
Abstract
Manfred Göthert, who had served Naunyn-Schmiedeberg’s Arch Pharmacol as Managing Editor from 1998 to 2005, deceased in June 2019. His scientific oeuvre encompasses more than 20 types of presynaptic receptors, mostly on serotoninergic and noradrenergic neurones. He was the first to identify presynaptic receptors for somatostatin and ACTH and described many presynaptic receptors, known from animal preparations, also in human tissue. In particular, he elucidated the pharmacology of presynaptic 5-HT receptors. A second field of interest included ligand-gated and voltage-dependent channels. The negative allosteric effect of anesthetics at peripheral nACh receptors is relevant for the peripheral clinical effects of these drugs and modified the Meyer-Overton hypothesis. The negative allosteric effect of ethanol at NMDA receptors in human brain tissue occurred at concentrations found in the range of clinical ethanol intoxication. Moreover, the inhibitory effect of gabapentinoids on P/Q Ca2+ channels and the subsequent decrease in AMPA-induced noradrenaline release may contribute to their clinical effect. Another ligand-gated ion channel, the 5-HT3 receptor, attracted the interest of Manfred Göthert from the whole animal via isolated preparations down to the cellular level. He contributed to that molecular study in which 5-HT3 receptor subtypes were disclosed. Finally, he found altered pharmacological properties of 5-HT receptor variants like the Arg219Leu 5-HT1A receptor (which was also shown to be associated with major depression) and the Phe124Cys 5-HT1B receptor (which may be related to sumatriptan-induced vasospasm). Manfred Göthert was a brilliant scientist and his papers have a major impact on today’s pharmacology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
On June 28, 2019, Professor Manfred Göthert, former Managing Editor of Naunyn-Schmiedeberg’s Arch Pharmacol, passed away in Hamburg, at the age of 79 years. Manfred Göthert was born in 1939 in Braunschweig. He started to study medicine at the University of Hamburg in 1959 and continued studies in Freiburg, Innsbruck, Vienna, and finally Göttingen where he graduated in 1965. In Göttingen, he also prepared his doctoral thesis and received his MD title (Dr. med.) in 1965. In 1967, he joined the Institute of Pharmacology of the University of Hamburg as a postdoctoral scholar where he completed his habilitation thesis in 1971 and received the title “Professor” in 1976. He was called to the University of Essen in 1978 (C3 Professor) and to the University of Bonn in 1985 (C4 Professor) where he was Head of the Institute of Pharmacology and Toxicology until his retirement in 2006. Manfred Göthert served Naunyn-Schmiedeberg’s Archives of Pharmacology as editor 1987-1995 and 2002-2003 and as Managing Editor 1995-2002. During his time as Managing Editor jointly with Karl Heinz Jakobs (Aktories et al. 2019), he guided the journal very well and initiated its change from a journal mainly recognized in the German Society for Experimental and Clinical Pharmacology and Toxicology to an internationally recognized platform to publish studies in experimental pharmacology. Naunyn-Schmiedeberg’s Archives of Pharmacology is extremely grateful to Manfred Göthert for his long-lasting service and will dearly miss his input.
The scientific work by Manfred Göthert encompasses 271 articles covered in pubmed, which appeared during the time period from 1968 (Göthert et al. 1968) to 2020 (Baranowska-Kuczko et al. 2020; Göthert et al. 2020). Since the titles of no less than 110 articles contain serotonin, 5-hydroxytryptamine, or 5-HT (encompassing eight 5-HT receptors; Tables 1 and 2), the title of this review and the headings of its chapters (“Serotonin” and “Beyond serotonin”) were chosen accordingly.
Serotonin
Manfred Göthert published his first papers dedicated to serotonin in 1972 and his interest in this monoamine lasted up to his death. His scientific activities directed towards serotonin may be differentiated into three periods (as reflected by the headings “Early studies”, “Presynaptic autoreceptors and heteroreceptors” and “Molecular vistas”) and roughly correspond to his time spent in Hamburg, Essen, and Bonn, respectively.
Early studies
Manfred Göthert performed some of his early studies on 5-HT in cooperation with the anatomist H.G. Baumgarten, who had described the 5-HT neurotoxins 5,6- and 5,7-dihydroxytryptamine (5,6- and 5,7-DHT) for the first time (for review, see Jonsson 1980). The question was whether the neurotoxins also affect noradrenergic neurones. In rodents, intraperitoneally administered 5,7-DHT destroyed the postganglionic sympathetic neurones with a potency comparable to that of 6-hydroxydopamine, the standard neurotoxin for noradrenergic neurones (Baumgarten et al. 1974), whereas 5,6-DHT showed such an effect at high doses only (Baumgarten et al. 1972a). Subsequently, the complexity of acute effects of 5-HT and its monohydroxylated and dihydroxylated analogs on cardiovascular parameters was studied in vitro, in situ, and in vivo. The compounds act (i) directly by activation of postsynaptic 5-HT receptors (Rs) (Table 1) and/or via effects on the noradrenergic system including (ii) activation of α-adrenoceptors (α-ARs), (iii) an indirect sympathomimetic effect, and/or (iv) activation of facilitatory presynaptic 5-HT-Rs (Baumgarten et al. 1972b; Göthert et al. 1973; Göthert and Klupp 1978). A masterpiece of this early phase is the paper by Göthert and Dührsen (1979) on rabbit atria (Table 2), in which the chronotropic effect and noradrenaline release were quantified. Infusion of 6-hydroxytryptamine (6-HT) led to a gradual increase in heart rate and noradrenaline release, whereas 5-HT itself and 5,7-DHT caused rapid increases in both parameters, followed by a fading down. Reserpine inhibited all effects, the inhibitor of the neuronal noradrenaline transporter, desipramine, selectively counteracted the effects of 6-HT, whereas the Ca2+ antagonist verapamil attenuated the effects of 5-HT and 5,7-DHT. The positive chronotropic effect of 5-HT was desensitized by prior exposure to 5-HT itself or 5,7-DHT. The data clearly revealed that mechanisms (iii) and (iv) are involved, respectively.
The facilitatory presynaptic receptor in the study by Göthert and Dührsen (1979) is a 5-HT3-R. This type of receptor can be regarded as a hub within the scientific work of Manfred Göthert (Table 2). One of his coworkers, interested in histamine H3-Rs, sometimes informed him about latest results obtained and he repeatedly responded: You mean 5-HT3? The 5-HT3-R will re-appear in many sections of this review, e.g. under the “Molecular vistas” section where molecular biological properties of this receptor will be considered. The reason why 5-HT3-Rs will also be discussed in the second chapter of this review is that they play a role beyond serotonin, i.e. the 5-HT3-R is just one example of ligand-gated ion channels (besides nicotinic acetylcholine (nACh) and/or N-methyl-D-aspartate (NMDA)-Rs) which are targeted by ethanol (“Ethanol, general anesthetics, gabapentinoids, and ion channels” section) and cannabinoids (“Cannabinoids” section).
Presynaptic autoreceptors and heteroreceptors
Presynaptic serotonin autoreceptors
Presynaptic receptors represent a mechanism by which a transmitter (or a locally formed mediator or a hormone) inhibits or increases the release of the same (autoreceptor) or of another transmitter (heteroreceptor). In 1971, the autoreceptors modulating the release of noradrenaline, acetylcholine, and γ-aminobutyric acid (GABA) from their respective neurones have been described for the first time (reviewed in Starke et al. 1989). It took until 1979 before the serotonin autoreceptor was identified, by the groups of Manfred Göthert in Essen (Göthert and Weinheimer 1979) and M. Raiteri in Genova (Cerrito and Raiteri 1979). Both groups examined the depolarization-induced release of tritium from brain preparations preloaded with 3H-serotonin. Göthert and Weinheimer (1979) used rat brain cortex slices, whereas Cerrito and Raiteri (1979) performed their study on synaptosomes (i.e., isolated nerve endings) from rat hypothalamus. In subsequent studies, Manfred Göthert, who was supported by E. Schlicker (since 1980) and K. Fink (since 1986) (see Fig. 1), further characterized the 5-HT autoreceptor, particularly the mechanism involved in its action (Göthert 1980a). In addition, a series of drug tools including agonists (Göthert and Schlicker 1983; Göthert et al. 1987; Schlicker et al. 1992a), antagonists (Schlicker and Göthert 1981; Schlicker et al. 1985a), and a 5-HT uptake inhibitor (important for performing superfusion studies; Classen et al. 1984) was examined. The autoreceptor retains its function in spontaneously hypertensive (SHR; Schlicker et al. 1988a) and even in senescent rats (Schlicker et al. 1989a). The 5-HT autoreceptor could also be identified in the human cerebral cortex (Schlicker et al. 1985b) and hippocampus (Schlicker et al. 1996a) and is likely to be involved in the pathogenesis of mood disorders and in the effect of antidepressant drugs (Groß et al. 1987; Starke et al. 1989); it might be a target for antihypertensive drugs (reviewed in Starke et al. 1989).

Manfred Göthert and his colleagues of the Institute of Pharmacology and Toxicology, University of Bonn. From left: Martin Barann (inset), Dieter Abbo Kalbhen, Karlfried Karzel, Ivar von Kügelgen, Kurt Racké, Gerhard J. Molderings, Manfred Göthert, Eberhard Schlicker, Michael Brüss, Klaus Fink, Markus Kathmann, and Heinz Bönisch. The photograph was taken on October 29, 2002 in front of the main door of the old institute building in Bonn-Poppelsdorf, Reuterstr. 2b
The major scientific topic in the research on the 5-HT autoreceptor was the determination of the 5-HT subtype. This task proved to be very exciting since 5-HT-R classification was still in its beginning at that time. Gaddum and Picarelli (1957) had proposed D-Rs and M-Rs on the basis of organ bath studies in the guinea-pig ileum. In the seventies, the radioligand binding technique was developed and allowed the rapid determination of receptor affinities of huge amounts of drugs. Peroutka and Snyder (1979) suggested 5-HT1-Rs and 5-HT2-Rs on the basis of their experiments with 3H-5-HT and 3H-spiroperidol, respectively. Both nomenclatures show partial overlap only, the D and the 5-HT2-R being very similar. To have a unified nomenclature, the D-Rs and M-Rs were re-named 5-HT2 and 5-HT3, respectively (Bradley et al. 1986).
To determine the pharmacological properties of the 5-HT autoreceptor in the rat brain, Manfred Göthert cooperated with G. Engel and D. Hoyer from Sandoz (now Novartis) in Basle, who contributed radioligand binding studies (see Fig. 2). Comparison of the potencies of agonists and antagonists at the 5-HT autoreceptor revealed identical properties with their affinities at 5-HT1 sites labeled with 3H-5-HT as opposed to their potencies at functional 5-HT2-Rs and 5-HT3-Rs (Engel et al. 1983). Since 5-HT1 sites are not homogeneous (Engel et al. 1983), its two components, termed 5-HT1A and 5-HT1B, were labeled by 3H-8-hydroxy-2-(di-n-propylamino)tetralin (3H-8-OH-DPAT) and 125I-cyanopindolol (in the presence of isoprenaline), respectively, and the autoreceptor could be sub-classified as 5-HT1B (Engel et al. 1986; Table 1). By the way, the latter article is the most frequently quoted original paper by Manfred Göthert (745 citations; Google Scholar, accessed on March 26, 2021).

Manfred Göthert and some colleagues. From left, first line: Jorge Gonçalves, Manfred Göthert, and Daniel Moura; second line: Mark Geyer, Daniel Hoyer, Ewan Mylecharane, David Nelson, Stephanie Watts, and Richard Green. The photograph was taken on occasion of the 1st EPHAR Serotonin Satellite Meeting in Porto (Portugal) in July 2004 organized by the International Society for Serotonin Research (formerly The Serotonin Club). Note that Moura (Molderings et al. 1993) and Hoyer (e.g., Engel et al. 1986) have cooperated with Manfred Göthert
Subsequent research revealed that 3H-5-HT binds to a third 5-HT1-R subtype, termed 5-HT1D, in the bovine brain (reviewed by Peroutka 1988). Since the 5-HT1B-R was found in the brain of rodents but not of other species (reviewed by Peroutka 1988), the pig brain was chosen as a model for the human brain (Fink et al. 1988). An additional cooperation study with D. Hoyer, based on superfusion and binding studies and a biochemical 5-HT1D-R model (inhibition of cAMP formation), revealed that the 5-HT autoreceptor in the pig brain can be classified as 5-HT1D-R (Schlicker et al. 1989b; Table 1).
With the advent of molecular biological methods, it became evident that 5-HT1D-Rs are heterogeneous; the two sub-subtypes were originally termed 5-HT1Dα and 5-HT1Dβ, respectively (Hoyer et al. 1994). Using ketanserin, which has a higher affinity for the former than for the latter receptor (Hoyer et al. 1994), the autoreceptor in not only the guinea-pig cerebral cortex (Bühlen et al. 1996), which served as a model for the human brain, but also in the human cerebral cortex itself could be classified as 5-HT1Dβ-R (Fink et al. 1995a; Table 1).
Comparison of the amino acid sequence of the human 5-HT1Dα-R and 5-HT1Dβ-R revealed an overall identity of 63% only although the pharmacological properties of both receptors are very similar. On the other hand, the amino acid sequence of the human 5-HT1Dβ-R shows an overall identity of 93% with that of the rat 5-HT1B-R; this is in marked contrast to the pronounced difference in the pharmacological properties (Price et al. 1997). Interesting enough, the exchange of one amino acid (Thr355Asn) conferred the pharmacological properties of the rat 5-HT1B to the human 5-HT1Dβ-R (Oksenberg et al. 1992). In other words, the human 5-HT1Dβ-R is the species homolog of the rat 5-HT1B-R and consequently was re-named h5-HT1B-R, whereas the human 5-HT1Dα-R was re-termed h5-HT1D-R (Hartig et al. 1996). In cooperation with D.N. Middlemiss and G.W. Price from SmithKline Beecham (now GlaxoSmithKline) in Harlow, who contributed two selective h5-HT1B-R antagonists (SB-216641, SB-236057) and one selective h5-HT1D-R antagonist (BRL-15572), the final proof that the 5-HT autoreceptor in the human (and guinea-pig) cerebral cortex is h5-HT1B was possible (Schlicker et al. 1997a; Middlemiss et al. 1999; Table 1).
The 5-HT5A-R, which was described for the first time in 1992/3, is Gi/o protein-coupled, like the 5-HT1-R subtypes (reviewed in Göthert et al. 2020). The possibility that it may serve as an additional inhibitory 5-HT autoreceptor was considered in a cooperation project with G. Groß from Abbott in Ludwigshafen. However, a 5-HT autoreceptor can be excluded at least for the mouse brain cortex and hippocampus since the highly selective 5-HT5A-R antagonist A-763079 did not increase 5-HT release nor did it shift the concentration-response curve of the unselective 5-HT-R agonist 5-CT to the right (Drescher et al. 2007).
Presynaptic serotonin heteroreceptors on noradrenergic neurones
The question whether 5-HT-Rs also serve as heteroreceptors on noradrenergic neurones has been studied as well and revealed different results in central and peripheral neurones. In rodent brain cortex slices, neither inhibitory (Göthert and Schlicker 1991) nor facilitatory (Schlicker et al. 1994a) 5-HT-Rs could be identified. By contrast, both inhibitory and facilitatory presynaptic 5-HT-Rs could be identified on sympathetic neurones innervating cardiovascular tissues. The facilitatory 5-HT3-Rs in the rabbit heart have already been discussed above (Göthert and Dührsen 1979).
Much emphasis was put on inhibitory presynaptic 5-HT-Rs. Although such a receptor had already been described in canine blood vessels (McGrath 1977), several new locations (human atrial appendages and saphenous vein; porcine coronary artery; rabbit pulmonary artery; rat vena cava) have been identified by Manfred Göthert; he was supported in this respect by G.J. Molderings (see Fig. 1) since 1986. The possibility that inhibitory 5-HT-Rs on the sympathetic neurones in the heart and in resistance vessels (identified in the pithed rat preparation; Göthert et al. 1986b) are involved in antihypertensive drugs targeting the 5-HT system had to be considered and again precise determination of the 5-HT-R subtype appeared mandatory. Presynaptic receptors were examined in superfused tissues preloaded with 3H-noradrenaline and the potencies of agonists and antagonists were, at least in some of the studies, correlated with their affinities in radioligand studies with native or recombinant 5-HT-Rs. The 5-HT-R in the rat vena cava (Göthert et al. 1986b) could be identified as r5-HT1B-R (Molderings et al. 1987; Table 1) and therefore resembles the autoreceptor in the brain of this species (see above Engel et al. 1986). On the other hand, the 5-HT-R in human atrial appendages (Molderings et al. 1996a; Schlicker et al. 1997a) and most probably also its counterpart in the human saphenous vein (Göthert et al. 1986a; Molderings et al. 1990) are h5-HT1D-Rs (Table 1); thus, they differ from the central autoreceptor, which is a h5-HT1B-R (see above Schlicker et al. 1997a; Middlemiss et al. 1999). One might have expected that the 5-HT-R in the pig coronary artery closely resembles the h5-HT1D-R but surprisingly it could not be ascribed to any of the 5-HT1-R subtypes and in pharmacological terms most closely resembles the 5-HT4-R (Molderings et al. 1989a; Table 1). Finally, the situation is particularly complicated in the rabbit pulmonary artery (Molderings et al. 2006). An inhibitory effect on noradrenaline release occurs indirectly via 5-HT4-Rs and directly via 5-HT1-Rs (Table 1). The 5-HT4-Rs are located presynaptically on cholinergic neurones where they increase acetylcholine release; acetylcholine in turn activates inhibitory muscarinic acetylcholine (mACh)-Rs on the postganglionic sympathetic neurones. By contrast, the inhibitory 5-HT1B-Rs or 5-HT1D-Rs (subtype not determined) are located on the sympathetic neurones themselves. The latter receptors decrease noradrenaline release in the presence of the mACh-R antagonist atropine only. The likely reason is that mACh activation abrogates the 5-HT1B/D-R-mediated effect (an analogous type of receptor interaction has been studied for the α2-AR and the 5-HT1B-R in the rat vena cava; see next paragraph).
The inhibitory effects mediated via presynaptic 5-HT-Rs were less pronounced than the α2-autoreceptor-mediated effects and sometimes were totally missing (human pulmonary artery; Freeman et al. 1981). This phenomenon, which casts some doubt on the physiological relevance of the presynaptic inhibitory 5-HT-Rs, is, however, at least partially related to the experimental conditions. Usually, the electrical stimulation used to evoke quasi-physiological 3H-noradrenaline release is extending over a time period of several minutes and for this reason, released noradrenaline can accumulate in the biophase of the axon terminals of the postganglionic sympathetic neurones; this phenomenon is even aggravated since the experiments are carried out in the presence of an inhibitor of noradrenaline re-uptake. Molderings and Göthert (1990) showed in the rat vena cava that the extent of the 5-HT-R-related inhibition of noradrenaline release was attenuated by α2-AR agonists and increased by antagonists of this receptor suggesting a receptor interaction between the α2-auto- and 5-HT1B-heteroreceptor (reviewed in Schlicker and Göthert 1998). Since α2-AR agonists and antagonists decrease and increase noradrenaline release, the possibility had to be considered that their modulatory effects on the 5-HT-R-related inhibition are related to their effects on noradrenaline release per se rather than to their effects on the α2-ARs. This possibility, however, could be excluded since the alteration of the inhibitory effect of 5-HT also occurred when noradrenaline release was adjusted by modification of the stimulation parameters. The study by Molderings and Göthert (1990) also explains findings in the rat vena cava that the extent of inhibition elicited by the 5-HT1-R agonist RU 24969 in the presence of an α-AR antagonist was much higher than the inhibitory effect of 5-HT in its absence (Schlicker et al. 1988d).
Presynaptic heteroreceptors on serotoninergic neurones
Finally, many efforts were dedicated to the identification of presynaptic heteroreceptors on the serotoninergic neurones in the brain. Manfred Göthert has examined inhibitory (Fig. 3a) and facilitatory heteroreceptors (Fig. 3b) in the rat brain cortex. In cortex slices, the excitatory amino acid glutamate evoked 5-HT release; its facilitatory effect was mimicked by agonists of the three types of ionotropic glutamate-Rs. The facilitatory effect of each of them was markedly inhibited by tetrodotoxin (which inhibits propagation of action potentials), suggesting that part of the AMPA-Rs, kainate-Rs, and NMDA-Rs are located presynaptically on the serotoninergic nerve endings (Fink et al. 1995b; Fig. 3b). Inhibitory presynaptic receptors were identified in slices and/or synaptosomes in which depolarization-induced 5-HT release was studied (Fig. 3a); the presynaptic receptors for histamine (Schlicker et al. 1988b), neuropeptide Y (Michel et al. 1990), and prostaglandins of the E series (Schlicker et al. 1987b) were identified for the first time. Although evidence for the existence of α-ARs on serotoninergic neurones has been presented by other authors before (Starke and Montel 1973; Farnebo and Hamberger 1974), final proof came from the study by Göthert and Huth (1980), in which the interaction of noradrenaline with an α-AR antagonist was studied. This receptor (i) belongs to the α2-AR subtype (Göthert et al. 1981a); (ii) may be subject to an endogenous tone, i.e. is also activated by endogenous noradrenaline (Göthert and Huth 1980; Schlicker et al. 1982; Feuerstein et al. 1993) although the evidence is not unequivocal (Göthert et al. 1981a; Schlicker et al. 1983); (iii) may be inhibitorily coupled to adenylate cyclase (Schlicker et al. 1987a); and (iv) also occurs in the human brain (shown in cooperation with the group of M. Raiteri; Raiteri et al. 1990). The α2-AR-mediated effect was increased when rats had been pretreated with 6-hydroxydopamine (to destroy the noradrenergic neurones) 3 weeks before the experiments and decreased when the animals had received desipramine in the drinking water for 3–4 weeks (Schlicker et al. 1982; Feuerstein et al. 1993). The latter finding might partially explain the delayed effect obtained with antidepressant drugs.

Inhibitory and facilitatory presynaptic heteroreceptors on serotoninergic neurones in rat brain cortex slices identified by Manfred Göthert. a The inhibitory effect of five transmitters or mediators leading to inhibition of the electrically (3 Hz) evoked 3H-5-HT release (the receptors are given in parentheses). The curves were re-drawn from Schlicker et al. (1991)—neuropeptide Y; Schlicker et al. (1987b)—prostaglandin E2; Göthert et al. (1983a)—noradrenaline; Schlicker et al. (1988b)—histamine; Schlicker et al. (1984a)—GABA. b Glutamate and the prototypical agonists at the three ionotropic glutamate receptors (AMPA, kainate, NMDA) facilitate 3H-5-HT release. Re-drawn from Fink et al. (1995b). In both panels, SEM values and statistics are not shown. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; GABA, γ-aminobutyric acid; NMDA, N-methyl-D-aspartate
Molecular vistas
Basic properties of ligand-gated 5-HT3 receptors
When in 1988 H. Bönisch moved from the University of Würzburg to the University of Bonn (to the Institute of Pharmacology and Toxicology, headed by Manfred Göthert since 1985; see Fig. 1), he introduced two important techniques, which led to a lively collaboration between his group (including M. Brüss and M. Barann) and Manfred Göthert. The culture of human or animal cell lines natively expressing a receptor of interest and, much more important, the establishment of molecular biology methods (such as cDNA cloning, site-directed mutagenesis, transfection of cells, and expression of receptors) enabled studies at the cellular and molecular level.
The first common project with Manfred Göthert was the characterization of 5-HT3-Rs in vitro (at the cellular or subcellular level). Altogether we used four different techniques to examine this receptor in rodent cell lines natively expressing the 5-HT3-R or in cells transfected with the cDNA of the mouse or human receptor, namely by measuring (i) the 5-HT-induced influx of a radioactively labeled cation through the cation channel of the receptor (Bönisch et al. 1993), (ii) the binding of a radioligand to the receptor protein (Barann et al. 2004), (iii) 5-HT-induced membrane potential changes in patches of cells by means of patch-clamp techniques (Barann et al. 1997), and (iv) aequorin bioluminescence changes caused by the 5-HT-induced Ca2+ influx in suspended cells expressing the human 5-HT3-R (Walstab et al. 2007). An overview of studies of M. Göthert at 5-HT3-Rs is given in Table 2.
