
Abstract
Purpose
To investigate the suitability of microdosed oral omeprazole for predicting CYP2C19 activity in vivo in combination with simultaneous assessment of CYP3A and CYP2D6 activity using both microdosed midazolam and yohimbine.
Methods
An open, fixed-sequence study was carried out in 20 healthy participants. Single microdosed (100 µg) and therapeutic (20 mg) doses of omeprazole were evaluated without comedication and after administration of established CYP2C19 perpetrators fluconazole (inhibition) and rifampicin (induction). To prevent degradation of the uncoated omeprazole microdose, sodium bicarbonate buffer was administered. The pharmacokinetics of omeprazole and its 5-hydroxy-metabolite were assessed as well as the pharmacokinetics of midazolam and yohimbine to estimate CYP3A4 and CYP2D6 activity.
Results
Calculated pharmacokinetic parameters after administration of 100 µg and 20 mg omeprazole in healthy subjects suggest dose proportionality. Omeprazole clearance was significantly decreased by fluconazole from 388 [95% CI: 266–565] to 47.2 [42.8–52.0] mL/min after 20 mg omeprazole and even further after 100 µg omeprazole (29.4 [24.5–35.1] mL/min). Rifampicin increased CYP2C19-mediated omeprazole metabolism. The omeprazole hydroxylation index was significantly related to omeprazole clearance for both doses. Both fluconazole and rifampicin altered CYP3A4 activity whereas no change of CYP2D6 activity was observed at all.
Conclusions
Microdosed oral omeprazole is suitable to determine CYP2C19 activity, also during enzyme inhibition and induction. However, the administration of sodium bicarbonate buffer also had a small influence on all victim drugs used.
Trial registration
EudraCT: 2017–004270-34.
Similar content being viewed by others


Introduction
Cytochrome P450 2C19 (CYP2C19) is an important enzyme of the cytochrome P450 enzyme family with large interindividual differences in the CYP2C19 activity due to genetic polymorphisms [1]. These differences in activity result in high interindividual variability of the metabolism of proton pump inhibitors (e.g., omeprazole), antiepileptic agents (e.g., brivaracetam, barbiturates, phenytoin), antiplatelet drugs (e.g., clopidogrel), and antidepressants (e.g., citalopram) [2,3,4]. Due to the individual allelic constellation, a population can be classified into 4 distinct CYP2C19 phenotypes, poor metabolizers (PM), intermediate metabolizers (IM), extensive metabolizers (EM), and ultrarapid metabolizers (UM) [5, 6]. Genetic differences also modify the propensity of the carrier for drug interactions and concurrent drug use can further modulate CYP2C19 activity resulting in a continuum of enzyme activities and potential discordances of phenotype and genotype [7]. Therefore, instead of the crude genetic classification, a more precise activity of the CYP2C19 isozyme can be determined by phenotyping with sensitive probe drugs.
Omeprazole as a commonly used proton pump inhibitor has previously been used to phenotype CYP2C19 activity using a therapeutic dose (20 or 40 mg single oral dose) [8]. A “hydroxylation index” is determined from a single 3 h blood sample, where the molar ratio of omeprazole to 5-OH-omeprazole plasma concentration is calculated [9]. This index reflects CYP2C19 activity and is related to the CYP2C19 genotype in the absence of interacting co-medication [10]. However, even a single oral dose of omeprazole can cause drug interactions by alteration of gastric pH (i.e., omeprazole reduces the AUC and Cmax of the protease inhibitor nelfinavir by approximately one third [11]), or by competitive inhibition of CYP2C19 (decrease of AUC and Cmax of active metabolite M8 of nelfinavir by 92% and 89%, respectively [11]). A microdose of omeprazole could avoid these interactions and could therefore be more suitable for CYP2C19 phenotyping because it does not cause autoinhibition of the target of interest (CYP2C19) and also does not cause gastrointestinal pH changes. Indeed, an exploratory study with some major limitations (small sample size, no CYP2C19 genotyping performed) was published in which 100 µg omeprazole was orally administered to explore the applicability of a microdose in a drug-drug interaction (DDI) trial [12]. However, microdosed intravenous omeprazole has been shown to represent hepatic CYP2C19 activity recently [13].
In this study, we investigated whether microdosed oral omeprazole is suitable to determine CYP2C19 activity in vivo in comparison to a therapeutic dose, also under conditions of CYP2C19 inhibition and induction. Since we have previously established midazolam and yohimbine microdosing to determine CYP3A and CYP2D6 activity [14,15,16,17], the additional simultaneous determination of CYP2C19 activity is a logical step to further develop a microdosed cocktail for phenotyping of the most important human CYP isozymes. Furthermore, the use of the hydroxylation index after the omeprazole microdose was evaluated in relation to the CYP2C19 genotype.
Materials and methods
The study was approved by the Competent Authority (BfArM, Bonn) in Germany (EudraCT No: 2017–004270-34) and the responsible Ethics Committee of the Medical Faculty of Heidelberg University. It was conducted at the DIN EN ISO9001-certified Clinical Research Unit (KliPS) of the Department of Clinical Pharmacology and Pharmacoepidemiology in accordance with the standards of Good Clinical Practice (as defined in the ICH E6 Guideline for Good Clinical Practice) and in agreement with the Declaration of Helsinki and all specific legal requirements in Germany.
