
Abstract
About 40% of the papers within the scientific oeuvre of Manfred Göthert (1939–2019) were dedicated to serotonin (5-hydroxytryptamine, 5-HT). He was not only the witness of the gradual definition of the fourteen 5-HT receptor subtypes but also was involved directly by identifying 5-HT1B, 5-HT1D and 5-HT3 receptors. Moreover, he identified presynaptic 5-HT receptors on central and/or peripheral serotoninergic, noradrenergic and/or cholinergic neurones. Two inhibitory (5-HT1B, 5-HT1D) and two facilitatory (5-HT3, 5-HT4) receptors were found, the 5-HT1B receptor representing a possible target for antidepressant drugs. Ten years earlier than electrophysiologists, he identified ligand-gated receptors like the 5-HT3 and the nicotinic acetylcholine (nACh) receptor as targets of halothane. Simultaneously with, but independent of, other authors he found that ethanol allosterically inhibits N-methyl-d-aspartate (NMDA) receptors, which are affected at an even lower concentration than 5-HT3 and nACh receptors. The latter two receptors were shown to be subject to allosteric inhibition also by cannabinoids via a mechanism unrelated to cannabinoid CB1 or CB2 receptors; cannabinoid inhibition of 5-HT3 receptors may represent a new target for the treatment of neuropathic pain.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
Introduction
Professor Manfred Göthert was invited to prepare an article for Pharmacological Reports after his honorary lecture given to the 17th International Congress of the Polish Pharmacological Society in Krynica Zdrój in September 2010 [1]. He was optimistic enough to plan a series of reviews about serotonin (5-hydroxytryptamine, 5-HT) discovery and stepwise disclosure of 5-HT receptor complexity. The first part, which covers the history of serotonin until the seventies, appeared in 2013 [2]. The second and final part, dedicated to some contributions of the late Manfred Göthert to 5-HT pharmacology, is based on his draft and has been completed by the three coauthors. Both articles represent a special type of review in a novel category of scientific papers, suggested to be named “contemporary witness report”, the properties of which have been described in detail previously [2].
Stepwise disclosure of 5-HT receptor subtypes
No less than 50 years elapsed between the first step to classify 5-HT receptors and the final classification (Fig. 1). The long journey, starting with Gaddum and Picarelli in 1957 [3] and ending with Niesler et al. in 2007 [4], will be described in 20 short paragraphs.

5-HT receptor classification. AC adenylate cyclase, PLC phospholipase C, (+) stimulation, (−) inhibition
- 1.
1957: Based on their organ bath experiments on the guinea-pig ileum, Gaddum and Picarelli [3] described two receptors named D and M after the initial letters of the discriminating antagonists dibenzyline (= phenoxybenzamine) and morphine, respectively.
- 2.
1979: Peroutka and Snyder [5] performed binding studies with three tritiated ligands in rat brain and found two distinct 5-HT binding sites, namely 5-HT1 sites labelled by 3H-5-HT and 5-HT2 sites labelled by 3H-spiroperidol (whereas 3H-lysergic acid diethylamide (3H-LSD) binds to both sites).
- 3.
1983–1986: Presynaptic inhibitory 5-HT autoreceptors in rat brain were first classified as 5-HT1 [6] and then as 5-HT1B [7].
- 4.
1983–1986: It has been shown that 5-HT1A receptors serve as somadendritic inhibitory 5-HT autoreceptors and as heteroreceptors on non-serotoninergic neurones; they can be labelled by 3H-8-hydroxy-2-(di-n-propylamino)tetralin (3H-8-OH-DPAT) [8] and are coupled inhibitorily to adenylate cyclase [9].
- 5.
1986: Bradley et al. [10] reconsidered the previous classification attempts and equated the D receptor with the 5-HT2 binding site and the M receptor with the 5-HT3 binding site. The functional counterpart of 5-HT1 sites is the 5-HT1-like receptors. “like” was used since in spite of some similarities with 5-HT1 binding sites, the receptor population is not homogeneous. Bradley et al. [10] stressed that the availability of selective antagonists is an important prerequisite for further identification of 5-HT1(-like) receptors.
- 6.
1987–1988: The “5-HT1C” receptor was cloned by Lübbert et al. [11] and characterised with respect to its transduction pathway by Hoyer [12].
- 7.
1987: The 5-HT1D receptor subtype has been identified, using 3H-5-HT binding to the bovine brain [13].
- 8.
1988–1990: The 5-HT4 receptor was characterised pharmacologically and biochemically [14, 15].
- 9.
1989–1993: Based on structure, operation and signal transduction, the 5-HT1C receptor has been re-named to become the 5-HT2C receptor [16, 17].
- 10.
1989–1993: The 5-HT1E receptor has been detected by 3H-5-HT binding [18] and the 5-HT1F receptor has been cloned [19].
- 11.
1992: The 5-HT2B receptor has been cloned and evidence has been presented that this receptor mediates the 5-HT-induced contraction of the rat stomach fundus [20, 21].
- 12.
1992–1993: The 5-HT2A receptor refers to the classical 5-HT2/D receptor, as agreed at the Serotonin Club Receptor Nomenclature Committee meeting in Houston in September 1992 (published in [17]).
- 13.
1992–1993: cDNAs encoding the 5-HT5A and 5-HT5B subtypes of the 5-HT5 receptor minifamily have been cloned [22, 23].
- 14.
1992–1996: Two human 5-HT1D receptors, termed 5-HT1Dα and 5-HT1Dβ, have been cloned [24] and have been re-termed h5-HT1D and h5-HT1B, respectively. The 5-HT1B receptor has been re-termed r5-HT1B [25].
- 15.
1993: Cloning and expression of the rat 5-HT6 receptor has been shown [26].
- 16.
1993–1996: Cloning of the 5-HT7 receptor has been described [27] and its distribution in terms of mRNA (in situ hybridization) and protein (autoradiography) has been analysed [28].
- 17.
1995: The human HTR3A gene has been cloned and it has been shown that the 5-HT3A subunits can form functional homopentameric receptors [29, 30].
- 18.
1999: The human HTR3B gene has been cloned. The 5-HT3B subunits alone cannot build functional homopentameric receptors, whereas the heteropentameric complexes of 5-HT3A and 5-HT3B subunits resemble the native receptor in their pharmacological properties [31].
- 19.
2003: The HTR3C, HTR3D and HTR3E genes have been termed “HTR3-like” genes based on comparative sequence analysis [32].
- 20.
2007: No functional homopentameric 5-HT3 complexes could be obtained from the putative 5-HT3C, 5-HT3D and 5-HT3E subunits. However, co-expression with 5-HT3A resulted in the formation of functional heteropentameric complexes with different 5-HT efficacies. The efficacy at the 5-HT3A/D and 5-HT3A/E complex was increased, whereas that at the 5-HT3A/C complex was decreased [4].