Basic properties of 5-HT3-Rs were initially examined at N1E-115 mouse neuroblastoma cells which natively express this receptor (Lummis et al. 1990). After Reiser and Hamprecht (1989) had shown that 14C-guanidinium is flowing through the open channel of 5-HT3-Rs (expressed in neuroblastoma x glioma hybrid cells), Manfred Göthert, in a collaboration with H. Bönisch (and his group), used this method to examine 5-HT3-Rs in more detail in mouse neuroblastoma N1E-115 cells (Bönisch et al. 1993). We could show that 5-HT and other 5-HT3-R agonists (e.g., phenylbiguanide, 2-methyl-5-HT) cause a concentration-dependent influx of this radioligand which, in contrast to the influx elicited by veratridine, was not inhibited by tetrodotoxin or 5-HT1-R, 5-HT2-R or 5-HT4-R antagonists but inhibited by ondansetron and other selective 5-HT3-R antagonists. All examined 5-HT3-R agonists caused bell-shaped concentration-response curves with slope factors of the ascending part of about 2, indicating rapid desensitization and positive cooperativity. The 5-HT-induced influx of the organic cation 14C-guanidinium was increased in the absence of Ca2+ indicating that Ca2+ accelerates desensitization kinetics. The 5-HT effect was potentiated by the neurokinin substance P and this potentiation was inhibited by ondansetron. This phenomenon had also been shown before by Reiser and Hamprecht (1989), and later Emerit et al. (1993) could demonstrate that in mouse NG108-15 cells, this potentiating effect was even more pronounced with substance P derivatives which are inactive at the various neurokinin-R classes. We additionally showed that substance P also potentiates the 5-HT3-R-mediated Bezold-Jarisch reflex (Malinowska et al. 1996; see the “Cannabinoids” section below and Table 2). In this study, Manfred Göthert concluded that substance P acts at an allosteric modulatory site of the 5-HT3-R, thus, producing an increase in cation flux through this channel, e.g. by affecting its open frequency or duration, without necessarily influencing its ligand recognition (orthosteric) site. We later identified ethanol (and other alcohols) as further positive allosteric modulators at 5-HT3-Rs of N1E-115 cells (Barann et al. 1995). Ethanol not only increased the 5-HT-induced cation influx (without affecting the 5-HT-induced inhibition of 3H-GR65630 binding) but it also abolished the descending part of the concentration-response curve for 5-HT3. The potentiating effect of alcohols (n-alkanols) showed the following rank order: methanol < ethanol < n-propanol, i.e., it increased with their lipophilicity. Interestingly, when in the presence of substance P the 5-HT-induced cation influx was already enhanced, the ability of ethanol to increase the 5-HT-induced influx was considerably diminished. Thus, alcohols (n-alkanols) by interacting with a modulatory hydrophobic region of the 5-HT3-R may either stabilize the open state or decrease desensitization as proposed by Davies (2011) for further positive allosteric modulators of the 5-HT3-R.
In N1E-115 mouse neuroblastoma cells, we studied the influence of sodium ion substitutes on the 5-HT-induced flux of 14C-guanidinium through the cation channel of the 5-HT3-R and on the competition of 5-HT with binding of the selective 5-HT3-R antagonist 3H-GR 65630 (Barann et al. 2004). Replacement of sodium by the organic cation choline caused both a rightward shift of the 5-HT concentration-response curve and an increase in the maximum effect of 5-HT, whereas replacement of Na+ by Li+ had no effect on the potency and maximal response of 5-HT. Replacement by Tris (tris(hydroxymethyl)aminomethane), tetramethylammonium (TMA), or N-methyl-D-glucamine (NMDG) caused an increase in the maximal response to 5-HT similar to that caused by choline. The potency of 5-HT was only slightly reduced by Tris, to a high degree decreased by TMA and choline, but not influenced by NMDG. The potency of 5-HT in inhibiting 3H-GR65630 binding to intact cells was much lower when sodium was replaced by choline, but remained unchanged after replacement by NMDG. These results indicate that NMDG, in contrast to choline, is a suitable sodium substituent for studies of 5-HT-evoked 14C-guanidinium flux through 5-HT3-R channels since it increases the signal-to-noise ratio without interfering with 5-HT binding (Barann et al. 2004). The results also suggest that choline might compete with 5-HT for binding to the 5-HT3-R and that the increased maximum response may be partly due to a choline-mediated delay of the 5-HT-induced desensitization.
Using the same techniques and cells, we examined several pharmacologically active compounds for their affinity to this receptor. In N1E-115 cells (and in rat brain cortical membranes), anpirtoline, a highly potent 5-HT1B-R agonist, behaved as 5-HT3-R antagonist (Göthert et al. 1995a). Both the 5-HT3-R channel and the voltage-gated sodium channel of N1E-115 cells were shown to be targets of steroids; however, their interaction is obviously due to a non-specific hydrophobic effect (Barann et al. 1999). Furthermore, imidazolines (e.g., idazoxane, cirazoline, or clonidine) as well as some σ ligands (e.g., ifenprodil) showed low inhibitory potencies at 5-HT3-Rs and it was suggested that they may exert their inhibitory effect on cation influx through the 5-HT3-R channel, at least in part, by interacting with σ2 binding sites (Molderings et al. 1996b).
By the installation of a patch-clamp workstation in his institute, Manfred Göthert initiated the establishment of the patch-clamp technique which finally was introduced by M. Barann. In superfused outside-out patches of N1E-115 cells, we examined further basic properties of the mouse 5-HT3-Rs in detail (Barann et al. 1997). We could show that at negative membrane potentials, 5-HT caused concentration-dependent inward currents which were characterized by a Hill coefficient of 1.8 and a peak current of about 21 pA at a high concentration of 5-HT (30 μM). We furthermore demonstrated that the currents induced by 30 μM 5-HT (for 2 s) were characterized by inward rectification, a monophasic onset, and a monophasic decay (desensitization), and that after a short washout period, fully desensitized patches completely recovered (Barann et al. 1997). In this study, we also demonstrated that pentobarbital causes inhibition of the 5-HT3-R through an open channel block. 5-HT-induced influx of 14C-guanidinium as well as patch-clamp techniques were used to characterize the effects of further anesthetics at the cation channel of the 5-HT3-R. By measuring the influx of the organic cation 14C-guanidinium induced by either veratridine or 5-HT, the influence of local and general anesthetics on cation influx through the fast, voltage-dependent sodium channel and through the 5-HT3-R cation channel was studied in N1E-115 mouse neuroblastoma cells (Barann et al. 1993). The 14C-guanidinium influx through both channels was inhibited by local and general anesthetics. With the exception of procaine and cocaine, which were equipotent at both channels, the local anesthetics were 4.4-fold (lidocaine) to 25-fold (tetracaine) more potent at the fast sodium channel than at the 5-HT3-R channel. The rank order of potency for general anesthetics was clearly different at the two channels. With the exception of ketamine, which was about equipotent at both channels, the general anesthetics were between 2.2 and 8.1-fold more potent at the 5-HT3-R channel than at the fast sodium channel and only at the fast sodium channels, their inhibitory potency was correlated with their lipophilicity. Thus, the relative high inhibitory potencies of the general anesthetics argue in favor of a specific interaction with the 5-HT3-R channel (Barann et al. 1993). Using the patch-clamp technique, we re-examined the abovementioned effects of ifenprodil (Molderings et al. 1996b) and we could show that it inhibited the peak currents evoked by 5-HT and that it also produced a concentration-dependent increase of the onset time constant (τON) of the 5-HT-induced currents and that ifenprodil accelerated current inactivation as reflected by a decrease of the current inactivation time constant (τOFF) (Barann et al. 1998).
Molecular biology of 5-HT3 receptors
In 1991, the first findings on the molecular biology of 5-HT3-Rs have been published (Maricq et al. 1991). Maricq and coworkers had cloned this receptor from mouse DNA and it was termed as 5-HT3A; thereafter, the human 5-HT3A cDNA was cloned by Miyake et al. (1995) and Belelli et al. (1995). All cloned 5-HT3A-R cDNAs show a high degree of amino acid identities of more than 80%. Hydrophobicity analysis of the deduced amino acid sequences predicts the receptor subunits to be integral membrane proteins with a large extracellular N-terminus, 4 transmembrane domains (TMs), a large intracellular loop between TM3 and TM4, and a short extracellular C-terminus (see Fig. 4). The ligand-binding domain is proposed at the N-terminal part (containing a Cys-loop, i.e. a cystine pair separated by 13 amino acids, conserved among all ligand-gated ion channels), and TM2 is the putative channel pore-forming domain of this homopentameric ion channel which is closely related to the α-subunit of the nACh-R (Ortells and Lunt 1995). When we cloned the human cDNA of the 5-HT3A-R from human amygdala, we amplified three cDNAs of different length, one corresponded to the already known cDNA, whereas the other two were a shorter and a longer alternative splice product (Brüss et al. 1998), and only the longer isoform with an insertion of 96 base pairs leading to the insertion of 32 amino acids into the extracellular loop between TM2 and TM3 was able to form an active receptor protein (Brüss et al. 1998). Both splice variants did not correspond to known mouse isoforms (Jackson and Yakel 1995). We cloned the short splice variant of the mouse 5-HT3-R (Hope et al. 1993), expressed the receptor protein in human embryonic kidney (HEK293) cells, and compared its pharmacological properties with those of the native mouse 5-HT3-R in N1E-115 neuroblastoma cells by means of 3H-GR65630 binding and 5-HT-induced 14C-guanidinium influx measurements (Brüss et al. 1999a). The differences between the two isoforms were, however, only marginal and may be due to cell-specific post-translational modifications of the receptor protein in the two cell types (Brüss et al. 1999a). To identify potential alternative exons, we sequenced all exons and introns, the length and positions of all introns of the coding region,and about 19 kb of the 5′-noncoding region of the human 5-HT3A-R gene (Brüss et al. 2000a). The human gene stretches over about 14.5 kb. From three published human 5-HT3A-R cDNAs, we could confirm only that reported by Miyake et al. (1995); the coding region of the human 5-HT3A-R gene is separated by eight introns located at positions nearly identical to those of the murine counterpart (Werner et al. 1994). The length of most introns differs markedly from those of the murine counterpart. Exon 1 encodes the membrane translocation, exons 2 to 6 encode the extracellular N-terminus, exon 7 encodes TM1, TM2, and the extracellular loop between TM2 and TM3, exon 8 codes for TM3, and exon 9 for the large intracellular loop between TM3 and TM4 as well as TM4 and the extracellular C-terminus (see Fig. 4).

a Genomic organization of the human 5-HT3A receptor gene (HTR3A) with exons (indicated by numbers), localization of the Cys-loop (cystein bond within the N-terminal region) and the four transmembrane regions (TM1-TM4), and the organization of the long (HTR3AL) and of the truncated (HTR3AT) splice variant. b The corresponding protein structure and the two naturally occurring 5-HT3A receptor variants Arg344His and Pro391Arg due to single-nucleotide polymorphisms of the HTR3A gene. c The protein structures of the human 5-HT3 receptor subtypes. UTR, untranslated region. All these variants and subtypes have been examined by Manfred Göthert (see text)
In outside-out patches of stably transfected HEK293 cells expressing the recombinant human 5-HT3A-R, we characterized basic properties of this receptor and we compared the effects of the barbiturate anesthetics methohexital and pentobarbital (which differ in their lipophilicity) on this receptor channel (Barann et al. 2000). Both anesthetics inhibited the 5-HT response with about equal potency but they clearly differed with respect to the kinetics of their effects indicating that lipophilicity may affect their access to an amphipathic site of action via both a hydrophilic and a hydrophobic pathway.
Of major scientific interest for Manfred Göthert was the exploration of functional consequences of genetic variations of human 5-HT-Rs. Concerning the human 5-HT3A-R, he was involved in the pharmacological characterization of two naturally occurring variants of the human 5-HT3A-R, a Pro391Arg and an Arg344His variant. Both had been detected in schizophrenic patients when the human 5-HT3A-R gene was screened for variations in schizophrenic patients and patients suffering from bipolar affective disorder (Niesler et al. 2001). Both missense mutations are located in the second intracellular loop of the receptor protein (see Fig. 4). The variant Pro391Arg receptor was examined in comparison to the wild-type form (each expressed in stably transfected HEK293 cells). In binding experiments with 3H-GR65630, the variant receptor exhibited no changes in receptor densities or affinities to diverse 5-HT3-R agonists and antagonists, and also the patch-clamp experiments showed no differences between the wild-type and variant receptor (Kurzwelly et al. 2004). Combrink et al. (2009) compared the other receptor variant (Arg344His) in transfected HEK293 cells and in comparison with the wild-type receptor. This comparison was performed using 3H-GR65630 binding and patch-clamp analyses including technically demanding single-channel analyses. In addition, 5-HT-induced Ca2+ currents through the 5-HT3A-R channel were measured by an aequorin luminescence-based Ca2+ assay which previously had been established in our group (Walstab et al. 2007). Compared to the wild-type receptor, the density of the variant receptor was decreased by nearly 50%, whereas the Ca2+ influx was unchanged. While the radioligand experiments revealed no differences for several agonists and antagonists between wild-type and variant receptor, single-channel analysis suggested an increase in channel open time; this increase appears to compensate for the reduction in variant receptor density.
In 1999, a further 5-HT3-R subunit, the 5-HT3B-R, was identified (Davies et al. 1999; Dubin et al. 1999), which, in contrast to the 5-HT3A-R, is not able to form a functional homopentameric receptor but which was able to cause, when co-expressed with the 5-HT3A subunit, subtle modifications in 5-HT3-R agonist and antagonist effects; in addition, heteromeric assemblies of human 5-HT3A and 5-HT3B subunits display larger single-channel conductance than homopentameric assemblies of 5-HT3A subunits (Dubin et al. 1999). Shortly after this report, we described a human short, truncated (5-HT3AT) and a long (5-HT3AL) splice variant of the human 5-HT3A-R subunit (Brüss et al. 2000b). The protein of the short isoform consists of only 238 amino acids with a single transmembrane domain (TM1), whereas the long isoform contains 32 additional amino acids within the extracellular loop between TM2 and TM3 (see Fig. 4). Both splice variants are co-expressed in the amygdala and hippocampus, whereas in the placenta, only the short splice variant is co-expressed (Brüss et al. 2000b). When expressed in transfected HEK293 cells, both splice variants are not able to form a functional receptor, but modify 5-HT responses at heteromeric 5-HT3A-Rs. Heteromeric assemblies of 5-HT3A and the 5-HT3AT subunit exhibit much larger 5-HT-induced cation fluxes than homomeric 5-HT3A-Rs, whereas heteromeric receptors containing the long splice variant display reduced cation fluxes (Brüss et al. 2000b). Thus, tissue-selective expression of 5-HT3A splice variants may contribute to the functional diversity of this receptor.
Using the aequorin luminescence-based Ca2+ assay, which had been shown to be a highly sensitive method for functional characterization of 5-HT3-Rs and which allows high-throughput screening (Walstab et al. 2007), we characterized three novel human 5-HT3-R subunits, 5-HT3C, 5-HT3D, and 5-HT3E (Niesler et al. 2007). The proteins of these novel genes, which had been isolated by Niesler et al. (2003), show the following structures: 5-HT3C and 5-HT3E present a huge N-terminal extracellular segment containing a cysteine loop, four hydrophobic TMs, a large intracellular loop between TM3 and TM4, and an extracellular C-terminus (see Fig. 4). The architecture of the 5-HT3D subunit (Fig. 4) is rather different, since it lacks the signal sequence and the large N-terminal region, including the ligand-binding site, indicating that it may not form a functional ion channel. Interestingly, the genes of the 5-HT3D and 5-HT3E subunits are predominantly or even exclusively (5-HT3E) expressed in the gastrointestinal tract (Niesler et al. 2003). Using immunofluorescence and immunoprecipitation of recombinantly expressed proteins, we explored whether they are able to form 5-HT3-Rs. Radioligand binding experiments and aequorin luminescence-based Ca2+ assays were performed to reveal whether they modulate 5-HT3-R function. We found that each of the respective candidates coassembled with 5-HT3A. The functional experiments revealed that the 5-HT3C, 5-HT3D, and 5-HT3E subunits alone cannot form functional receptors. Co-expression with 5-HT3A, however, results in the formation of functional heteromeric 5-HT3-Rs, which exhibit quantitatively different properties compared with homomeric 5-HT3A-Rs (Niesler et al. 2007). An excellent review on genetics, molecular biology, physiology, and pharmacology of 5-HT3-Rs has been published by Walstab et al. (2010). It should be noted that M. Brüss (together with M. Göthert and H. Bönisch) had an intense collaboration on 5-HT3-Rs with B. Niesler (Department of Human Genetics, University of Heidelberg). The PhD student, J. Walstab, involved in this project was later working as postdoc in Niesler’s laboratory when this collaboration between the universities of Bonn and Heidelberg was continued after the sudden and unexpected death of M. Brüss.
Variants of metabotropic 5-HT receptors
Within a collaborative research center (SFB 400: Molecular Basis of CNS Disorders) at the University of Bonn, Manfred Göthert was involved in the characterization of naturally occurring variants of the human metabotropic 5-HT1A-Rs, 5-HT1B-Rs, 5-HT2C-Rs, and 5-HT7-Rs. Genetic variations in 5-HT-Rs might contribute not only to genetics of diseases but also to changes in pharmacological properties of these receptors (for a short review, see Göthert et al. 1998). Table 1 shows an overview of all studies of M. Göthert at metabotropic 5-HT-Rs.
Central 5-HT1A-Rs act as somadendritic autoreceptors on serotoninergic neurones and in many brain regions, this receptor has been identified in high density, e.g. in the hippocampus and amygdala where it has been assumed to be involved in the regulation of mood and anxiety. This receptor is a target for anxiolytic and antidepressant drugs (Hamon 1997; Blier and De Montigny 1997; Kaufman et al. 2016). 5-HT1A-Rs are preferentially coupled to Gi/o proteins to inhibit adenylate cyclase but can also be coupled to inwardly rectifying potassium channels mediating hyperpolarization (Andrade and Nicoll 1986; Albert and Vahid-Ansari 2019). In a systematic screening for mutations in the promoter and coding regions of the human 5-HT1A gene, Erdmann et al. (1994) identified two naturally occurring receptor variants with either Ile28Val (in the N-terminus) or Arg219Leu (in the third intracellular loop) substitutions (Table 1, Fig. 5); the allele frequency of the Ile28Val and the Arg219Leu variants is about 8% and <1%, respectively. In transfected COS-7 cells, we could show that the Ile-28-Val exchange had no effect on receptor expression or on the affinities (measured in 3H-8-OH-DPAT binding experiments) of a series of agonists or antagonists at 5-HT1A-Rs (Brüss et al. 1995). However, the Arg-219-Leu exchange examined later in transfected HEK293 cells by 3H-8-OH-DPAT and 35S-GTPγS binding (a measure of G protein coupling) to membranes as well as inhibition of forskolin-stimulated 3H-cAMP formation by agonists (in whole cells) revealed an impairment of signal transduction (Brüss et al. 2005a). While the variant receptor did not differ from the wild-type receptor with respect to receptor density or potencies of agonists or antagonists, the ability of 5-HT to stimulate 35S-GTPγS binding to the variant receptor and of agonists to inhibit forskolin-stimulated cAMP accumulation was decreased by 60-90% (Brüss et al. 2005a). Interestingly, in an association study of major depression with this Arg219Leu variant, we could show that this receptor variant is associated with major depression and that it may play a role in the pathogenesis of depression (Haenisch et al. 2009).

Schematic diagram of a human metabotropic 5-HT receptor in which amino acid exchanges and their position in naturally occurring variants of the 5-HT1A, 5-HT1B, 5-HT2C, and 5-HT7 receptor are indicated. Manfred Göthert has explored all shown variants (see text)
A further naturally occurring 5-HT-R variant examined by Manfred Göthert was the human 5-HT1B-R in which Phe in position 124 (within the third transmembrane domain) is substituted by Cys (Table 1, Fig. 5). The allele frequency of this variation is 2%. In transfected COS-7 cells, the Phe124Cys variant, in comparison to the wild-type receptor, showed a reduction by 70% of surface expression (Bmax) and two to three times higher affinity for several agonists (e.g., 5-CT or 5-HT) in radioligand binding experiments with 3H-5-carboxamidotryptamine (3H-5-CT) (Brüss et al. 1999b). This result was confirmed in a second study using transfected C6 glioma cells; this study additionally showed an about 50-65% lower efficacy of agonists (such as 5-CT, 5-HT, or sumatriptan) in stimulating 35S-GTPγS binding to membranes of cells expressing the Phe124Cys receptor variant (Kiel et al. 2000). In whole cells expressing the variant receptor, 5-CT and sumatriptan inhibited the forskolin-stimulated cAMP accumulation 3.2-fold more potently than in cells expressing the wild-type receptor. Thus, the Phe-124-Cys mutation modifies the pharmacological properties of the 5-HT1B receptor and may account for pharmacogenetic differences in the action of 5-HT1B-R ligands (Kiel et al. 2000).
Manfred Göthert proposed that the sumatriptan-induced vasospasm, which occurs at low incidence as a side effect in migraine therapy, may at least partly be related to the expression of the Phe124Cys variant of the h5-HT1B-R in patients with additional pathogenetic factors such as coronary heart disease. This proposal was later tested in human temporal arteries from patients undergoing neurosurgery. These arteries were used to examine whether in vivo expression of the Phe124Cys 5-HT1B-R variant (Cys/Phe genotype) modifies 5-HT-induced constriction (mediated not only by 5-HT1B but also by co-expressed 5-HT2A-Rs). It was shown that in arteries from Cys/Phe individuals, the contribution of 5-HT1B-Rs to the mediation of the effects of 5-HT was increased (Verheggen et al. 2006; Table 1).
5-HT2C-Rs are widely expressed in the central nervous system and appear to play an important role in psychiatric disorders and drug dependence (Giorgetti and Tecott 2004; Chagraoui et al. 2016). The pre-mRNA of the 5-HT2C-R undergoes post-transcriptional editing resulting in diversity among RNA transcripts and 5-HT2C-Rs are heterogeneous due to alternative splicing (Werry et al. 2008; Bass 2002; Wang et al. 2000). In addition, a single-nucleotide polymorphism (SNP) in the 5-HT2C-R gene, leading to substitution of cysteine 23 to serine (Cys23Ser) in the N-terminal domain of the 5-HT2C-R (Table 1, Fig. 5), had been found to be associated with neuropsychological diseases (Lerer et al. 2001) and to alter the response to clozapine (Segman et al. 1997). An allele frequency of about 13% has been found for this variant. Since published results concerning the functional properties of the two isoforms were inconsistent, Manfred Göthert examined, in more detail, the wild-type and the Cys23Ser variant of the 5-HT2C-R in transiently transfected HEK293 cells with respect to function (by an aequorin luminescence-based Ca2+ assay) and to surface expression (by means of 3H-mesulergine binding) (Walstab et al. 2011). Surface expression of the Cys23Ser variant was found to be 116% of that of the wild-type receptor. No difference was observed between wild-type and variant receptor concerning 5-HT-induced increase in cytosolic Ca2+ and its inhibition by the inverse agonist SB206553. Furthermore, no difference between wild-type and variant receptor was observed in the time-dependent reduction of 5-HT-induced increase in cytosolic Ca2+, i.e. of the rapid and strong receptor desensitization due to preexposure of the cells to 5-HT. On the other hand, prolonged preexposure to SB206553 caused resensitization of the receptor, i.e., elevation of the Ca2+ response. However, at the variant receptor, this elevation was seen already within 1 h, whereas at the wild-type receptor, a preexposure time of 4.5 h was needed for this effect to occur. The different time course of SB206553-induced resensitization of the two isoreceptors might be therapeutically relevant for some atypical antipsychotics (such as clozapine) and certain antidepressants (such as mirtazapine) acting as inverse agonists at 5-HT2C-Rs. Prolonged preexposure to an inverse agonist is assumed to reduce the constitutive activity of the 5-HT-R in vivo, thereby increasing receptor responsiveness to classical agonists (Walstab et al. 2011).