Study population
All participants were mentally and physically healthy as confirmed by the medical history, a physical examination, a 12-lead electrocardiogram, and appropriate laboratory analyses. The participants had to undergo a urine drug screening and women were tested for pregnancy. Participants could be included in the trial if none of the following exclusion criteria were present: intake of any continuous medication other than oral contraceptives or any other substance including grapefruit juice known to interact with drug metabolizing enzymes or drug transporters, any condition, which could potentially modify absorption, distribution, metabolism, or excretion of the study drugs, allergies (except for mild forms of hay fever) or history of hypersensitivity reactions, smoking, excessive alcohol drinking, blood donation or participation in a clinical trial within the last month, positive drug screening or known or admitted drug abuse, and inability to communicate well with the investigator. Neither pregnant nor lactating women were included. All participants were required to consent to the use of two appropriate contraception methods and gave their written informed consent before any study measures were carried out.
All participants had a known CYP2C19 genotype previously determined in a genotyping study where common cytochrome isozyme polymorphisms were examined. CYP2C19 genotyping was performed for CYP2C19*2 (rs4244285), CYP2C19*3 (rs4986893), and for CYP2C19*17 (rs12248560) as previously described [13] and the presence of two wild type alleles was assumed if none of the tested polymorphisms was present.
Study design and blood sampling
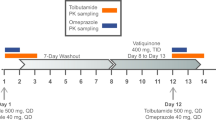
This was an open-label clinical trial with fixed sequence design consisting of a total of seven inpatient study days and eight short visits for each participant. The study was conducted in 3 phases separated by 2 washout periods in which omeprazole microdose, omeprazole regular dose, CYP2C19 inhibition, and CYP2C19 induction were tested (Fig. 1).

Study design of the clinical trial. MDZ = 10 µg midazolam; YOH = 50 µg yohimbine; µOME = 100 µg omeprazole solution; OME = 20 mg omeprazole capsule
Omeprazole was administered orally as a regular dose of 20 mg (OMEP® 20 mg, gastro-resistant capsule, HEXAL AG, Holzkirchen, Germany). The microdose of 100 µg omeprazole was freshly prepared using powder for solution for infusion (OMEP® 40 mg, powder for solution for infusion, HEXAL) that had been dissolved in 5 mL of NaCl (0.9% w/v); 12.5 µL (= 100 µg omeprazole) of this solution was diluted in 100 mL sodium bicarbonate (4.2% w/v). Ten minutes prior to omeprazole dosing, 50 mL sodium bicarbonate buffer (4.2% w/v) was administered to increase gastric pH to prevent degradation of the uncoated omeprazole.
Midazolam and yohimbine were used as a biomarker for CYP3A and CYP2D6 activity. Simultaneously with the omeprazole dose, each participant received a freshly prepared solution of 10 µg midazolam on every inpatient study day (Dormicum® V 5 mg/5 mL solution for injection, Roche, Grenzach-Wyhlen, Germany) and an oral dose of 50 µg yohimbine (Yohimbinum hydrochloricum D4®; Deutsche Homöopathie Union, Karlsruhe, Germany, containing 25 µg per tablet).
The CYP2C19 perpetrator fluconazole (Fluconazol HEXAL® 200 mg, HEXAL) was given for 5 days in oral doses of 400 mg on day 1 and 200 mg on days 2 to 5. After a washout period of at least 7 days, rifampicin was given for 7 days using a daily dose of 600 mg (Eremfat® 600 mg, RIEMSER Pharma GmbH, Greifswald, Germany). A diary was used to ensure correct intake of fluconazole and rifampicin at home.
On each pharmacokinetic study day, 16 blood samples of 4.9 mL each were collected to determine omeprazole, midazolam, and yohimbine concentrations. In the first study phase with midazolam and yohimbine, only 5 blood samples of 2.7 mL each were obtained (limited sampling pre-dose, 2, 2.5, 3, and 4 h after administration of study medication). In the 3 subsequent phases, samples were taken before pre-dose and 5, 10, 15, 20, 30, 45 min and 1, 1.5, 2, 2.5, 3, 4, 6, 8, and 24 h after administration of study medication. Blood samples were centrifuged for 10 min at 2500 g and 4 °C, and separated plasma was distributed into 4 aliquots, each stored at − 20 °C until analysis.
Quantification of omeprazole, 5-OH-omeprazole, midazolam, and yohimbine
Plasma concentrations of omeprazole and 5-OH-omeprazole after the 20 mg and 100 µg dose were analyzed with 2 different methods using high-performance and ultra-high performance liquid chromatography-tandem mass spectrometry (LC–MS/MS and UPLC-MS/MS) [13]. Calibration ranges were between 1–1000 ng/mL (LC–MS/MS) and 10–10,000 pg/mL (UPLC-MS/MS), respectively. The lower limits of quantification (LLOQ) for therapeutic doses were 1 ng/mL for omeprazole and 5-OH-omeprazole with within-batch and between-batch accuracies of 98–104% and precision of ≤ 7.0%. The LLOQ for the microdose were 10 pg/mL for both, omeprazole and 5-OH-omeprazole. Plasma concentrations could be quantified with within-batch and between-batch accuracies of 99–109% and precision of ≤ 13.2% [13].
Midazolam concentrations in plasma were determined by UPLC-MS/MS. The assay’s LLOQ was 0.37 pg/mL, and the accuracy/precision values were 96–99%/ ≤ 12.0%, respectively [18]. Yohimbine plasma concentrations were determined using an UPLC-MS/MS assay with an LLOQ of 5 pg/mL; accuracies ranged between 88 and 96% and precision was ≤ 11.7% [19].
