
Abstract
Background and Objective
Although polypharmacy is a particular challenge in daily rheumatological practice, clinical research on the effects of hydroxychloroquine (HCQ), a commonly used drug for patients with rheumatic diseases, is sparse on cytochrome P450 (CYP)-mediated metabolism. We have shown that pre-treatment with pantoprazole does not alter HCQ absorption in healthy volunteers. In this paper, we report the effects of a single 400 mg dose of HCQ on specific CYP3A and CYP2D6 substrates in healthy volunteers.
Methods
In the trial, participants were randomized into two groups (HCQ plus a 9-day course of pantoprazole, or HCQ only). As a secondary endpoint, the effects of a single oral dose of HCQ on the exposure of the oral microdosed CYP3A probe drug midazolam (30 μg) and the oral microdosed CYP2D6 probe drug yohimbine (50 μg) were studied in 23 healthy volunteers (EudraCT no. 2020-001470-30, registered 31 March 2020).
Results
The exposure of the probe drugs after intake of HCQ compared with baseline values was quantified by the partial area under the plasma concentration–time curve 0–6 h after administration (AUC0–6 h) for yohimbine and the partial AUC2–4 h for midazolam. Under HCQ, yohimbine AUC0–6 h was unchanged, independent of CYP2D6 genotypes and pantoprazole exposure. Midazolam AUC2–4 h was 25% higher on the day of HCQ administration than at baseline (p = 0.0007). This significant increase was driven by the pantoprazole subgroup, which showed a 46% elevation of midazolam AUC2–4 h as compared with baseline (p < 0.0001). The ratio of midazolam to 1-OH-midazolam partial AUC2–4 h significantly increased from 3.03 ± 1.59 (baseline) to 3.60 ± 1.56 (HCQ) in the pantoprazole group (p = 0.0026).
Conclusion
In conclusion, we observed an increased midazolam exposure most likely related to pantoprazole.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
HCQ did not inhibit the metabolism by CYP2D6 and CYP3A in the short term. |
We found evidence that pantoprazole can act as an inhibitor of CYP3A and moderately changes midazolam exposure. |
1 Introduction
Hydroxychloroquine (HCQ) is still an important medicine for patients with rheumatic diseases and has a long, albeit declining, tradition in the treatment of malaria. In addition, there was much interest in HCQ when searching for a remedy in the early coronavirus disease 2019 (COVID-19) pandemic [1]. Polypharmacy, reflecting comorbidity, is common in patients with rheumatic diseases [2,3,4]. In a survey among rheumatologists, interfering comorbidities and pharmacological management were identified as clinically relevant situations in the management of difficult-to-treat rheumatoid arthritis not covered by current guideline recommendations [5].
In rheumatic arthritis and systemic lupus erythematosus, cardiovascular diseases or mood disorders are common comorbidities and present a challenge to medical management [4,5,6]. Research into drug–drug interactions (DDIs) is important for the co-treatment of rheumatic diseases and their comorbidities. Common cardiovascular drugs and anti-thrombotic medicines are cytochrome P450 (CYP) 3A4 substrates: examples include atorvastatin, simvastatin, dronedarone, eplerenone and ticagrelor. The β1-selective beta blockers metoprolol and nebivolol are CYP2D6 substrates. Also, many antidepressants belong to the group of CYP2D6 substrates, such as venlafaxine, amitriptyline, doxepin, nortriptyline and selective serotonin re-uptake inhibitors, such as fluvoxamine, fluoxetine and paroxetine [7].
Although it is assumed that HCQ may have clinically relevant interactions with other drugs metabolized by CYP isozymes, there is surprisingly little evidence on HCQ’s interaction potential from clinical trials. In 2020, several reviews of the potential DDIs of HCQ were published, which largely extrapolated their recommendations from chloroquine trial data (e.g. [8]). From the COVID-19 pandemic, there is an episodic report on elevated clarithromycin blood concentrations under treatment with HCQ in two patients, but in vitro assessments by the same authors did not support the hypothesis that the parent drug HCQ inhibits CYP3A4 activity [9]. However, the inhibitory potential of HCQ metabolites was not analysed in this study. Later, in vitro evidence showed that HCQ metabolites inhibit CYP3A4 [10]: only in 2023, a detailed in vitro study showed that HCQ is metabolized by CYP2D6, CYP3A4 and CYP2C8, and also suggested that HCQ and its metabolites might inhibit CYP2D6 [10].