The human 5-HT7-R, first described by Bard et al. (1993), was the most recently identified member of the 5-HT-R family. This receptor, which is coupled to Gs protein to stimulate cAMP formation, is expressed in the central nervous system, e.g., in the thalamus, hypothalamus, hippocampus, cerebral cortex, amygdala, and dorsal raphe; it is involved in circadian rhythm by acting at the suprachiasmatic nucleus (Lovenberg et al. 1993) and seems to play a role in the action of antipsychotics and antidepressants (Matthys et al. 2011). Alternative splicing at the second intron, located at the C-terminal end of the 5-HT7-Rs, gives rise to three splice variants (5-HT7(a,b,d), Heidmann et al. 1997) which show the same pharmacological properties; among these splice variants, the 5-HT7(a) is the most abundant isoform (Gellynck et al. 2013). All three splice variants have very similar abilities to stimulate adenylyl cyclase in HEK293 cells (Krobert et al. 2001), indicating that the C-terminal tail does not influence ligand binding or G protein coupling. Systematic mutation screening in patients suffering from schizophrenia or bipolar affective disorder revealed an SNP leading to the exchange of proline against leucine in position 279 in the third intracellular loop of the receptor protein (Table 1, Fig. 5; Erdmann et al. 1996); interestingly, both control individuals and patients exhibited the same allele frequency (1%). Manfred Göthert has studied this Pro279Leu variant in comparison to the wild-type receptor in transfected HEK293 cells by means of binding of 3H-5-CT to membranes and stimulation of 3H-cAMP formation in whole cells evoked by 5-HT-R agonists (Kiel et al. 2003). All agonists and antagonists examined exhibited no difference in affinity between the variant receptor and the wild-type receptor. However, in cells expressing the Pro279Leu variant, the intrinsic activity of all agonists examined in stimulating 3H-cAMP accumulation was almost abolished and their potency was 2.9–4.3-fold lower.
The mutation screening study of Erdmann et al. (1996; see above) additionally revealed an SNP leading to the exchange of threonine against lysine in position 92 located in the first transmembrane domain of the receptor protein (Fig. 5). The allele frequency of this variant was <1% for both control individuals and patients (Erdmann et al. 1996). In HEK293 cells transfected either with the wild-type cDNA or that of the Thr92Lys variant, we determined binding of 3H-5-CT to membranes and stimulated 3H-cAMP accumulation in whole cells (Brüss et al. 2005b). The variant receptor exhibited 3–11 times lower binding affinity to the agonists 5-HT, 5-CT, 8-OH-DPAT, sumatriptan, and RU24969 compared to the wild-type receptor. The variant receptor, however, did not differ from the wild type with respect to the binding properties of antagonists such as risperidone, mesulergine, clozapine, and SB-269970. In agreement with the decreased binding affinity of 5-HT, 5-CT, RU24969, and 8-OH-DPAT for the variant receptor, these agonists were less potent in stimulating 3H-cAMP accumulation in cells expressing the variant receptor. Sumatriptan did not stimulate cAMP accumulation in spite of its affinity for both receptor isoforms pointing to a weak antagonistic property of this drug at the 5-HT7-R. SB-269970 and clozapine were equipotent at both the variant and the wild-type receptor in producing a rightward shift of the 5-HT concentration-response curve for its stimulant effect on 3H-cAMP accumulation. Thus, the results of our two studies (Kiel et al. 2003; Brüss et al. 2005b; Table 1) may have relevance for drugs acting on 5-HT7-Rs which affect circadian rhythm.
Beyond serotonin
Carbon monoxide toxicology
When Manfred Göthert joined the Institute of Pharmacology at the University of Hamburg in 1967, he first did not deal with serotonin and not even with pharmacology. As a member of the group of G. Malorny, then head of department, the first scientific studies of Manfred Göthert were dedicated to carbon monoxide toxicology. Experiments on animals exposed to carbon monoxide revealed that the partial pressure of CO in the pneumoperitoneum (which served to quantify the partial pressure of CO in tissue) is the lower, the higher its affinity for hemoglobin is. This was shown for not only rats (high), guineapigs (high), and rabbits (low hemoglobin affinity) (Göthert et al. 1968, 1970) but also for single rabbits, which for unknown reasons markedly differed in their hemoglobin affinity (Göthert and Malorny 1969). Moreover, the partial pressure of CO in the pneumoperitoneum of rabbits exposed to air containing 1000 parts per million (ppm) of CO at normal (1 bar) or increased pressure (3 or 4 bar) was higher in the former than in the latter groups (Gerhardt et al. 1972). Although these findings may appear strange at first glance, they can easily be explained by the law of mass action; carbon monoxide and oxygen compete for the same receptor (hemoglobin).
CO toxicology was also studied in humans. In volunteers who breathed air containing 50 ppm CO under normal (1 bar) and hyperbaric (3 bar) pressure over a time period of 2 h, the percentage of CO hemoglobin in the group with normal pressure (7%) was higher than in the group with increased pressure (5%) (Gerhardt et al. 1971). This result is practically relevant since the possibility had to be considered that Caisson workers might suffer from an increased CO-hemoglobin concentration. On the basis of this study, a reduction of the threshold limit value (maximale Arbeitsplatzkonzentration, MAK) for CO in Caisson workers did not appear to be necessary. In another study (Bender et al. 1971), human volunteers exposed to air containing 100 ppm CO over a time period of 2.5 h showed a CO concentration of 55 ppm in the alveolar air and exhibited reduced visual perception, manual dexterity, and ability to learn and to perform certain intellectual tasks. This study clearly confirmed that the reduction of the MAK for CO in Germany from 100 to 50 ppm in 1966 was really justified.
Ethanol, general anesthetics, gabapentinoids, and ion channels
General anesthetics, alcohols, and ligand-gated cation channels
In parallel to the toxicological studies on CO, Manfred Göthert became interested in the mode of action of general anesthetics. Although this work is apparently unrelated to 5-HT-Rs, it was an important period of his work and Manfred Göthert remained life-long interested in the cardiovascular and neuronal effects of general anesthetics and alcohols. Specifically, this early work was continued in Bonn 15 years later with K. Fink on ionotropic glutamate-Rs and with H. Bönisch, M. Brüss, and M. Barann on 5-HT3-Rs. In the early 70s, not much was known on the mode of action of general anesthetics; while it is even today not completely clear for inhalational anesthetics, for the injectable anesthetics propofol and etomidate, the GABAA-R with a specific subunit composition has been found as the predominant target. According to the current understanding, all ligand-gated ion channels are targets of anesthetic drugs, however, in a more specific way than general hydrophobic interaction with cell membrane or membrane proteins according to the Meyer-Overton correlation (for review, see Franks 2006; Lynch 2008). Most attention for all anesthetics has been attracted by not only interactions with the GABAA-R but also the other ligand-gated ion channels as well as Na+ and K+ channels (Rudolph and Antkowiak 2004; Hemmings et al. 2019). After diethylether had been widely administered as an anesthetic since Morton’s demonstration in the Massachusetts General Hospital in Boston in 1846, halothane was introduced in Germany in 1956, methoxyflurane in 1960, and enflurane in 1966. These inhalative anesthetics were frequently used in anesthesiology around 1970 when Manfred Göthert started to work on it; obviously other inhalative anesthetics such as isoflurane, desflurane, or sevoflurane that were less metabolized followed later.
Manfred Göthert presented first results of this work at the 10th Spring Meeting of the German Pharmacological Society in Mainz 1969 (Göthert and Benthe 1969). In diethylether-anesthetized guinea-pigs or rats, noradrenaline and adrenaline concentrations increased in the heart but decreased in the adrenal medulla, whereas in halothane anesthesia, noradrenaline only initially decreased in the heart and in chloroform anesthesia, noradrenaline decreased in the heart for the entire duration of application (Göthert 1971). It explained the overall less pronounced cardiosuppressive effects of diethylether vs. chloroform or halothane but did not reveal the underlying mode of action of these general anesthetics. In a series of further studies, he provided an in-depth analysis of the differential effects of diethylether, chloroform, halothane, enflurane, or methoxyflurane on catecholamine release in the adrenal medulla and myocardium and the resulting cardiovascular effects (Göthert and Tuchinda 1973; Benthe et al. 1973; Göthert et al. 1974a, b; Schmoldt and Göthert 1974). The overall negative chronotropic effect of halothane could be attributed to the inhibition of catecholamine release from adrenal medulla (30%), the inhibition of noradrenaline release from sympathetic nerve terminals in the heart (25%; Table 3), a direct effect on the myocardium (25%), and a stimulatory effect on parasympathetic nerves (20%; Göthert and Tuchinda 1973). In vitro in cat heart, the order of negative ionotropic effects, i.e., halothane > chloroform > diethylether, was confirmed (Benthe et al. 1973). In 1974, he showed the inhibition of ACh-stimulated catecholamine release from isolated bovine adrenal medulla by inhalation anesthetics (halothane (Table 3), methoxyflurane, chloroform) and aliphatic alcohols (n-propanol, n-butanol, n-pentanol, n-hexanol) and concluded, as mode of action, a hydrophobic interaction with membranes correlating to the membrane-buffer partition coefficients (Göthert et al. 1974a). The catecholamine release from cat adrenal medulla after splanchnic nerve stimulation was largely reduced by chloroform and ether (Göthert et al. 1975). However, this could not be attributed at in vivo applied anesthetic concentrations to a reduced catecholamine synthesis in the adrenal medulla (Schmoldt and Göthert 1974). The spontaneous release of catecholamines from the adrenal medulla was concentration-dependently decreased by halothane (Göthert and Dreyer 1973) and, similarly, by methoxyflurane (Dreyer et al. 1974). Halothane decreased the coronary flow in the isolated rabbit heart perfused at constant pressure (Göthert and Guth 1975) presumably by reducing the vasoconstrictor effect of noradrenaline on coronary arteries. As only the nACh-R-mediated noradrenaline release from myocardial sympathetic nerve terminals was inhibited by halothane and not the release induced by high K+, he concluded that halothane might cause a conformational change most potently of the nACh-R protein (Göthert et al. 1974b; Göthert 1974; Table 3), an idea which is still assumed to be a major part of the anesthetics’ mechanism. In the adrenal medulla, he located the effect of halothane to the cell membrane of chromaffin cells or, more specifically, to membrane proteins such as the nACh-R or, at lower potency, GABA-Rs and 5-HT-Rs (Göthert et al. 1976a; Table 3). An investigation of the mode of action of the cardiovascular effects of enflurane resulted in the same conclusion, i.e. that enflurane interacted with hydrophobic regions of the nACh-R while mACh-Rs were much less sensitive to enflurane (Göthert and Wendt 1977a, b).
Besides the work on inhalative anesthetics, Manfred Göthert also studied the mode of action of the injectable anesthetic pentobarbital where he also identified as the most potent effect the inhibition of nACh-R-mediated noradrenaline release from rabbit cardiac sympathetic nerves which is still in line with current understanding of barbiturate action (Ye and Ewing 2018); the inhibition of high K+ or electrically evoked noradrenaline release occurred at 10-30 times higher pentobarbital concentrations (Göthert and Rieckesmann 1978; Table 3).
In 1976, he extended the idea of a hydrophobic interaction of the inhalative anesthetics with the nACh-R to the alcohols ethanol, 1-propanol, 1-butanol, and 1-pentanol using dimethylphenylpiperazine-induced or ACh-induced noradrenaline release from isolated perfused rabbit heart; he suspected an altered agonist-receptor interaction (Göthert and Kennerknecht 1975; Göthert et al. 1976b). In these experiments, he found a threshold concentration of 36 mM ethanol for the inhibitory effect on nACh-Rs which may be achieved in in vivo intoxications (Göthert and Thielecke 1976; Table 3). After Manfred Göthert had become Professor of Pharmacology in Bonn, this work was continued on another ligand-gated ion channel, the N-methyl-D-aspartate (NMDA)-R. K. Fink and Manfred Göthert established and characterized the model of NMDA-R-mediated noradrenaline release in brain slices (Fink et al. 1989, 1990c) and supported it by radioligand binding studies (Fink et al. 1992c). They used the functional approach of NMDA-evoked noradrenaline release to demonstrate the inhibitory effect of ethanol on NMDA-Rs. The threshold concentration was between 10 and 32 mM which was definitely in the range of clinical ethanol intoxications (Göthert and Fink 1989; Table 3, Fig. 6). The experiments were repeated with the aliphatic alcohols methanol, ethanol, 1-propanol, 1-butanol, 1-pentanol, and 1-hexanol and demonstrated a strong correlation of the alcohols’ membrane-buffer partition coefficients and their inhibitory potencies (Fink and Göthert 1990, 1991); ethanol turned out slightly more potent than the overall correlation between hydrophobicity and potency indicating a more specific interaction. As soon as they identified presynaptic NMDA-Rs on noradrenergic axonal terminals in rat neocortex using a synaptosomal preparation (Fink et al. 1990b; Göthert and Fink 1991), they could also demonstrate the inhibitory effect of ethanol on presynaptic NMDA-Rs (Fink and Göthert 1992). The existence of the ionotropic glutamate-Rs NMDA, kainate, and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) was also shown in the human neocortex, including the inhibitory effects of ethanol at concentrations that were observed in human intoxications in vivo (Fink et al. 1992a, b; Table 3). All these data of Manfred Göthert’s group contributed strong evidence to the idea that general anesthetics and alcohols have rather individual effects on different neuronal target structures as compiled in box 2 of the review by Rudolph and Antkowiak (2004).

Effect of ethanol and ifenprodil on the NMDA-evoked release of various transmitters from rat cerebral cortex and/or striatal slices. a and b The concentration-response curves (CRCs, SEM values not shown). The negative logarithms of the concentrations causing the half-maximum effect, i.e. an inhibition by 50% (ethanol, pIC50%) and by 20% (ifenprodil, pIC20%), were correlated with each other as shown in c, yielding a correlation coefficient close to 1. This suggests that ethanol acts on NMDA receptors containing an N2B subunit which is inhibited by ifenprodil in noradrenergic, serotoninergic, and GABAergic neurones. Closed circles, cortical slices preincubated with 3H-noradrenaline; closed squares, cortical slices, 3H-5-HT; closed rhomboids, cortical slices, 3H-GABA; open circles, striatal slices, 3H-dopamine; open triangles, striatal slices, 3H-choline; open squares, striatal slices, 3H-5-HT (CRCs not shown); open rhomboids, striatal slices 3H-GABA (CRCs not shown). Re-drawn from Fink and Göthert (1996). d The structure of NMDA receptors (which occur as di-heteromeric or tri-heteromeric tetramers) and the site of action of ifenprodil. GABA, γ-aminobutyric acid; NMDA, N-methyl-D-aspartate
Although the interaction may be partially or even predominantly hydrophobic by nature, it still depends on size, type, and exposition of the hydrophobic patches or uncharged sequences of the individual receptor protein. In order to explain the differing sensitivities to ethanol of NMDA-Rs on noradrenergic, serotonergic, GABAergic, dopaminergic, and cholinergic neurones, K. Fink and Manfred Göthert compared the ethanol effects to the respective effects of the NR2B (current nomenclature GluN2B) subunit preferential receptor antagonist ifenprodil. NMDA-Rs are tetramerically composed of two obligatory GluN1 and two GluN2 or GluN3 subunits; GluN2 exists in four subtypes A-D and GluN3 in two subtypes A and B (Paoletti et al. 2013) while the specific role of different NMDA-R assemblies is not completely clear. They found a strong correlation (0.96; p<0.001) of ethanol and ifenprodil potency and concluded that ethanol predominantly inhibits NR2B-assembled NMDA-Rs on noradrenergic, serotoninergic, and GABAergic neurones (Fink and Göthert 1996) (Fig. 6).
During these experiments, the potential effect of hyperosmolarity was discussed within the group and, as few agents e.g. sugars, urea, and ethanol can cause serious hyperosmolar disorders in patients, the question was addressed with D-glucose. Interestingly, high K+-evoked GABA release in neocortex was strongly increased by D-glucose ≥32 mM, that of acetylcholine was unaffected, and that of noradrenaline and 5-HT was decreased (Fink and Göthert 1993a). The increased GABA release remained elusive but might result from the blockade of ATP-sensitive K+ channels by increased ATP levels. It was concluded that an interaction between GABAergic interneurones and other neurones downstream was the underlying mechanism that increased GABA release and that this phenomenon could explain some of the symptoms in hyperosmolar diabetic coma (Fink et al. 1994a). The group also proved the modulation of NMDA-R-mediated noradrenaline release by presynaptic α2-ARs and H3-Rs (Fink and Göthert 1993b; Fink et al. 1994b; Table 4) as well as the modulation of NMDA R-mediated 5-HT release (Fig. 3) by 5-HT autoreceptors (Fink et al. 1996) and presynaptic α2-heteroreceptors (Fink et al. 1995b).
Gabapentinoids and voltage-gated Ca2+ channels
A further chapter of work focussed on presynaptic voltage-gated Ca2+ channels. Using Ca2+ fluorometry on synaptosomes, the PhD student W. Meder identified the P/Q-type (meanwhile referred to as CaV2.1 α1 subunit) voltage-gated Ca2+ channel as the major and the N-type (CaV2.2 α1 subunit) and the Na+/Ca2+ exchanger as minor contributors to presynaptic Ca2+ entry in rat and human neocortex (Meder et al. 1997, 1999; Fink et al. 2002a). In the same paradigm, the mode of action of the gabapentinoids gabapentin and pregabalin was discovered, which inhibit P/Q-type (CaV2.1) voltage-gated Ca2+ channels by binding to its α2δ subunit resulting downstream in attenuated glutamate/aspartate release and, thus, less activation of AMPA-R input on noradrenergic terminals (Fink et al. 2000, 2002b) (Fig. 7). The latter work has been cited by 549 articles (Google Scholar, accessed on March 26, 2021), 3 patents since it appeared (ResearchGate.net). It was again the result of a close collaboration with, in this case, Parke-Davis and, after its acquisition, with Pfizer; the work was initially triggered by Feuerstein’s finding that ω-conotoxin GVIA inhibited noradrenaline and acetylcholine release in the human neocortex (Feuerstein et al. 1990), an effect, which could now be explained.

Chain of events involved in the inhibitory effect of gabapentin on noradrenaline (NA) release in rat brain cortex. Gabapentin inhibits the K+-induced a Ca2+ influx via P/Q-type (but not N-type) Ca2+ channels, b glutamate and aspartate release, and c NA release via AMPA (but not NMDA) receptors. Experiments were performed on slices or synaptosomes (dotted columns) and results are expressed as means ± SEM (*p < 0.05, **p < 0.01, based on the t-test for paired (B) or unpaired (A, C) data). The fact that the inhibitory effect of gabapentin on NA release (C) was not retained in isolated nerve endings (synaptosomes) demonstrates that it is not related to a direct effect on the noradrenergic neurone. The effect of gabapentin occurred in the range of therapeutically relevant plasma concentrations of 10–100 μM. Re-drawn from Fink et al. (2000). The experiments were further elaborated in the study by Fink et al. (2002b), which shows that the mechanisms also occur in human cortical slices and also extend to pregabalin, another gabapentinoid, but not to its enantiomer R-(-)-3-isobutylgaba. The schematic drawing in d shows that gabapentin (and pregabalin) (i) inhibit Ca2+ influx into glutamatergic neurones via P/Q-type (CaV2.1) voltage-gated Ca2+ channels by binding to its α2δ subunit, subsequently leading to (ii) decreased glutamate release, (iii) reduced activation of excitatory AMPA receptors on noradrenergic neurones, and (iv) eventually to a decrease in NA release. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; EAA, excitatory amino acids; NMDA, N-methyl-D-aspartate
Various presynaptic receptors, imidazolines, and agmatine
Various presynaptic receptors
Noradrenaline attracted the attention of Manfred Göthert even earlier than 5-HT did. In a series of studies, structure-activity relationships were determined for compounds provided by the Beiersdorf company (Hamburg) with respect to their noradrenaline-depleting effect (Benkert et al. 1975) or their antagonistic effects at α1-ARs and/or α2-ARs (Benthe et al. 1972; Göthert et al. 1983b; Schlicker et al. 1984b). The α1-AR antagonist BE 2254 played some role as drug tool in own studies (e.g., Göthert et al. 1981b) or studies from other groups (e.g., 125I-BE 2254 in Engel and Hoyer 1981). However, Manfred Göthert became particularly interested in presynaptic autoreceptors and heteroreceptors on noradrenergic neurones. In the same year, in which he entered this field (Göthert 1977), long reviews about numerous types and sites of presynaptic receptors written by key players appeared (Langer 1977; Starke 1977; Westfall 1977); nonetheless, he became one of the major scientists in this area of research.
In the central nervous system, α2-autoreceptors, e.g. in the rat (Göthert et al. 1979b) and mouse cerebral cortex (Schlicker et al. 1992b), and particularly heteroreceptors attracted his attention (Table 4); he was supported in this respect by E. Schlicker and K. Fink and later by M. Kathmann. In detail, he dealt with histamine, opioid, somatostatin, and cannabinoid receptors (the latter ones will be described in the “Cannabinoids” section; Table 4). Presynaptic histamine H3-Rs, originally identified as autoreceptors by Arrang et al. (1983), were identified for the first time on serotoninergic, noradrenergic, and dopaminergic neurones of the brain (Schlicker et al. 1988b, 1989c, 1993; Fink et al. 1990a; Fig. 3, Table 4). Although most studies were carried out on rodent brain, the H3-R was also identified on the noradrenergic neurones of the human cerebral cortex (Schlicker et al. 1994c). Its counterpart in the mouse brain cortex was examined in detail. (i) In a mechanistic study, the receptor-mediated inhibitory effect was the more pronounced the lower the Ca2+ concentration in the medium or the stimulation frequency was; moreover, experiments with N-ethylmaleimide (Schlicker et al. 1994b) and pertussis toxin (Schlicker et al. 1994c) suggested that this receptor is Gi/o protein-coupled. The latter findings were interesting since the H3-R was cloned 5 years later only (Lovenberg et al. 1999). (ii) A receptor interaction, like that shown for the r5-HT1B-R in the rat vena cava (Molderings and Göthert 1990; see the “Presynaptic serotonin heteroreceptors on noradrenergic neurones” section), was also shown for the H3-R in the mouse brain. Pre-activation of the α2-autoreceptor decreases the inhibitory effect mediated via the H3-R; the reverse is true as well (Schlicker et al. 1992b). Since α2-autoreceptor blockade consequently increases the extent of the H3-R-mediated effect, subsequent experiments were usually performed in the presence of the α2-AR antagonist rauwolscine. (iii) The potencies of new H3-R antagonists/inverse agonists were compared to their affinities in radioligand studies in the mouse brain (Nickel et al. 2001). The compounds had been synthesized by W. Schunack and H. Stark (Berlin), who later developed the H3-R inverse agonist pitolisant together with J.C. Schwartz (Paris), which has been marketed as a novel drug against narcolepsy in 2016 (Ganellin et al. 2018). A typical property of H3-Rs is their constitutive activity (Rouleau et al. 2002) and accordingly the H3-R inverse agonist pitolisant tended to increase noradrenaline in mouse brain cortex slices (reviewed in Schlicker and Kathmann 2016). The possibility has to be considered that this effect (provided that it also occurs in human brain) contributes to the main action and/or the side effects of pitolisant.
Somatostatin not only serves as an inhibitor of the release of a series of hormones including growth hormone but also occurs in several brain regions (Günther et al. 2018). In a paper that appeared in Nature, Göthert (1980b) was the first to show that somatostatin also acts via presynaptic inhibitory receptors. This effect, which was examined in rat brain slices, is selective in two respects. First, somatostatin inhibits noradrenaline release in the hypothalamus but not in the cerebral cortex. Second, the inhibitory effect of somatostatin does not extend to 5-HT release.
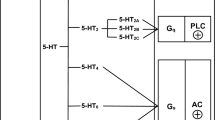
Peripheral presynaptic receptors on postganglionic sympathetic neurones attracted the attention of Manfred Göthert already in Hamburg and later in Essen (supported by F. Hentrich) and Bonn (supported by G. Molderings). Unlike in the brain, the peripheral noradrenergic neurones are equipped with a series of facilitatory receptors. Six of them have been considered, including two ligand-gated ion channels (5-HT3, Tables 2 and 3 and nACh-R, Tables 3 and 4), one Gq-coupled-receptor (AT1; Table 4) and three Gs-coupled receptors (β2, MC2, and DP; Table 4). For each of the four G protein-coupled receptors, at least one human model has been described.