Presynaptic 5-HT receptors
Presynaptic 5-HT receptors are possible targets for new therapeutic drugs against human diseases (including depressive disorders [33]). In this context, an answer is sought to the question whether the presynaptic receptor under study belongs to the same 5-HT receptor subtype in the mammalian species (including humans) investigated here. If this should not hold true, preclinical in vitro and in vivo investigations within the development of a new drug should be carried out in a species in which the same 5-HT receptor subtype is expressed as the presynaptic receptor in humans. In fact, such ideas concerning species differences were relevant since—as shown schematically in Fig. 2—different 5-HT receptor subtypes (5-HT1B, 5-HT1D and 5-HT4) are expressed as presynaptic 5-HT heteroreceptors in blood vessels. In the rabbit heart, a fourth 5-HT receptor type, i.e., facilitatory 5-HT3 receptors, has been identified on the sympathetic nerve endings (see next section).

Schematic representation of species-dependent differences in 5-HT receptor subtypes expressed as inhibitory presynaptic 5-HT heteroreceptors on the unshielded axon terminal of a postganglionic sympathetic nerve in a blood vessel. This terminal, symbolised by a varicosity from a chain of such structures, forms a synapse with a smooth muscle cell within the wall of arterial and venous blood vessels. The unshielded axon terminal varicosities are the sites of noradrenaline (NA) release upon depolarization by invading action potentials (APs), evoked by transmural electrical stimulation. This presynaptic 5-HT receptor belongs to subtype 1B in the rat vena cava [36], to subtype 1D in the human saphenous vein [34], to subtype 1B or 1D in the rabbit pulmonary artery [74] and to an unknown (orphan) receptor type in the pig coronary artery [75]. “(–)”: inhibition of NA release; “(+)”: amplification of the α-adrenoceptor-mediated smooth muscle contraction by a 5-HT2 (more precisely: 5-HT2A) receptor subtype. The inset shows the vicinity of a smooth muscle cell in the rabbit pulmonary artery in lower magnification. In this vessel, even two 5-HT receptors influence NA release. The 5-HT1B or 5-HT1D receptor on the sympathetic nerve directly inhibits NA release whereas the 5-HT4 receptor on the parasympathetic nerve facilitates the release of acetylcholine (ACh) which in turn inhibits NA release via the muscarinic M2 ACh receptor. (−), inhibitory effect; (+), stimulatory effect
Classification of the presynaptic receptor also provides an answer to the question whether, in a given species, the presynaptic 5-HT autoreceptors in the brain and the presynaptic 5-HT receptors on sympathetic nerves generally belong to different 5-HT receptor subtypes. If so, it would be possible to modify transmitter release by 5-HT auto- and heteroreceptors independent of each other. This was actually observed in the case of the human [34] and guinea-pig [34, 35] but not rat [7, 36] auto- and heteroreceptors.
The most reliable method for functional determination of the 5-HT receptor subtype mediating a given effect is dependent on the availability of selective antagonists of the multiple 5-HT receptor subtypes [10]. The antagonists, by their ability to partly or completely inhibit the modulating effect of serotonin, disclosed the receptor subtypes involved in the brain and in the periphery [34]. An early approach on rat brain cortex slices [7] was based on the measurement of the inhibitory potencies (half-maximum inhibitory effects on transmitter release) of multiple non-selective 5-HT receptor agonists.
Both agonist potencies (Fig. 3a) and discriminative antagonists (Fig. 3b) were considered in the case of the human atrium. The maximum effect exerted via the presynaptic 5-HT receptor was an inhibition of noradrenaline release by 30%. Accordingly, an inhibition by 15%, i.e., the IC15 value, represented the half-maximum effect. Furthermore, that approach comprised determination of the dissociation constants (KDs; obtained from radioligand binding studies) of the agonists at least at those 5-HT receptor subtypes, hypothesised to be involved in the function under study. In the next step, the logarithms of the dissociation constants and of the potencies were used to calculate, by regression analyses, the correlation coefficients of the agonists for the various 5-HT receptors under consideration.

a Functional identification of the 5-HT1 receptor subtype in the human atrium by regression analyses, relating the negative logarithms of the potencies of five non-selective 5-HT receptor agonists for their inhibition of the electrically stimulated noradrenaline release (pIC15) to the negative logarithms of their inhibition constants (pKi), determined in ligand binding experiments, yielding significant correlation for the non-rodent (human) 5-HT1B and the 5-HT1D receptor (but not for the rodent 5-HT1B receptor; not shown here). Re-drawn from [37]. b, c Functional identification of the 5-HT1 receptor subtype by using antagonists with discriminative properties, i.e., the non-rodent (human) 5-HT1B receptor antagonist SB-216,641 and the 5-HT1D receptor antagonists BRL-15,572 and ketanserin. The 5-HT heteroreceptor in the human atrium (b) was compared with the 5-HT autoreceptor in human cerebral cortex synaptosomes preincubated with 3H-5-HT (stimulation with K+ 25 mM) (c). The data unequivocally identify the heteroreceptor as 5-HT1D and the autoreceptor as 5-HT1B. *P < 0.05, compared to the respective value without antagonist. Re-drawn from [34, 37, 76]
If a significant correlation for only one 5-HT receptor subtype occurred, this result unequivocally would disclose the 5-HT receptor involved in modulation. If there were more than one 5-HT receptor subtypes exhibiting significant correlation—in the example of Fig. 3a, the non-rodent 5-HT1B and the 5-HT1D receptor (previously termed 5-HT1Dβ and 5-HT1Dα, respectively)—the highest correlation coefficient may be suggested to point to the receptor involved. In the example of the human atrium, shown in Fig. 3a, about the same correlation coefficient was observed for the non-rodent 5-HT1B and for the 5-HT1D receptor [36]. This might mean that both receptors are involved in the modulation of NA release.
To solve the dilemma, antagonists with discriminative properties have been identified by the pharmaceutical industry, the group of Manfred Göthert having contributed the evidence in favour of their selectivity of action [34, 38]. Two compounds, SB-216,641 and SB-236,057, proved to exhibit antagonistic properties at the non-rodent 5-HT1B receptor [34, 38] whereas BRL-15,572 was found to be a selective 5-HT1D antagonist [34]. In addition, ketanserin had been shown to possess some preference for the 5-HT1D receptor [39]. Figure 3b shows that the inhibitory effect was attenuated by BRL-15,572 and ketanserin but not affected by SB-216,641. These data prove that the 5-HT1D but not the non-rodent 5-HT1B receptor is involved [34, 37]. Figure 3c shows the data for the 5-HT autoreceptor in the human cerebral cortex, for which series with agonists could not be run. The inhibitory effect of the 5-HT receptor agonist 5-carboxamidotryptamine was attenuated by SB-216,641 but not affected by BRL-15,572 or ketanserin. These data demonstrate that the autoreceptor is a non-rodent 5-HT1B receptor. This conclusion is re-inforced by the effectiveness of SB-236,057 to act as an antagonist in the human (and guinea-pig) brain [38].
