes:
1) construction and cloning of the cassette [(φ80-attL) - KmR - (φ80-attR)] in the plasmid;
2) using the obtained plasmid, pMWattphi, as the template for PCR amplification of φ80-removable KmR-marker flanked by the 36 bp arms homologous to the desired loci on the MG1655 chromosome;
3) Red integration of the markers into the chromosome of MG-Δ(φ80-attB) for construction of the desired library of "marked" strains;
4) each obtained strain was cured from the marker by φ80-Int/Xis-system [30]. In this way the "unmarked" part of the library was constructed;
5) new strains and MG1655 (as a control) were tested for φ80-driven integration/excision of the "conditionally-replicated integrative and modular (CRIM) plasmid" pAH162 carrying φ80-attP site [30], using φ80-Int- or φ80-Int/Xis-helper plasmids, respectively. Here, the efficiencies of integration/excision In or Out of the artificial points were practically the same as for In/Out of the native site.
Although the efficiency of CRIM plasmids excision from all tested sites in our experiments was 15–30% (not 100%, as reported in [30]), nevertheless, selection the marker-less clones was not the problem. We noticed, as well that, in our hands, the efficiency of λ-Int/Xis-driven excision under the same conditions was significantly higher and exceeded 80%.
Construction of φ80-integrative CRIM plasmid with λ-removable "vector part"
The φ80-cognate CRIM plasmids with different selective markers can be used for integration of cassettes in the obtained strains that differ in location of φ80-attB. A recombinant strain that contains multiple insertions can be constructed by P1 transduction. However the presence of plasmids' markers in the bacterial chromosome cannot satisfy "marker-less" criteria for the practical application of the engineered strain. CRIM plasmids constructed by Haldimann and Wanner [30], only allow the site-specific excision of the entire recombinant structure initially inserted in the chromosome. It is useful to modify CRIM plasmids to allow excising of the vector part after site-specific integration of recombinant DNA. In this way, the new φ80-cognate CRIM plasmid (Fig. 2) with a λ-removable vector part was obtained (for details see Methods).

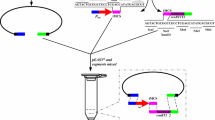
New φ80-cognate CRIM plasmids with λ-removable part. A. Map of pAH162-λattL-TcR-λattR. This plasmid could be used as a vector for molecular cloning of the genes of interest followed by φ80-Int-dependent integration of the recombinant plasmid in bacterial chromosome and λ-Int/Xis-dependent excision of the selective marker-carrier vector part. B. Map of pAH162-λattL-TcR-λattR-CmR. The recombinant plasmid constructed on the basis of new CRIM-vector, that contains cat-gene under the transcriptional control of phage T7 A2-promoter, as a model gene for integration in the chromosome according to Dual-In/Out strategy.
The new plasmid pAH162-λattL-TcR-λattR (Fig. 2) retains from its progenitor pAH162 the non-modified fragment carrying φ80-attP and the multi-cloning site flanked by bacterial (rgnB) and phage λ (tL3) transcription terminators. This fragment is bracketed by λ-attL/R sites which allow λ-Int/Xis excision of the vector part, including conditionally-replicative origin of R6K (oriRγ) and the selective marker, TcR (from Tn10). We have confirmed the expected properties of pAH162-λattL-TcR-λattR by its integration into the native φ80-attB of MG1655 chromosome, followed by λ-Int/Xis excision of the marked vector part flanked by λ-attL/attR. Of 80 tested clones cures from λ-Int/Xis-helper plasmid, more than 80% have lost the TcR marker of integrated CRIM plasmid, as well.
Exploiting the new system for multiple integration of cat-gene into E. colichromosome
To test the Dual-In/Out strategy of marker-less strain construction, we used the efficiently expressed variant of cat-gene as the model cassette. This experiment includes several steps:
1) cloning of cat-gene into pAH162-λattL-TcR-λattR, using E. coli CC118 (λpir+) strain as the recipient;
2) φ80-driven integration of the recombinant CRIM plasmid into the strains that differ in location of φ80-attB;
3) construction of "marker-less" strains with single cat-cassette by λ-Int/Xis excision of TcR-containing DNA fragment flanked by λ-attL/R-sites, followed by determination of cat expression levels in these strains;
4) combining the set of cat-cassettes in one "marker-less" strain by sequential P1-duction of the "marked" cassettes followed by curing the TcR-marker from the recipient strain before the next stage of transduction.