In a clinical trial, initiated during the COVID-19 pandemic, we investigated HCQ as a victim drug of a suspected proton-pump inhibitor-mediated increase in gastric pH in healthy volunteers but found no significant effect of pantoprazole on HCQ exposure [11]. To our knowledge, there is only one other DDI trial with HCQ in humans. That trial evaluated the interaction of HCQ with metoprolol and dextromethorphan in healthy volunteers and indicated that HCQ weakly inhibits CYP2D6 [12].
In this paper, we report the effects of a single 400 mg dose of HCQ on specific CYP3A and CYP2D6 substrates in healthy volunteers. As part of a clinical DDI trial that explored the effect of pantoprazole on HCQ absorption as a primary endpoint [13], healthy volunteers were administered microdoses of the CYP3A substrate midazolam and the CYP2D6 substrate yohimbine at baseline and after a single dose of HCQ [14, 15]. The secondary endpoint of the trial was to evaluate HCQ as a potential inhibitor of CYP3A and CYP2D6 activity in humans.
2 Methods
2.1 Trial Protocol
The trial protocol has been published in detail separately [13] (EudraCT no. 2020-001470-30, registered 31 March 2020; DRKS00021573). In brief, we conducted a phase I DDI trial in healthy volunteers aged 18–60 years. The trial took place in the ISO 9001:2015-certified Early Phase Clinical Trial Unit (Klinisch-Pharmakologisches Studienzentrum, KliPS) of Heidelberg University Hospital in 2020. The trial was conducted according to the guidelines of Good Clinical Practice, the ethical principles expressed in the Declaration of Helsinki, and all legal requirements for clinical trials in Germany. The trial was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University, Germany (ethical approval number AFmo-265/2020), and by the competent authority (BfArM, Bonn, Germany). Participants were fully informed and gave their written consent prior to any trial-related procedures. Main exclusion criteria were relevant abnormalities in the medical history, physical examination or laboratory evaluation; use of any medication (except hormonal contraception or thyroid hormones); intake of HCQ, chloroquine or travel to malaria risk regions within the last 3 months; intake of quinine or consumption of quinine-containing drinks (bitter lemon, tonic water, bitter orange); consumption of citrus fruits or products of these fruits within 7 days prior to the first dose of trial medication; prolonged QTc time (defined as QTcF > 460 ms in females and QTcF > 440 ms in males). Participants separately consented to genotyping for drug-metabolizing enzymes according to a pre-defined protocol (genotyping biobank, ethical approval number S-026/2004).
2.2 Endpoints
The evaluation of HCQ as a perpetrator drug on CYP3A4 and CYP2D6 was a predefined secondary objective of this trial. Endpoints were the ratio of area under the plasma concentration–time curve 2–4 h after administration (AUC2–4 h) of microdosed midazolam and the ratio of AUC0–6 h of microdosed yohimbine at baseline and under a single dose of HCQ.
The primary objective of this trial was to evaluate the effect of the proton-pump inhibitor pantoprazole on the absorption of HCQ; the results of the primary endpoint have been published previously [11].
2.3 Drug Administration and Blood Sampling
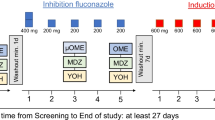
First, CYP3A and CYP2D6 baseline activities were evaluated with microdosed midazolam (30 µg; Dormicum® V 5 mg/5 ml, Cheplepharm Arzneimittel GmbH, Greifswald, Germany, in 100 ml water) and yohimbine (50 µg; Yohimbinum hydrochloricum D4®, Deutsche Homöopathie-Union-Arzneimittel GmbH and Co. KG, Karlsruhe, Germany). The drugs were administered 3 h after a standardized meal. The timing of the meal was the same on trial day 6 when HCQ was administered with a meal prior to the microdosed drugs. Starting after the baseline assessments, participants were 1:1 randomized either to a 9-day course of oral pantoprazole 40 mg (Pantoprazol HEXAL®, Holzkirchen, Germany) once daily (to reach a steady state) or to control (without pantoprazole) for the evaluation of the primary objective of the trial. At day 6, a single oral dose of 400 mg HCQ (two tablets of Quensyl® 200 mg; Sanofi-Aventis, Frankfurt, Germany) was administered to all participants with a standardized meal. Pantoprazole intake on this day took place 1 h before the meal (Fig. 1). A total of 3 h after the intake of HCQ (i.e. 4 h after pantoprazole), participants received the microdosed oral probe drugs. Plasma samples (lithium heparin) were collected for 6 h (pre-dose and 0.5, 1, 2, 2.5, 3, 4, and 6 h after administration). CYP phenotyping using microdosed midazolam [14, 16, 17] and yohimbine [15] has been published previously.