Interaction of halothane with 5-HT3 and nACh receptors
When Manfred Göthert got in touch with general anaesthesia for the first time of his life, he was a 5- and 6-year-old boy who, under inhalation anaesthesia with diethyl ether, underwent surgery for hernia in 1944 and torticollis in 1945, respectively. As usual in those days, an experienced nurse administered the volatile anaesthetic via a mask for both induction and maintenance of unconsciousness. During induction, he felt tortured by symptoms of the excitation stadium such as laryngo- and bronchospasms and intensive skeletal muscle activity in attempts to “escape” from being physically fixed to the ground by strong human arms. After the end of “ether” administration, he experienced again an excitation stadium with painful movements of the “surgically impaired” muscles and long-lasting nausea and vomiting. Almost 12 years later, on the occasion of appendectomy, his memories of previous anaesthesia and surgery were still dominated by those very unpleasant symptoms. However, he was surprised that, this time, he did not suffer from any of those adverse effects because anaesthesia was induced with an intravenous anaesthetic, probably a barbiturate, and maintained with an inhalation anaesthetic with more favourable properties than diethyl ether, most likely halothane. At the latest from this new experience onward his fear from anaesthesia mutated to fascination by the phenomenon of drug-induced unconsciousness. Later, as a young medical doctor, this led him to work on general anaesthetics.
Until the fifties of the twentieth century, the Meyer–Overton “theory of anaesthesia” was generally accepted. Meyer based his ideas concerning the sites and mechanisms of action of anaesthetics on their physicochemical properties, in particular, the correlation of their potency as anaesthetics with their solubility in water and lipoids, i.e., their water/lipoid partition coefficients. The same conclusion was drawn by Overton from his biological approach to this problem, namely simple experimental models such as anaesthesia of tadpoles by addition of such drugs to their water environment. According to this concept, lipid components of the plasma membrane were believed to be the sites of action [40]. However, doubts were raised against this concept from the thirties of the previous century onward. Instead, lipophilic proteins or lipophilic domains of proteins were suggested to be critically involved in their depressant effect on neuronal systems [40]. The pre- and postsynaptic plasma membrane is generally accepted to host the site underlying the depressant effect of ethanol. In particular, functional proteins such as neurotransmitter transporters and receptors come into play. An important step forward in this direction was the finding by Ueda and Kamaya [41] that general anaesthetics are capable of inhibiting firefly luciferase by direct interaction of the drugs with its enzymatic property.
In his studies dedicated to elucidate the molecular targets involved in the acute effect of anaesthetics, Manfred Göthert studied catecholamine-secreting cells of the perfused bovine adrenal by increasing the K+ concentration (leading to membrane depolarization), by activation of G protein-coupled receptors such as histamine H1 and muscarinic acetylcholine (mACh) receptors and by ionotropic receptors such as nicotinic ACh (nACh) and 5-HT3 receptors. Only the release evoked by nACh and 5-HT3 receptor activation was antagonised by halothane at clinically relevant concentrations (Fig. 4a [42, 43]). The degree of inhibition by halothane and other anaesthetics and alcohols correlated with their hydrophobic property, suggesting that the neuronal mechanism underlying the acute action of these drugs is based on their interaction with hydrophobic regions of the respective receptors which exhibit the common property to be ligand-gated ion channels. Halothane was also examined in the sympathetic nerve terminals of the isolated rabbit heart, in which noradrenaline release was stimulated by electrical impulses delivered to the intact postganglionic nerves attached to the hearts, by K+ depolarization or by a nACh receptor agonist. Only noradrenaline release evoked by nACh receptor activation was affected (Fig. 4b [44]).

Effect of halothane on evoked catecholamine release in bovine adrenal medulla (a) and on evoked noradrenaline release in rabbit heart (b). Only the release evoked by acetylcholine (ACh), γ-aminobutyric acid (GABA) and 5-HT, acting via ionotropic nACh, GABAA and 5-HT3 receptors, respectively, was inhibited whereas the release evoked by pilocarpine and histamine, acting via metabotropic mACh and H1 receptors, respectively, and by K+ depolarization and electrical stimulation (ES) was not. Note that the vertical broken line corresponds to the concentration of halothane reached in saline solution when the anaesthetic is used at the minimum alveolar concentration (MAC). SEM values and statistics are not shown. Re-drawn from [42, 43] (a) and [44] (b)
Manfred Göthert wrote in retrospect [45]: “Thus, we were the first to identify the nACh and the 5-HT3 receptor, two ligand-gated ion channels, as sites of action of halothane. These results were obtained about one decade earlier than the same conclusions from electrophysiological data. My biochemical models obviously were unknown systems used by an unknown author, who published so far unknown results—a typical constellation for not being cited in the relevant literature. I was very disappointed by this outcome of my experimental investigations. However, on the other hand, it was Klaus Starke who pointed at their importance in an article which appeared in Trends in Pharmacological Sciences [46]. He identified my publications as progress beyond the hypothesis of Overton and Meyer, who had speculated lipid constituents of the cell membrane to be sites of action of general anaesthetics and alcohols. The results obtained by my group suggested hydrophobic regions of nACh and 5-HT3 receptors to be their sites of action. By inducing a conformational change of the receptor protein structure, they mediate a decrease in function underlying their acute depressant effect on the CNS.”
Interaction of ethanol with 5-HT3, nACh and NMDA receptors
Many of the points discussed above for halothane also hold true for ethanol. The approach of Manfred Göthert to the identification of relevant sites underlying the effect of moderate doses of ethanol was guided by the consideration that any neuronal function contributing to the systemic depressant effect of ethanol should become manifest at such a concentration range relevant at social drinking. His studies dedicated to the elucidation of the mechanism(s) of ethanol action were performed in three steps. First, the effects of ethanol on nACh and 5-HT3 receptors were analysed in the model of the postganglionic sympathetic axon terminals of the rabbit heart (already described above). Second, the potential involvement of ionotropic glutamate receptors (of the N-methyl-d-aspartate (NMDA) and kainate type) was investigated on noradrenergic axon terminals of the rat brain. Third, the selectivity of the effect of ethanol towards 5-HT3 receptors was examined in an in vivo model.
- 1.
In the rabbit heart, ethanol inhibited noradrenaline release evoked by nACh and 5-HT3 receptor activation more markedly than that evoked by high K+ or electrical stimulation (Fig. 5a [47, 48]). The inhibitory effect of ethanol on the nACh receptor-evoked noradrenaline release occurred at concentrations compatible with moderate intoxication.