The previously constructed [19] pMW118-(λattL-CmR-λattR) plasmid was chosen as a template for PCR amplification of cat-gene. In this plasmid the structural part of cat-gene with its native RBS from E. coli Tn9 is under the transcriptional control of rather strong phage T7 A2 promoter [33] that is recognized by E. coli RNA polymerase with σ70 in a constitutive manner. This cat-gene was amplified by PCR with the primers that carry restriction sites for cloning in pAH162-λattL-TcR-λattR. The transformants of E. coli CC118 (λpir+) carrying the recombinant plasmid of interest, were selected on the medium supplemented with Cm. The expected structure of the plasmid was verified by restriction analysis and PCR.
At the next stage this plasmid was integrated by φ80-Int system into MG1655-derived strains with a different location of φ80-attB, using TcR-marker for selection (Fig. 3). The correct integration was confirmed by PCR, and the corresponding strains were entitled MG-TcR-cat-(i), where (i) varied from 1 to 6 depending on the number of φ80-attB sites. Subsequently, the vector part of the integrated plasmids in all MG-TcR-cat-(i) strains was excised by λ-Int/Xis site-specific recombination. After checking by PCR, the corresponding recombinant strains retaining the cat-gene in their chromosomes, was entitled MG-cat-(i).

Integration of plasmid pAH162-λ attL -Tc R -λ attR -Cm R into MG1655-derived strains with different location of φ80- attB .
The efficiency of cat-gene expression in MG-cat-(i)-strains was evaluated by determination of Cat enzymatic activity (Fig. 4, Table 1). As expected (see explanation above and reference [31]), the level of cat-gene expression correlates with the distance between the integration point and oriC: the strains with the cat-gene position closer to oriC have higher Cat activity.

The cat -gene expression in MG- cat -(i)-strains. The dependence of the expression level of the cat-carrier cassetes (T7A2-cat) on their positions in the chromosome is indicated by arrows. Cat-activity of the MG-cat-(6) strain in which Cassette (T7A2-cat) is located in native φ80-attB was taken as 100%.
Several cat-cassettes were combined in one strain by sequential P1-duction of Tc-marked fragments from MG-TcR-cat-(i) strains into MG-cat-(j) (where i ≠ j), followed by λ-Int/Xis-driven curing of TcR-marker that gives an MG-cat-(i + j) "marker-less" strain carrying two cat-cassettes in (i) and (j) positions; a third cassette was added by an analogous procedure. In addition, all steps used in increasing the number of cat-cassettes in the chromosome of recombinant "marker-less" strain were controlled by determination of Cat activity. The data presented in Table 1 show that the total Cat activity of multi-integrants correlates with the expected sum of the Cat activities of the corresponding single-integrants.
Discussion
In this paper we have described the development of a new strategy for the integration of genes/operons of interest during plasmid-less marker-less recombinant strain construction, which we have named Dual-In/Out. It combines Red-mediated insertion of the artificial φ80-attB site into desired point of bacterial genome, φ80-Int-dependent site-specific integration of recombinant DNA of interest constructed on the basis of specially constructed CRIM plasmid followed by λ-Int/Xis-mediated excision of the plasmid's vector part flanked by λattL/R, out of the chromosome.
In this study a library composed of five strains that differ in the position of inserted φ80-removable KmR-marker and five isogenic marker-less strains for the possible φ80-dependent integration of a cassette has been obtained. (In fact, the sixth strain from this library is the MG1655 wild type with the native φ80-attB site). The "marked" part of this library can be used to select, in advance, the desirable points for the cassette integration that can be useful for construction (modification) of the producer strain. This is achieved by checking that transduction of the marker to a particular location does not affect the producer features (growth rate, desirable product yield, etc.). After preliminary selection of the members of the library based on testing "marked" strains, the corresponding "unmarked" variants can be directly used for the φ80-driven integration of cassette(s).