Trial design. Adapted from the trial protocol published in [13]. At baseline, cytochrome P450 (CYP) 3A and CYP2D6 activity were phenotyped with a microdose of midazolam and yohimbine. Thereafter, one group was started on a 9-day course of once daily (qd) pantoprazole, while the other group served as a control group. On the 6th day, a single dose of hydroxychloroquine 400 mg was administered, after which the CYP3A and CYP2D6 phenotyping was repeated. Cmax plasma peak concentration
2.4 Quantification of Midazolam and Yohimbine
Midazolam and yohimbine plasma concentrations were quantified using validated ultra-high performance liquid chromatography coupled to tandem mass spectrometry (UPLC–MS/MS) as described previously [14, 16, 18]. The methods were developed and validated according to the International Council for Harmonisation (ICH) M10 guideline on bioanalytical method validation and fulfilled the ICH validation criteria [19]. The lower limit of quantification for midazolam, 1-hydroxy-midazolam (1-OH-midazolam), and yohimbine was 1 pg/ml. Inter-day accuracy (precision) varied from +9.56% (1.25%) to +12.9% (2.63%) for midazolam, from −13.1% (1.51%) to −8.64% (3.81%) for 1-OH-midazolam, and from −4.69% (4.87%) to +2.91% (10.0%) for yohimbine.
2.5 Evaluation of CYP2D6 Genotype
Genotyping of CYP2D6 was performed using a single-base primer extension method as described previously [15]. CYP2D6 activity was scored on the basis of the sum of the allele activity scores assigned by PharmGKB (https://www.pharmgkb.org/) and CPIC (https://cpicpgx.org/) as follows: 0 poor metabolizer; 0.5 and 1 intermediate metabolizer; and 1.25, 1.5 and 2 normal metabolizer.
2.6 In Vitro Determination of CYP3A4 Inhibition by Pantoprazole
Inhibition of CYP3A4 by pantoprazole was quantified with the P450-Glo™ CYP3A4 Assay (Promega Corporation, Madison, WI, USA) using a membrane preparation containing recombinant human CYP3A4 according to the manufacturer’s instructions. This assay is based on the conversion of the luminogenic CYP3A4 substrate luciferin-IPA to luciferin by CYP3A4 and was conducted as described previously [20]. Eight concentrations of pantoprazole (0.05–100 µM) were investigated in triplicate, and the experiment was conducted in quadruplicate. Data were analysed and half maximal inhibitory concentrations (IC50) were calculated with GraphPad Prism Version 10.0 (GraphPad Software, San Diego, CA, USA) using the four-parameter fit (sigmoidal concentration–response curves with variable slope).
2.7 Pharmacokinetic and Statistical Analysis
The non-compartmental pharmacokinetic analysis was performed using Phoenix WinNonlin Version 8.3.5 (Certara, Inc., Princeton, NJ, USA). For yohimbine, AUC0–6 h and plasma peak concentration (Cmax) were calculated [15], and for midazolam and 1-OH-midazolam, AUC2–4 h was calculated [17]. Geometric means and associated confidence intervals were calculated for all pharmacokinetic parameters. A ratio-paired t-test with a 90% confidence interval was applied, and an acceptance interval of 80–125% was assumed on the basis of the range used in bioequivalence trials [21]. Between-group analyses were carried out with unpaired t-tests. A p value < 0.1 was considered statistically significant. The statistical analysis was conducted with GraphPad Prism 10.0 (GraphPad Software, San Diego, CA, USA).