Data analysis
Standard non-compartmental pharmacokinetic parameters of omeprazole and 5-OH-omeprazole were calculated using Kinetica 5.0 (Thermo Fisher Scientific, Waltham, MA, USA): maximum plasma concentration (Cmax), time to reach Cmax (tmax), terminal elimination half-life (t1/2), area under the plasma concentration–time curve extrapolated to infinity (AUCtot), apparent volume of distribution at steady-state (Vss), apparent volume of distribution (Vz), and apparent oral clearance (Cl/F). In addition, as a metric for CYP2C19 activity, the hydroxylation index (HI) was calculated as the molar ratio of the concentrations of omeprazole and 5-OH-omeprazole at 3 h [8].
For CYP3A activity evaluation, a limited sampling strategy based on midazolam AUC from 2 to 4 h was used to calculate the metabolic clearance as described previously [20]. To evaluate the activity of CYP2D6, the yohimbine clearance (Cl/F) was calculated by non-compartmental analysis using Kinetica 5.0 [17]. Data are presented as geometric mean and 95% confidence intervals (95% CI) unless stated otherwise.
To analyze the differences of pharmacokinetic parameters between microdosed and regular dosed omeprazole as well as the influence of CYP2C19 perpetrators, analysis of variance after logarithmic transformation and respective post hoc tests were used. A p value < 0.05 was considered significant. In addition, the geometric mean ratios and their respective 90% CIs have been calculated for Cmax, AUC and Cl/F with no interaction concluded if the 90% confidence interval includes 1. The statistical analysis was performed using Prism 8.4 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Twenty healthy non-smoking participants (15 females) were eligible for this clinical trial. They were between 20 and 48 years of age with an average of 29.4 ± 8.2 years. Seven were UM (CYP2C19*17/*17, *1/*17), 6 EM (*1/*1), 6 IM (*1/*2, *2/*17), and one was PM (*2/*2).
Omeprazole
The plasma concentration–time profiles after a single oral dose of a 20 mg commercial capsule differed between the study conditions (Fig. 2, upper panel). Inhibition of CYP2C19 using fluconazole significantly increased Cmax 3.25-fold with unchanged tmax, induction of CYP2C19 by rifampicin significantly decreased Cmax by 85% with unchanged tmax (Table 1). CYP2C19 induction and inhibition were reflected in all other pharmacokinetic parameters, with omeprazole clearance decreasing significantly by 88% during fluconazole and increasing 11-fold during rifampicin (Table 1). Compared to baseline, the AUC of the metabolite 5-OH-omeprazole was slightly decreased during inhibition and profoundly decreased during induction, while the terminal elimination half-life of the metabolite was prolonged during inhibition and reduced during induction (Table 1).

Geometric mean ± 95% CI plasma concentration vs. time curves for normal dosed omeprazole (upper panel) and microdosed omeprazole (bottom panel) during baseline condition (black) and during concomitant intake of fluconazole (blue) or rifampicin (red)
The plasma concentration–time profiles after a single oral 100 µg dose of omeprazole as a solution differed substantially from the corresponding profiles after administration of the 20-mg omeprazole gastro-resistant capsule, which was also true for conditions of inhibition and induction of CYP2C19 (Fig. 2). Cmax occurred much earlier after administration of the solution (tmax = 14.8 min) (Table 2) than after the commercial capsule (tmax = 178 min) (Table 1). As for the regular dose, all pharmacokinetic parameters of the microdose obtained during induction and inhibition significantly differed from the corresponding baseline values (Table 2). Induction and inhibition of CYP2C19 was reflected in omeprazole clearance, which was significantly decreased during fluconazole to 7.8% of baseline clearance and increased 5.8-fold during rifampicin (Table 2). The AUC of 5-OH-omeprazole was significantly decreased during inhibition and induction compared to baseline, while terminal elimination half-life was increased during inhibition and reduced during induction (Table 2). These effects are comparable to those observed after 20 mg omeprazole (Table 1).
In Table 3, the geometric mean ratios of Cmax, AUC and the apparent oral omeprazole clearances are listed for the 2 doses and the 3 conditions. No dose-normalized AUC and clearance differences were observed between the omeprazole doses given alone indicating dose proportionality. A significant influence of both perpetrators (fluconazole and rifampicin) on omeprazole clearance was observed for both doses used but during either perpetrator drug omeprazole clearance after 100 µg was almost twofold lower than after 20 mg (Table 3).
Hydroxylation index (HI)
The hydroxylation index of omeprazole as the molar ratio of omeprazole to 5-OH-omeprazole plasma concentration from the single 3 h blood sample varied considerably by a factor of 278 (range: 0.1660–46.22) after 20 mg and by 419 (range: 0.2600–108.9) after 100 µg omeprazole (alone and during fluconazole and rifampicin) (Table 4). For both omeprazole doses used, the relationship between the HI and the apparent oral clearance is shown in Fig. 3. For both doses a log–log regression revealed an adjusted r2 > 0.9 and a slope close to − 1 (20 mg omeprazole: − 1.034; 100 µg omeprazole: − 0.8061). As the regression analysis mixes intra- and interindividual variability, the regression lines are not shown and the results are for illustration purposes only.