This library can be extended by the Red-mediated insertion of φ80-attB sites into new points on MG1655 chromosome with deleted native φ80-attB. The plasmid pMWattphi can be used as the template for PCR with the primers for this purpose. The designing of the primers can be based on the known "native" insertions in the E. coli genome, as described in this paper. On the other hand, φ80-attB integration can be used for simultaneous inactivation of an "undesirable" gene whose expression decreases the performance of a producer strain. It is important to keep in mind that the integrated cassette "ter rgnB – (genes of interest) – tL3" could be downstream from a native transcription unit in the chromosome. In this case, terminator tL3 prevents transcription of chromosomal genes originating in the cassette, and ter rgnB protects the cassette from the bacterial transcription read through.
In earlier techniques, the library of E. coli strains with different positions for site-specific (Flp-dependent) integration was constructed by random insertions of corresponding recombinogenic (FRT) sites by the Tn5-system [3]. At first glance, this library may have the same applications as the library described in this paper. Moreover, its improvement requires only determination of the FRT integration points for already obtained several hundreds of independent Tn5-driven integrants [3]. On the other hand, the expansion of our library depends on the separate Red-driven integrations, each leading to construction of only one new member. However, Red-driven integration is now used as a routine procedure and, according to our experience, can be provided in a quantity of several tens per week. At the same time our library has two advantages: (i) due to the initially designed points of the φ80-attB insertion we can exclude the interference between sequentially introduced cassettes, while random insertions more often localize near oriC due to a gene dosage effect produced by replication of the bacterial chromosome [29]; (ii) it is possible to combine distribution of site-specific integration points simultaneously with deletion of some "undesirable" genes.
As already mentioned, the Dual-In/Out strategy allows construction of marker-less strain carrying different cassettes and/or several copies of the same cassette. The presence of several copies might be necessary to increase the level of the cassette expression. However, transduction of additional copies into the same strain may lead to possible chromosome rearrangements due to general recombination events between repeated sequences.
These questions of general recombination can be divided in two groups. The first one is the question of the additional insertion of the new "marked" cassette during the transduction process, not into the point corresponding to its location in the donor genome, but into the "unmarked" cassette in the recipient chromosome. This will lead to substitution of the new cassette for the previous one. Theoretically, this process can not be excluded, but its probability is extremely low due to the small distance between φ80-attP and λ-attR in the plasmid pAH162-λattL-TcR-λattR which is used as the vector for the cassettes cloning (Fig. 2), and so, the size of the possible "arm" for envisaged general recombination would be too small. At least, we have never detected substitutions instead of expected amplification in tested clones. In any case, verifying the amplification of the integrated cassettes by PCR is desirable.
Concerning the stability of maintaining identical cassettes in the genome, direct or inverted repeats should be considered. In case of directly repeated cassettes the general recombination between them would lead to the deletion of part of the bacterial genome. According to the proposed design of the integration points, the distance between the cassettes will exceed the size of the transducing phage genome (about 100 kb in case of P1 phage). Only a few parts of the E. coli MG1655 chromosome more than 100 kb in length, do not contain any of the ca. 230 absolutely essential genes. If the designed points of integration are outside these few parts, the strains with the deleted regions between directly repeated Cassettes will not survive. General recombination between long inverted repeats leads to the chromosomal inversions that, in turn, can change the properties of the strain [34–36]. Although these events are rather rare, it could be recommended to avoid construction of the strains with inversely repeated copies of the cassette. This is not a difficult task, because the developed Dual-In/Out strategy of strain construction allows the determination, in advance, not only of location, but also of the orientation of the desired insertions.
Conclusion
Summing up, the developed Dual-In/Out strategy is rather straightforward, but convenient combination of previously developed methods. Previous approaches of integration of rather large DNA fragments, usually, use only one high-performance site-specific recombination system. When the site-specific recombination is used for insertion of the fragment, the selective marker remains in the chromosome [30]. When it is used for excision of the selective marker, the initial integration of the cassette is carried out by general recombination [3]. The combination of Red/ET-driven and two site-specific recombination systems in one strategy for integration cassettes carrying several genes/operons, during construction of marker-less strains with desired structure is rather obvious, and, probably, it will be useful for fundamental and applied fields of microbiology and biotechnology.