3 Results
Of the original 24 healthy volunteers, one participant from the control group withdrew consent for personal reasons and was therefore excluded. The median age of the 23 participating volunteers (12 in the pantoprazole group/11 in the control group) was 26 years (range 21–60 years), with 26 years in the pantoprazole group and 27 years in the control group. A total of 12 (52%) participants were female, and of those, 7 were in the pantoprazole group. The body mass index was 25.1 kg/m2 [standard deviation (SD) 3.7], with 24.0 kg/m2 (SD 3.1) in the pantoprazole group and 26.4 kg/m2 (SD 4.0) and in the control group.
3.1 Yohimbine—CYP2D6
The extent of yohimbine exposure varied, as expected, according to the CYP2D6 genotype: in total, there were two poor metabolizers (of whom one was in the control group and one was in the pantoprazole group), ten intermediate metabolizers (seven in the control group, three in the pantoprazole group), and eleven normal metabolizers of CYP2D6 (four in the control group, seven in the pantoprazole group) (Table 1 and Fig. 2). There was a significant difference in yohimbine AUC0–6 h at baseline between the normal and the intermediate metabolizers (p = 0.04); the group of the poor metabolizers was too small for statistical comparisons. Baseline versus HCQ showed no significant change in yohimbine AUC0–6 h after administration of HCQ. Cmax increased by 21% (Table 1 and Fig. 2). We observed no significant change in yohimbine exposure in any subgroup (pantoprazole or control; genotypes) under HCQ.

Partial area under the concentration-time curve from 0 to 6 h (AUC0–6 h) of yohimbine at baseline and after intake of hydroxychloroquine (HCQ) with or without pantoprazole in 23 healthy volunteers. Geometric mean with 95% confidence interval for normal metabolizers of CYP2D6 (NM; n = 11), intermediate metabolizers (IM; n = 10) and poor metabolizers (PM; n = 2)
3.2 Midazolam—CYP3A
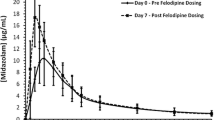
Baseline versus HCQ: Midazolam AUC2–4 h was 25% higher than at baseline, and the AUC2–4 h of 1-OH-midazolam increased by 17% (Table 2 and Fig. 3) in all study participants after HCQ intake. However, the increase in midazolam AUC2–4 h was driven by the subgroup with concomitant intake of pantoprazole. In the pantoprazole subgroup, there was a significant increase in AUC2–4 h of 46% and an increase of 17.5% in the AUC2–4 h of 1-OH-midazolam, whereas there was no change in the control subgroup. The ratio of midazolam AUC2–4 h to 1-OH-midazolam AUC2–4 h increased significantly in the pantoprazole subgroup (Table 2). Pantoprazole versus control group: At baseline, there was no significant difference between the randomized groups (pantoprazole or control) in midazolam AUC2–4 h, 1-OH-midazolam AUC2–4 h or their ratios. After the intake of HCQ, there was a difference between the control group and the pantoprazole group in midazolam AUC2–4 h and its ratio to 1-OH-midazolam AUC2–4 h (p = 0.0597 and p = 0.0527, respectively) but not for 1-OH-midazolam AUC2–4 h.

Partial area under the concentration–time curve from 2 to 4 h (AUC2–4 h) of midazolam at baseline and after intake of hydroxychloroquine (HCQ) with or without pantoprazole in 23 healthy volunteers. *p ≤ 0.0001 for baseline versus HCQ + pantoprazole. Without the outlier in the pantoprazole group, the change in partial AUC2–4 h was still significant: geometric mean ratio (GMR) 1.44 [90% confidence interval 1.30–1.60], p < 0.0001 for the pantoprazole group, and GMR 1.24 [90% confidence interval 1.20–1.37], p = 0.0016 for all participants. ns non significant
CYP3A4 inhibition by pantoprazole (IC50 = 23.8 ± 10.9 µM) was verified in vitro (Fig. 4) confirming previous data [24].