The presynaptic β2-AR was examined with respect to its location, mechanism, and physiological role. (i) In two in vitro models and in one in situ model (Table 4), the effect of a β-AR agonist was counteracted by inhibitors of the angiotensin-converting enzyme and/or angiotensin AT1-R antagonists. These data suggest that at least part of the β2-ARs is not located directly on the postganglionic sympathetic nerve endings but rather in the wall of blood vessels. When activated by a β2-AR agonist, the receptors lead to an increased formation of angiotensin II, which in turn facilitates noradrenaline release via presynaptic AT1-R. In the rat vena cava and human saphenous vein, the occurrence of AT1-Rs has been shown directly (Göthert and Kollecker 1986; Molderings et al. 1988b). (ii) Hentrich et al. (1985) showed in the human pulmonary artery that a stimulator of cAMP formation, an inhibitor of its degradation, and a lipid-soluble cAMP analog increase noradrenaline release. The facilitatory effect of the β-AR agonist isoprenaline on noradrenaline release was markedly enhanced in the presence of a low concentration of the stimulator of cAMP formation, suggesting that the β2-AR is coupled to adenylate cyclase. (iii) The possibility that the β2-AR, reached by endogenous adrenaline, is involved in a positive feedback loop and may even be implicated in the development of essential hypertension had to be considered (for review, see Rand and Majewski 1984). If this were true also for the human saphenous vein, a β-AR antagonist should decrease noradrenaline release. Such an effect, however, did not occur, even if the veins were preincubated with 3H-adrenaline instead of 3H-noradrenaline (Molderings et al. 1988a).
Göthert (1981) was the first to identify a presynaptic ACTH-R. In experiments similar to those described in the previous paragraph, evidence was presented that also the presynaptic ACTH-R, which was identified on the postganglionic neurones of the rabbit pulmonary artery, is positively coupled to adenylate cyclase (Göthert and Hentrich 1984). Although evidence for a facilitatory receptor for prostaglandin D2 (DP-R) was first presented by Nakajima and Toda (1984) in canine mesenteric arteries, final proof for their existence based on an appropriate antagonist was given for four human tissues in the lab of Manfred Göthert (Table 4). Interesting enough, in each of the four tissues, both facilitatory (DP) and inhibitory (EP3) prostaglandin-Rs could be identified (Table 4).
In addition to the EP3-R, other types of inhibitory heteroreceptors and the inhibitory α2-autoreceptor have been identified on peripheral noradrenergic neurones (most receptor types also in human tissues; Table 4). For the α2-autoreceptor in the human saphenous vein and the rabbit pulmonary artery, the potencies of α2-AR antagonists in functional experiments were correlated with their affinities in radioligand binding studies on tissues or cells expressing one α2-subtype only (Molderings and Göthert 1995a). The study clearly showed that the autoreceptor is α2A both in the human and in the rabbit vessel. These results conform to two general observations, namely (i) that presynaptic α2-ARs belong to the α2A-subtype in species like humans and rabbits, whereas in guinea pigs and rodents, the α2D-subtype (which turned out to be the species homolog of α2A) is involved instead (Trendelenburg et al. 1997) and (ii) that, compared to the α2A/D-subtype, the α2B-subtype, and/or α2C-subtype, plays no or a smaller role only (Brede et al. 2004). Nonetheless, the α2A-autoreceptors in humans and rabbits do not possess completely identical properties since rilmenidine and oxymetazoline behave as antagonists in humans but as agonists in rabbits (Molderings et al. 2000a, 2003a). This is also reflected by marked differences in the amino acid sequence of the two species homologs (Molderings et al. 2000a; Brüss et al. 2003).
Imidazolines
For the identification of α2-ARs, ligands with imidazoline structure like the agonist clonidine and the antagonist BDF 6143 have been used frequently. In addition to their effects on α2-ARs, imidazolines appear to possess α2-AR-independent effects and in this context, (i) imidazoline recognition sites (Göthert et al. 1983a), (ii) presynaptic imidazoline receptors, and (iii) imidazoline I1 and I2 binding sites attracted the attention of Manfred Göthert. The latter two topics, which were elaborated together with G.J. Molderings and reviewed repeatedly (Göthert et al. 1995b, 1999; Molderings and Göthert 1999; Molderings et al. 1995b, 1999a), will be considered here in more detail.
The study of Docherty et al. (1982), a cooperation project between Manfred Göthert and K. Starke, was an early hint to the occurrence of presynaptic imidazoline-Rs in addition to α2-ARs in rabbit aorta and pulmonary artery. BDF 6143, an α2-AR antagonist with imidazoline structure, concentration-dependently facilitated, did not affect or even inhibited noradrenaline release, whereas a pure inhibitory effect occurred when the α2-ARs had been blocked by rauwolscine, an α2-AR antagonist devoid of imidazoline structure. Göthert and Molderings et al. (1991) showed that imidazolines like the α1-AR agonist cirazoline, the α2-AR agonist clonidine, and the α2-AR antagonists idazoxan and phentolamine inhibited noradrenaline release in the presence of rauwolscine. The latter proved to be antagonistic not only against α2-ARs (high pA2 > 8) but also against imidazoline-Rs (low pA2 < 7; Molderings et al. 1991). Further studies revealed that the potencies of the imidazoline-R agonists are not correlated with their lipophilicity (log P) and their affinities at the imidazoline I1 and I2 binding sites discussed below (Molderings and Göthert 1995b). Presynaptic imidazoline-Rs, although not found in the rabbit or rat brain (Schlicker et al. 1997c), were also identified in human, guinea-pig, and rat cardiovascular tissues (Table 4). In addition to high concentrations of rauwolscine, also high concentrations of the cannabinoid CB1-R antagonists rimonabant (former name SR141716) and LY320135 showed an antagonistic effect at the presynaptic imidazoline-Rs (Molderings et al. 1999b).
Presynaptic imidazoline-Rs were also found in the rabbit heart (Fuder and Schwarz 1993), the rat kidney (Bohmann et al. 1994), and, using an electrophysiological technique, in rat superior cervical ganglion neurones (Chung et al. 2010). Nonetheless, the evidence is not unequivocal; Gaiser et al. (1999) re-investigated the effects of 10 α-AR and/or imidazoline-R agonists in the rabbit pulmonary artery using conditions under which an endogenous α2-AR-mediated auto-inhibition of noradrenaline release does not occur. In their study, rauwolscine revealed the same potency against each agonist (pA2 ~ 8) leaving no place for presynaptic imidazoline-Rs. However, Molderings et al. (2002a, b) found another example of a functional imidazoline-R in a cell line, i.e. pheochromocytoma 12 cells (PC12) of rats, which possess many properties of sympathetic neurones but are devoid of α2-AR and CB1-R mRNA. The inhibitory effect of imidazolines was shared by 1-oleoyl-lysophosphatidic acid which activates LPA1-Rs and LPA3-Rs (former designation edg2 and edg7, respectively; Chun et al. 2010). mRNA of the latter two receptors, which are the only G protein-coupled receptors with a significant homology (of ~ 40%) to α2-Rs and CB1-Rs, could indeed be detected in PC12 cells (Molderings et al. 2002a). LPA1-Rs and/or LPA3-Rs may represent the molecular entities of the presynaptic imidazoline-Rs; unfortunately, final proof based on knockout mice is so far missing.
Imidazoline binding sites, which are pharmacologically different from α-ARs and presynaptic imidazoline-Rs, were identified first in bovine ventrolateral medulla (I1) and human fat cells (I2); a third type of imidazoline binding site (I3) was suggested as well (reviewed in Regunathan and Reis 1996). In collaboration with G.J. Molderings, Manfred Göthert further characterized imidazoline sites in a threefold manner. (i) The occurrence of imidazoline sites in various preparations was shown by radioligand binding studies. Using 3H-clonidine and 3H-idazoxan, both I1 (Molderings et al. 1993) and I2 sites (Molderings et al. 1994b) were found in bovine adrenal medulla, i.e. in a tissue which is devoid of α2-ARs. Using the same radioligands, I1 sites were also identified in the aforementioned PC12 cells (Molderings et al. 2007a) and I2 sites in rat and human stomach (Molderings et al. 1998b). Experiments dedicated to further disclose the properties of the I1 sites in PC12 cells showed that the ligands under study had a similar affinity for 3H-clonidine and 3H-lysophosphatidic acid which labels sphingosine (S1P)-Rs; in subsequent experiments, binding to both ligands was abolished by short interfering RNA (siRNA) directed towards S1P1-Rs and S1P3-Rs (Molderings et al. 2007a). This finding is very interesting since the latter receptors and the LPA-Rs (the putative molecular entities of the presynaptic imidazoline receptors above) represent the two subgroups of the lysophospholipid receptors (Chun et al. 2010). Although 3H-idazoxan labels I2 sites both in the rat stomach (Molderings et al. 1998b) and bovine adrenal medulla (Molderings et al. 1994b), 3H-clonidine binds to different entities in either tissue. As opposed to I1 sites in the adrenal medulla, 3H-clonidine binds to σ-like receptors in the rat stomach (Molderings et al. 1995a), i.e. to receptors which were originally considered an opioid receptor subtype but later turned out to be two entities (σ1 and σ2) with entirely different pharmacological properties (reviewed in Kim and Pasternak 2017). Nonetheless, the σ-like receptors labeled with 3H-clonidine in the rat stomach are not identical with the σ2 sites labeled by 3H-1,2-di-(2-tolyl)guanidine in this tissue and were designated as non-I1-non-I2 sites instead (Molderings et al. 1998b). “True” σ2 sites, labeled by 3H-1,2-di-(2-tolyl)guanidine, could also be identified in human stomach and in N1E-115 neuroblastoma cells (Molderings et al. 1998b).
(ii) In several papers, efforts were made to define principal mechanistic properties of the imidazoline and σ2 sites. I1 binding (3H-clonidine; bovine adrenal medulla) was inhibited by the stable GTP analog 5′-guanylylimidodiphosphate (Gpp(NH)p), whereas binding to I2 (3H-idazoxan; bovine adrenal medulla) and non-I1-non-I2 sites (3H-clonidine; rat stomach) was not, suggesting that only I1 sites are G protein-coupled (Molderings et al. 1993, 1994b, 1995a). The coupling of I1 sites to G proteins was further supported by the finding that I1-Rs on PC12 cells can be classified as S1P-Rs (Molderings et al. 2007a), which belong to the G protein-coupled receptors (Chun et al. 2010). When S1P3-Rs (which most efficiently bind to Gq protein; Chun et al. 2010) were transiently expressed in human embryonic kidney (HEK) 293 cells, imidazolines consequently led to an increase in intracellular Ca2+ (Molderings et al. 2007a). For σ2 sites, an entirely different mechanism is very likely. Thus, imidazoline and σ ligands inhibited the 5-HT3-R-induced 14C-guanidinium influx in N1E-115 cells (a model of cation influx); the results suggest that this effect is, at least in part, related to an interaction with σ2 sites (Molderings et al. 1996b).
(iii) Finally, studies were carried out to determine functional effects associated with imidazoline and related receptors. Molderings et al. (2007b) found that ligands at imidazoline/S1P-Rs inhibited the protein content (which represents an estimate for cell numbers) in PC12 cells; this effect could be reduced by siRNA species directed towards S1P1-Rs, S1P2-Rs, and S1P3-Rs. Imidazolines and/or σ ligands did not affect the tone of rat gastric strips and acid secretion, suggesting that I2, non-I1-non-I2, or σ2 sites are not implicated in the latter two functional effects (Molderings et al. 1998b). Despite their lack of effect on acid secretion, imidazolines led to an increase in histamine release in the rat stomach (Molderings et al. 1999c), which, most probably, is related to the inhibition of KATP channels, i.e. the mechanism known for the increase in insulin release elicited by imidazolines (Chan 1998) or sulfonylureas (Rorsman et al. 1990). The facilitatory effect on histamine release was shared by agmatine which was identified in human gastric juice in high concentrations and showed an even higher concentration in the stomach of Helicobacter pylori-positive subjects, pointing to a pathophysiological role in gastroduodenal ulcer (Molderings et al. 1999a). Apart from the latter effect in the stomach, imidazolines were suggested to have various effects also on cardiovascular functions (Molderings and Göthert 1999). Endogenous ligands at imidazoline-Rs (and α-ARs) may play a role in vascular smooth muscle proliferation and blood pressure regulation and the imidazoline moxonidine and the oxazoline rilmenidine appear to exert their antihypertensive effect at least partially via I1-Rs (Schäfer et al. 1995). Exogenously added imidazolines may also possess antiarrhythmic effects implicating central and peripheral and α2-AR-dependent and α2-AR-independent sites of action (Molderings and Göthert 1999). They may, however, also interfere with the beneficial cardiac effects of compounds activating KATP channels due to their known antagonistic effect at this mechanism.
Agmatine
Agmatine, decarboxylated arginine, is an aminoguanidine that was discovered by the Nobel laureate A. Kossel (Kossel 1910) in bacteria and plants. More than 80 years later, Reis and coworkers purified agmatine from bovine brain and they discovered that agmatine is an endogenous ligand at imidazoline binding sites (Li et al. 1994). Thereafter, a variety of agmatine-mediated effects in mammals has been described (for recent reviews, see Laube and Bernstein 2017; Xu et al. 2018). In the late 1990s, Manfred Göthert and G.J. Molderings started to examine agmatine (Fig. 8). In a study on the expression of imidazoline binding sites in rat and human stomach, they demonstrated that Helicobacter pylori is able to form and to release the endogenous imidazoline-R ligand agmatine and that considerable amounts of agmatine are present in human gastric juice, especially in that from H. pylori-positive patients (Molderings et al. 1999a, d). Shortly thereafter, they showed that at the α2D-AR of rat vena cava and brain cortex (the α2D-AR is the species homolog of the α2A-AR) agmatine acts as a competitive antagonist at the ligand recognition site and that it enhances the effects of agonists probably by binding to an allosteric site which seems to be labeled by agmatine (Molderings et al. 2000b).

The many faces of agmatine. Aspects studied by Manfred Göthert are marked with red color
Since agmatine had been shown to be degraded in mammalian tissues not only to urea but also to the polyamine putrescine, Manfred Göthert examined whether both polyamines are taken up by the same or by different transport systems in the human glioma cell line SK-MG-1 (Molderings et al. 2001). This study demonstrated the existence of a specific uptake system for agmatine which is not identical with that for putrescine. In addition, agmatine uptake was not due to the activity of organic cation transporters such as OCT1, OCT2, OCT3, OCTN1, or OCTN2 neither in human SK-MG-1 glioma cells nor in six human intestinal tumor cells (Heinen et al. 2003; Molderings et al. 2003b). Since agmatine is present in human gastric juice, it was of interest to prove whether exogenous agmatine is taken up in the stomach. By in vitro exposure of rat isolated stomach to 14C-agmatine and by oral administration to rats in vivo, Manfred Göthert showed that this polyamine is accumulated in the stomach wall and distributed in various tissues and that the accumulation of agmatine was dose-dependently decreased by simultaneous administration of putrescine (Molderings et al. 2002b). The results indicated a transport system for agmatine and they were compatible with the idea of an entero-hepatic recirculation of agmatine (Molderings et al. 2002b; Fig. 8). In a further in vivo study with rats, administration of agmatine after partial hepatectomy was shown to reduce liver regeneration indicating a potential contribution of agmatine to the development of liver diseases (Molderings et al. 2003c). Agmatine, on the other hand, was shown to inhibit concentration-dependently the proliferation of six human intestinal tumor cell lines; in addition, the agmatine content in colon carcinoma tissue from patients who underwent surgery was much lower than in adjacent normal tissue which was interpreted to indicate an antineoplastic action of agmatine (Molderings et al. 2004). In a subsequent investigation, Manfred Göthert also demonstrated an antiproliferative effect of agmatine in rat and human hepatoma cells, indicating an involvement of agmatine in liver cell growth (Kribben et al. 2004). In a study in tumor cells of colonic, hepatic, and neuronal origin, Manfred Göthert examined the molecular basis for the antiproliferative effect of agmatine (Wolf et al. 2007). Agmatine inhibited the proliferation of all examined cells. At the human hepatoma cell line HepG2, it was demonstrated that the antiproliferative effect is due to an interaction with neither the NO synthases, the polyamine-dependent hypusination of the translation factor elF5a, nor an agmatine-induced reduction in availability of intracellular arginine but it may be due to an increase in intracellular caspase-3 activity, indicating a promotion of apoptosis (Wolf et al. 2007; Fig. 8). Finally, regulatory mechanisms underlying agmatine homeostasis in humans have been explored in human colon resectates by measuring mRNA encoding ornithine decarboxylase (ODC), diamine oxidase (DAO), and arginine decarboxylase (ADC), from the production of agmatine by 10 cultured bacterial strains of the residual intestinal microflora and from measurement of agmatine in portal venous blood plasma (Haenisch et al. 2008). The study showed that (i) the level of mRNA was lower (ADC and DAO) or higher (ODC) in neoplastic tissue than in the adjacent normal tissue, that (ii) bacteria strains considerably differed in agmatine production, and (iii) a substantial hepatic agmatine removal from blood occurred. Thus, a perturbation of agmatine homeostasis has been proven to be involved in the regulation of malignant cell proliferation, and agmatine available for intestinal absorption may differ considerably depending on the composition of the bacterial flora, and finally, the liver plays a crucial role in the maintenance of agmatine homeostasis in the human organism (Haenisch et al. 2008).
Cannabinoids
Cannabinoids have been attracting the attention of Manfred Göthert since 1995. He became interested (i) in presynaptic CB1-Rs (and was supported in this respect by E. Schlicker, K. Fink, and G.J. Molderings), (ii) in cardiovascular effects of the endocannabinoid anandamide, and (iii) in CB1-R-independent effects of cannabinoids on 5-HT3-Rs and nACh-Rs (supported by B. Malinowska; see Fig. 9). Part of the latter studies was carried out in the laboratory of B. Malinowska when Manfred Göthert was awarded an Alexander von Humboldt Polish Honorary Research Fellowship after his retirement (2006). This fourth part of his professional career (following Hamburg, Essen, and Bonn) lasted from 2006 to 2009 and was split into several periods.

The ceremony of awarding the title of Doctor honoris causa of the Medical University of Białystok (Poland) to Manfred Göthert (12 December 2003). First line: Barbara Malinowska, Manfred Göthert, and Maciej Kaczmarski. Second line: Włodzimierz Buczko, Edmund Przegaliński, Zbigniew Herman, Jacek Nikliński, and Jan Górski. Note that B. Malinowska and W. Buczko (e.g., Malinowska et al. 1995) and E. Przegaliński (Przegaliński et al. 2005) cooperated with Manfred Göthert
(i) CB1-Rs are typically located presynaptically on neurones and their activation leads to the inhibition of the release of the respective neurotransmitter (Schlicker and Kathmann 2001; Szabo and Schlicker 2005). Examples of such receptors could be identified on noradrenergic neurones of the hippocampus and of blood vessels (Table 4) and on the dopaminergic amacrine cells of the retina (Schlicker et al. 1996b). Presynaptic CB1-Rs were found in human and guinea pig but not rat and mouse hippocampus (Schlicker et al. 1997b). This finding stresses once again that one has to be careful when using rodent tissues as a model for humans. The CB1-R inverse agonist rimonabant facilitated (or tended to facilitate) transmitter release in the human and guinea-pig hippocampus (Schlicker et al. 1997b) and in the guinea-pig retina (Schlicker et al. 1996b). Although this phenomenon may be related to an increased formation of endocannabinoids (e.g., anandamide or 2-arachidonoylglycerol), the alternative explanation, i.e. that the CB1-Rs are constitutively active, is at least as likely (Pertwee 2005; Szabo and Schlicker 2005). It is of interest in this context that the density of CB1-Rs in some brain regions exceeds 1 pmol/mg and is higher than that of any other G protein-coupled receptor (Baker et al. 2003).
It is tempting to assume that the CB1-Rs identified in human hippocampus also contribute to the effects of hashish/marijuana on cognitive functions although one has to consider that presynaptic inhibitory CB1-Rs also occur on hippocampal glutamatergic neurones which prevail when compared to noradrenergic neurones (Szabo and Schlicker 2005). A similar reasoning may hold true for rimonabant, which was available from 2006 to 2009 as an anti-obesity agent and was withdrawn from the market due to its potential of serious psychiatric disorders (Ioannides-Demos et al. 2011). Provided that the retinal CB1-Rs also occur in humans, the possibility has to be considered that decreased retinal dopamine release is the biological substrate for the use of cannabis by Caribbean fishermen to ameliorate their night vision (West 1991). It has been shown for mice that the decrease in retinal dopamine occurring by night is associated with an increased rod electrical coupling (Jin et al. 2015). In that study, the rod coupling could be further increased by a D2-R antagonist and it would be plausible that the same may hold true for the inhibition of dopamine release.
(ii) Anandamide induces complex cardiovascular effects. In urethane-anesthetized mice and rats, rapid intravenous (i.v.) injection of anandamide elicits a triphasic response (e.g., Malinowska et al. 2001, 2010, 2012; Kwolek et al. 2005): phase I—a rapid, pronounced bradycardia and a transient drop in blood pressure; phase II—a brief pressor response, and phase III—a more prolonged, marked decrease in blood pressure. Since similar triphasic changes were also obtained after i.v. administration of methanandamide, a stable analog of anandamide, one can exclude the possibility that anandamide acts indirectly via its arachidonic acid metabolites (Malinowska et al. 2001). In conscious rodents, phase II is the most evident one, phase I is induced only by the higher doses of anandamide, and phase III is absent. As early as in 1996, it was shown that CB1-Rs are involved in the stimulation of phase III, since it was diminished by their antagonist rimonabant (for review, see Malinowska et al. 2012). Three years later, anandamide was recognized as endogenous ligand of vanilloid TRPV1-Rs, since both it and methanandamide were shown to induce vasodilation of rat isolated mesenteric and hepatic arteries and the guinea-pig basilar artery in a manner sensitive to the TRPV1-R antagonist capsazepine (for literature, see Malinowska et al. 2001). Phase I resembled the so-called Bezold-Jarisch reflex, which can be induced by the activation of TRPV1-Rs and 5-HT3-Rs located on vagal afferent C-fibers in the heart. Thus, M. Göthert decided to use this model in order to check whether anandamide activated TRPV1-Rs under in vivo conditions and we were the first demonstrating this property of anandamide. The working hypothesis of M. Göthert confirmed experiments in which the anandamide and/or methanandamide-induced phase I (i) was abolished by bilateral vagotomy and in pithed rats (the latter model offers the opportunity to study drug effects on the peripheral cardiovascular system only); (ii) was diminished by capsazepine and by the non-selective TRPV1-R antagonist ruthenium red but not by rimonabant; (iii) similarly to in vitro experiments, the TRPV1-R agonist capsaicin was more potent than anandamide and methanandamide in stimulation of phase I (Malinowska et al. 2001; Kwolek et al. 2005). Interesting enough, we found that acute myocardial ischemia enhances the vanilloid TRPV1-R-mediated Bezold-Jarisch reflex induced by low doses of anandamide (Lupiński et al. 2011).
M. Göthert had also decided to deal with mechanism(s) underlying phase II in urethane-anesthetized rats. Rimonabant and bilateral vagotomy failed to modify phase II excluding the involvement of CB1-Rs and the possibility that it was the simple response to the preceding hypotension. The additional use of pithed rats allowed us to determine peripheral and central components responsible for the anandamide-induced and/or methanandamide-induced phase II. The peripheral component (also observed in pithed rats; most probably located in blood vessels) was sensitive to nifedipine, ruthenium red, and pentobarbitone and, hence, probably represents a Ca2+-dependent mode of action. The central one (absent in pithed rats) was reduced by some β-AR antagonists (the non-selective propranolol and the β2-selective antagonist ICI118551 but not by the β1-AR antagonist CGP20712), by an NMDA-R (MK-801) and by thromboxane A2 (TP)-R antagonists (sulotroban, daltroban, and SQ 29548). In addition, all above compounds decreased the pressor response to intracerebroventricular (i.c.v.) injection of anandamide (studied in the presence of CB1-R and TRPV1-R antagonists). Anandamide and methanandamide failed to bind to TP-Rs on washed rat platelets and an inhibitor of thromboxane A2 synthase furegrelate i.c.v. reduced the pressor effect of anandamide i.v. suggesting that anandamide causes an increase in thromboxane A2 synthesis in the brain (Kwolek et al. 2005; Malinowska et al. 2010). We later identified the paraventricular nucleus as the possible central site of anandamide action (Grzęda et al. 2017).