Methods
Strains and plasmids
Escherichia coli K-12 MG1655, a wild-type strain with sequenced genome [32], was used as the recipient for the insertion of the artificial φ80-attB. CRIM plasmids were propagated in CC118 λpir+ strain [37]. E. coli W3350(80) lysogenic for phage φ80 was obtained from GosNIIGenetika collection and used as a template for PCR-driven amplification of φ80-attL/R sites.
pKD46 was used as a donor of λRed-genes for providing Red-dependent recombination according to the described procedure [8].
pAH123 and pAH129-helper plasmids [30], GenBank accession numbers AY048726 and AY048727 respectively. These helper plasmids were used for φ80-dependent integration/excision procedures.
pMWts-λInt/Xis-helper plasmid is similar to the plasmid pMP955A described in [1]. It has low-copy-number thermo-sensitive replicon pSC101, genes xis and int of phage λ under the control of λPR, thermo-sensitive repressor cIts857. It is used for λ-Int/Xis-mediated excision of the DNA fragments flanked by λattL/R.
pMWattphi – this recombinant plasmid was constructed on the base of pMW118 (GenBank accession number AB005475). This plasmid is used as the template for PCR amplification of fragment (φ80-attL) - KmR - (φ80-attR) flanked with 36 bp arms homologous to targeted site in MG1655 DNA. Hybrid φ80-attL and φ80-attR sites were obtained by PCR amplification from purified chromosome of E. coli W3350(80) using primers: P1 – P2 for attL and P3 – P4 for attR. Amplified fragments were restricted with EcoRI-BamHI and XbaI-PstI restrictases and cloned into corresponding sites flanking kan on plasmid to give pMWattphi (Fig. 3).
P1 5'-atagaattcgaaaggtcatttttcctgaatatgc-3'
P2 5'-ataggatccatcattgaatgggtacacatttttg-3'
P3 5'-atattctagagatttgaatagcgagcgtaccttag-3'
P4 5'-atactgcagtcgtttgttgacagctggtccaatg-3'
pAH162-λattL-TcR-λattR – integrative plasmid. Construction of this plasmid included several steps with isolation and analysis of recombinant DNA intermediates. The structure of this plasmid is shown in Fig. 2. Sequence landmarks: 1) from 6 to 1031 – fragment from pAH162 (GenBank accession number AY048738) which contains conditional-replication origin oriRγ; 2) from 1038 to1145 – fragment contains attL of phage λ from plasmid pMW118-(λattL-tetA-tetR-λattR) which structural similar to the plasmid pMW118-(λattL-CmR-λattR) [19]; 3) from 1153 to 2274 – fragment from pAH162 which contains bacterial terminator rgnB, multiple cloning sites MCS, phage λ terminator tL3, phage attachment attP phi80; 4) from 2281 to 2462 – fragment contains attR of phage λ from plasmid pMW118-(λattL-tetA-tetR-λattR); 5) from 2456 to 4463 – fragment from plasmid pMW118-(λattL-tetA-tetR-λattR) which contains the Tn10-encoded tetracycline resistance gene tetA and the repressor gene tetR.
pAH162-λattL-TcR-λattR-CmR was constructed by cloning of the SphI-SacI cat-carrier DNA fragment amplified from pMW118-(λattL-CmR-λattR) [19] with primers P5 – P6.
P5 5'-cagtaagcatgcgcggccgcccggataagtagacagcctgataag-3'
P6 5'-cagtaagagctcgcggccgcttacgccccgccctgccactc-3'
Molecular biology methods
Restriction analysis of the recombinant plasmids and Ca2+-dependent transformation of E. coli cells were performed in accordance with the routine experimental protocols [38]. Commercially available preparations of restrictases, T4 DNA ligase and the Klenow fragment of E. coli DNA polymerase I (Fermentas, Lithuania) were used. PCR fragment for cloning were generated by using AccuTaq DNA polymerase (Sigma, USA). Sigma (USA) products were used for the isolation of plasmid DNA, extraction of DNA fragments from agarose gels.