Fig. 5 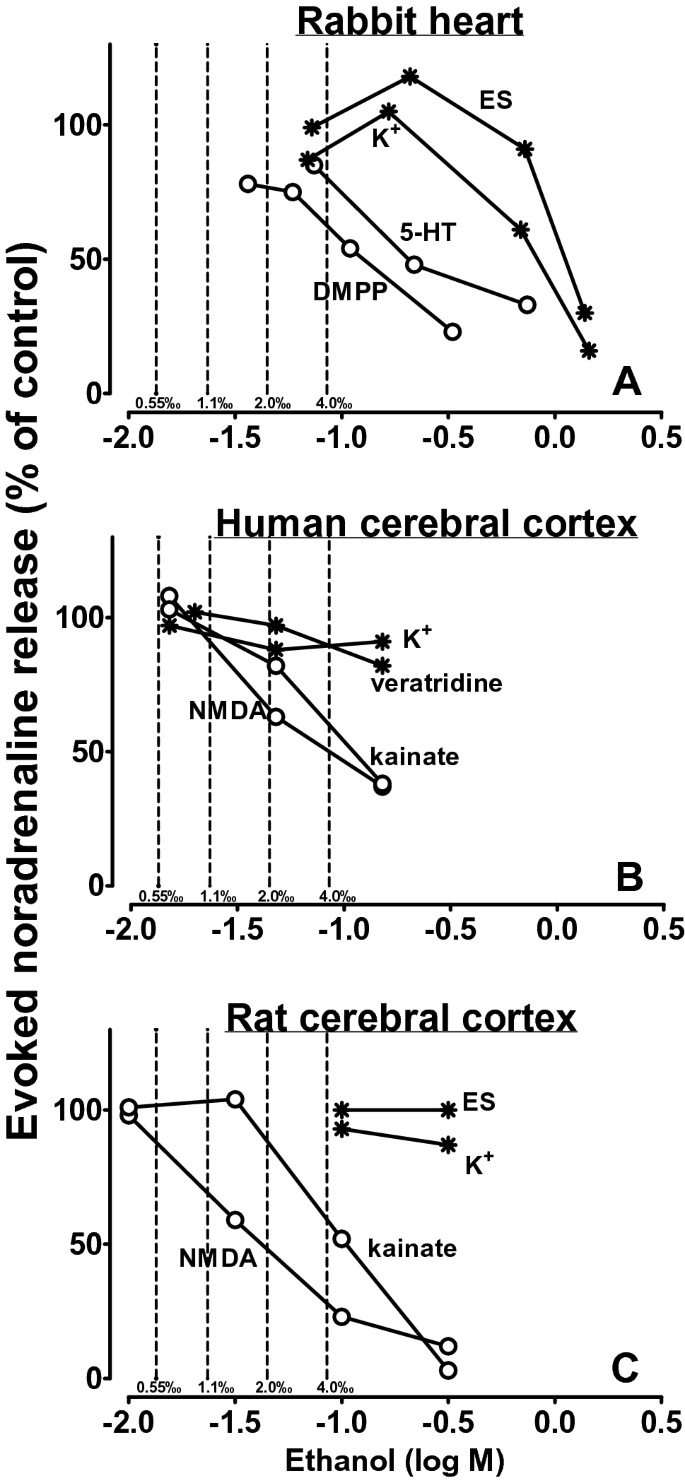
Effect of ethanol on evoked noradrenaline release in the rabbit heart (a), in human (b) and rat cerebral cortex slices (c). Only the release evoked by dimethylphenylpiperazinium (DMPP), 5-HT, N-methyl-d-aspartate (NMDA) and kainate acting via ionotropic nACh, 5-HT3, NMDA and kainate receptors, respectively, was inhibited whereas the release evoked by veratridine, K+ depolarization and electrical stimulation (ES) was not affected (or at extremely high concentrations of ethanol only). SEM values and statistics are not shown. Re-drawn from [47, 48] (a), [51] (b) and [49, 50] (c)
- 2.
The studies on cerebral cortex slices from rat (Fig. 5c) and human brain (Fig. 5b) revealed that noradrenaline release evoked by NMDA and kainate was inhibited by ethanol at even lower ethanol concentrations compared to the release evoked by nACh and 5-HT3 receptor activation; again, the release evoked by high K+, veratridine and/or electrical pulses was hardly affected [49,50,51]. Indeed the inhibitory effect of ethanol on NMDA receptors was found by Göthert and Fink [49] simultaneously with, but independent of, Lovinger et al. [52] (who used an electrophysiological technique).

Since NMDA-evoked noradrenaline release in the brain was a new scientific area in the end-eighties, a thorough pharmacological and functional characterization of the model was carried out. The effect of NMDA was competitively antagonised by 2-amino-5-phosphonovaleric acid and non-competitively by dizocilpine (MK 801) [53]. The NMDA-evoked noradrenaline release was abolished by tetrodotoxin suggesting dependence of release on action potentials depolarizing the cell membrane of axon terminals and also by the absence of Ca2+ ions in the superfusion fluid, involving abolishment of influx of Ca2+ ions into the terminals, probably via voltage-gated Ca2+ channels; presence of the physiological Mg2+ concentration of 1.2 mM in the superfusion fluid also abolished the NMDA-evoked noradrenaline release [53]. Several strategies proved useful to overcome the inhibitory effect of Mg2+, including an increase in the K+ concentration to 20 or 25 mM or the addition of veratridine 1 µM or 3,4-diamino-pyridine 200 µM [54].
NMDA also increases 5-HT release in the rat brain [55]. Further series of experiments provided clear-cut evidence that the NMDA receptor undergoes desensitisation [53]—an effect which is typical of ligand-gated ion channels. The phenomenon of desensitisation may also explain why Manfred Göthert could not establish nACh and 5-HT3 receptor models in the brain. He wrote [45]: “My first attempt failed to identify an analogous model of a central nervous system cell system. Desensitization of nicotinic and 5-HT receptors on noradrenaline, dopamine or serotonin nerve terminals in superfused cerebral slices appeared to be too fast as to be applicable for stimulation of release of radioactively labelled neurotransmitter in response to addition of a nicotinic or 5-HT3 receptor agonist.”
- 3.
The inhibitory effect of ethanol on a 5-HT3 receptor-related paradigm became the first subject of cooperation between Manfred Göthert’s institute in Bonn and B. Malinowska’s group in Białystok. In detail, the Bezold–Jarisch reflex (reflex bradycardia, see Fig. 6a) was examined, which can be evoked via various receptors located on the terminals of afferent cardiopulmonary C fibres, including 5-HT3 receptors and Transient Receptor Potential V1 (TRPV1) receptors (former designation Vanilloid Receptors 1, VR1). The 5-HT3 receptor-evoked Bezold–Jarisch reflex was inhibited by ethanol at moderate doses whereas the TRPV1 receptor-evoked Bezold–Jarisch reflex was not affected, suggesting that the 5-HT3 receptor on the afferent vagal nerve fibres is the site of action of ethanol (Fig. 6b [56]). In a further common investigation carried out in both university departments, substance P facilitated the 5-HT3 receptor-mediated Bezold–Jarisch reflex, whereas (+)-tubocurarine produced an inhibition [57].