(iii) The pioneering electrophysiological experiments on rat nodose ganglion neurones demonstrating that cannabinoid receptor agonists inhibited the 5-HT3-R-mediated currents (Fan 1995) allowed M. Göthert again to study the function of his favorite 5-HT3-Rs, this time in connection with cannabinoid pharmacology. Indeed, non-competitive inhibitory effects of cannabinoid-R agonists, mainly anandamide, CP55940, WIN55212-2 (but not its inactive S-(-)-enantiomer WIN55212-3), which were resistant to the CB1-R antagonist rimonabant, were determined in vitro on the 5-HT-induced current in HEK 293 cells expressing recombinant human 5-HT3A-Rs (Barann et al. 2002) and in vivo on the Bezold-Jarisch reflex induced by the 5-HT3-R agonist phenylbiguanide (but not by the TRPV1-R antagonist capsaicin) in urethane-anaesthetized rats (Godlewski et al. 2003; Table 2). The cannabinoids act probably at an allosteric modulatory site of the 5-HT3-R itself because of (i) the slow development of the inhibition (about 3 min in vitro and 10-20 min in vivo); (ii) the failure of cannabinoids to inhibit binding of the 5-HT3-R radioligand 3H-GR65630 to membranes of HEK 293 cells stably transfected with human 5-HT3A-Rs; and (iii) the lack of an inhibition of the 5-HT-induced current when the cannabinoids were administered to the patches exclusively during, but not before, stimulation with 5-HT. The necessity of stimulation of the 5-HT3-Rs at their orthosteric site for the inhibitory effect of anandamide exerted via its allosteric binding site was confirmed in double CB1/CB2-R knockout mice, in which the anandamide-induced analgesia (but not catalepsy) was reduced in the presence of the 5-HT3-R antagonist ondansetron preventing 5-HT tonically released from the adjacent serotoninergic nerve terminals from binding to the orthosteric site (Rácz et al. 2008). The inhibitory influence of cannabinoids on 5-HT3-Rs may be important in 5-HT3-R-mediated responses like analgesia and emesis.
One should keep in mind that the function of 5-HT3-Rs is modulated by numerous substances (e.g., Al Kury et al. 2018). We found that (+)-tubocurarine (but not another non-depolarizing neuromuscular blocking agent, pipecuronium) inhibited and substance P (but not mastoparan, a peptide from wasp venom that shares the property of substance P to activate G proteins) potentiated the 5-HT3-R (but not the TRPV1-R)-mediated Bezold-Jarisch reflex (Malinowska et al. 1996).
Anandamide and methanandamide allosterically inhibit the nicotine-evoked currents through recombinant homopentameric α7 nACh-Rs in Xenopus oocytes (for literature, see Baranowska et al. 2008). Allosteric sites on a transmitter-gated ion channel may be considered potential targets of new classes of therapeutic drugs. Thus, Manfred Göthert decided to check whether methanandamide inhibits the function of the above receptors under in vivo conditions in urethane-anesthetized pithed rats treated with atropine. Urethane anesthesia makes the potential involvement of presynaptic CB1-Rs unlikely (Kurz et al. 2009). We found that methanandamide (similarly to the subunit-non-selective nACh-R antagonist hexamethonium and the selective α7 nACh-R antagonist methyllycaconitine) reduced the nicotine-induced tachycardia (maximally by 40% in each case). Non-additivity of their inhibitory effects suggested that methanandamide acts probably at an allosteric site of α7 subunit-containing nACh-Rs (Baranowska et al. 2008).
Conclusions
Manfred Göthert has published no less than 271 papers, 110 of which are related to serotonin; his h index amounts to 57 (Google Scholar, accessed on March 26, 2021). Some of his major scientific achievements and their clinical implications will be heralded below (“Major scientific achievements” and “Clinical implications” sections).
Major scientific achievements
The mechanism of action of general anesthetics was unclear over a long time period. The Meyer-Overton rule (1899-1901) only describes that there is an excellent correlation between their potency as anesthetics and their hydrophobicity (Fig. 10). There was general belief that the anesthetics act via unspecific hydrophobic interactions with membrane lipids or lipoproteins. By contrast, Manfred Göthert showed in 1974 that anesthetics are negative allosteric modulators of nACh-Rs (later extended to 5-HT3-Rs, another group of ligand-gated cation channels), i.e. that they exhibit specific hydrophobic properties. Using electrophysiological techniques, Franks and Lieb (1984) showed that anesthetics act on central NMDA-Rs, a third type of ligand-gated cation channels (Fig. 10). Subsequent studies revealed that they also act as positive allosteric modulators at GABAA-Rs, another example of ligand-gated ion channels which, however, lead to hyperpolarization as opposed to depolarization obtained with nACh-Rs, 5-HT3-Rs, and NMDA-Rs. Today, the concept of a specific interaction of anesthetics with GABAA-Rs is widely acknowledged as their site of action (Fig. 10). Unfortunately, the pioneering work of Manfred Göthert was not appreciated by the scientific community and, deeply disappointed, he wrote in retrospect (Göthert 2014): Thus, we were the first to identify the nACh and the 5-HT3 receptor, two ligand-gated ion channels, as sites of action of halothane. These results were obtained about one decade earlier than the same conclusions from electrophysiological data. My biochemical models obviously were unknown systems used by an unknown author, who published so far unknown results – a typical constellation for not being cited in the relevant literature.

Milestones in the elucidation of the mode of action of general anesthetics and Manfred Göthert’s contributions (seminal papers are given in the boxes). Manfred Göthert showed that the general anesthetic halothane is a negative allosteric modulator at periperal nACh und 5-HT3 receptors; in other words, anesthetics have a much more specific effect than suggested by the Meyer-Overton hypothesis. According to Franks and Lieb (1984, 1997), anesthetics inhibit nACh and 5-HT3 receptors which are also present in the brain (see figure) and NMDA receptors solely occurring in the brain (see figure) and stimulate central GABAA receptors (see figure). All receptors are ligand-gated ion channels; only the GABAA receptors (today believed to be the major target of anesthetic action) are inhibitory. Manfred Göthert also showed that ethanol inhibits peripheral nACh and 5-HT3 receptors at concentrations obtained under moderate intoxication. Simultaneously with, but independent from, Lovinger et al. (1989), he found that ethanol inhibits NMDA receptors at concentrations occurring under social drinking. Note that the action of general anesthetics and ethanol is very selective: E.g., voltage-dependent cation channels (NaV, CaV; see figure) are affected at extremely high concentrations only
Manfred Göthert also observed that ethanol, like the general anesthetics, inhibits peripheral nACh-Rs and 5-HT3-Rs and that the effect on the nACh-Rs occurred at ethanol concentrations compatible with moderate intoxication (Göthert and Thielecke 1976). More than 10 years later, he examined the effect of ethanol also on central NMDA-Rs (Fig. 10). Again, an inhibitory effect could be shown and the potency of ethanol at NMDA-Rs was even higher than that at peripheral nACh-Rs (Göthert and Fink 1989; see Fig. 5 in Göthert et al. 2020). The study by Göthert and Fink et al. (1989), together with that by Lovinger et al. (1989) based on an electrophysiological technique, demonstrates that the NMDA-R is a major site of action of ethanol.
For the sake of comparison, Manfred Göthert also considered noradrenaline release evoked by activation of Gq protein-coupled receptors, K+ depolarization, and electrical stimulation (Table 3). The fact that noradrenaline release evoked by the latter methods was affected by very high concentrations of halothane and ethanol only (if at all) shows again that the two compounds possess a highly specific mode of action. Electrical stimulation, via activation of voltage-dependent Na+ channels, eventually leads to opening of voltage-dependent Ca2+ channels (VDCC). It was tempting to assume that the VDCCs in the rabbit heart are blocked by the classical Ca2+ antagonists like verapamil but this held true for extremely high concentrations only, excluding that L-type VDCCs (present nomenclature: CaV1.x) are involved (Göthert et al. 1979c). In subsequent years when more appropriate drug tools had become available, Manfred Göthert could show that the VDCCs in the rat and human neocortex belong to the P-/Q-type (CaV2.1; major part) and N-type (CaV2.2; minor part) (see the “Gabapentinoids and voltage-gated Ca2+ channels” section), whereas the VDCCs in the human atrium belong to the N-type (CaV2.2; Molderings et al. 2000c). The L-type, P-/Q-type, and N-type VDCCs differ in their α1 subunit; another part of the VDCCs, the α2δ subunit, is inhibited by the gabapentinoids and Manfred Göthert could disclose a chain of events involved in their therapeutic action in rat and human brain (see the “Clinical implications” section).
Part of the nACh-Rs, 5-HT3-Rs, and NMDA-Rs mentioned above are presynaptic receptors which represent another research field in which Manfred Göthert excelled. Examples of presynaptic receptors have been described decades ago but systematic studies started around 1970 only (Fig. 11). Manfred Göthert became interested in this topic from 1974 on and in the subsequent three decades, there was a lively competition between the groups of E. Muscholl in Mainz, K. Starke in Freiburg, and Manfred Göthert in Essen and since 1986 in Bonn; many of the numerous papers dedicated to this topic appeared in Naunyn-Schmiedeberg’s Arch Pharmacol. Manfred Göthert studied 23 different types of presynaptic receptors (Fig. 11); he was most interested in presynaptic receptors on central serotoninergic and noradrenergic neurones and on peripheral noradrenergic neurones. He was the first to identify presynaptic somatostatin and ACTH (MC2)-Rs (Göthert 1980b, 1981) and (together with Cerrito and Raiteri 1979) serotonin autoreceptors (Göthert and Weinheimer 1979). Twelve receptors were also examined in human tissues (Tables 1 and 4). Although the mere identification of presynaptic receptors was important per se (since this area was terra incognita at that time), Manfred Göthert provided more than just functional anatomy. Thus, his studies were helpful with respect to the elucidation of receptor classification (e.g., of serotonin receptor subtypes, see next paragraph). Moreover, he disclosed the inhibitory interaction of different types of Gi/o protein-coupled presynaptic receptor types with each other or revealed the mechanisms behind the receptor level (e.g., by increasing cAMP levels or inhibiting Gi/o protein by N-ethylmaleimide or pertussis toxin).

Milestones in the identification of presynaptic receptors and contributions of Manfred Göthert (seminal papers are given in the boxes). The figure shows that he studied the modulation of noradrenaline release from postganglionic sympathetic neurones by 15 types of presynaptic receptors. Activation of ligand-gated ion channels (LGICs), Gq-coupled and Gs-coupled receptors increases noradrenaline release (+; see vesicles fusing with the cell membrane and releasing noradrenaline molecules into the synaptic cleft); activation of Gi/o protein-coupled receptors decreases noradrenaline release (−). Signaling following activation of G protein-coupled receptors as described by Kubista and Boehm (2006). The types of presynaptic receptors studied by Manfred Göthert are given next to the yellow boxes; in the case of the 5-HT4-R, a parasympathetic neurone is interpolated and the increased release of ACh eventually leads to inhibition of noradrenaline release (for details, see the “Presynaptic serotonin heteroreceptors on noradrenergic neurones” section). On noradrenergic and serotoninergic neurones of the brain, only LGICs and Gi/o protein-coupled presynaptic receptors occur (not shown) and 13 types of presynaptic receptors were identified by Manfred Göthert (see table on the right hand side). Altogether, 23 different types of presynaptic receptors were examined. ACh, acetylcholine; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C
Manfred Göthert is identified with serotonin by many colleagues and indeed research dedicated to this monoamine accompanied him from the early seventies of the previous century until his death. When his interest for serotonin was kindled for the first time, there was only the simple classification of Gaddum and Picarelli (1957), encompassing D (5-HT2A)-Rs and M (5-HT3)-Rs. Using organ bath studies and experiments on pithed rats and on anesthetized animals, he identified new models of those receptors or refined the available ones. Moreover, he showed effects of serotonin itself or of its derivatives on postsynaptic α-adrenoceptors or examined their indirect sympathomimetic effect. In cooperation with H.G. Baumgarten, he studied the selectivity of the neurotoxins 5,6-DHT and 5,7-DHT in terms of serotoninergic and noradrenergic neurones (“Early studies” section). Few years later, a new serotonin receptor classification (5-HT1 and 5-HT2), based on radioligand binding studies, was proposed by Peroutka and Snyder (1979). Manfred Göthert also switched to methods based on radioligands, i.e. superfusion studies on tissues preincubated with 3H-serotonin, and identified presynaptic serotonin autoreceptors and heteroreceptors as well as presynaptic heteroreceptors on serotoninergic neurones (“Presynaptic serotonin heteroreceptors on noradrenergic neurones” and “Presynaptic heteroreceptors on serotoninergic neurones” sections, Tables 1 and 2). Again some years later, serotonin receptor subclassification was no longer based on native but on cloned receptors (starting with Lübbert et al. 1987). Manfred Göthert took the opportunity to further refine his methodological armamentarium and compared the properties of naturally occurring mutants of human serotonin receptors with their respective wild types both of which were expressed in cell lines. Differences were obtained for almost all receptors under study and at least some of them are clinically relevant (see the “Clinical implications” section).
When Manfred Göthert was retired in 2006, the number of serotonin receptor families had increased to seven and the complete number of subtypes had reached at least 14 entities (Göthert et al. 2020). As a matter of fact, he dealt with 6 of the 7 receptor families (except for 5-HT6-Rs) but 5-HT3-Rs fascinated him most (Fig. 12). Three aspects will be briefly discussed. First, simultaneously with, but independently from, Fozard et al. (1979), he identified the 5-HT3-R leading to noradrenaline release in the rabbit heart; this presynaptic receptor serves as one of the peripheral targets of anesthetics (Tables 2 and 3). Second, he provided an in-depth analysis of the human 5-HT3-R expressed in a cell line, using four elegant methods (Table 2, Fig. 12). Third, he studied splice variants and naturally occurring variants of this receptor and was involved in the delineation of the 5-HT3C, 5-HT3D, and 5-HT3E subtypes (Figs. 4 and 12).

Milestones in determination of 5-HT3-R structure and function and some methods and results of the work of Manfred Göthert (seminal papers are given in the boxes)
Clinical implications
The oeuvre of Manfred Göthert not only is important from the viewpoint of science but also has numerous clinical implications some of which will be highlighted here.
-
His discovery that the inhibitory effect of halothane on nACh-R-mediated noradrenaline release in cardiovascular tissues occurs in a concentration range obtained under general anesthesia was an early hint that this anesthetic has a much more specific site of action than believed before (Göthert 1974).
-
The inhibitory effect of ethanol on NMDA-R-mediated transmitter release in the brain represents one of its major sites of action (Göthert and Fink 1989).
-
The inhibitory effect of gabapentin and pregabalin (inhibitors of the α2δ subunit of voltage-dependent Ca2+ channels) on AMPA-induced noradrenaline release occurred under clinically relevant concentrations of these drugs (Fink et al. 2002b).
Manfred Göthert has identified numerous sites of presynaptic receptors involved in the main action or the side effects of drugs.
-
The serotonin autoreceptor in the brain may serve as an example. Many antidepressants, including the selective serotonin re-uptake inhibitors (SSRIs), have an indirect effect on the autoreceptor, i.e. the increased serotonin concentration in the synaptic cleft downregulates the density of the autoreceptor via which the monoamine restricts its own release; as a consequence, serotonin release is gradually increasing. This phenomenon explains why the effect of such antidepressants is developing with a time lag of some days or few weeks (Göthert and Schlicker 1993). Although it is an attractive hypothesis that 5-HT1B-R antagonists, by interrupting the negative feedback loop, may elicit an immediate increase in serotonin release associated with an instantaneous onset of antidepressant activity, little evidence for such antidepressants is currently available (Tiger et al. 2018). Nonetheless, interesting data exist for the β-blocker pindolol, which also blocks presynaptic 5-HT1B autoreceptors and, at even lower concentrations, somadendritic 5-HT1A autoreceptors. In some but not all clinical studies, pindolol accelerated and/or increased the antidepressant activity of SSRIs when given in combination (Artigas et al. 2018).
Manfred Göthert studied the molecular properties of naturally occurring serotonin-R-variants in cell lines transfected with the respective cDNAs. Two examples for which differences between mutant and wild type exist and additional studies in humans or in human tissue were performed will be heralded here.
-
First, the Arg219Leu variant of the 5-HT1A-R is associated with major depression and may play a role in the pathogenesis of depression (Haenisch et al. 2009).
-
Second, occurrence of the Phe124Cys variant of the 5-HT1B-R led to a more marked contraction of human vessels to serotonin and may explain the increased liability of some individuals to sumatriptan-induced vasospasm (Verheggen et al. 2006).
The aforementioned examples are related to ligand-gated and voltage-dependent cation channels, to presynaptic receptors and to serotonin receptors, the classical topics of Manfred Göthert, but one should not overlook important clinical implications from another two areas of his research.
-
At the very beginning of his scientific career, Manfred Göthert dealt with carbon monoxide toxicology. His studies revealed that lowering of the general threshold limit value (MAK) for CO from 100 to 50 ppm was justified but that a special adjustment for Caisson workers was not necessary (“Carbon monoxide toxicology” section).
-
Towards the end of his career, Manfred Göthert became interested in agmatine. He was involved in studies in which agmatine formation by bacteria in the human gastrointestinal tract, its handling by the human body (including a special carrier), and its putative antiproliferative effect in human tissue was described (Fig. 8).
Personal remarks
Aspects apart from scientific issues have not been considered here. Thus, Manfred Göthert served as highly estimated academic teacher or as dedicated Dean of the Medical Faculty of the University of Bonn (1998-2002) and President of the German Society for Experimental and Clinical Pharmacology and Toxicology (DGPT; 1997-1999) and the Federation of European Pharmacological Societies (EPHAR; 2004-2006). Manfred Göthert excelled through all aspects of his professional career. We remember him “as a constantly smiling man, an eternal optimist, determined in his views, kind, straightforward and spontaneous in dealing with other people, who did not care about maintaining a distance between him as a boss and colleagues and for whom being a scientist was not only a profession but a lifestyle” (obituary by Malinowska et al. 2020). We dearly miss him.