Construction of strains with different locations of the φ80-attBsite
The deletion of the native φ80-attB was carried out by Red-dependent integration of "λ-excisable" CmR-marker which has been amplified by PCR from pMW118-(λattL-CmR-λattR) [19] with primers P7 – P8. DNA locus modification was verified by PCR with primers P9 – P10.
P7 5'-gtaatcaaaggatttgagcgagcaactgtacctcagcgctcaagttagtataaaaaagctgaac-3'
P8 5'-acatttagcacgtttacagttactgcatgatgaaggtgaagcctgcttttttatactaagttgg-3'
P9 5'-tgcagcgcgtgaatgtgtta-3'
P10 5'-ctcaagacaaagctgatagcc-3'
The obtained marker-less strain, MG-Δ(φ80-attB), was used as the recipient for the insertion of the artificial φ80-attB. pMWattphi was used as the template for PCR amplification of fragment (φ80-attL) - KmR - (φ80-attR) which integrated into the chromosome of MG-Δ(φ80-attB) to the desired loci – the set of genes disrupted by IS5-elements insertion. For these purposes the following primers were used:
IS5.7 P11 5'-tcctaaagaaagtatctattctgatacggttgttgagaaaggtcatttttcctgaatatg-3'
P12 5'-aagccatttacacgcacaaaatctgaaaaacgtacctcgtttgttgacagctggtccaatg-3'
P13 5'-gtcttctcacgggaacggtt-3'
IS5.8 P14 5'-gagggtatcagtacattgaaatgaatggcgccgcaggaaaggtcatttttcctgaatatg-3'
P15 5'-tctggtttgccgcgccacccatttgaacaatttgattcgtttgttgacagctggtccaatg-3'
P16 5'-cctcccttttcgatagcgacaa-3'
IS5.9 P17 5'-gggcgtattaccgcgcaaatagataccttgcaccgcgaaaggtcatttttcctgaatatg-3'
P18 5'-ctgcggatcatcaatggcgtcaatcatgccgaaatg-tcgtttgttgacagctggtccaatg-3'
P19 5'-gttcaatatgcgcggcatacca-3'
IS5.10 P20 5'-tatcaattgacgttaaggtgactctggaagctgcaggaaaggtcatttttcctgaatatg-3'
P21 5'-tattgactgaatgactaccgaagttaacaactccgctcgtttgttgacagctggtccaatg-3'
P22 5'-ttccggtggtcatactatccattc-3'
IS5.11 P23 5'-attattaaccattaatgacaaccttttacgagcaaagaaaggtcatttttcctgaatatg-3'
P24 5'-tatgaaagattggttatcctggcctctaaaaatttatcgtttgttgacagctggtccaatg-3'
P25 5'-ctttttcattaggcagtggcctc-3'
The integration of fragment was verified by PCR with primers P13, 16 – P26 and P19, 22, 25 – P27.
P26 5'-tgtttcgggcggaccaaatgata-3'
P27 5'-gccatggcagaatctgctccatgcggg-3'
Plasmid integration and excision of the vector part of integrated plasmid
For testing the new MG1655 strains with artificial φ80-attB sites and CRIM plasmid integration/excision, procedures were driven by standard protocols using helper plasmids pAH123 (φ80-Int) and pAH129 (φ80-Int/Xis) respectively [30]. The vector part of CRIM plasmids was excised by λInt/Xis – site-specific recombination using helper plasmids pMWts-λInt/Xis by standard protocols [1]. The integration of plasmid was verified by PCR with primers P13, 16, 19, 22, 25, 28 – P30 and P 26, 27, 29 – P31. P28 and P29 are primers for native φ80-attB [19]; P30 is a primer annealing on vector part of integrated plasmid [19] and P31 is annealing on tetR gene.