Relationship between omeprazole hydroxylation index and the omeprazole apparent oral clearance for normal dosed omeprazole (upper panel) and microdosed omeprazole (bottom panel) during baseline condition (black) and during concomitant intake of fluconazole (blue) or rifampicin (red). During induction with rifampicin in 5 participants, omeprazole was below LLOQ at 3 h after microdosed omeprazole; no hydroxylation index was calculated. This also occurred in 2 participants after normal dosed omeprazole
Midazolam
The calculated metabolic clearance of midazolam as a marker of CYP3A activity during CYP2C19 inhibition and induction conditions was analyzed using repeated measures ANOVA and post hoc testing of every condition against baseline and between omeprazole microdose and normal dose. Compared to baseline without omeprazole, midazolam CLmet was unchanged irrespective of the administered omeprazole dose although the geometric mean ratios are different from 1 (Fig. 4). However, because both fluconazole and rifampicin also affect CYP3A activity, this resulted in a significant reduction of CLmet during fluconazole and significant increase of CLmet during rifampicin (Fig. 4). Comparison of midazolam CLmet between microdose and normal dose of omeprazole showed significantly lower clearances after omeprazole microdosing, irrespective of the condition (inhibition or induction) (Fig. 4).

Calculated metabolic clearance of midazolam (geometric mean ± 95% confidence interval) in relation to the different study conditions (baseline condition (black), concomitant intake of fluconazole (blue), rifampicin (red)). The results of the repeated measures ANOVA after logarithmic transformation with Sidak’s multiple comparisons test. In addition, the geometric mean ratios (GMR) with their 90% confidence intervals (90% CI) are listed. ns = not significant; µOME = 100 µg omeprazole solution; OME = 20 mg omeprazole capsule. ***p < 0.005
Yohimbine
Fluconazole and rifampicin did not modify yohimbine pharmacokinetics a marker of CYP2D6 activity (suppl Table 1). However, apparent yohimbine clearance was significantly lower after omeprazole microdoses than after regular doses both at baseline (2528 mL/min vs. 3274 mL/min) and during fluconazole (2224 mL/min vs. 2884 mL/min). Also, during rifampicin, yohimbine clearance was lower with microdosed omeprazole than with normal dose omeprazole but this difference did not reach significance (p = 0.0506; suppl Table 1).
Safety and tolerability
All study drugs were well tolerated and no serious adverse events occurred. Most adverse events occurred after intake of fluconazole and rifampicin (mainly headaches and nausea), all were mild and transient, and no actions had to be taken.
Discussion
Microdose vs. therapeutic dose
We evaluated the suitability of the pharmacokinetics an oral omeprazole microdose (100 µg) to predict changes in CYP2C19 activity and compared it with the pharmacokinetics of a therapeutic dose (20 mg). Under baseline conditions, there were no significant differences of dose-normalized AUC, elimination half-life, and clearance between microdosed and therapeutic dosed omeprazole suggesting dose proportionality, which offers the possibility of replacing the therapeutic omeprazole dose by a microdose as phenotyping measure. However, significant differences regarding dose-normalized Cmax and tmax were observed, which can be caused by the different dosage forms. Commercial gastro-resistant capsules (20 mg) were used with delayed release (tmax 178 min with Cmax 327.7 ng/mL). For the omeprazole microdose, we prepared an oral solution with 75 mmol sodium bicarbonate and omeprazole to prevent the acid labile omeprazole from being destroyed in the stomach prior to absorption. In contrast, absorption of this omeprazole solution was fast (tmax 15 min) with correspondingly higher dose-normalized Cmax (896 ng/mL). The amount of sodium bicarbonate administered with omeprazole seems to be important for absorption. Park and co-workers administered 10 mmol of sodium bicarbonate resulting in a relative bioavailability (Frel) of 0.35 of the microdose [13]. Bioequivalence studies showed that uncoated omeprazole in combination with 48 mmol sodium bicarbonate was bioequivalent to an encapsulated formulation [21]. In our study, 75 mmol sodium bicarbonate resulted in Frel of 1.02, indicating successful prevention of acid destruction of the microdosed omeprazole solution.
Omeprazole microdose vs. therapeutic dose during fluconazole
Fluconazole, a strong CYP2C19 inhibitor, increased Cmax and AUC of the 20 mg omeprazole dose threefold and eightfold and reduced clearance to 12% of the baseline value. This confirms the already known strong inhibition of fluconazole [12]. Using 100 µg omeprazole, an even more pronounced inhibitory effect on CYP2C19 was observed with AUC increasing 12-fold and the clearance decreasing to 7.8% of baseline. Omeprazole has been reported to have non-linear pharmacokinetics after a single high dose and also repetitive administration due to autoinhibition of CYP2C19 [22, 23]. When using an omeprazole microdose, this autoinhibition disappears and the effect of an inhibitor is more pronounced. Although a time-dependent inhibitory effect of fluconazole has not been investigated in a clinical trial, it seems likely that there is no further increase of inhibition with time, since on day 5 of fluconazole, there was less inhibition (normal omeprazole dose) than on day 3 with the omeprazole microdose.
Because fluconazole is a competitive CYP2C19 inhibitor [24], higher doses and thereby higher concentrations of fluconazole will cause stronger inhibition. After a low dose of fluconazole (8 days of 50 mg fluconazole daily), a 2.7–2.8-fold increase of omeprazole AUC was found [12]. Doubling the fluconazole dose (100 mg fluconazole daily for 5 days) showed a sixfold increase of omeprazole AUC in healthy volunteers [25]. In our study, the dose regimen for fluconazole was according to the Summary of Product Characteristics [26]. With oral administration of 400 mg fluconazole on day 1 and 200 mg fluconazole on day 2 to 5, eightfold (20 mg omeprazole) and 12-fold (100 µg omeprazole) increases of omeprazole AUC were reached. A pH-mediated influence of omeprazole or sodium bicarbonate on fluconazole pharmacokinetics is unlikely since changes in gastric pH did not affect fluconazole Cmax, tmax, and AUC [27].