Fig. 6 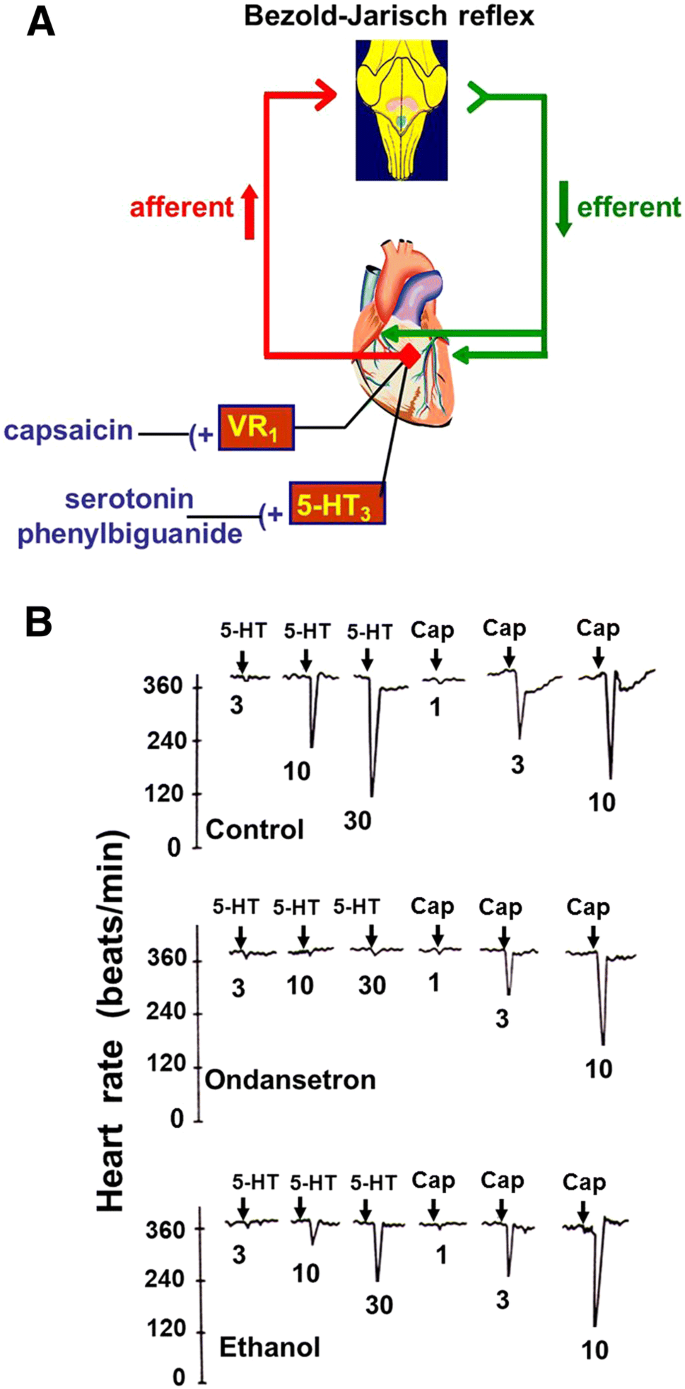
Effect of ethanol on the Bezold–Jarisch reflex elicited by 5-HT3 and V1 (TRPV1) receptor activation in the anaesthetized rat. a shows that the Bezold–Jarisch reflex encompasses a sensory vagal nerve projecting to the brain stem and an efferent cholinergic nerve projecting to the heart; reflex activation leads to an extreme bradycardia. b shows that the 5-HT3 receptor antagonist ondansetron 10 µg/kg i.v. abolished and ethanol 2 g/kg i.p. attenuated the bradycardia elicited by 5-HT (doses in µg/kg i.v.) via 5-HT3 receptors but did not affect the bradycardia elicited by capsaicin (CAP; doses in µg/kg i.v.) via V1 receptors. Re-drawn from [56]

Interaction of cannabinoids with 5-HT3 and nACh receptors
Cannabinoids act via cannabinoid CB1 and CB2 receptors [58] and via a multitude of other mechanisms [59]. Although Manfred Göthert was involved in the identification of presynaptic CB1 receptors (reviewed in [60]), he was mainly interested in CB1 receptor-independent effects of cannabinoids on 5-HT3 receptors. Both in vitro and in vivo studies were published to this topic.
The possibility that cannabinoid receptor agonists might directly act at 5-HT3 receptors was derived from an electrophysiological study on rat nodose ganglion [61]. In that investigation on whole cells, Δ9-tetrahydrocannabinol (THC), the endocannabinoid anandamide and other cannabinoids including the enantiomeric pair (−)-CP-55,940 and (+)-CP-56,667 stereoselectively inhibited the 5-HT-evoked current, the latter compound being less potent by a factor of more than 10 compared to its enantiomer. The anandamide-induced inhibition was noncompetitive. These results, although compatible with an action at the 5-HT3 receptor itself, did not rule out the alternative that the nodose ganglion cells might be endowed with CB1 receptors as well. However, experiments with a CB1 receptor antagonist like rimonabant were not carried out in that study [61].
In spite of other plausible possibilities of explanation, the presence of an allosteric site on the 5-HT3 receptor channel was an attractive hypothesis when considering that allosterically modulatory sites are typical features of this and other ligand-gated ion channels such as the GABA and nicotinic acetylcholine receptors and of G protein-coupled receptors as well [59]. To shed some light in this matter, patch-clamp experiments were carried out on excised outside-out patches from HEK (human embryonic kidney) 293 cells expressing the recombinant human 5-HT3A receptor [62]. In particular, exogenous cannabinoids and anandamide have been used to examine whether these compounds directly influence the human 5-HT system in a cannabinoid receptor-independent manner. This study was Manfred Göthert’s last scientific project; it was supported by a grant of the Deutsche Forschungsgemeinschaft (German Research Council; DFG). For several reasons, not all aspects addressed in that application could be fully elaborated, but a clear-cut positive answer to the basic question was possible.