Abbreviations
- 2-Methyl-5-HT:
-
2-Methyl-5-hydroxytryptamine
- 5-CT:
-
5-Carboxamidotryptamine
- 5-HT:
-
5-Hydroxytryptamine (serotonin)
- 5,6-DHT:
-
5,6-Dihydroxytryptamine
- 5,7-DHT:
-
5,7-Dihydroxytryptamine
- 6-HT:
-
6-Hydroxytryptamine
- 8-OH-DPAT:
-
8-Hydroxy-2-(di-n-propylamino)tetralin
- ACTH:
-
Adrenocorticotropic hormone
- ACh:
-
Acetylcholine
- ADC:
-
Arginine decarboxylase
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AR:
-
Adrenoceptor
- COS:
-
Cells being CV-1 (simian) in origin, and carrying the SV40 genetic material
- CRC:
-
Concentration-response curve
- DAO:
-
Diamine oxidase
- edg:
-
Endothelial differentiation gene
- GABA:
-
γ-Aminobutyric acid
- GppNHp:
-
Guanosine-5′-[β,γ-imido]triphosphate
- GTPγS:
-
Guanosine-5′-[γ-thio]triphosphate
- HEK:
-
Human embryonic kidney
- i.v.:
-
Intravenous
- LGIC:
-
Ligand-gated ion channel
- LPA:
-
Lysophosphatidic acid
- MAC:
-
Minimal alveolar concentration
- mACh:
-
Muscarinic acetylcholine
- MAK:
-
Maximale Arbeitsplatzkonzentration (maximum concentration in the work place)
- nACh:
-
Nicotinic acetylcholine
- NMDA:
-
N-Methyl-D-aspartate
- NMDG:
-
N-Methyl-D-glucamine
- OCT, OCTN:
-
Organic cation transporters
- ODC:
-
Ornithine decarboxylase
- PC12:
-
Pheochromocytoma 12
- ppm:
-
Parts per million
- R:
-
Receptor
- SEM:
-
Standard error of the mean
- SHR:
-
Spontaneously hypertensive rat
- siRNA:
-
Short interfering RNA
- SNP:
-
Single-nucleotide polymorphism
- SSRI:
-
Selective serotonin re-uptake inhibitor
- TM:
-
Transmembrane domain
- TMA:
-
Tetramethylammonium
- Tris:
-
Tris(hydroxymethyl)aminomethane
- TRPV :
-
Transient receptor potential vanilloid
- UTR:
-
Untranslated region
- VDCC:
-
Voltage-dependent calcium channels
- τOFF :
-
Inactivation time constant
- τON :
-
Onset time constant
References
Aktories K, Gierschik P, Meyer zu Heringdorf D, Schmidt M, Schultz G, Wieland T (2019) cAMP guided his way: a life for G protein-mediated signal transduction and molecular pharmacology–tribute to Karl H. Jakobs. Naunyn Schmiedeberg's Arch Pharmacol 392:887–911
Al Kury LT, Mahgoub M, Howarth FC, Oz M (2018) Natural negative allosteric modulators of 5-HT3 receptors. Molecules 23:E3186
Albert PR, Vahid-Ansari F (2019) The 5-HT1A receptor: signaling to behavior. Biochimie 161:34–45
Andrade R, Nicoll RA (1986) Pharmacological distribution of serotonin receptors on single pyramidal neurones of the rat hippocampus recorded in vitro. J Physiol 394:99–124
Arrang JM, Garbarg M, Schwartz JC (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302:832–837
Artigas F, Bortolozzi A, Celada P (2018) Can we increase speed and efficacy of antidepressant treatments? Part I: General aspects and monoamine-based strategies. Eur Neuropsychopharmacol 28:445–456
Baker D, Pryce G, Giovannoni G, Thompson AJ (2003) The therapeutic potential of cannabis. Lancet Neurol 2:291–298
Barann M, Göthert M, Fink K, Bönisch H (1993) Inhibition by anaesthetics of 14C-guanidinium flux through the voltage-gated sodium channel and the cation channel of the 5-HT3 receptor of N1E-115 neuroblastoma cells. Naunyn Schmiedeberg's Arch Pharmacol 347:125–132
Barann M, Ruppert K, Göthert M, Bönisch H (1995) Increasing effect of ethanol on 5-HT3 receptor-mediated 14C-guanidinium influx in N1E-115 neuroblastoma cells. Naunyn Schmiedeberg's Arch Pharmacol 352:149–156
Barann M, Göthert M, Bönisch H, Dybek A, Urban BW (1997) 5-HT3 receptors in outside-out patches of N1E-115 neuroblastoma cells: basic properties and effects of pentobarbital. Neuropharmacology 36:655–664
Barann M, Bönisch H, Urban BW, Göthert M (1998) Inhibition of 5-HT3 receptor cation channels by ifenpropdil in excised patches of N1E-115 cells. Naunyn Schmiedeberg's Arch Pharmacol 358:145–152
Barann M, Göthert M, Brüss M, Bönisch H (1999) Inhibition by steroids of [14C]-guanidinium flux through the voltage-gated sodium channel and the cation channel of the 5-HT3 receptor of N1E-115 neuroblastoma cells. Naunyn Schmiedeberg's Arch Pharmacol 360:234–241
Barann M, Meder W, Dorner Z, Brüss M, Bönisch H, Göthert M, Urban BW (2000) Recombinant human 5-HT3A receptors in outside out patches of HEK293 cells: basic properties and barbiturate effects. Naunyn Schmiedeberg's Arch Pharmacol 362:255–265
Barann M, Molderings G, Brüss M, Bönisch H, Urban BW, Göthert M (2002) Direct inhibition by cannabinoids of human 5-HT3A receptors: probable involvement of an allosteric modulatory site. Br J Pharmacol 137:589–596
Barann M, Schmidt K, Göthert M, Urban B, Bönisch H (2004) Influence of sodium substitutes on 5-HT-mediated effects at mouse 5-HT3 receptors. Br J Pharmacol 142:501–508
Baranowska U, Göthert M, Rudź R, Malinowska B (2008) Methanandamide allosterically inhibits in vivo the function of peripheral nicotinic acetylcholine receptors containing the α7-subunit. J Pharmacol Exp Ther 326:912–919
Baranowska-Kuczko M, Kozłowska H, Schlicker E, Göthert M, MacLean MR, Kozłowski M, Kloza M, Sadowska O, Malinowska B (2020) Reduction of the serotonin 5-HT1B and 5-HT2A receptor-mediated contraction of human pulmonary artery by the combined 5-HT1B receptor antagonist and serotonin transporter inhibitor LY393558. Pharmacol Rep 72:756–762
Bard JA, Zgombick J, Adham N, Vaysse P, Branchek TA, Weinshank RL (1993) Cloning of a novel human serotonin receptor (5-HT7) positively linked to adenylate cyclase. J Biol Chem 268:23422–23426
Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71:817–846
Baumgarten HG, Göthert M, Holstein AF, Schlossberger HG (1972a) Chemical sympathectomy induced by 5.6-dihydroxytryptamine. Z Zellforsch 128:115–134
Baumgarten HG, Göthert M, Schlossberger HG, Tuchinda P (1972b) Mechanism of pressor effect of 5,6-dihydroxytryptamine in pithed rats. Naunyn Schmiedeberg's Arch Pharmacol 274:375–384
Baumgarten HG, Groth HP, Göthert M, Manian AA (1974) The effect of 5,7-dihydroxytryptamine on peripheral adrenergic nerves in the mouse. Naunyn Schmiedeberg's Arch Pharmacol 282:245–254
Belelli D, Balcarek JM, Hope AG, Peters JA, Lambert JJ, Blackburn TP (1995) Cloning and functional expression of a human 5-hydroxytryptamine type 3AS receptor subunit. Mol Pharmacol 48:1054–1062
Bender W, Göthert M, Malorny G, Sebbesse P (1971) Effects of low carbon monoxide concentrations in man. Arch Toxikol 27:142–158
Benkert B, Benthe HF, Göthert M, Schmoldt A (1975) Relationships between structure and the noradrenaline depleting effects of guanidine and amidine derivatives. Arzneimittelforschung 25:1404–1408
Benthe HF, Göthert M, Tuchinda P (1972) Noradrenaline antagonistic effects of some phenylethylamine and phenoxyethylamine derivatives. Arzneimittelforschung 22:1468–1474
Benthe HF, Göthert M, von Klinggräff G (1973) Negative inotropic effect of inhalation anaesthetics and the compensation of this effect by cardiac glycosides. Anaesthesist 22:62–68
Blier P, De Montigny C (1997) Current psychiatric uses of drugs acting on the serotonin system. In: Baumgarten HG, Göthert M (eds) Handbook of experimental pharmacology, Vol. 129: Serotoninergic Neurons and 5-HT Receptors in the CNS. Springer-Verlag, Berlin, pp 727–750
Bohmann C, Schollmeyer P, Rump LC (1994) Effects of imidazolines on noradrenaline release in rat isolated kidney. Naunyn Schmiedeberg's Arch Pharmacol 349:118–124
Bönisch H, Barann M, Graupner J, Göthert M (1993) Characterization of 5-HT3 receptors of N1E-115 cells by use of the influx of the organic cation [14C]guanidinium. Br J Pharmacol 108:436–442
Bradley PB, Engel G, Feniuk W, Fozard JR, Humphrey PP, Middlemiss DN, Mylecharane EJ, Richardson BP, Saxena PR (1986) Proposals for the classification and nomenclature of functional receptors for 5-hydroxytryptamine. Neuropharmacology 25:563–576
Brede M, Philipp M, Knaus A, Muthig V, Hein L (2004) α2-Adrenergic receptor subtypes–novel functions uncovered in gene-targeted mouse models. Biol Cell 96:343–348
Brickley SG, Mody I (2012) Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73:23–34
Brüss M, Bühlen M, Erdmann J, Göthert M, Bönisch H (1995) Binding properties of the naturally occuring human 5-HT1A receptor variant with the Ile28Val substitution in the extracellular domain. Naunyn Schmiedeberg's Arch Pharmacol 352:455–458
Brüss M, Göthert M, Hayer M, Bönisch H (1998) Molecular cloning of alternatively spliced human 5-HT3 receptor cDNAs. Ann N Y Acad Sci 861:234–235
Brüss M, Molderings GJ, Bönisch H, Göthert M (1999a) Pharmacological differences and similarities between the native mouse 5-HT3 receptor in N1E-115 cells and of a cloned short splice variant of the mouse 5-HT3 receptor expressed in HEK293 cells. Naunyn Schmiedeberg's Arch Pharmacol 360:225–233
Brüss M, Bönisch H, Bühlen H, Nöthen MM, Propping P, Göthert M (1999b) Modified ligand binding to the naturally occurring Cys-124 variant of the human serotonin 5-HT1B receptor. Pharmacogenetics 9:95–102
Brüss M, Eucker T, Göthert M, Bönisch H (2000a) Exon-intron organization of the human 5-HT3 receptor gene. Neuropharmacology 39:308–315
Brüss M, Barann M, Hayer-Zillgen M, Eucker T, Göthert M, Bönisch H (2000b) Modified 5-HT3A receptor function by co-expression of alternatively spliced human 5-HT3A receptor isoforms. Naunyn Schmiedeberg's Arch Pharmacol 362:392–401
Brüss M, Bönisch H, Göthert M, Molderings GJ (2003) Molecular structure of the rabbit α2-adrenoceptor: a contribution to the α2-adrenoceptor versus I1 imidazoline receptor controversy. Naunyn Schmiedeberg's Arch Pharmacol 367:328–331
Brüss M, Kostanian A, Bönisch H, Göthert M (2005a) The naturally occurring Arg219Leu variant of the human 5-HT1A receptor: impairment of signal transduction. Pharmacogenetics 15:257–264
Brüss M, Kiel S, Bönisch H, Kostanian A, Göthert M (2005b) Decreased agonist, but not antagonist, binding to the naturally occurring Thr92Lys variant of the h5-HT7(a) receptor. Neurochem Int 47:196–203
Bühlen M, Fink K, Böing C, Göthert M (1996) Evidence for presynaptic location of inhibitory 5-HT1Dβ-like autoreceptors in the guinea-pig brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 353:281–289
Cerrito F, Raiteri M (1979) Serotonin release is modulated by presynaptic autoreceptors. Eur J Pharmacol 57:427–430
Chagraoui A, Thibaut F, Skiba M, Thuillez C, Bourin M (2016) 5-HT2C receptors in psychiatric disorders: a review. Prog Neuro-Psychopharmacol Biol Psychiatry 66:120–135
Chan SLF (1998) Clonidine-displacing substance and its putative role in control of insulin secretion: a minireview. Gen Pharmacol 31:525–529
Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH (2010) International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 62:579–587
Chung S, Ahn DS, Kim YH, Kim YS, Joeng JH, Nam TS (2010) Modulation of N-type calcium currents by presynaptic imidazoline receptor activation in rat superior cervical ganglion neurons. Exp Physiol 95:982–993
Classen K, Göthert M, Schlicker E (1984) Effects of DU 24565 (6-nitroquipazine) on serotoninergic and noradrenergic neurones of the rat brain and comparison with the effects of quipazine. Naunyn Schmiedeberg's Arch Pharmacol 326:198–202
Combrink S, Kostanian A, Walstab J, Barann M, Brüss M, Göthert M, Bönisch H (2009) Characterization of the naturally occurring Arg344His variant of the human 5-HT3A receptor. Pharmacol Rep 61:785–797
Davies PA (2011) Allosteric modulation of the 5-HT3 receptor. Curr Opin Pharmacol 11:75–80
Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF (1999) The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature 397:359–363
Docherty JR, Göthert M, Dieckhöfer C, Starke K (1982) Effects of 4-chloro-2-(2-imidazolin-2-ylamino)-isoindoline hydrochloride (BE 6143) at pre- and postsynaptic α-adrenoceptors in rabbit aorta and pulmonary artery. Arzneimittelforschung 32:1534–1540
Drescher KU, Amberg W, Kling A, Wicke K, Gross G, Schoemaker H, Sullivan JP, Żelaszczyk D, Schlicker E, Göthert M, Garcia-Ladona FJ (2007) Pharmacological characterization of 5-HT5A antagonists: effect on serotonin autoreceptors and acetylcholine release. Naunyn Schmiedeberg's Arch Pharmacol 375(Suppl 1):25–26
Dreyer C, Bischoff D, Göthert M (1974) Effects of methoxyflurane anesthesia on adrenal medullary catecholamine secretion: inhibition of spontaneous secretion and secretion evoked by splanchnic-nerve stimulation. Anesthesiology 41:18–26
Dubin AE, Huvar R, D’Andrea MR, Pyati J, Zhu JY, Joy KC, Wilson SJ, Galindo JE, Glass CA, Luo L, Jackson MR, Lovenberg TW, Erlander MG (1999) The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J Biol Chem 274:30799–30810
Emerit MB, Riad M, Fattaccini CM, Hamon M (1993) Characteristics of [14C]guanidinium accumulation in NG 108-15 cells exposed to serotonin, 5-HT3 receptor ligands and substance P. J Neurochem 60:2059–2067
Engel G, Hoyer D (1981) [125I]BE 2254, a new high affinity radioligand for α1-adrenoceptors. Eur J Pharmacol 73:221–224
Engel G, Göthert M, Müller-Schweinitzer E, Schlicker E, Sistonen L, Stadler PA (1983) Evidence for common pharmacological properties of [3H]5-hydroxytryptamine binding sites, presynaptic 5-hydroxytryptamine autoreceptors in CNS and inhibitory presynaptic 5-hydroxytryptamine receptors on sympathetic nerves. Naunyn Schmiedeberg's Arch Pharmacol 324:116–124
Engel G, Göthert M, Hoyer D, Schlicker E, Hillenbrand K (1986) Identity of inhibitory presynaptic 5-hydroxytryptamine (5-HT) autoreceptors in the rat brain cortex with 5-HT1B binding sites. Naunyn Schmiedeberg's Arch Pharmacol 332:1–7
Erdmann J, Shimron-Abarbanell D, Cichon S, Albus M, Meier W, Lichtermann D, Minges J, Reuner U, Franzek E, Ertl MA, Hebebrand J, Remschmidt H, Lehmkuhl G, Poustka F, Schmidt M, Fimmers R, Körner J, Rietschel M, Propping P, Nöthen MM (1994) Systematic screening for mutations in the promoter and the coding region of the 5-HT1A gene. Am J Med Genet 60:393–399
Erdmann J, Nöthen MM, Shimron-Abarbanell D, Rietschel M, Albus M, Borrmann M, Maier W, Franzek E, Körner J, Weigelt B, Fimmers R, Propping P (1996) The human serotonin 7 (5-HT7) receptor gene: genomic organization and systematic mutation screening in schizophrenia and bipolar affective disorder. Mol Psychiatry 1:392–397
Fan P (1995) Cannabinoid agonists inhibit the activation of 5-HT3 receptors in the rat nodose ganglion neurons. J Neurophysiol 73:907–910
Farnebo LO, Hamberger B (1974) Regulation of [3H]5-hydroxytryptamine release from rat brain cortex slices. J Pharm Pharmacol 26:642–644
Farrant M, Nusser Z (2005) Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6:215–229
Feuerstein TJ, Dooley DJ, Seeger W (1990) Inhibition of norepinephrine and acetylcholine release from human neocortex by ω-conotoxin GVIA. J Pharmacol Exp Ther 252:778–785
Feuerstein TJ, Mutschler A, Lupp A, Van Velthoven V, Schlicker E, Göthert M (1993) Endogenous noradrenaline activates α2-adrenceptors on serotonergic nerve endings in human and rat neocortex. J Neurochem 61:474–480
Fink K, Göthert M (1990) Inhibition of N-methyl-D-aspartate-induced noradrenaline release by alcohols is related to their hydrophobicity. Eur J Pharmacol 191:225–229
Fink K, Göthert M (1991) Ethanol inhibits the N-methyl-D-aspartate (NMDA)-induced attenuation of the NMDA-evoked noradrenaline release in the rat brain cortex: interaction with NMDA-induced desensitization. Naunyn Schmiedeberg's Arch Pharmacol 344:167–173
Fink K, Göthert M (1992) Presynaptic site of action underlying the ethanol-induced inhibition of norepinephrine release evoked by stimulation of N-methyl-D-aspartate (NMDA) receptors in rat cerebral cortex. Brain Res 572:27–32
Fink K, Göthert M (1993a) High D-glucose concentrations increase GABA release but inhibit release of norepinephrine and 5-hydroxytryptamine in rat cerebral cortex. Brain Res 618:220–226
Fink K, Göthert M (1993b) Modulation of N-methyl-D-aspartate (NMDA)-stimulated noradrenaline release in rat brain cortex by presynaptic α2-adrenoceptors. Naunyn Schmiedeberg's Arch Pharmacol 348:372–378
Fink K, Göthert M (1996) Both ethanol and ifenprodil inhibit NMDA-evoked release of various neurotransmitters at different, yet proportional potency: potential relation to NMDA receptor subunit composition. Naunyn Schmiedeberg's Arch Pharmacol 354:312–319
Fink K, Schlicker E, Betz R, Göthert M (1988) Identification of presynaptic 5-HT1 autoreceptors in pig brain cortex synaptosomes and slices. Naunyn Schmiedeberg's Arch Pharmacol 338:14–18
Fink K, Göthert M, Molderings G, Schlicker E (1989) N-methyl-D-aspartate (NMDA) receptor-mediated stimulation of noradrenaline release, but not release of other neurotransmitters, in the rat brain cortex: receptor location, characterization and desensitization. Naunyn Schmiedeberg's Arch Pharmacol 339:514–521
Fink K, Schlicker E, Neise A, Göthert M (1990a) Involvement of presynaptic H3 receptors in the inhibitory effect of histamine on serotonin release in the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 342:513–519
Fink K, Bönisch H, Göthert M (1990b) Presynaptic NMDA receptors stimulate noradrenaline release in the cerebral cortex. Eur J Pharmacol 185:115–117
Fink K, Göthert M, Schlicker E (1990c) Veratridine and other depolarizing agents counteract the inhibitory effect of Mg2+ ions on N-methyl-D-aspartate (NMDA)-induced noradrenaline release in vitro. Naunyn Schmiedeberg's Arch Pharmacol 342:53–60
Fink K, Schultheiss R, Göthert M (1992a) Inhibition of N-methyl-D-aspartate- and kainate-evoked noradrenaline release in human cerebral cortex slices by ethanol. Naunyn Schmiedeberg's Arch Pharmacol 345:700–703
Fink K, Schultheiss R, Göthert M (1992b) Stimulation of noradrenaline release in human cerebral cortex mediated by N-methyl-D-aspartate (NMDA) and non-NMDA receptors. Br J Pharmacol 106:67–72
Fink K, Blohm M, Molderings G, Bönisch H, Göthert M (1992c) Preferential location of N-methyl-D-aspartate (NMDA) receptors on postsynaptic membranes and on non-noradrenergic nerve terminals of the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 345:633–638
Fink K, Zentner J, Göthert M (1994a) Increased GABA release in the human brain cortex as a potential pathogenetic basis of hyperosmolar diabetic coma. J Neurochem 62:1476–1481
Fink K, Schlicker E, Göthert M (1994b) N-Methyl-D-aspartate (NMDA)-stimulated noradrenaline release in rat brain cortex is modulated by presynaptic H3-receptors. Naunyn Schmiedeberg's Arch Pharmacol 349:113–117
Fink K, Zentner J, Göthert M (1995a) Subclassification of presynaptic 5-HT autoreceptors in the human cerebral cortex as 5-HT1Dβ receptors. Naunyn Schmiedeberg's Arch Pharmacol 352:451–454
Fink K, Schmitz V, Böing C, Göthert M (1995b) Stimulation of serotonin release in the rat brain cortex by activation of ionotropic glutamate receptors and its modulation via α2-heteroreceptors. Naunyn Schmiedeberg's Arch Pharmacol 352:394–401
Fink K, Böing C, Göthert M (1996) Presynaptic 5-HT autoreceptors modulate N-methyl-D-aspartate-evoked 5-hydroxytryptamine release in the guinea-pig brain cortex. Eur J Pharmacol 300:79–82
Fink K, Meder W, Dooley DJ, Göthert M (2000) Inhibition of neuronal Ca2+ influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br J Pharmacol 130:900–906
Fink K, Meder WP, Clusmann H, Göthert M (2002a) Ca2+ entry via P/Q-type Ca2+ channels and the Na+/Ca2+ exchanger in rat and human neocortical synaptosomes. Naunyn Schmiedeberg's Arch Pharmacol 366:458–463
Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, Göthert M (2002b) Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology 42:229–236
Fozard JR, Mobarok Ali ATM, Newgrosh G (1979) Blockade of serotonin receptors on autonomic neurones by (-)-cocaine and some related compounds. Eur J Pharmacol 59:195–210
Franks NP (2006) Molecular targets underlying general anaesthesia. Br J Pharmacol 147:S72–S81
Franks NP, Lieb WR (1984) Do general anaesthetics act by competitive binding to specific receptors? Nature 310:599–601
Franks NP, Lieb WR (1997) Selectivity of general anesthetics: a new dimension. Nat Med 3:377–378
Freeman WK, Rorie DK, Tyce GM (1981) Effects of 5-hydroxytryptamine on neuroeffector junction in human pulmonary artery. J Appl Physiol Respir Environ Exerc Physiol 51:693–698
Fuder H, Schwarz P (1993) Desensitization of inhibitory prejunctional α2-adrenoceptors and putative imidazoline receptors on rabbit heart sympathetic nerves. Naunyn Schmiedeberg's Arch Pharmacol 348:127–133
Gaddum JH, Picarelli ZP (1957) Two kinds of tryptamine receptor. Br J Pharmacol 12:323–328
Gaiser EG, Trendelenburg AU, Starke K (1999) A search for presynaptic imidazoline receptors at rabbit and rat noradrenergic neurones in the absence of α2-autoinhibition. Naunyn Schmiedeberg's Arch Pharmacol 359:123–132
Ganellin CR, Schwartz JC, Stark H (2018) Discovery of pitolisant, the first marketed histamine H3-receptor inverse agonist/antagonist for treating narcolepsy. In: Fischer J, Klein C, Childers WE (eds) Successful drug discovery, vol 3. Wiley-VCH, Weinheim, pp 359–381
Gellynck E, Heyninck K, Andressen KW, Haegeman G, Levy FO, Vanhoenacker P, Van Craenenbroeck K (2013) The serotonin 5-HT7 receptors: two decades of research. Exp Brain Res 230:555–568
Gerhardt T, Göthert M, Malorny G, Wilke H (1971) Carbon monoxide toxicity during the breathing of CO and air mixtures under hyperbaric pressure. Int Arch Arbeitsmed 28:127–140
Gerhardt T, Göthert M, Ko KD, Malorny G (1972) The partial pressure of carbon monoxide and oxygen in tissues of rabbits breathing a mixture of CO and air at increased pressure. Int Arch Arbeitsmed 30:161–172
Giorgetti M, Tecott LH (2004) Contributions of 5-HT2C receptors to multiple actions of central serotonin systems. Eur J Pharmacol 488:1–9
Godlewski G, Göthert M, Malinowska B (2003) Cannabinoid receptor-independent inhibition by cannabinoid agonists of the peripheral 5-HT3 receptor-mediated von Bezold-Jarisch reflex. Br J Pharmacol 138:767–774
Göthert M (1971) Effects of various inhalation narcotics on the catecholamine concentration in heart and suprarenal glands. Anaesthesist 20:135–140
Göthert M (1972) Die Sekretionsleistung des Nebennierenmarks unter dem Einfluß von Narkotica und Muskelrelaxantien. Springer, Berlin
Göthert M (1974) Effects of halothane on the sympathetic nerve terminals of the rabbit heart. Differences in membrane actions of halothane and tetracaine. Naunyn Schmiedeberg's Arch Pharmacol 286:125–143
Göthert M (1977) Effects of presynaptic modulators on Ca2+-induced noradrenaline release from cardiac sympathetic nerves. Naunyn Schmiedeberg's Arch Pharmacol 300:267–272
Göthert M (1980a) Serotonin-receptor-mediated modulation of Ca2+-dependent 5-hydroxytryptamine release from neurones of the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 314:223–230
Göthert M (1980b) Somatostatin selectively inhibits noradrenaline release from hypothalamic neurones. Nature 288:86–88
Göthert M (1981) ACTH1-24 increases stimulation-evoked noradrenaline release from sympathetic nerves by acting on presynaptic ACTH receptors. Eur J Pharmacol 76:295–296
Göthert M (1984) Facilitatory effect of adrenocorticotropic hormone and related peptides on Ca2+-dependent noradrenaline release from sympathetic nerves. Neuroscience 11:1001–1009
Göthert M (2014) Life in movement as a scientist. In: Philippu A (ed) Geschichte und Wirken der pharmakologischen, klinisch-pharmakologischen und toxikologischen Institute im deutschsprachigen Raum. Berenkamp, Wattens, pp 185–210
Göthert M, Benthe HF (1969) Influence of ether-, halothane- and chloroform-anesthesia on the catecholamine content of the heart. Naunyn Schmiedebergs Arch Pharmakol 264:237–238