Microdose vs. therapeutic dose during rifampicin
Rifampicin is well characterized as an inducer of multiple cytochrome P450 isozymes, among them also CYP2C19 [28]. As expected, rifampicin increased omeprazole clearance irrespective of the omeprazole dose. However, when a microdose was used, omeprazole clearance increased 5.8-fold, whereas omeprazole clearance increased 11-fold after 20 mg omeprazole. CYP2C19 induction by rifampicin is mediated by an activation of CYP2C19 transcription, which is a time-dependent process. In our study, we administered rifampicin 600 mg daily for 7 days. It has been reported that maximal induction of intestinal and hepatic CYP3A4 was achieved in > 90% of participants within 5 and 10 days [29]. Other studies found that the maximum effect of rifampicin induction on CYP2C and CYP3A4 was achieved after 7 to 11 days [28, 30, 31]. Because the microdose of omeprazole was administered after 5 days of rifampicin and the 20 mg dose after 7 days, this might be the reason for the observed difference in the magnitude of induction. This is in line with the induction of the CYP3A4 substrate midazolam with an increase of metabolic clearance by 10.1-fold on day 5 and 11.9-fold on day 7 (Fig. 4), suggesting that induction was still evolving. The inducing effect of a low dose rifampicin (150 mg) on omeprazole pharmacokinetics decreased omeprazole AUC to 22% after a microdose and 20% after a 20 mg dose after 8 and 9 days of rifampicin treatment, respectively [12]. There was no significant difference between the 2 omeprazole doses, probably due to the long rifampicin treatment. Likely due to the lower rifampicin dose, the induction was not as pronounced compared to the observed AUC reduction to 17% (day 5, microdose) and 8% (day 7, 20 mg dose). In another study investigating the influence of rifampicin (6 days, 450 mg daily) on 20 mg omeprazole, omeprazole AUC decreased to 12.9% [32]. Comparing these three studies, a dose-dependent and time-dependent effect of rifampicin induction on omeprazole elimination is observed confirming the dependency of rifampicin-induced induction reported earlier [33].
Plasma concentrations of the metabolite 5-OH-omeprazole were also reduced during induction. Increased metabolite exposure would result during induction as the formation rate increases; however, further metabolism of 5-OH-omeprazole is partly catalyzed by CYP3A4 to omeprazole hydroxysulfone [34]. Therefore, with CYP3A4 also induced by rifampicin the exposure of 5-OH-omeprazole can be reduced due to enhanced secondary metabolism, which is also reflected in the decreased metabolite half-life.
CYP2C19 genotype
Because the aim of this investigation was to test whether microdosed oral omeprazole can also be used to assess CYP2C19 activity in vivo, the CYP2C19 genotypes of the study participants are also important. Therefore, study participants were divided into four groups according to their CYP2C19 genotype: UM, EM, IM, and PM. No significant differences of omeprazole (20 mg) pharmacokinetics have been observed between UM, IM, and EM under baseline conditions as well as during fluconazole and also rifampicin. The same resulted using microdosed omeprazole (suppl. Figure 1).
In ultrarapid metabolizers with one or two CYP2C19*17 alleles CYP2C19 activity is increased and homozygous carriers of CYP2C19*17 had significantly lower exposure with the CYP2C19 substrates omeprazole and escitalopram than carriers of the wild-type allele [35, 36]. In our study, omeprazole clearance after 20 mg was 515 mL/min (95% CI: 260–1021 mL/min; n = 7) in ultrarapid metabolizers and 367 mL/min (95% CI: 188–720 mL/min; n = 6) in extensive metabolizers, which was significantly different. Similar clearances were observed after 100 µg omeprazole with 476 mL/min (95% CI: 238–952 mL/min) in UMs and 367 mL/min (95% CI: 194–693 mL/min) in EMs. However, the clearances showed a large overlap between the CYP2C19 genotypes (suppl. Figure 1). Only the single poor metabolizer of this study was easily detected under baseline conditions since threefold and 6.5-fold higher Cmax and AUC were observed (after 20 mg omeprazole in comparison with the EM group) with omeprazole clearance being as low as 57 mL/min.
During fluconazole, omeprazole clearance was reduced to 49 mL/min (UM), 45 mL/min (EM), 47 mL/min (IM), and 44 mL/min (PM), respectively (suppl. Figure 1), confirming that fluconazole is a potent CYP2C19 inhibitor. Fluconazole is listed as a moderate CYP3A4 inhibitor by the FDA [37], which is confirmed by our data using midazolam. Since CYP3A4 plays a minor role in omeprazole metabolism, which can be confirmed by the small clearance reduction in the PM by fluconazole, the observed clearance reduction in CYP2C19 metabolizers can be attributed to CYP2C19 inhibition.
Enzyme induction using rifampicin profoundly increased omeprazole clearance to 6321 mL/min (UM), 3527 mL/min (EM), 4903 mL/min (IM), and 649 mL/min (PM) (suppl. Figure 1). This large difference between the PM and the other genotypes demonstrates the induction of CYP2C19. A more than tenfold increase of omeprazole clearance is observed in the one PM participant, which suggests induction of CYP3A and confirms previously observed data [32]. Similar results for 100 µg omeprazole were observed during fluconazole as well as rifampicin.