In detail [62], the potencies of five cannabinoid receptor agonists as inhibitors of 5-HT-induced current in HEK 293 cells were determined to compare the rank order of their potencies with that of the Ki values as inhibitors of radioligand binding to human cannabinoid CB1 and CB2 receptors in membrane preparations. The rank orders of potencies and affinities in the three paradigms were clearly different from each other, arguing against an involvement of the cannabinoid receptors in the inhibition of 5-HT-induced current. For various reasons (such as a relatively high potency), the synthetic cannabinoid CB1 receptor agonist, WIN-55,212-2, was used as a representative drug in more detailed and sophisticated investigations into the site(s) and mechanism(s) underlying the inhibitory effect on 5-HT-induced current. In support of the conclusion drawn from the experiment with the five cannabinoid receptor agonists, the inhibition caused by WIN-55,212-2 was not altered by the antagonist rimonabant. WIN-55,212-3, the enantiomer of WIN-55,212-2, did not affect the 5-HT-induced current, suggesting that a second stereoselective cannabinoid receptor-independent binding site is involved in the inhibition of 5-HT-induced current by the active enantiomer. Analysis of the influence of WIN-55,212-2 on the concentration–response curve of 5-HT revealed that the cannabinoid did not change the EC50 value of 5-HT in stimulating current but reduced the maximum effect, suggesting noncompetitive inhibition. In fact, these results conform to the involvement of an allosteric mechanism in the function of the 5-HT3 receptor: binding of an appropriate cannabinoid to the allosteric site inhibits the current induced by activation of the orthosteric site with a 5-HT3 receptor agonist. At a given 5-HT3 receptor agonist concentration, the extent of inhibition is increasing as the cannabinoid concentration is being raised.
Whether or not the drugs produce an open channel block could be derived from experiments in which the time schedule of drug application was modified and in which the voltage dependence of the inhibitory effect of WIN-55,212-2 was investigated [62]. WIN-55,212-2 was applied at three different modes: (1) exclusive co-application of the drug with the stimulatory 5-HT pulse of 2 s duration (no preexposure to the drug); (2) application of the drug 3 min before, during and after stimulation with 5-HT; (3) application exclusively for 3 min prior to and after, but not together with, 5-HT. The site involved in the inhibition of channel function was found not to be easily accessible, since 3 min, i.e., a rather long equilibrium period, before stimulation was necessary to establish the effect. Furthermore, the drug failed to produce an inhibition when applied exclusively during, but not before, stimulation with 5-HT. However, location in trans-membrane or cytosolic domains of the receptor protein rather than within the open pore is compatible with the necessity of the presence of the cannabinoids before stimulation in such an equilibrium time and with the lack of voltage dependence of the WIN-55,212-2-induced inhibition of 5-HT-evoked current.
The location in or close to the transmembrane domain or in the cytosolic domain should make this site easily accessible to the endogenous ligand anandamide. Accordingly, the allosteric site of the 5-HT3 receptor may be tonically activated by anandamide. Thus, this compound and/or other endocannabinoids may play a physiological role by mediating a regulatory effect on the functional state of the 5-HT receptor. The novel alternative pathway may be assumed to gain importance when CB1 and CB2 receptors are blocked. It is an attractive hypothesis to postulate that the human 5-HT3 receptor is endowed with a motif which in human 5-HT3 receptors is recognised by all CB1 and CB2 receptor agonists examined in our study [62]. Simultaneously with us other researchers studied the effect of anandamide on the 5-HT-induced current through the 5-HT3A receptor channel in Xenopus oocytes expressing the cloned mouse 5-HT3 receptor [63].
In subsequent studies of Manfred Göthert, a strategy was developed for the identification of the binding epitope of the cannabinoid allosteric site of the human 5-HT3 receptor. Unfortunately, this project could only be initiated, but nevertheless will be briefly described here. Site-directed mutagenesis and a chimera of the 5-HT3 receptor and another receptor which is highly related with respect to the amino acid sequence, such as the human α7-nACh receptor, were the backbone of our strategy. Prerequisites for its application were that the affinities for cannabinoids of the two ligand-gated ion channels are high and different enough. Candidates in this respect were THC, WIN-55,212-2 and last but not least anandamide.
On the basis of these considerations, the first step of our strategy was to succeed in inducing ion flux through recombinant human α7-nACh receptors and to determine at which potency the allosteric site ligands inhibit the ACh-induced ion flux through the receptor channel. In fact, the cDNA for the human α7-nACh receptor has been cloned and stably expressed in HEK 293 cells. However, for large series of such experiments, the electrophysiological technique is too complex and time consuming. Therefore, a luminometric assay of Ca2+ influx into whole cells was developed [64]. At this stage, we had to finish the project which basically looked very promising.
In appropriate in vivo studies, further support has been sought for the suggestion that the orthosteric site of the 5-HT3 receptor is under the inhibitory control by cannabinoids acting at the allosteric site. Results which are at least compatible with this hypothesis have been obtained in three in vivo studies, one of them involving peripheral 5-HT3 receptors, the other two models dealing with central 5-HT3 receptors.
The first of these papers deals with the cannabinoid receptor-independent inhibitory effect of cannabinoid receptor agonists on the 5-HT3 receptor-mediated Bezold–Jarisch reflex [65]. In rats pretreated with the CB1 receptor antagonist rimonabant, the Bezold–Jarisch reflex was stimulated by activation either of the 5-HT3 receptor with 5-HT or phenylbiguanide or of the TRPV1 receptor with capsaicin. The phenylbiguanide-, but not the capsaicin-, induced Bezold–Jarisch reflex was inhibited by CP-55,940 or WIN-55,212-2, i.e., agonists at the CB1 receptor whereas, in agreement with the in vitro results [62], WIN-55,212-3 (the enantiomeric partner of the latter drug) was inactive.
The second report was devoted to the putative role of 5-HT3 receptors in the cannabinoid receptor-independent decrease in cocaine hyperlocomotion in rats [66]. Their basal locomotor activity expressed as distance travelled in centimetre per hour was increased by cocaine. The hyperlocomotion caused by cocaine was abolished by WIN 55,212-2, an effect which was not altered by rimonabant. WIN-55,212-3, the enantiomer of WIN-55,212-2, was ineffective (Fig. 7a). The 5-HT3 receptor antagonist ondansetron shared the inhibitory effect of WIN 55,212-2 on cocaine-evoked hyperlocomotion (Fig. 7b).

Effect of cannabinoid receptor ligands and ondansetron on the cocaine (COC; 10 mg/kg i.p.)-induced hyperlocomotion in rats. The cannabinoid receptor agonist WIN-55,212-2 (WIN-2) at 6 mg/kg i.p. (but not the same dose of its enantiomer WIN-55,212-3; WIN-3) abolished the effect of COC; this effect was not affected by the CB1 receptor antagonist rimonabant (RIM) 5 mg/kg i.p. (a). The 5-HT3 receptor antagonist ondansetron (OND; doses in mg/kg i.p.) attenuated the effect of COC dose-dependently (b). **P < 0.01, compared to control; #P < 0.05, ##P < 0.01, compared to COC. Re-drawn from [66]
In both in vivo models considered so far, the pattern of active and inactive drugs determined in the respective models mimicked in principal that of the findings in vitro. Therefore, the data are in agreement and, thus, add some evidence to the suggestion that the function of the 5-HT3 receptor is controlled by an allosteric site.