P28 5'-taaggcaagacgatcagg-3'
P29 5'-ctgcttgtggtggtgaat-3'
P30 5'-acgagtatcgagatggca-3'
P31 5'-gtaaactcgcccagaagctagg-3'
Cat activity assays
The Cat activity was assayed using a spectrophotometric method (UVmini 1240; Shumadzu, Japan). Log-phase cells harvested at OD595 = 0.8 were resuspended in potassium phosphate buffer (50 mM; pH 7.5). Cell lysates were prepared by sonication. The quantity of protein was determined by the Bradford method [39]. Assays were performed in 1 ml (1 cm light path) cuvettes at room temperature. The reaction mixture in each cuvette contained 100 μl of 1 M Tris-hydrochloride, pH 7.5, 100 μl of 1 mM acetyl CoA (Sigma, USA), 100 μl of 10 mM 5, 5'-dithio-bis-2-nitrobenzoic acid (DTNB; Sigma, USA), 0.05 mg of protein, H2O for a total volume of 0.99 ml. 10 μl of 10 mM Cm (Sigma, USA) was added to start the reaction, and thionitrobenzoic acid (TNB) production was followed at 412 nm. Enzyme activity was calculated in terms of nmol of thionitrobenzoic acid produced per min per mg of protein. The results were averaged over three independently grown cultures for each clone; the scatter was no more than 15%.
Abbreviations
- PCR:
-
polymerase chain reaction
- Cm:
-
chloramphenicol
- Km:
-
kanamycin
- Tc:
-
tetracyclin.
References
Peredelchuk MY, Bennet GN: A method for construction of E. coli strains with multiple DNA insertions in the chromosome. Gene. 1997, 187 (2): 231-238.
Boyd D, Weiss DS, Chen JC, Beckwith J: Towards single-copy gene expression systems making gene cloning physiologically relevant: lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J Bacteriol. 2000, 182: 842-847.
Huang LC, Wood EA, Cox MM: Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J Bacteriol. 1997, 179: 6076-6083.
Martinez-Morales F, Borges AC, Martinez K, Shanmugam KT, Ingram LO: Chromosomal integration of heterologous DNA in Escherichia coli with precise removal of markers and replicons used during construction. J Bacteriol. 1999, 181: 7143-7148.
Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR: Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999, 27: 4409-4415.
Murphy KC: Use of bacteriophage λ recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998, 180: 2063-2071.
Zhang Y, Buchholtz F, Muyrers JPP, Stewart AF: A new logic for DNA engineering using recombination in Escherichia coli. Nature Genetics. 1998, 20: 123-128.
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K12 using PCR products. Proc Natl Acad Sci USA. 2000, 97: 6640-6645.
Murphy KC, Campellone KG, Poteete AR: PCR-mediated gene replacement in Escherichia coli. Gene. 2000, 246: 321-330.
Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL: An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000, 97: 5978-5983.
Zhang Y, Muyrers JP, Testa G, Stewart AF: DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol. 2000, 18: 1314-1317.
Copeland NG, Jenkins NA, Court DL: Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet. 2001, 2: 769-779.
Court DL, Sawitzke JA, Thomason LC: Genetic engineering using homologous recombination. Annu Rev Genet. 2002, 36: 361-388.
Zhou JG, Hong X, Huang CF: Recombineering and its application. Yi Chuan Xue Bao. 2003, 30: 983-988.
Murray NE: The impact of phage lambda: from restriction to recombineering. Biochem Soc Trans. 2006, 34: 203-207.
Meynial-Salles I, Cervin MA, Soucaille P: New tool for metabolic pathway engineering in Escherichia coli: one-step method to modulate expression of chromosomal genes. Appl Environ Microbiol. 2005, 71: 2140-2144.
Gulevich AYu, Biryukova IV, Scorokhodova AYu, Krylov AD, Belareva AV, Mashko SV: Method for producing L-amino acid using bacterium having enhanced expression of pckA gene. WO/2004/090125
Katashkina JI, Lunts MG, Doroshenko VG, Fomina SA, Skorokhodova AYu, Ivanovskaya LV, Mashko SV: Optimization of bacterial sucB expression by promoter mutagenesis for efficient L-glutamic acid production. WO/2004/080386
Katashkina JI, Skorokhodova AYu, Zimenkov DV, Gulevich AYu, Minaeva NI, Doroshenko VG, Biryukova IV, Mashko SV: Tuning of expression level of the genes of interest located in the bacterial chromosome. Mol Biol (Mosk). 2005, 39 (5): 823-831.