Hydroxylation index (HI)
The omeprazole hydroxylation index (HI) has previously been used as marker for CYP2C19 phenotyping [8,9,10, 38]. After calculation of the HI, however, there were no significant differences between the CYP2C19 genotypes (Table 4). Only the one PM was clearly distinguished. Probably due to the low sample size of each genotype our results are in contrast to published data where 160 participants (113 EM, 40 IM, and 7 PM) [8] and 300 participants (124 EM, 129 IM, and 47 PM) [38] were studied. Since the HI is calculated from a single blood sample collected 3 h after omeprazole dosing, we observed differences between the 100 µg and 20 mg dose due to the different formulation with different tmax values.
Midazolam
Microdosed midazolam as a CYP3A4 marker [14] in combination with a limited sampling methodology [20, 39] was able to assess the influence of inhibition and induction of CYP3A4 by fluconazole and rifampicin in this study. The metabolic clearance of midazolam during fluconazole decreased to 28% and 26% of the baseline value with 100 µg and 20 mg omeprazole, respectively. This corresponds well to the reported 3.6-fold increase of midazolam AUC after oral administration of 7.5 mg midazolam during fluconazole treatment [40]. Rifampicin as a non-selective inducer of multiple CYP enzymes increased the metabolic clearance of midazolam 10–12-fold. This is in accordance with the midazolam AUC decrease to 4.1% of the baseline value during treatment with 600 mg rifampicin daily over 5 days [41].
Because the effect of the perpetrators was studied with the 100 µg and 20 mg omeprazole dose, the design was suitable to also study the influence of omeprazole dose. The metabolic clearance of midazolam after 20 mg omeprazole was significantly higher than after administration of 100 µg omeprazole during baseline, fluconazole, and rifampicin. An auto-inhibitory effect of 20 mg omeprazole can be ruled out since this would have been the opposite effect, especially during baseline. Because the omeprazole microdose was always combined with sodium bicarbonate, an enhanced absorption of midazolam might have occurred [42, 43]. Midazolam alters its structure in solutions of pH > 6 and gets more lipophilic thus improving membrane permeability [44, 45].
Yohimbine
Although there was no effect on CYP2D6 expected by the substances used in this study, we included microdosed yohimbine as a marker substance mainly to generate more data on a potential microdosed CYP cocktail. Both fluconazole and rifampicin as perpetrators did not alter the clearance of yohimbine. Decreased yohimbine clearance was observed when 100 µg omeprazole was administered instead of 20 mg, which was significant during baseline and fluconazole, but did not reach significance during rifampicin (suppl. Table 1). This is quite similar to the observations with midazolam, which likewise might be caused by the administration of sodium bicarbonate resulting in the same clearance alteration. So far, there are no data published on yohimbine regarding pH-dependent alteration of gastrointestinal absorption.
Limitations
There are two major limitations in this study, one being the small sample size of the different CYP2C19 genotypes and the second the effect of the use of sodium bicarbonate in order to stabilize the liquid omeprazole microdose. We have included all known genotypes of CYP2C19, which actually resulted in a large clearance variability (> 14-fold) of omeprazole. The use of sodium bicarbonate altered yohimbine and midazolam pharmacokinetics when combining them with microdosed omeprazole for a microdosed CYP cocktail. This limits the usability and comparability of the cocktail approach with microdosed omeprazole. A gastro-resistant formulation of microdosed omeprazole (e.g., a capsule) might be necessary to eliminate the influence of the sodium bicarbonate solution.
In summary, this clinical study assessed the CYP2C19 activity using microdosed oral omeprazole in humans in comparison to a therapeutic 20 mg dose. Omeprazole microdoses appear similarly suitable for phenotyping of CYP2C19 as therapeutic doses. The extent of drug interactions with CYP2C19 inhibitors and inducers are in a similar order of magnitude as after therapeutic doses. The use of a microdosed CYP cocktail (omeprazole, midazolam, and yohimbine) was only slightly impacted by co-administering sodium bicarbonate to prevent disintegration of omeprazole in the peptic milieu but needs further investigation.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zhou HH (2001) CYP2C19 genotype determines enzyme activity and inducibility of S-mephenytoin hydroxylase. Clin Chim Acta 313(1–2):203–208. https://doi.org/10.1016/s0009-8981(01)00696-9
Li-Wan-Po A, Girard T, Farndon P, Cooley C, Lithgow J (2010) Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol 69(3):222–230. https://doi.org/10.1111/j.1365-2125.2009.03578.x
Shah BS, Parmar SA, Mahajan S, Mehta AA (2012) An insight into the interaction between clopidogrel and proton pump inhibitors. Curr Drug Metab 13(2):225–235. https://doi.org/10.2174/138920012798918390