This statement could also be supported by the results obtained in the third in vivo approach towards such a unifying interpretation [67]. In that model which was based on experiments with THC and anandamide in cannabinoid receptor knockout mice, the antinociceptive effect (“analgesia”) by these drugs was determined as latency in seconds until the first sign of pain in the hot plate test. In wild-type but not in cannabinoid CB1 and CB2 receptor double knockout mice, THC caused antinociception which was resistant to blockade by ondansetron indicating that “analgesia” by the phytocannabinoid is mediated exclusively by cannabinoid receptors, i.e., is not related to the 5-HT3 receptor. By contrast, both in wild-type and double knockout animals, the endocannabinoid anandamide induced “analgesia”. When, however, 5-HT3 receptors were subjected to a “pharmacological knockout” with a high dose of ondansetron, the “analgesia” did no longer occur. These data exclude the involvement of CB1 and CB2 receptors in the “analgesic” effect of anandamide and suggest a role of the 5-HT3 receptor system in this action.
The explanation of the ability of ondansetron to induce “analgesia” is based on two properties: (i) 5-HT3 receptors mediate a pronociceptive function [68]. (ii) Descending 5-HT neurones [69] target 5-HT3 receptors in the spinal cord—receptors which are involved in the expression of pronociceptive behaviour. One may conclude that the removal of pronociception by blockade of the orthosteric site of the 5-HT3 receptor by ondansetron may become manifest in “analgesia”. In fact, evidence was presented for an analgesic effect of ondansetron in neuropathic pain [70]. Binding of anandamide at sufficiently high amount to the allosteric site of the 5-HT3 receptor to completely eliminate the function of the receptor abolishes ion current through the receptor channel, thus mimicking the antagonism of ondansetron at the orthosteric site of the receptor.
The final project within this series was dedicated to the nACh receptor, which like the 5-HT3 receptor is a member of the Cys-loop family within the superfamily of ligand-gated ion channels [71]. Remember that both receptors behaved similarly against halothane and ethanol, as described in the two preceding sections. Interesting enough, Oz et al. [72] found that R-methanandamide (MAEA), the metabolically stable analogue of anandamide, inhibited the ACh-induced current through the chick α7-nACh receptor channel expressed in Xenopus oocytes and that this inhibition was noncompetitive. Although own in vitro studies were not performed, the possibility that this mechanism may also occur in vivo was considered in a project for which Manfred Göthert received an Alexander von Humboldt Polish Honorary Research Fellowship from the Fundacja na Rzecz Nauki Polskiej (Foundation for Polish Science; FNP) after his retirement. Indeed, a CB1 receptor-independent noncompetitive inhibitory effect of R-methanandamide against the nicotine-induced tachycardia was obtained in rats in vivo [73].
Conclusions
The late Professor Manfred Göthert contributed to 5-HT pharmacology and beyond in several aspects. He was involved in the stepwise classification of the 5-HT receptor subtypes, particularly of 5-HT1B, 5-HT1D and 5-HT3 receptors. He identified four types of presynaptic receptors on central and/or peripheral serotoninergic, noradrenergic and/or cholinergic neurones, i.e., inhibitory 5-HT1B and 5-HT1D and facilitatory 5-HT3 and 5-HT4 receptors. Ten years in advance of other authors, he found that ionotropic receptors, including 5-HT3 and nACh receptors, are targeted by halothane and other general anaesthetics. The latter two receptors were also allosterically inhibited by ethanol but, as shown simultaneously but independent of other authors, another type of ionotropic receptors, namely NMDA receptors, were affected at even lower concentrations. Finally, he found that 5-HT3 and nACh receptors were allosterically inhibited also by cannabinoids via a CB1 and CB2 receptor-independent mechanism. It has to be stressed that only part of the about 265 papers by Manfred Göthert (about 40% of which refer to 5-HT) was covered in this article. The three coauthors, who have been working with this eminent pharmacologist over a time period of decades, felt honoured to complete the draft of his last manuscript.
References
Göthert M. Development of 5-HT receptor complexity within 39 years—from drugs as tools to new therapeutics. Pharmacol Rep. 2010;62(Suppl 1):3.
Göthert M. Serotonin discovery and stepwise disclosure of 5-HT receptor complexity over four decades. Part I. General background and discovery of serotonin as a basis for 5-HT receptor identification. Pharmacol Rep. 2013;65(4):771–86.
Gaddum JH, Picarelli ZP. Two kinds of tryptamine receptor. Br J Pharmacol. 1957;12(3):134–9.
Niesler B, Walstab J, Combrink S, Möller D, Kapeller J, Rietdorf J, Bönisch H, Göthert M, Rappold G, Brüss M. Characterization of the novel human serotonin receptor subunits 5-HT3C, 5-HT3D, and 5-HT3E. Mol Pharmacol. 2007;72(1):8–17.
Peroutka SJ, Snyder SH. Multiple serotonin receptors: differential binding of [3H]5-hydroxytryptamine, [3H]lysergic acid diethylamide and [3H]spiroperidol. Mol Pharmacol. 1979;16(3):687–99.
Engel G, Göthert M, Müller-Schweinitzer E, Schlicker E, Sistonen L, Stadler PA. Evidence for common pharmacological properties of [3H]5-hydroxytryptamine binding sites, presynaptic 5-hydroxytryptamine autoreceptors in CNS and inhibitory presynaptic 5-hydroxytryptamine receptors on sympathetic nerves. Naunyn-Schmiedeberg’s Arch Pharmacol. 1983;324(2):116–24.
Engel G, Göthert M, Hoyer D, Schlicker E, Hillenbrand K. Identity of inhibitory presynaptic 5-hydroxytryptamine (5-HT) autoreceptors in the rat brain cortex with 5-HT1B binding sites. Naunyn-Schmiedeberg’s Arch Pharmacol. 1986;332(1):1–7.
Gozlan H, El Mestikawy S, Pichat L, Glowinski J, Hamon M. Identification of presynaptic serotonin autoreceptors using a new ligand: 3H-PAT. Nature. 1983;305(5930):140–2.
De Vivo M, Maayani S. Characterization of the 5-hydroxytryptamine1a receptor-mediated inhibition of forskolin-stimulated adenylate cyclase activity in guinea pig and rat hippocampal membranes. J Pharmacol Exp Ther. 1986;238(1):248–53.
Bradley PB, Engel G, Feniuk W, Fozard JR, Humphrey PP, Middlemiss DN, Mylecharane EJ, Richardson BP, Saxena PR. Proposals for the classification and nomenclature of functional receptors for 5-hydroxytryptamine. Neuropharmacology. 1986;25(6):563–76.