In vitro inhibition of cytochrome P450 (CYP) 3A4 by pantoprazole. Depicted are the results of four experiments conducted with each concentration in triplicate. Data are expressed as mean ± standard deviation (pooled data, n = 12)
4 Discussion
We detected no significant increase of yohimbine exposure by a single dose of HCQ, indicating that HCQ is not a clinically relevant CYP2D6 inhibitor. In vitro, HCQ and its metabolites, mainly desethylchloroquine, have been reported to competitively and reversibly inhibit CYP2D6 activity, albeit only mildly [10]. In the only clinical trial on the CYP2D6-inhibiting properties of HCQ so far, CYP2D6-inhibiting properties of HCQ were evaluated after an 8-day course of 400 mg HCQ twice daily in seven healthy volunteers [12]: after administration of HCQ, the AUC of the CYP2D6 substrate metoprolol increased by 65% and Cmax by 72%. Interestingly, the urinary metabolic ratio of dextromethorphan did not show any significant change. The effect on metoprolol was consistent for the six homozygous extensive (normal) CYP2D6 metabolizers, while the heterozygous extensive (intermediate) metabolizer did not show any relevant change in exposure. In our trial including more participants with a greater variation of genotypes, we did not observe an effect in the CYP2D6 genotype subgroups. Because of the large variation of yohimbine exposure due to the genotype, also a large effect would have been visible. We administered 400 mg HCQ as a single dose on the day of CYP phenotyping. Assuming a low affinity of HCQ to CYP2D6, this single dose of HCQ may have resulted in too low blood concentrations to exert perpetrator properties on yohimbine.
The exposure of the CYP3A substrate midazolam increased by 25% in all participants after the intake of HCQ. However, this observation was driven by the subgroup receiving pantoprazole, in which the AUC of midazolam was increased by as much as 46%. The partial AUC2–4 h for 1-OH-midazolam, the active metabolite of midazolam, increased by 17% under HCQ in all. This tended to be the case in the pantoprazole group, while there was clearly no effect in the control group. Concurrently, the ratio between the partial AUC of midazolam and 1-OH-midazolam also increased significantly after the combined intake of HCQ and pantoprazole (indicating reduced production of 1-OH midazolam by CYP3A), while it remained unchanged in the control group receiving only HCQ. The increased ratio strongly suggests that the metabolism of midazolam was inhibited by pantoprazole (leading to increased absorption due to reduced first-pass metabolism or to slower elimination) and that the exposure change was not primarily caused by gastric pH changes although a contribution cannot be completely excluded.
From clinical drug interaction trials with various CYP3A4 substrates, pantoprazole is not known as a perpetrator [22, 23]. However, in vitro pantoprazole inhibits CYP3A4-mediated midazolam metabolism at relatively high concentrations [24], which we confirmed with another CYP3A4 inhibition assay also in vitro.
Midazolam is a well-absorbed compound with moderate to high pre-systemic extraction ratio; administered in therapeutic or microdoses, it has an oral bioavailability of approximately 25% [25]. Intestinal first-pass metabolism contributes approximately 50% to the overall first-pass metabolism of oral midazolam [26], indicating that modulation of intestinal CYP3A can substantially increase midazolam bioavailability. In our trial, 40 mg pantoprazole was administered with approximately 200 ml of fluid, yielding local concentrations in the gut that likely exceeded 500 µM. In vitro, pantoprazole reduced CYP3A activity by 75% at concentrations of 100 µM, suggesting that in the first hours after oral administration, local pantoprazole concentrations may be high enough to substantially inhibit CYP3A in the gut. Because pantoprazole peak concentrations are reached approximately 2–3 h after oral administration [27] and may be considerably delayed by food [28], it may well be that at the time of midazolam administration, i.e. 4 h after pantoprazole administration, local intestinal concentrations were still in the inhibitory range. Peak serum concentrations were much lower (2.5 µg/ml equalling 6.5 µM [29]) and associated with less CYP3A inhibition, explaining that pantoprazole is not known as a CYP3A inhibitor from daily practice.
Limitations: Because HCQ steady state is only reached after 4 months [30], our trial was designed with a single-dose administration of HCQ. Although we did not find a short-term inhibitory effect of HCQ on CYP3A, we cannot exclude the possibility that time-dependent CYP3A inhibition develops with longer-term HCQ treatment. The active metabolites desethylchloroquine, didesethylchloroquine,and desethylhydroxychloroquine (DHCQ) are time-dependent inhibitors of CYP3A4 in vitro [10]. This could explain the elevated clarithromycin exposure that was reported for two patients with COVID-19 treated with HCQ [9]. In this context, the CYP2D6 genotype could theoretically indirectly influence time-dependent CYP3A4 inhibition in longer-term HCQ treatment via the formation of DHCQ: A Korean trial found that the DHCQ:HCQ ratio is lower in carriers of CYP2D6*10 polymorphisms who have reduced CYP2D6 activity [31].