Shirasaka Y, Sager JE, Lutz JD, Davis C, Isoherranen N (2013) Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug-drug interactions. Drug Metab Dispos 41(7):1414–1424. https://doi.org/10.1124/dmd.113.051722
Hicks JK, Swen JJ, Thorn CF, Sangkuhl K, Kharasch ED, Ellingrod VL, Skaar TC, Muller DJ, Gaedigk A, Stingl JC (2013) Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther 93(5):402–408. https://doi.org/10.1038/clpt.2013.2
Wedlund PJ (2000) The CYP2C19 enzyme polymorphism. Pharmacology 61(3):174–183. https://doi.org/10.1159/000028398
Bahar MA, Setiawan D, Hak E, Wilffert B (2017) Pharmacogenetics of drug-drug interaction and drug-drug-gene interaction: a systematic review on CYP2C9, CYP2C19 and CYP2D6. Pharmacogenomics 18(7):701–739. https://doi.org/10.2217/pgs-2017-0194
Chang M, Dahl ML, Tybring G, Gotharson E, Bertilsson L (1995) Use of omeprazole as a probe drug for CYP2C19 phenotype in Swedish Caucasians: comparison with S-mephenytoin hydroxylation phenotype and CYP2C19 genotype. Pharmacogenetics 5(6):358–363. https://doi.org/10.1097/00008571-199512000-00004
Kimura M, Ieiri I, Wada Y, Mamiya K, Urae A, Iimori E, Sakai T, Otsubo K, Higuchi S (1999) Reliability of the omeprazole hydroxylation index for CYP2C19 phenotyping: possible effect of age, liver disease and length of therapy. Br J Clin Pharmacol 47(1):115–119. https://doi.org/10.1046/j.1365-2125.1999.00858.x
Niioka T, Uno T, Sugimoto K, Sugawara K, Hayakari M, Tateishi T (2007) Estimation of CYP2C19 activity by the omeprazole hydroxylation index at a single point in time after intravenous and oral administration. Eur J Clin Pharmacol 63(11):1031–1038. https://doi.org/10.1007/s00228-007-0331-3
Fang AF, Damle BD, LaBadie RR, Crownover PH, Hewlett D Jr, Glue PW (2008) Significant decrease in nelfinavir systemic exposure after omeprazole coadministration in healthy subjects. Pharmacotherapy 28(1):42–50. https://doi.org/10.1592/phco.28.1.42
Park GJ, Bae SH, Park WS, Han S, Park MH, Shin SH, Shin YG, Yim DS (2017) Drug-drug interaction of microdose and regular-dose omeprazole with a CYP2C19 inhibitor and inducer. Drug Des Devel Ther 11:1043–1053. https://doi.org/10.2147/DDDT.S131797
Mahmoudi M, Foerster KI, Burhenne J, Weiss J, Mikus G, Haefeli WE (2021) Application of Microdosed Intravenous Omeprazole to Determine Hepatic CYP2C19 Activity. J Clin Pharmacol 61(6):789–798. https://doi.org/10.1002/jcph.1789
Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE (2013) A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther 93(6):564–571. https://doi.org/10.1038/clpt.2013.27
Geist M, Bardenheuer H, Burhenne J, Mikus G (2019) Alteration of drug-metabolizing enzyme activity in palliative care patients: microdosed assessment of cytochrome P450 3A. Palliat Med 33(7):850–855. https://doi.org/10.1177/0269216319843629
Geist MJP, Ziesenitz VC, Bardenheuer HJ, Burhenne J, Skopp G, Mikus G (2019) Minor contribution of cytochrome P450 3A activity on fentanyl exposure in palliative care cancer patients. Sci Rep 9(1):14635. https://doi.org/10.1038/s41598-019-51279-6
Vay M, Meyer MJ, Blank A, Skopp G, Rose P, Tzvetkov MV, Mikus G (2020) Oral Yohimbine as a New Probe Drug to Predict CYP2D6 Activity: Results of a Fixed-Sequence Phase I Trial. Clin Pharmacokinet 59(7):927–939. https://doi.org/10.1007/s40262-020-00862-6
Burhenne J, Halama B, Maurer M, Riedel KD, Hohmann N, Mikus G, Haefeli WE (2012) Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1’-hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem 402(7):2439–2450. https://doi.org/10.1007/s00216-011-5675-y
Vay M, Sauter M, Mikus G, Burhenne J (2019) Quantification of microdosed oral yohimbine and its major metabolite in human plasma in the picogram range. Bioanalysis 11(16):1459–1467. https://doi.org/10.4155/bio-2019-0129
Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G (2011) Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin Pharmacol Ther 90(5):666–673. https://doi.org/10.1038/clpt.2011.164
Pilbrant A, Cederberg C (1985) Development of an oral formulation of omeprazole. Scand J Gastroenterol Suppl 108:113–120. https://doi.org/10.3109/00365528509095824
Cederberg C, Rohss K, Lundborg P, Olbe L (1993) Effect of once daily intravenous and oral omeprazole on 24-hour intragastric acidity in healthy subjects. Scand J Gastroenterol 28(2):179–184. https://doi.org/10.3109/00365529309096067
Hassan-Alin M, Andersson T, Niazi M, Rohss K (2005) A pharmacokinetic study comparing single and repeated oral doses of 20 mg and 40 mg omeprazole and its two optical isomers, S-omeprazole (esomeprazole) and R-omeprazole, in healthy subjects. Eur J Clin Pharmacol 60(11):779–784. https://doi.org/10.1007/s00228-004-0841-1
Wienkers LC, Wurden CJ, Storch E, Kunze KL, Rettie AE, Trager WF (1996) Formation of (R)-8-hydroxywarfarin in human liver microsomes. A new metabolic marker for the (S)-mephenytoin hydroxylase, P4502C19. Drug Metab Dispos 24(5):610–614
Kang BC, Yang CQ, Cho HK, Suh OK, Shin WG (2002) Influence of fluconazole on the pharmacokinetics of omeprazole in healthy volunteers. Biopharm Drug Dispos 23(2):77–81. https://doi.org/10.1002/bdd.291
Hexal AG (2017) Fachinformation: Fluconazol HEXAL® 200 mg Hartkapseln