Rivero-Müller A, Lajic S, Huhtaniemi I: Assisted large fragment insertion by Red/ET-recombination (ALFIRE) – an alternative and enhanced method for large fragment recombineering. Nucl Acids Res. 2007, 35: e78-
de Lorenzo V, Herrero M, Jakubzik U, Timmis KN: Mini-Tn5 transposons for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol. 1990, 172: 6568-6572.
de Lorenzo V, Timmis KN: Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 1994, 235: 386-405.
Rode CK, Obreque VH, Bloch CA: New tools for integrated genetic and physical analyses of the Escherichia coli chromosome. Gene. 1995, 166: 1-9.
Hamilton CH, Aldea M, Washburn BK, Babitzke P, Kushner SR: New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989, 171: 4617-4622.
Kangia K, Delor I, Cornelis GR: A wide host suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. J Bacteriol. 1991, 109: 137-141.
Le Borge S, Palmeros B, Valle F, Bolivare F, Gosset G: pBRINT-Ts: a plasmid family with a temperature-sensitive replicone, designed for chromosomal integration into lacZ gene of Escherichia coli. Gene. 1998, 223: 213-219.
Link AJ, Phillips D, Church GM: Methods for generating precise deletions and insertion in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 1997, 179: 6228-6237.
Posfai G, Koob MD, Kirkpatrick HA, Blattner : Versatile insertion plasmids for targeting genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J Bacteriol. 1997, 179: 4426-4428.
Zimenkov DV, Skorokhodova AYu, Katashkina JI, Minaeva NI, Savrasova EA, Biryukova IV, Doroshenko VG, Akhverdyan VZ, Mashko SV: E. coli chromosome regions that are more preferable for gene insertion, when the phage Mu-driven system is used for integration. Biotechnology in Russia. 2004, 6: 1-22.
Haldimann A, Wanner BL: Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001, 183: 6384-6393.
Sousa C, de Lorenzo V, Cebolla A: Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology. 1997, 143: 2071-2078.
Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y: The complete genome sequence of Escherichia coli K-12. Science. 1997, 277: 1453-1474.
Deuschle U, Kammerer W, Gentz R, Bujard H: Promoters of Escherichia coli: a hierarchy of in vivo strength indicates alternate structures. EMBO J. 1986, 5: 2987-2994.
McClain MS, Blomfield IC, Eisestein BI: Roles of fimB and fimE in site-specific DNA inversion associated with phase variation of type 1 fimbriae in Escherichia coli. J Bacteriol. 1991, 173: 5308-5314.
Komoda Y, Enomoto M, Tominaga A: Large inversion in Escherichia coli K-12 1485IN between inversely oriented IS3 elements near lac and cdd. Genetics. 1991, 129: 639-645.
Schofield MA, Agbunag R, Miller JH: DNA inversions between short inverted repeats in Escherichia coli. Genetics. 1992, 132: 295-302.
Herrero M, de Lorenzo V, Timmis KN: Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol. 1990, 172: 6557-6567.
Sambrook J, Fitsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbor, Cold Spring Harbor Press
Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976, 72: 248-254.
Acknowledgements
The authors are indebted to Prof. B.L. Wanner (Dept. of Biological Sciences, Purdue Univ., West Lafayette, Indiana 47907, USA) who kindly provided us with plasmids pKD46, pAH162, pAH123, pAH129. We are grateful to Dr. J. I. Katashkina ("Ajinomoto-Genetika Research Institute", Moscow 117545, Russia) for the gift of pMWts-λInt/Xis plasmid.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
NIM obtained the library of strains with different locations of the φ80-attB site, performed the integration of plasmid pAH162-λattL-TcR-λattR-CmR to these strains, carried out Cat-assay experiments and edited the manuscript. ERG and DVZ designed the primers and the construction scheme and constructed pMWattphi and pAH162-λattL-TcR-λattR plasmids. AYS and IVB participated in the design of the study and helped draft the manuscript. SVM supervised and coordinated the work and edited the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Minaeva, N.I., Gak, E.R., Zimenkov, D.V. et al. Dual-In/Out strategy for genes integration into bacterial chromosome: a novel approach to step-by-step construction of plasmid-less marker-less recombinant E. coli strains with predesigned genome structure. BMC Biotechnol 8, 63 (2008). https://doi.org/10.1186/1472-6750-8-63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6750-8-63