Göthert M, Dreyer C (1973) Inhibitory effect of halothane anaesthesia on catecholamine release from the adrenal medulla. Naunyn Schmiedeberg's Arch Pharmacol 277:253–266
Göthert M, Dührsen U (1979) Effects of 5-hydroxytryptamine and related compounds on the sympathetic nerves of the rabbit heart. Naunyn Schmiedeberg's Arch Pharmacol 308:9–18
Göthert M, Fink K (1989) Inhibition of N-methyl-D-aspartate (NMDA)- and L-glutamate-induced noradrenaline and acetylcholine release in the rat brain by ethanol. Naunyn Schmiedeberg's Arch Pharmacol 340:516–521
Göthert M, Fink K (1991) Stimulation of noradrenaline release in the cerebral cortex via presynaptic N-methyl-D-aspartate (NMDA) receptors and their pharmacological characterization. J Neural Transm Suppl 34:121–127
Göthert M, Guth M (1975) Influence of halothane on the effects of endogenous and exogenous noradrenaline on the isolated rabbit heart perfused at constant pressure. Anaesthesist 24:27–31
Göthert M, Hentrich F (1984) Role of cAMP for regulation of impulse-evoked noradrenaline release from the rabbit pulmonary artery and its possible relationship to presynaptic ACTH receptors. Naunyn Schmiedeberg's Arch Pharmacol 328:127–134
Göthert M, Hentrich F (1985) Identification of presynaptic β2-adrenoceptors on the sympathetic nerve fibres of the human pulmonary artery. Br J Pharmacol 85:933–941
Göthert M, Huth H (1980) α-Adrenoceptor-mediated modulation of 5-hydroxytryptamine release from rat brain cortex slices. Naunyn Schmiedeberg's Arch Pharmacol 313:21–26
Göthert M, Kennerknecht E (1975) Influence of aliphatic alcohols and inhalation anaesthetics on the sympathetic nerve terminals of the rabbit heart. Naunyn Schmiedeberg's Arch Pharmacol 287(Suppl):R5
Göthert M, Klupp N (1978) Cardiovascular effects of neurotoxic indolethylamines. Ann NY Acad Sci 305:457–477
Göthert M, Kollecker P (1986) Subendothelial β2-adrenoceptors in the rat vena cava: facilitation of noradrenaline release via local stimulation of angiotensin II synthesis. Naunyn Schmiedeberg's Arch Pharmacol 334:156–165
Göthert M, Malorny G (1969) On the distribution of carbon monoxide between blood and tissue. Arch Toxikol 24:260–270
Göthert M, Molderings GJ (1991) Involvement of presynaptic imidazoline receptors in the α2-adrenoceptor-independent inhibition of noradrenaline release by imidazoline receptors. Naunyn Schmiedeberg's Arch Pharmacol 343:271–282
Göthert M, Rieckesmann JM (1978) Inhibition of noradrenaline release from sympathetic nerves by pentobarbital. Experientia 34:282–284
Göthert M, Schlicker E (1983) Autoreceptor-mediated inhibition of 3H-5-hydroxytryptamine release from rat brain cortex slices by analogues of 5-hydroxytryptamine. Life Sci 32:1183–1191
Göthert M, Schlicker E (1991) Regulation of serotonin release in the central nervous system by presynaptic heteroreceptors. In: Feigenbaum J, Hanani M (eds) Presynaptic regulation of neurotransmitter release: a handbook. Freund Publishing House, Tel Aviv, pp 845–876
Göthert M, Schlicker E (1993) Relevance of 5-HT autoreceptors for psychotropic drug action. In: Gram LF, Balant LP, Meltzer HY, Dahl SG (eds) Clinical pharmacology in psychiatry-strategies in psychotropic drug development. Psychopharmacology series, vol 10. Springer, Berlin, pp 38–51
Göthert M, Thielecke G (1976) Inhibition by ethanol of noradrenaline output from peripheral sympathetic nerves: possible interaction of ethanol with neuronal receptors. Eur J Pharmacol 37:321–328
Göthert M, Tuchinda P (1973) On the mechanism of the negative-chronotropic effect of halothane. Anaesthesist 22:334–338
Göthert M, Wehking E (1980) Inhibition of Ca2+-induced noradrenaline release from central noradrenergic neurons by morphine. Experientia 36:239–241
Göthert M, Weinheimer G (1979) Extracellular 5-hydroxytryptamine inhibits 5-hydroxytryptamine from rat brain cortex slices. Naunyn Schmiedeberg's Arch Pharmacol 310:93–96
Göthert M, Wendt J (1977a) Inhibition of adrenal medullary catecholamine secretion by enflurane: I. Investigations in vivo. Anesthesiology 46:400–403
Göthert M, Wendt J (1977b) Inhibition of adrenal medullary catecholamine secretion by enflurane: II. Investigations in isolated bovine adrenals-site and mechanism of action. Anesthesiology 46:404–410
Göthert M, Lutz F, Malorny G (1968) Carbon monoxide partial pressure in pneumoperitoneum of rabbits, guinea pigs and rats in relation to carbonmonoxyhemoglobin content. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol 260:122–123
Göthert M, Lutz F, Malorny G (1970) Carbon monoxide partial pressure in tissue of different animals. Environ Res 3:303–309
Göthert M, Tuchinda P, Baumgarten HG (1973) Pressor effect of indolethylamines. Eur J Pharmacol 21:242–245
Göthert M, Schmoldt A, Thielecke G (1974a) On the mechanism of action of inhalation anaesthetics in the catecholamine liberation from the adrenal marrow. Anaesthesist 23:137–141
Göthert M, Guth M, Bille U (1974b) Proceedings: influence of halothane on peripheral sympathetic nerves and on the effects of noradrenaline on myocardium. Naunyn Schmiedeberg's Arch Pharmacol 282(Suppl):R26
Göthert M, Bischoff D, Dreyer C (1975) The influence of inhalation anaesthetics on catecholamine secretion from the adrenal medulla in vivo. Anaesthesist 24:19–26
Göthert M, Dorn W, Loewenstein I (1976a) Inhibition of catecholamine release from the adrenal medulla by halothane. Site and mechanism of action. Naunyn Schmiedeberg's Arch Pharmacol 294:239–249
Göthert M, Kennerknecht E, Thielecke G (1976b) Inhibition of receptor-mediated noradrenaline release from the sympathetic nerves of the isolated rabbit heart by anaesthetics and alcohols in proportion to their hydrophobic property. Naunyn Schmiedeberg's Arch Pharmacol 292:145–152
Göthert M, Dührsen U, Rieckesmann JM (1979a) Ethanol, anaesthetics and other lipophilic drugs preferentially inhibit 5-hydroxytryptamine- and acetylcholine-induced noradrenaline release from sympathetic nerves. Arch Int Pharmacodyn Ther 242:196–209
Göthert M, Pohl IM, Wehking E (1979b) Effects of presynaptic modulators on Ca2+-induced noradrenaline release from central noradrenergic neurons. Noradrenalin and enkephalin inhibit release by decreasing depolarization-induced Ca2+ influx. Naunyn Schmiedeberg's Arch Pharmacol 307:21–27
Göthert M, Nawroth P, Neumeyer H (1979c) Inhibitory effects of verapamil, prenylamine and D600 on Ca2+-dependent noradrenaline release from the sympathetic nerves of isolated rabbit hearts. Naunyn Schmiedeberg's Arch Pharmacol 310:11–19
Göthert M, Huth H, Schlicker E (1981a) Characterization of the receptor subtype involved in alpha-adrenoceptor-mediated modulation of serotonin release from rat brain cortex slices. Naunyn Schmiedeberg's Arch Pharmacol 317:199–203
Göthert M, Nolte J, Weinheimer G (1981b) Preferential blockade of postsynaptic α-adrenoceptors by BE 2254. Eur J Pharmacol 70:35–42
Göthert M, Schlicker E, Köstermann F (1983a) Relationship between transmitter uptake inhibition and effects of α-adrenoceptor agonists on serotonin and noradrenaline release in the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 322:121–128
Göthert M, Dieckhöfer C, Nolte J (1983b) Preferential blockade of α1-adrenoceptors in the rabbit pulmonary artery by derivatives of β-phenylethylamine chemically related to BE 2254. J Cardiovasc Pharmacol 5:12–18
Göthert M, Schlicker E, Hentrich F, Rohm N, Zerkowski HR (1984) Modulation of noradrenaline release in human saphenous vein via presynaptic α2-adrenoceptors. Eur J Pharmacol 102:261–267
Göthert M, Kollecker P, Rohm N, Zerkowski HR (1986a) Inhibitory presynaptic 5-hydroxytryptamine (5-HT) receptors on the sympathetic nerves of the human saphenous vein. Naunyn Schmiedeberg's Arch Pharmacol 332:317–323
Göthert M, Schlicker E, Kollecker P (1986b) Receptor-mediated effects of serotonin and 5-methoxytryptamine on noradrenaline release in the rat vena cava and in the heart of the pithed rat. Naunyn Schmiedeberg's Arch Pharmacol 332:124–130
Göthert M, Schlicker E, Fink K, Classen K (1987) Effects of RU 24969 on serotonin release in rat brain cortex: further support for the identity of serotonin autoreceptors with 5-HT1B sites. Arch Int Pharmacodyn Ther 288:31–42
Göthert M, Hamon M, Barann M, Bönisch H, Gozlan H, Laguzzi R, Metzenauer P, Nickel B, Szelenyi I (1995a) 5-HT3 receptor antagonism by anpirtoline, a mixed 5-HT1 receptor agonist/5-HT3 receptor antagonist. Br J Pharmacol 114:269–274
Göthert M, Molderings GJ, Fink K, Schlicker E (1995b) α2-Adrenoceptor-independent inhibition by imidazolines and guanidines of noradrenaline release from peripheral, but not central noradrenergic neurones. Ann N Y Acad Sci 763:405–419
Göthert M, Propping P, Bönisch H, Brüss M, Nöthen MM (1998) Genetic variation in human 5-HT receptors: potential pathogenetic and pharmacological role. Ann N Y Acad Sci 861:26–30
Göthert M, Brüss M, Bönisch H, Molderings GJ (1999) Presynaptic imidazoline receptors. New developments in characterization and classification. Ann N Y Acad Sci 881:171–184
Göthert M, Bönisch H, Malinowska B, Schlicker E (2020) Serotonin discovery and stepwise disclosure of 5-HT receptor complexity over four decades. Part II. Some contributions of Manfred Göthert. Pharmacol Rep 72:271–284
Groß G, Hante K, Göthert M (1987) Effect of antidepressant and neuroleptic drugs on the electrically evoked release of serotonin from rat cerebral cortex. Psychopharmacology 91:175–181
Grzęda E, Schlicker E, Toczek M, Zalewska I, Baranowska-Kuczko M, Malinowska B (2017) CB1 receptor activation in the rat paraventricular nucleus induces bi-directional cardiovascular effects via modification of glutamatergic and GABAergic neurotransmission. Naunyn Schmiedeberg's Arch Pharmacol 390:25–35
Günther T, Tulipano G, Dournaud P, Bousquet C, Csaba Z, Kreienkamp HJ, Lupp A, Korbonits M, Castano JP, Wester HJ, Culler J, Melmed S, Schulz S (2018) International Union of Basic and Clinical Pharmacology. CV. Somatostatin receptors: structure, function, ligands, and new nomenclature. Pharmacol Rev 70:763–835
Haenisch B, von Kügelgen I, Bönisch H, Göthert M, Sauerbruch T, Schepke M, Marklein G, Höfling K, Schröder D, Molderings GJ (2008) Regulatory mechanisms underlying agmatine homeostasis in humans. Am J Physiol Gastrointest Liver Physiol 295:G1104–G1110
Haenisch B, Linsel K, Brüss M, Gilsbach R, Propping P, Nöthen MM, Rietschel M, Fimmers R, Maier W, Zobel A, Höfels S, Guttenthaler V, Göthert M, Bönisch H (2009) Association of major depression with rare functional variants in norepinephrine transporter and serotonin1A receptor genes. Am J Med Genet B Neuropsychiatr Genet 150B:1013–1016
Hamon M (1997) The main features of central 5-HT1A receptors. In: Baumgarten HG, Göthert M (eds) Handbook of experimental pharmacology, Vol. 129: Serotoninergic Neurons and 5-HT Receptors in the CNS. Springer-Verlag, Berlin, pp 239–268
Hartig PR, Hoyer D, Humphrey PPA, Martin GR (1996) Alignment of receptor nomenclature with the human genome: classification of 5-HT1B and 5-HT1D receptor subtypes. Trends Pharmacol Sci 17:103–105
Heidmann DE, Metcalf MA, Kohen R, Hamblin MW (1997) Four 5-hydroxytryptamine7 (5-HT7) receptor isoforms in human and rat produced by alternative splicing: species differences due to altered intron-exon organization. J Neurochem 68:1372–1381
Heinen A, Brüss M, Bönisch H, Göthert M, Molderings GJ (2003) Pharmacological characteristics of the specific transporter for the endogenous cell growth inhibitor agmatine in six tumor cell lines. Int J Color Dis 18:314–319
Hemmings HC, Riegelhaupt PM, Kelz MB, Solt K, Eckenhoff RG, Orser BA, Goldstein PA (2019) Towards a comprehensive understanding of anesthetic mechanisms of action: a decade of discovery. Trends Pharmacol Sci 40:464–481
Hentrich F, Göthert M, Greschuchna D (1985) Involvement of cAMP in modulation of noradrenaline release in the human pulmonary artery. Naunyn Schmiedeberg's Arch Pharmacol 330:245–247
Hentrich F, Göthert M, Greschuchna D (1986) Noradrenaline release in the human pulmonary artery is modulated by presynaptic α2-adrenoceptors. J Cardiovasc Pharmacol 8:539–544
Hope AG, Downie DL, Sutherland L, Lambert JJ, Peters JA, Burchell B (1993) Cloning and functional expression of an apparent splice variant of the murine 5-HT3 receptor A subunit. Eur J Pharmacol 245:187–192
Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP (1994) International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev 46:157–203
Ioannides-Demos LL, Piccenna L, McNeill JJ (2011) Pharmacotherapies for obesity: past, current, and future therapies. J Obes 2011:179674
Jackson MB, Yakel JL (1995) The 5-HT3 receptor channel. Annu Rev Physiol 57:447–468
Jin NG, Chuang AZ, Masson PJ, Ribelayga CP (2015) Rod electrical coupling is controlled by a circadian clock and dopamine in mouse retina. J Physiol 593:1597–1631
Jonsson G (1980) Chemical neurotoxins as denervation tools in neurobiology. Annu Rev Neurosci 3:169–187
Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J 17:250–252
Kathmann M, Schlicker E, Göthert M (1994) Intermediate affinity and potency of clozapine and low affinity of other neuroleptics and of antidepressants at H3 receptors. Psychopharmacology 116:464–468
Kathmann M, Bauer U, Schlicker E, Göthert M (1999) Cannabinoid CB1 receptor-mediated inhibition of NMDA- and kainate-stimulated noradrenaline and dopamine release in the brain. Naunyn Schmiedeberg's Arch Pharmacol 359:466–470
Kaufman J, DeLorenzo C, Choudhury S, Parsey RV (2016) The 5-HT1A receptor in major depressive disorder. Eur Neuropsychopharmacol 26:397–410
Kiel S, Brüss M, Bönisch H, Göthert M (2000) Pharmacological properties of the naturally occurring Phe-124-Cys variant of the human 5-HT1B receptor: changes in ligand binding, G-protein coupling and second messenger formation. Pharmacogenetics 10:655–666
Kiel S, Bönisch H, Brüss M, Göthert M (2003) Impairment of signal transduction in response to stimulation of the naturally occurring Pro279Leu variant of the h5-HT7(a) receptor. Pharmacogenetics 13:119–126
Kilpatrick GJ, Jones BJ, Tyers MB (1987) Identification and distribution of 5-HT3 receptors in rat brain using radioligand binding. Nature 330:746–748
Kim FJ, Pasternak GW (2017) Cloning the sigma2 receptor: wandering 40 years to find an identity. Proc Natl Acad Sci 114:6888–6890
Kossel A (1910) Über das Agmatin. Z Physiol Chem 66:257–261
Kribben B, Heller J, Trebicka J, Sauerbruch T, Brüss M, Göthert M, Molderings GJ (2004) Agmatine (decarboxylated arginine), a modulator of liver cell homeostasis and proliferation. Naunyn Schmiedeberg's Arch Pharmacol 369:160–165
Krobert KA, Bach T, Syversveen T, Kvingedal AM, Levy FO (2001) The cloned human 5-HT7 receptor splice variants: a comparative characterization of their pharmacology, function and distribution. Naunyn Schmiedeberg's Arch Pharmacol 363:620–632
Kubista K, Boehm S (2006) Molecular mechanisms underlying the modulation of exocytotic noradrenaline release via presynaptic receptors. Pharmacol Ther 112:213–242
Kurz C, Baranowska U, Lupiński S, Göthert M, Malinowska B, Schlicker E (2009) Urethane, but not pentobarbitone, attenuates presynaptic receptor function in rats: a contribution to the choice of anaesthetic. Br J Pharmacol 157:1474–1482
Kurzwelly D, Barann M, Kostanian A, Bönisch H, Göthert M, Brüss M (2004) Pharmacological and electrophysiological properties of the naturally occurring Pro391Arg variant of the human 5-HT3A receptor. Pharmacogenetics 14:165–172
Kwolek G, Zakrzeska A, Schlicker E, Göthert M, Godlewski G, Malinowska B (2005) Central and peripheral component of the pressor effect of anandamide in urethane-anaesthetized rats. Br J Pharmacol 145:567–575
Langer SZ (1977) Sixth Gaddum memorial lecture, National Institute for Medical Research, Mill Hill, January 1977. Presynaptic receptors and their role in the regulation of transmitter release. Br J Pharmacol 60:481–497
Laube G, Bernstein HG (2017) Agmatine: multifunctional arginine metabolite and magic bullet in clinical neuroscience? Biochem J 474:2619–2640
Lerer B, Macciardi F, Segman RH, Adolfsson R, Blackwood D, Blairy S, Del Favero J, Dikeos DG, Kaneva R, Lilli R, Massat I, Milanova V, Muir W, Noethen M, Oruc L, Petrova T, Papadimitriou GN, Rietschel M, Serretti A, Souery D, Van Gestel S, Van Broeckhoven C, Mendlewicz J (2001) Variability of 5-HT2C receptor cys23ser polymorphism among European populations and vulnerability to affective disorder. Mol Psychiatry 6:579–585
Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, Reis DJ (1994) Agmatine: an endogenous clonidine-displacing substance in the brain. Science 263:966–969
Likungu J, Molderings GJ, Göthert M (1996) Presynaptic imidazoline receptors and α2-adrenoceptors in the human heart: discrimination by clonidine and moxonidine. Naunyn Schmiedeberg's Arch Pharmacol 354:689–692
Lindmar R, Löffelholz K, Muscholl E (1968) A muscarinic mechanism inhibiting the release of noradrenaline from peripheral adrenergic nerve fibres by nicotinic agents. Br J Pharmacol Chemother 32:280–294
Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, Foye PE, Racke M, Slone AL, Siegel BW, Danielson PE, Sutcliffe JG, Erlander MG (1993) A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms. Neuron 11:449–458
Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG (1999) Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 55:1101–1107
Lovinger DM, White G, Weight FF (1989) Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243:1721–1724
Lübbert H, Hoffman BJ, Snutch TP, van Dyke T, Levine AJ, Hartig PR, Lester HA, Davidson N (1987) cDNA cloning of a serotonin 5-HT1C receptor by electrophysiological assays of mRNA-injected Xenopus oocytes. Proc Natl Acad Sci 84:4332–4336
Lummis SC, Kilpatrick GJ, Martin IL (1990) Characterization of 5-HT3 receptors in intact N1E- 115 neuroblastoma cells. Eur J Pharmacol 189:223–227
Lupiński SŁ, Schlicker E, Pędzińska-Betiuk A, Malinowska B (2011) Acute myocardial ischemia enhances the vanilloid TRPV1 and serotonin 5-HT3 receptor-mediated Bezold-Jarisch reflex in rats. Pharmacol Rep 63:1450–1459
Lynch C (2008) Meyer and Overton revisited. Anesth Analg 107:864–867
Malinowska B, Göthert M, Godlewski G, Wróbel B, Bönisch H, Buczko W (1995) Inhibitory effect of ethanol on the 5-hydroxytryptamine-induced Bezold-Jarisch reflex–involvement of peripheral 5-HT3 receptors. Eur J Pharmacol 293:71–76
Malinowska B, Godlewski G, Buczko W, Göthert M (1996) Facilitation by substance P and inhibition by (+)-tubocurarine of the 5-HT3 receptor-mediated Bezold-Jarisch reflex in rats. Eur J Pharmacol 315:159–164
Malinowska B, Kwolek G, Göthert M (2001) Anandamide and methanadamide induce both vanilloid VR1- and cannabinoid CB1 receptor-mediated changes in heart rate and blood pressure in anaesthetized rats. Naunyn Schmiedeberg's Arch Pharmacol 364:562–569
Malinowska B, Zakrzeska A, Kurz CM, Göthert M, Kwolek G, Wielgat P, Braszko JJ, Schlicker E (2010) Involvement of central β2-adrenergic, NMDA and thromboxane A2 receptors in the pressor effect of anandamide in rats. Naunyn Schmiedeberg's Arch Pharmacol 381:349–360
Malinowska B, Baranowska-Kuczko M, Schlicker E (2012) Triphasic blood pressure responses to cannabinoids: do we understand the mechanism? Br J Pharmacol 165:2073–2088
Malinowska B, Przegaliński E, Schlicker E (2020) Prof. Dr. med. Dr. h. c. mult. Manfred Göthert (1939-2019). Pharmacol Rep 72:267–270
Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D (1991) Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 254:432–437
Matthys A, Haegeman G, Van Craenenbroeck K, Vanhoenacker P (2011) Role of the 5-HT7 receptor in the central nervous system: from current status to future perspectives. Mol Neurobiol 43:228–253
McGrath MA (1977) 5-Hydroxytryptamine and neurotransmitter release in canine blood vessels. Inhibition by low and augmentation by high concentrations. Circ Res 41:428–435
Meder W, Fink K, Göthert M (1997) Involvement of different calcium channels in K+- and veratridine-induced increases of cytosolic calcium concentration in rat cerebral cortical synaptosomes. Naunyn Schmiedeberg's Arch Pharmacol 356:797–805
Meder W, Fink K, Zentner J, Göthert M (1999) Calcium channels involved in K+- and veratridine-induced increase of cytosolic calcium concentration in human cerebral cortical synaptosomes. J Pharmacol Exp Ther 290:1126–1131
Meyer H (1899) Welche Eigenschaft der Anästhetica bedingt ihre narkotische Wirkung? Arch Exp Pathol Pharmakol 42:109–118
Michel MC, Schlicker E, Fink K, Boublik JH, Göthert M, Willette RN, Daly RN, Hieble JP, Rivier JE, Motulsky HJ (1990) Distinction of NPY receptor subtypes in vitro and in vivo. I. NPY18-36 discriminates between NPY receptor subtypes in vitro. Am J Phys 259:E131–E139
Middlemiss DN, Göthert M, Schlicker E, Scott CM, Selkirk JV, Watson J, Gaster LM, Wyman P, Riley G, Price GW (1999) SB-236057, a selective 5-HT1B receptor inverse agonist, blocks the human terminal 5-HT autoreceptor. Eur J Pharmacol 375:359–365
Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL (1997) Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 389:385–389
Miyake A, Mochizuki S, Takemoto Y, Akuzawa S (1995) Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol Pharmacol 48:407–416
Molderings GJ, Göthert M (1990) Mutual interaction between presynaptic α2-adrenoceptors and 5-HT1B receptors on the sympathetic nerve terminals of the rat vena cava. Naunyn Schmiedeberg's Arch Pharmacol 341:391–397
Molderings GJ, Göthert M (1995a) Subtype determination of presynaptic α2-autoreceptors in the rabbit pulmonary artery and human saphenous vein. Naunyn Schmiedeberg's Arch Pharmacol 352:483–490
Molderings GJ, Göthert M (1995b) Inhibitory presynaptic imidazoline receptors on sympathetic nerves in the rabbit aorta differ from I1- and I2-imidazoline binding sites. Naunyn Schmiedeberg's Arch Pharmacol 351:507–516
Molderings GJ, Göthert M (1998) Presynaptic imidazoline receptors mediate inhibition of noradrenaline release from sympathetic nerves in rat blood vessels. Fundam Clin Pharmacol 12:388–397
Molderings GJ, Göthert M (1999) Imidazoline binding sites and receptors in cardiovascular tissue. Gen Pharmacol 32:17–22
Molderings G, Fink K, Schlicker E, Göthert M (1987) Inhibition of noradrenaline release in the rat vena cava via 5-HT1B receptors. Naunyn Schmiedeberg's Arch Pharmacol 336:245–250
Molderings G, Likungu J, Zerkowski HR, Göthert M (1988a) Presynaptic β2-adrenoceptors on the sympathetic nerve fibres of the human saphenous vein: no evidence for involvement in adrenaline-mediated positive feedback loop regulating noradrenergic transmission. Naunyn Schmiedeberg's Arch Pharmacol 337:408–414
Molderings GJ, Likungu J, Hentrich F, Göthert M (1988b) Facilitatory presynaptic angiotensin receptors on the sympathetic nerves of the human saphenous vein and pulmonary artery. Potential involvement in β-adrenoceptor-mediated facilitation of noradrenaline release. Naunyn Schmiedeberg's Arch Pharmacol 338:228–233
Molderings GJ, Göthert M, Fink K, Roth E, Schlicker E (1989a) Inhibition of noradrenaline release in the pig coronary artery via a novel serotonin receptor. Eur J Pharmacol 164:213–222
Molderings GJ, Göthert M, van Ahlen H, Porst H (1989b) Noradrenaline release in human corpus cavernosum and its modulation via presynaptic α2-adrenoceptors. Fundam Clin Pharmacol 3:497–504