Zimmermann T, Yeates RA, Riedel KD, Lach P, Laufen H (1994) The influence of gastric pH on the pharmacokinetics of fluconazole: the effect of omeprazole. Int J Clin Pharmacol Ther 32(9):491–496
Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT (2003) Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 42(9):819–850. https://doi.org/10.2165/00003088-200342090-00003
Kapetas AJ, Sorich MJ, Rodrigues AD, Rowland A (2019) Guidance for rifampin and midazolam dosing protocols to study intestinal and hepatic cytochrome P450 (CYP) 3A4 induction and de-induction. AAPS J 21(5):78. https://doi.org/10.1208/s12248-019-0341-y
Ohnhaus EE, Breckenridge AM, Park BK (1989) Urinary excretion of 6 beta-hydroxycortisol and the time course measurement of enzyme induction in man. Eur J Clin Pharmacol 36(1):39–46. https://doi.org/10.1007/BF00561021
Fromm MF, Busse D, Kroemer HK, Eichelbaum M (1996) Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology 24(4):796–801. https://doi.org/10.1002/hep.510240407
Kamiya C, Inui N, Hakamata A, Miyakawa S, Tanaka S, Uchida S, Namiki N, Odagiri K, Watanabe H (2019) Effect of co-administered inducer or inhibitor on omeprazole pharmacokinetics based on CYP2C19 genotype. J Pharmacol Sci 139(4):361–366. https://doi.org/10.1016/j.jphs.2019.03.001
Kanebratt KP, Diczfalusy U, Backstrom T, Sparve E, Bredberg E, Bottiger Y, Andersson TB, Bertilsson L (2008) Cytochrome P450 induction by rifampicin in healthy subjects: determination using the Karolinska cocktail and the endogenous CYP3A4 marker 4beta-hydroxycholesterol. Clin Pharmacol Ther 84(5):589–594. https://doi.org/10.1038/clpt.2008.132
Andersson T, Miners JO, Veronese ME, Birkett DJ (1994) Identification of human liver cytochrome P450 isoforms mediating secondary omeprazole metabolism. Br J Clin Pharmacol 37(6):597–604. https://doi.org/10.1111/j.1365-2125.1994.tb04310.x
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79(1):103–113. https://doi.org/10.1016/j.clpt.2005.10.002
Rudberg I, Mohebi B, Hermann M, Refsum H, Molden E (2008) Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin Pharmacol Ther 83(2):322–327. https://doi.org/10.1038/sj.clpt.6100291
FDA (2017) Drug development and drug interactions: table of substrates, inhibitors and inducers. In: ed
Rosemary J, Adithan C, Padmaja N, Shashindran CH, Gerard N, Krishnamoorthy R (2005) The effect of the CYP2C19 genotype on the hydroxylation index of omeprazole in South Indians. Eur J Clin Pharmacol 61(1):19–23. https://doi.org/10.1007/s00228-004-0833-1
Hohmann N, Kocheise F, Carls A, Burhenne J, Haefeli WE, Mikus G (2015) Midazolam microdose to determine systemic and pre-systemic metabolic CYP3A activity in humans. Br J Clin Pharmacol 79(2):278–285. https://doi.org/10.1111/bcp.12502
Olkkola KT, Ahonen J, Neuvonen PJ (1996) The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg 82(3):511–516. https://doi.org/10.1097/00000539-199603000-00015
Backman JT, Olkkola KT, Neuvonen PJ (1996) Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther 59(1):7–13. https://doi.org/10.1016/S0009-9236(96)90018-1
Elliott P, Dundee JW, Elwood RJ, Collier PS (1984) The influence of H2 receptor antagonists on the plasma concentrations of midazolam and temazepam. Eur J Anaesthesiol 1(3):245–251
Jo JH, Kim SJ, Nam WS, Seung EJ, Lee S (2016) Decreased absorption of midazolam in the stomach due to low pH induced by co-administration of Banha-sasim-tang. Environ Health Toxicol 31:e2016016. https://doi.org/10.5620/eht.e2016016
Andersin R (1991) Solubility and acid-base behaviour of midazolam in media of different pH, studied by ultraviolet spectrophotometry with multicomponent software. J Pharm Biomed Anal 9(6):451–455. https://doi.org/10.1016/0731-7085(91)80246-6
Odou P, Barthelemy C, Robert H (1998) Development of midazolam sublingual tablets: in vitro study. Eur J Drug Metab Pharmacokinet 23(2):87–91. https://doi.org/10.1007/BF03189320
Acknowledgements
The authors are grateful for excellent assistance of Marlies Stützle-Schnetz, MSc, RN with the study conduct, and Magdalena Longo and Andrea Deschlmayr for technical support during analytical procedures.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AE, AB, WEH, and GM designed the study. AE, PR, GM, and AB conducted the study and data collection. KF and JB performed the analytical quantifications. AE and GM conducted the pharmacokinetic and statistical analysis and wrote the first draft of the paper. All authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study was conducted at a single center (KliPS) at the University Hospital Heidelberg, Germany. The study protocol was approved by the Ethics Committee of the Medical Faculty of Heidelberg University and the German competent authority (EudraCT: 2017–004270-34). The study was performed in accordance with Good Clinical Practice and the Declaration of Helsinki.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elbe, A., Foerster, K.I., Blank, A. et al. Evaluation of CYP2C19 activity using microdosed oral omeprazole in humans. Eur J Clin Pharmacol 78, 975–987 (2022). https://doi.org/10.1007/s00228-022-03304-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-022-03304-3