Yohimbine AUC0–6 h was highly variable due to the different CYP2D6 genotypes. This makes it difficult to find a statistically significant change between baseline and intervention. To mitigate this problem, we analysed the genotype subgroups, but those were small, which presents another statistical challenge. We cannot exclude that there might have been an effect in a larger group.
Furthermore, the microdosing method for CYP phenotyping using probe drugs has been validated in fasting condition. In the present trial, HCQ tablets had to be swallowed with a meal, so CYP microdosing could not take place in a fasting state but only 3 h after the intake of HCQ and the standardized meal. Administering an oral standard dose of midazolam 1 h after a meal delays the peak plasma concentration by 0.9 h as compared with the fasting state, and decreases midazolam exposure [32]. Midazolam AUC2–4 h in this trial was at the lower end of the range known from previous trials in our department, and we are aware of a mild food effect from another trial [33]. However, the identical food–drug interval was also followed at baseline. So, even though the absorption might have been slightly different from the fasting state, comparability between the trial parts was ensured.
In conclusion, HCQ did not inhibit metabolism by CYP2D6 and CYP3A in the short term. Whether or not longer treatment, which leads to higher concentrations of parent compound and potentially interacting metabolites, modulates these isozymes differently still needs to be shown. Concurrently, we found evidence that pantoprazole can act as an inhibitor of CYP3A4 and moderately changes midazolam exposure. This was unexpected, because no clinically relevant perpetrator properties with other CYP3A substrates have been described for pantoprazole so far. However, CYP3A inhibitor characteristics have repeatedly been described in vitro. Whether this is due to the particular timing and is only observed with CYP3A substrates with quantitatively relevant intestinal metabolism remains to be investigated.
Data availability
The datasets generated during and/or analysed during the current trial are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Axfors C, et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun. 2021;12(1):2349.
Seguin DJ, et al. Polypharmacy and potentially inappropriate medication use in older adults with systemic lupus erythematosus. Arthritis Care Res (Hoboken). 2023;75:356–364.
Gomides APM, et al. High levels of polypharmacy in rheumatoid arthritis—a challenge not covered by current management recommendations: data from a large real-life study. J Pharm Pract. 2021;34(3):365–71.
Jack JD, et al. Polypharmacy in Middle-European rheumatoid arthritis-patients: a retrospective longitudinal cohort analysis with systematic literature review. Front Med (Lausanne). 2020;7: 573542.
Roodenrijs NMT, et al. Characteristics of difficult-to-treat rheumatoid arthritis: results of an international survey. Ann Rheum Dis. 2018;77(12):1705–9.
Zhang L, et al. Prevalence of depression and anxiety in systemic lupus erythematosus: a systematic review and meta-analysis. BMC Psychiatry. 2017;17(1):70.
FDA. Drug Development and Drug Interactions. 05/12/2022. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table3-1. Accessed 20 Nov 2023.
Rendic S, Guengerich FP. Metabolism and interactions of chloroquine and hydroxychloroquine with human cytochrome P450 enzymes and drug transporters. Curr Drug Metab. 2020;21(14):1127–35.
Li X, et al. The parent drugs chloroquine and hydroxychloroquine do not inhibit human CYP3A activity in vitro. Eur J Clin Pharmacol. 2020;76(10):1481–2.
Paludetto MN, et al. Hydroxychloroquine is metabolized by CYP2D6, CYP3A4, and CYP2C8, and inhibits CYP2D6, while its metabolites also inhibit CYP3A in vitro. Drug Metab Dispos. 2023;51:293–305.
Stoll F, et al. Effect of pantoprazole on the absorption of hydroxychloroquinea a randomized drug-drug interaction trial in healthy adults. Clin Pharmacol Drug Dev. 2022;11(2):285–90.
Somer M, et al. Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br J Clin Pharmacol. 2000;49(6):549–54.
Stoll F, et al. Impact of pantoprazole on absorption and disposition of hydroxychloroquine, a drug used in Corona Virus Disease-19 (Covid-19): a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):584.