Molderings GJ, Werner K, Likungu J, Göthert M (1990) Inhibition of noradrenaline release from the sympathetic nerves of the human saphenous vein via presynaptic 5-HT receptors similar to the 5-HT1D subtype. Naunyn Schmiedeberg's Arch Pharmacol 342:371–377
Molderings GJ, Hentrich F, Göthert M (1991) Pharmacological characterization of the imidazoline receptor which mediates inhibition of noradrenaline release in the rabbit pulmonary artery. Naunyn Schmiedeberg's Arch Pharmacol 344:630–638
Molderings GJ, Weißenborn G, Schlicker E, Likungu J, Göthert M (1992a) Inhibition of noradrenaline release from the sympathetic nerves of the human saphenous vein by presynaptic histamine H3 receptors. Naunyn Schmiedeberg's Arch Pharmacol 346:46–50
Molderings GJ, van Ahlen H, Göthert M (1992b) Modulation of noradrenaline release in human corpus cavernosum by presynaptic prostaglandin receptors. Int J Impot Res 4:19–25
Molderings GJ, Moura D, Fink K, Bönisch H, Göthert M (1993) Binding of [3H]clonidine to I1-imidazoline sites in bovine adrenal medullary membranes. Naunyn Schmiedeberg's Arch Pharmacol 348:70–76
Molderings GJ, Colling E, Likungu J, Jakschik J, Göthert M (1994a) Modulation of noradrenaline release from the sympathetic nerves of the human saphenous vein and pulmonary artery by presynaptic EP3- and DP-receptors. Br J Pharmacol 111:733–738
Molderings GJ, Kundt L, Göthert M (1994b) [3H]Idazoxan binding to bovine adrenal medullary membranes: identification and pharmacological characterization of I2-imidazoline sites. Naunyn Schmiedeberg's Arch Pharmacol 350:252–257
Molderings GJ, Donecker K, Göthert M (1995a) Characterization of non-adrenergic [3H]clonidine binding sites in rat stomach: high affinity of imidazolines, guanidines and sigma ligands. Naunyn Schmiedeberg's Arch Pharmacol 351:561–564
Molderings GJ, Ruppert K, Bönisch H, Göthert M (1995b) No relationship of I1- and I2-imidazoline binding sites to inhibitory effects of imidazolines on ligand-gated ion channels. An investigation in the adrenal medulla and in neuroblastoma cells. Ann N Y Acad Sci 763:420–432
Molderings GJ, Frölich D, Likungu J, Göthert M (1996a) Inhibition of noradrenaline release via presynaptic 5-HT1Dα receptors in human atrium. Naunyn Schmiedeberg's Arch Pharmacol 353:272–280
Molderings GJ, Schmidt K, Bönisch H, Göthert M (1996b) Inhibition of 5-HT3 receptor function by imidazolines in mouse neuroblastoma cells: potential involvement of σ2 binding sites. Naunyn Schmiedeberg's Arch Pharmacol 354:245–252
Molderings GJ, Likungu J, Jakschik J, Göthert M (1997) Presynaptic imidazoline receptors and non-adrenoceptor [3H]-idazoxan binding sites in human cardiovascular tissues. Br J Pharmacol 122:43–50
Molderings GJ, Likungu J, Göthert M (1998a) Modulation of noradrenaline release from the sympathetic nerves of human right atrial appendages by presynaptic EP3- and DP-receptors. Naunyn Schmiedeberg's Arch Pharmacol 358:440–444
Molderings GJ, Donecker K, Burian M, Simon WA, Schröder DW, Göthert M (1998b) Characterization of I2 imidazoline and σ binding sites in the rat and human stomach. J Pharmacol Exp Ther 285:170–177
Molderings GJ, Burian M, Menzel S, Donecker K, Homann J, Nilius M, Göthert M (1999a) Imidazoline recognition sites and stomach function. Ann N Y Acad Sci 881:332–343
Molderings GJ, Likungu J, Göthert M (1999b) Presynaptic cannabinoid and imidazoline receptors in the human heart and their potential relationship. Naunyn Schmiedeberg's Arch Pharmacol 360:157–164
Molderings GJ, Menzel S, Göthert M (1999c) Imidazoline derivatives and agmatin induce histamine release from the rat stomach. Naunyn Schmiedeberg's Arch Pharmacol 360:711–714
Molderings GJ, Burian M, Homann J, Nilius M, Göthert M (1999d) Potential relevance of agmatine as a virulence factor of Helicobacter pylori. Dig Dis Sci 44:2397–2404
Molderings GJ, Bönisch H, Brüss M, Likungu J, Göthert M (2000a) Species-specific pharmacological properties of human α2A-adrenoceptors. Hypertension 36:405–410
Molderings GJ, Menzel S, Kathmann M, Schlicker E, Göthert M (2000b) Dual interaction of agmatine with the rat alpha2D-adrenoceptor: competitive antagonism and allosteric activation. Br J Pharmacol 130:1706–1712
Molderings GJ, Likungu J, Göthert M (2000c) N-type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation 101:403–407
Molderings GJ, Bönisch H, Göthert M, Brüss M (2001) Agmatine and putrescine uptake in the human glioma cell line SK-MG-1. Naunyn Schmiedeberg's Arch Pharmacol 363:671–679
Molderings GJ, Bönisch H, Hammermann R, Göthert M, Brüss M (2002a) Noradrenaline release-inhibiting receptors on PC12 cells devoid of α2- and CB1 receptors: similarities to presynaptic imidazoline and edg receptors. Neurochem Int 40:157–167
Molderings GJ, Heinen A, Menzel S, Göthert M (2002b) Exposure of rat isolated stomach and rats in vivo to [14C]agmatine: accumulation in the stomach wall and distribution in various tissues. Fundam Clin Pharmacol 16:219–225
Molderings GJ, Bönisch H, Brüss M, Göthert M (2003a) alpha2A-Adrenergic versus imidazoline receptor controversy in rilmenidine’s action: alpha2A-antagonism in humans versus alpha2A-agonism in rabbits. Ann N Y Acad Sci 1009:279–282
Molderings GJ, Brüss M, Bönisch H, Göthert M (2003b) Identification and pharmacological characterization of a specific agmatine transport system in human tumor cell lines. Ann N Y Acad Sci 1009:75–81
Molderings GJ, Heinen A, Menzel S, Lübbecke F, Homann J, Göthert M (2003c) Gastrointestinal uptake of agmatine: distribution in tissues and organs and pathophysiologic relevance. Ann N Y Acad Sci 1009:44–51
Molderings GJ, Kribben B, Heinen A, Schröder D, Brüss M, Göthert M (2004) Intestinal tumor and agmatine (decarboxylated arginine): low content in colon carcinoma tissue specimens and inhibitory effect on tumor cell proliferation in vitro. Cancer 101:858–868
Molderings GJ, Brüss M, Göthert M (2006) Functional and molecular identification of 5-hydroxytryptamine receptors in rabbit pulmonary artery: involvement in complex regulation of noradrenaline release. Pharmacol Rep 58:188–199
Molderings GJ, Bönisch H, Brüss M, Wolf C, von Kügelgen I, Göthert M (2007a) S1P receptors in PC12 and transfected HEK293 cells: molecular targets of hypotensive imidazoline I1 receptor ligands. Neurochem Int 51:476–485
Molderings GJ, Göthert M, von Kügelgen I (2007b) Characterization of an antiproliferative effect of imidazoline receptor ligands on PC12 cells. Pharmacol Rep 59:789–794
Nakajima M, Toda N (1984) Neuroeffector actions of prostaglandin D2 on isolated dog mesenteric arteries. Prostaglandins 27:407–419
Nelson L, Guo TZ, Lu J, Saper CB, Franks NP, Maze M (2002) The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci 5:979–984
Nickel T, Bauer U, Schlicker E, Kathmann M, Göthert M, Sasse A, Stark H, Schunack W (2001) Novel histamine H3-receptor antagonists and partial agonists with a non-aminergic structure. Br J Pharmacol 132:1665–1672
Niesler B, Weiss B, Fischer C, Nöthen MM, Propping P, Bondy B, Rietschel M, Maier W, Albus M, Franzek E, Rappold GA (2001) Serotonin receptor gene HTR3A variants in schizophrenic and bipolar affective patients. Pharmacogenetics 11:21–27
Niesler B, Frank B, Kapeller J, Rappold GA (2003) Cloning, physical mapping and expression analysis of the human 5-HT3 serotonin receptor-like genes HTR3C, HTR3D and HTR3E. Gene 310:101–111
Niesler B, Walstab J, Combrink S, Möller D, Kapeller J, Rietdorf J, Bönisch H, Göthert M, Rappold G, Brüss M (2007) Characterization of the novel human serotonin receptor subunits 5-HT3C,5-HT3D, and 5-HT3E. Anal Biochem 368:185–192
Oksenberg D, Marsters SA, O’Dowd BF, Jin H, Havlik S, Peroutka SJ, Ashkenazi A (1992) A single amino-acid difference confers major pharmacological variation between human and rodent 5-HT1B receptors. Nature 360:161–163
Ortells MO, Lunt GG (1995) Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci 18:121–127
Overton E (1901) Studien über die Narkose, zugleich ein Beitrag zur allgemeinen Pharmakologie. Gustav Fischer, Jena
Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400
Peroutka SJ (1988) 5-Hydroxytryptamine receptor subtypes. Annu Rev Neurosci 11:45–60
Peroutka SJ, Snyder SH (1979) Multiple serotonin receptors: differential binding of [3H]-5-hydroxytryptamine, [3H]lysergic acid diethylamide and [3H]-spiroperidol. Mol Pharmacol 16:687–699
Pertwee RG (2005) Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci 76:1307–1324
Price GW, Burton MJ, Collin LJ, Duckworth M, Gaster L, Göthert M, Jones BJ, Roberts C, Watson JM, Middlemiss DN (1997) SB-216641 and BRL-15572–compounds to pharmacologically discriminate h5-HT1B and h5-HT1D receptors. Naunyn Schmiedeberg's Arch Pharmacol 356:312–320
Przegaliński E, Göthert M, Frankowska M, Filip M (2005) WIN 55,212-2-induced reduction of cocaine hyperlocomotion: possible inhibition of 5-HT3 receptor function. Eur J Pharmacol 517:68–73
Rácz I, Bilkei-Gorzo A, Markert A, Stamer F, Göthert M, Zimmer A (2008) Anandamide effects on 5-HT3 receptors in vivo. Eur J Pharmacol 596:98–101
Raiteri M, Maura G, Folghera S, Cavazzani P, Andrioli GC, Schlicker E, Schalnus R, Göthert M (1990) Modulation of 5-hydroxytryptamine release by presynaptic inhibitory α2-adrenoceptors in the human cerebral cortex. Naunyn Schmiedeberg's Arch Pharmacol 342:508–512
Rand MJ, Majewski H (1984) Adrenaline mediates a positive feedback loop in noradrenergic transmission: its possible role in development of hypertension. Clin Exp Hypertens 6:347–370
Regunathan S, Reis DJ (1996) Imidazoline receptors and their endogenous ligands. Annu Rev Pharmacol Toxicol 36:511–564
Reiser G, Hamprecht B (1989) Substance P and serotonin act synergistically to activate a cation permeability in a neuronal cell line. Brain Res 479:40–48
Reynolds DS, Rosahl TW, Cirone J, O’Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter AR, Atack J, Macaulay AJ, Hadingham KL, Hutson PH, Belelli D, Lambert JJ, Dawson GR, McKernan R, Whiting PJ, Wafford KA (2003) Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J Neurosci 23:8608–8617
Richardson BP, Engel G, Donatsch P, Stadler PA (1985) Identification of serotonin M-receptor subtypes and their specific blockade by a new class of drugs. Nature 316:126–131
Riker WF, Roberts F, Standaert G, Fujimori H (1957) The motor nerve terminal as the primary focus for drug-induced facilitation of neuromuscular transmission. J Pharmacol Exp Ther 121:286–312
Robinson DH, Toledo AH (2012) Historical development of modern anesthesia. J Investig Surg 25:141–149
Rorsman P, Berggren PO, Bokvist K, Efendic S (1990) ATP-regulated K+ channels and diabetes mellitus. News Physiol Sci 5:143–147
Rouleau A, Ligneau X, Tardivel-Lacombe J, Morisset J, Gbahou F, Schwartz JC, Arrang JM (2002) Histamine H3-receptor-mediated [35S]GTPγS binding: evidence for constitutive activity of the recombinant and native rat and human H3 receptors. Br J Pharmacol 135:383–392
Rudolph U, Antkowiak B (2004) Molecular and neuronal substrates for general anesthetics. Nat Rev Neurosci 5:709–720
Salmoiraghi GC, Page IH, McGubbin JW (1956) Cardiovascular and respiratory response to intravenous serotonin in rats. J Pharmacol Exp Ther 118:477–481
Schäfer SG, Kaan EC, Christen MO, Löw-Kröger A, Mest HJ, Molderings GJ (1995) Why imidazoline receptor modulator in the treatment of hypertension? Ann N Y Acad Sci 763:659–672
Schlicker E, Göthert M (1981) Antagonistic properties of quipazine at presynaptic serotonin receptors and α-adrenoceptors in rat brain cortex slices. Naunyn Schmiedeberg's Arch Pharmacol 317:204–208
Schlicker E, Göthert M (1998) Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res Bull 47:129–132
Schlicker E, Kathmann M (2001) Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22:565–572
Schlicker E, Kathmann M (2016) Role of the histamine H3 receptor in the central nervous system. Handb Exp Pharmacol 241:277–299
Schlicker E, Göthert M, Clausing R (1982) Acute or chronic changes of noradrenergic transmission do not affect the α-adrenoceptor-mediated inhibition of 3H-serotonin release. Naunyn Schmiedeberg's Arch Pharmacol 320:38–44
Schlicker E, Göthert M, Köstermann F, Clausing R (1983) Effects of α-adrenoceptor antagonists on the release of serotonin and noradrenaline from rat brain cortex slices. Influence of noradrenaline uptake inhibition and determination of pA2 values. Naunyn Schmiedeberg's Arch Pharmacol 323:106–113
Schlicker E, Classen K, Göthert M (1984a) GABAB receptor-mediated inhibition of serotonin release in the rat brain. Naunyn Schmiedeberg's Arch Pharmacol 326:99–105
Schlicker E, Brodde OE, Göthert M, Schaperdoth M (1984b) Increased affinity and preference of halogenated derivatives of BE 2254 for α1-adrenoceptors demonstrated by functional and binding experiments. J Cardiovasc Pharmacol 6:1238–1244
Schlicker E, Göthert M, Hillenbrand K (1985a) Cyanopindolol is a highly potent and selective serotonin autoreceptor antagonist in the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 331:398–401
Schlicker E, Brandt F, Classen K, Göthert M (1985b) Serotonin release in human cerebral cortex and its modulation via serotonin receptors. Brain Res 331:337–341
Schlicker E, Fink K, Classen K, Göthert M (1987a) Facilitation of serotonin (5-HT) release in the rat brain cortex by cAMP and probable inhibition of adenylate cyclase in 5-HT nerve terminals by presynaptic α2-adrenoceptors. Naunyn Schmiedeberg's Arch Pharmacol 336:251–256
Schlicker E, Fink K, Göthert M (1987b) Influence of eicosanoids on serotonin release in the rat brain: inhibition by prostaglandins E1 and E2. Naunyn Schmiedeberg's Arch Pharmacol 335:646–651
Schlicker E, Classen K, Göthert M (1988a) Presynaptic serotonin receptors and α-adrenoceptors on central serotoninergic and noradrenergic neurones of normotensive and spontaneously hypertensive rats. J Cardiovasc Pharmacol 11:518–528
Schlicker E, Betz R, Göthert M (1988b) Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 337:588–590
Schlicker E, Erkens K, Göthert M (1988c) Probable involvement of vascular angiotensin II formation in the ß2-adrenoceptor-mediated facilitation of the neurogenic vasopressor response in the pithed rat. Naunyn Schmiedeberg's Arch Pharmacol 338:536–542
Schlicker E, Molderings G, Betz R, Göthert M (1988d) Antagonistic properties of RU 24969, a preferential 5-HT1 receptor agonist, at presynaptic α2-adrenoceptors of central and peripheral neurones. Pharmacol Toxicol 63:281–285
Schlicker E, Betz R, Göthert M (1989a) Investigation into the age-dependence of release of serotonin and noradrenaline in the rat brain cortex and of autoreceptor-mediated modulation of release. Neuropharmacology 28:811–815
Schlicker E, Fink K, Göthert M, Hoyer D, Molderings G, Roschke I, Schoeffter P (1989b) The pharmacological properties of the presynaptic serotonin autoreceptor in the pig brain cortex conform to the 5-HT1D receptor subtype. Naunyn Schmiedeberg's Arch Pharmacol 340:45–51
Schlicker E, Fink K, Hinterthaner M, Göthert M (1989c) Inhibition of noradrenaline release in the rat brain cortex via presynaptic H3 receptors. Naunyn Schmiedeberg's Arch Pharmacol 340:633–638
Schlicker E, Schunack W, Göthert M (1990) Histamine H3 receptor-mediated inhibition of noradrenaline release in pig retina discs. Naunyn Schmiedeberg's Arch Pharmacol 342:497–501
Schlicker E, Groß G, Fink K, Glaser T, Göthert M (1991) Serotonin in the rat brain cortex is inhibited by neuropeptide Y but not affected by ACTH1-24, angiotensin II, bradykinin and delta-sleep-inducing peptide. Naunyn Schmiedeberg's Arch Pharmacol 343:117–122
Schlicker E, Werner U, Hamon M, Gozlan H, Nickel B, Szelenyi I, Göthert M (1992a) Anpirtoline, a novel, highly potent 5-HT1B receptor agonist with antinociceptive/antidepressant-like actions in rodents. Br J Pharmacol 105:732–738
Schlicker E, Behling A, Lümmen G, Malinowska, Göthert M (1992b) Mutual interaction of histamine H3-receptors and α2-adrenoceptors on noradrenergic terminals in mouse and rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 345:639–646
Schlicker E, Behling A, Lümmen G, Göthert M (1992c) Histamine H3A receptor-mediated inhibition of noradrenaline release in the mouse brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 345:489–493
Schlicker E, Fink K, Detzner M, Göthert M (1993) Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm Gen Sect 93:1–10
Schlicker E, Kathmann M, Exner HJ, Detzner M, Göthert M (1994a) The 5-HT3 receptor agonist 1-(m-chlorophenyl)-biguanide facilitates noradrenaline release by blockade of α2-adrenoceptors in the mouse brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 349:20–24
Schlicker E, Kathmann M, Detzner M, Exner HJ, Göthert M (1994b) H3 receptor-mediated inhibition of noradrenaline release: an investigation into the involvement of Ca2+ and K+ ions, G protein and adenylate cyclase. Naunyn Schmiedeberg's Arch Pharmacol 350:34–41
Schlicker E, Malinowska B, Kathmann M, Göthert M (1994c) Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam Clin Pharmacol 8:128–137
Schlicker E, Fink K, Zentner J, Göthert M (1996a) Presynaptic inhibitory serotonin autoreceptors in the human hippocampus. Naunyn Schmiedeberg's Arch Pharmacol 354:393–396
Schlicker E, Timm J, Göthert M (1996b) Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn Schmiedeberg's Arch Pharmacol 354:791–795
Schlicker E, Fink K, Molderings GJ, Price GW, Duckworth M, Gaster L, Middlemiss DN, Zentner J, Likungu J, Göthert M (1997a) Effects of selective h5-HT1B (SB-216641) and h5-HT1D (BRL-15572) receptor ligands on guinea-pig and human 5-HT auto- and heteroreceptors. Naunyn Schmiedeberg's Arch Pharmacol 356:321–327
Schlicker E, Timm J, Zentner J, Göthert M (1997b) Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn Schmiedeberg's Arch Pharmacol 356:583–589
Schlicker E, Fink K, Kathmann M, Molderings GJ, Göthert M (1997c) Effects of imidazolines on noradrenaline release in brain: an investigation into its relationship to imidazoline, α2 and H3 receptors. Neurochem Int 30:73–83
Schmoldt A, Göthert M (1974) The influence of anaesthetic agents on the catecholamine synthesis in the adrenal medulla. Anaesthesist 23:10–13
Schultheiß T, Flau K, Kathmann M, Göthert M, Schlicker E (2005) Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in guinea-pig vessels, but not in rat and mouse aorta. Naunyn Schmiedeberg's Arch Pharmacol 372:139–146
Segman RH, Ebstein RP, Heresco-Levy U, Gorfine M, Avnon M, Gur E, Nemanov L, Lerer B (1997) Schizophrenia, chronic hospitalization and the 5-HT2C receptor gene. Psychiatr Genet 7:75–78
Starke K (1977) Regulation of noradrenaline release by presynaptic receptor systems. Rev Physiol Biochem Pharmacol 77:1–124
Starke K, Montel H (1973) Involvement of α-receptors in clonidine-induced inhibition of transmitter release from central monoamine neurones. Neuropharmacology 12:1073–1080
Starke K, Göthert M, Kilbinger H (1989) Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev 69:864–989
Szabo B, Schlicker E (2005) Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol 168:327–365
Szelenyi I, Herold H, Göthert M (1994) Emesis induced in domestic pigs: a new experimental tool for detection of antiemetic drugs and for evaluation of emetogenic potential of new anticancer agents. J Pharmacol Toxicol Methods 32:109–116
Tiger M, Varnäs K, Okubo Y, Lundberg J (2018) The 5-HT1B receptor–a potential target of antidepressant treatment. Psychopharmacology 235:1317–1334
Trendelenburg P (1917) Physiologische und pharmakologische Versuche über die Dünndarmperistaltik. Arch Exp Pathol Pharmakol 81:55–129
Trendelenburg AU, Sutej I, Wahl CA, Molderings GJ, Rump LC, Starke K (1997) A re-investigation of questionable subclassifications of presynaptic α2-autoreceptors: rat vena cava, rat atria, human kidney and guinea-pig urethra. Naunyn Schmiedeberg's Arch Pharmacol 356:721–737
Verheggen R, Werner I, Lücker A, Brüss M, Göthert M, Kaumann AJ (2006) 5-Hydroxytryptamine-induced contraction of human temporal arteries coexpressing 5-HT2A receptors and wild-type or variant (Phe124Cys) 5-HT1B receptors: increased contribution of 5-HT1B receptors to the total contractile amplitude in arteries from Phe124Cys heterozygous individuals. Pharmacogenet Genomics 16:601–607
Walstab J, Combrink S, Brüss M, Göthert M, Niesler B, Bönisch H (2007) Aequorin luminescence-based assay for 5-hydroxytryptamine (serotonin) type 3 receptor characterization. Anal Biochem 368:185–192
Walstab J, Rappold G, Niesler B (2010) 5-HT3 receptors: role in disease and target of drugs. Pharmacol Ther 128:146–169
Walstab J, Steinhagen F, Brüss M, Göthert M, Bönisch H (2011) Differences between human wild-type and C23S variant 5-HT2C receptors in inverse agonist-induced resensitization. Pharmacol Rep 63:45–53
Wang Q, O’Brien PJ, Chen CX, Cho DS, Murray JM, Nishikura K (2000) Altered G protein-coupling functions of RNA editing isoform and splicing variant serotonin2C receptors. J Neurochem 74:1290–1300
Werner P, Kawashima E, Reid J, Hussy N, Lundström K, Buell G, Humbert Y, Jones KA (1994) Organization of the mouse 5-HT3 receptor gene and functional expression of two splice variants. Brain Res Mol Brain Res 26:233–241
Werry TD, Loiacono R, Sexton PM, Christopoulos A (2008) RNA editing of the serotonin 5HT2C receptor and its effects on cell signalling, pharmacology and brain function. Pharmacol Ther 119:7–23
West ME (1991) Cannabis and night vision. Nature 351:703–704
Westfall TC (1977) Local regulation of adrenergic neurotransmission. Physiol Rev 57:659–728
Wolf C, Brüss M, Hänisch B, Göthert M, von Kügelgen I, Molderings GJ (2007) Molecular basis for the antiproliferative effect of agmatine in tumor cells of colonic, hepatic, and neuronal origin. Mol Pharmacol 71:276–283
Xu W, Gao L, Li T, Shao A, Zhang J (2018) Neuroprotective role of agmatine in neurological diseases. Curr Neuropharmacol 16:1296–1305
Ye D, Ewing A (2018) On the action of general anesthetics on cellular function: barbiturate alters the exocytosis of catecholamines in a model cell system. Chem Phys Chem 19:1173–1179
Acknowledgements
We would like to thank J. Walstab for drawing Fig. 4 and B. Niesler who supported this help and M. Baranowska-Kuczko, A. Kicman, A. Pędzińska-Betiuk, and J. Weresa for drawing Figs. 3, 6, 7, 11, and 12.
Availability of data and materials
The content of this review is exclusively based on the papers quoted in the text.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
“Early studies,” “Presynaptic autoreceptors and heteroreceptors,” “Carbon monoxide toxicology,” “Major scientific achievements,” and “Clinical implications” sections—ES; “Molecular vistas” section—HB; “Ethanol, general anesthetics, gabapentinoids, and ion channels” section—KBF; “Various presynaptic receptors, imidazolines, and agmatine” section—ES and GJM; “Cannabinoids” section—BM, HB and ES; “Personal remarks” section—ES and BM. All authors read and approved the manuscript; ghost writers were not involved and a paper mill was not used.
Corresponding author
Ethics declarations
Ethical approval
Not applicable since original data from humans or animals have not been used.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bönisch, H., Fink, K.B., Malinowska, B. et al. Serotonin and beyond—a tribute to Manfred Göthert (1939-2019). Naunyn-Schmiedeberg's Arch Pharmacol 394, 1829–1867 (2021). https://doi.org/10.1007/s00210-021-02083-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-021-02083-5