Halama B, et al. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93(6):564–71.
Vay M, et al. Oral yohimbine as a new probe drug to predict CYP2D6 activity: results of a fixed-sequence phase I trial. Clin Pharmacokinet. 2020;59(7):927–39.
Katzenmaier S, Markert C, Mikus G. Proposal of a new limited sampling strategy to predict CYP3A activity using a partial AUC of midazolam. Eur J Clin Pharmacol. 2010;66(11):1137–41.
Katzenmaier S, et al. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin Pharmacol Ther. 2011;90(5):666–73.
Vay M, et al. Quantification of microdosed oral yohimbine and its major metabolite in human plasma in the picogram range. Bioanalysis. 2019;11(16):1459–67.
EMA. ICH guideline M10 on bioanalytical method validation and study sample analysis. 2022. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf. Accessed 20 Nov 2023.
Weiss J, et al. Does the circulating ketoconazole metabolite N-deacetyl ketoconazole contribute to the drug-drug interaction potential of the parent compound? Eur J Pharm Sci. 2022;169: 106076.
EMA. Guideline on the investigation of bioequivalence. 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 20 Nov 2023.
Bliesath H, et al. Pantoprazole does not interact with nifedipine in man under steady-state conditions. Int J Clin Pharmacol Ther. 1996;34(2):51–5.
Steinijans VW, et al. Lack of pantoprazole drug interactions in man: an updated review. Int J Clin Pharmacol Ther. 1996;34(1 Suppl):S31-50.
Li XQ, et al. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32(8):821–7.
Breithaupt MH, et al. Oral bioavailability of microdoses and therapeutic doses of midazolam as a 2-dimensionally printed orodispersible film in healthy volunteers. Eur J Clin Pharmacol. 2022;78(12):1965–72.
Thummel KE, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59(5):491–502.
Calabresi L, et al. Pharmacokinetic interactions between omeprazole/pantoprazole and clarithromycin in health volunteers. Pharmacol Res. 2004;49(5):493–9.
Ochoa D, et al. Effect of food on the pharmacokinetics of omeprazole, pantoprazole and rabeprazole. BMC Pharmacol Toxicol. 2020;21(1):54.
Summary of Product Characteristics, Pantoprazol 40 mg magensaftresistente Tabletten. Hexal, Holzkirchen. 2021.
Summary of Product Characteristics, Quensyl® 200 mg Filmtabletten. Sanofi-Aventis, Frankfurt. 2022.
Lee JY, et al. Association of polymorphisms of cytochrome P450 2D6 with blood hydroxychloroquine levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2016;68(1):184–90.
Bornemann LD, et al. Influence of food on midazolam absorption. J Clin Pharmacol. 1986;26(1):55–9.
Huppertz A, et al. Differential effect of a continental breakfast on tacrolimus formulations with different release characteristics. Clin Pharmacol Drug Dev. 2021;10(8):899–907.
Acknowledgements
We thank the staff of the Early Clinical Trial Unit and the Analytical-Chemical Laboratory, Department of Clinical Pharmacology and Pharmacoepidemiology at Heidelberg University Hospital, Germany, for their great assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization was performed by GM and WEH; methodology (trial protocol) was performed by AB, DC, FS, GM and WEH; investigation (execution of trial) was performed by AB, FS and KSG; investigation (analysis of samples) was performed by JB, JW, MMT and MT; formal analysis was performed by FS, DC and JW; supervision was carried out by WEH; writing—original draft was performed by FS; and writing—review and editing was carried out by all authors.
Corresponding author
Ethics declarations
Conflict of interest
No author reports a conflict of interest.
Ethical approval
The trial was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University, Germany (ethical approval number AFmo-265/2020).
Informed consent
Participants were fully informed and gave their written consent prior to any trial-related procedures.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Stoll, F., Blank, A., Mikus, G. et al. Evaluation of Hydroxychloroquine as a Perpetrator on Cytochrome P450 (CYP) 3A and CYP2D6 Activity with Microdosed Probe Drugs in Healthy Volunteers. Eur J Drug Metab Pharmacokinet 49, 101–109 (2024). https://doi.org/10.1007/s13318-023-00872-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-023-00872-2