Lübbert H, Hoffman BJ, Snutch TP, van Dyke T, Levine AJ, Hartig PR, Lester HA, Davidson N. cDNA cloning of a serotonin 5-HT1C receptor by electrophysiological assays of mRNA-injected Xenopus oocytes. Proc Natl Acad Sci. 1987;84(12):4332–6.
Hoyer D. Molecular pharmacology and biology of 5-HT1C receptors. Trends Pharmacol Sci. 1988;9(3):89–94.
Heuring RE, Peroutka SJ. Characterization of 3H-5-HT binding in bovine caudate. J Neurosci. 1987;7(3):894–903.
Dumuis A, Bouhelal R, Sebben M, Cory R, Bockaert J. A nonclassical 5-hydroxytryptamine receptor positively coupled with adenylate cyclase in the central nervous system. Mol Pharmacol. 1988;34(6):880–7.
Bockaert J, Sebben M, Dumuis A. Pharmacological characterization of 5-hydroxytryptamine4 (5-HT4) receptors positively coupled to adenylate cyclase in adult guinea pig hippocampal membranes: effect of substituted benzamide derivatives. Mol Pharmacol. 1990;37(3):408–11.
Hoyer D, Waeber C, Schoeffter P, Palacios JM, Dravid A. 5-HT1C receptor-mediated stimulation of inositol phosphate production in pig choroid plexus. A pharmacological characterization. Naunyn-Schmiedeberg’s Arch Pharmacol. 1989;339(3):252–8.
Humphrey PPA, Hartig P, Hoyer D. A proposed new nomenclature for 5-HT receptors. Trends Pharmacol Sci. 1993;14(6):233–6.
Leonhardt S, Herrick-Davis K, Titeler M. Detection of a novel serotonin receptor subtype (5-HT1E) in human brain: interaction with a GTP-binding protein. J Neurochem. 1989;53(2):465–71.
Adham N, Kao HT, Schechter LE, Bard J, Olsen M, Urquhart D, Durkin M, Hartig PR, Weinshank RL, Branchek TA. Cloning of another human serotonin receptor (5-HT1F): a fifth 5-HT1 receptor subtype coupled to the inhibition of adenylate cyclase. Proc Natl Acad Sci. 1993;90(2):408–12.
Foguet M, Hoyer D, Pardo LA, Parekh A, Kluxen FW, Kalkman HO, Stühmer W, Lübbert H. Cloning and functional characterization of the rat stomach fundus serotonin receptor. EMBO J. 1992;11(9):3481–7.
Kursar JD, Nelson DL, Wainscott DB, Cohen ML, Baez M. Molecular cloning, functional expression, and pharmacological characterization of a novel serotonin receptor (5-hydroxytryptamine2F) from rat stomach fundus. Mol Pharmacol. 1992;42(4):549–57.
Plassat JL, Boschert U, Amlaiky N, Hen R. The mouse 5-HT5 receptor reveals a remarkable heterogeneity within the 5-HT1D family. EMBO J. 1992;11(13):4779–86.
Matthes H, Boschert U, Amlaiky N, Grailhe R, Plassat JL, Muscatelli F, Mattei MG, Hen R. Mouse 5-hydroxytryptamine5A and 5-hydroxytryptamine5B receptors define a new family of serotonin receptors: cloning, functional expression, and chromosomal location. Mol Pharmacol. 1993;43(3):313–9.
Weinshank RL, Zgombick JM, Macchi MJ, Branchek TA, Hartig PR. Human serotonin 1D receptor is encoded by a subfamily of two distinct genes: 5-HT1Dα and 5-HT1Dβ. Proc Natl Acad Sci. 1992;89(8):3630–4.
Hartig PR, Hoyer D, Humphrey PPA, Martin GR. Alignment of receptor nomenclature with the human genome: classification of 5-HT1B and 5-HT1D receptor subtypes. Trends Pharmacol Sci. 1996;17(3):103–5.
Monsma FJ Jr, Shen Y, Ward RP, Hamblin MW, Sibley DR. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol. 1993;43(3):320–7.
Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, Foyer PE, Racke M, Slone AL, Siegel BW, Danielson PE, Sutcliffe JF, Erlander MG. A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms. Neuron. 1993;11(3):449–58.
Gustafson EL, Durkin MM, Bard JA, Zgombick J, Branchek TA. A receptor autoradiographic and in situ hybridization analysis of the distribution of the 5-ht7 receptor in rat brain. Br J Pharmacol. 1996;117(4):657–66.
Miyake A, Mochizuki A, Takemoto Y, Akuzawa S. Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol Pharmacol. 1995;48(3):407–16.
Belelli D, Balczarek JM, Hope AG, Peters JA, Lambert JJ, Blackburn TP. Cloning and functional expression of a human 5-hydroxytryptamine type 3AS receptor subunit. Mol Pharmacol. 1995;48(6):1054–62.
Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397(6717):359–63.
Niesler B, Frank B, Kapeller J, Rappold GA. Cloning, physical mapping and expression analysis of the human 5-HT3 serotonin receptor-like genes HTR3C, HTR3D and HTR3E. Gene. 2003;310:101–11.
Tiger M, Varnäs K, Okubo Y, Lundberg J. The 5-HT1B receptor—a potential target for antidepressant treatment. Psychopharmacology. 2018;235(5):1317–34.
Schlicker E, Fink K, Molderings GJ, Price GW, Duckworth M, Gaster L, Middlemiss DN, Zentner J, Likungu J, Göthert M. Effects of selective h5-HT1B (SB-216641) and h5-HT1D (BRL-15572) receptor ligands on guinea-pig and human 5-HT auto- and heteroreceptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 1997;356(3):321–7.
Schoeffter P, Sahin-Erdemli I. Further characterization of the 5-hydroxytryptamine 5-HT1-like receptor mediating contraction of guinea-pig iliac artery. Eur J Pharmacol. 1992;219(2):295–301.
Molderings GJ, Fink K, Schlicker E, Göthert M. Inhibition of noradrenaline release via presynaptic 5-HT1B receptors in the rat vena cava. Naunyn-Schmiedeberg’s Arch Pharmacol. 1987;336(3):245–50.
Molderings GJ, Frölich D, Likungu J, Göthert M. Inhibition of noradrenaline release via presynaptic 5-HT1Dα receptors in human atrium. Naunyn-Schmiedeberg’s Arch Pharmacol. 1996;353(3):272–80.
Middlemiss DN, Göthert M, Schlicker E, Scott CM, Selkirk JV, Watson J, Gaster LM, Wyman P, Riley G, Price GW. SB-236057, a selective 5-HT1B receptor inverse agonist, blocks the 5-HT human terminal autoreceptor. Eur J Pharmacol. 1999;375(1–3):359–65.
Zgombick JM, Schechter LE, Kucharewicz SA, Weinshank RL, Branchek TA. Ketanserin and ritanserin discriminate between recombinant human 5-HT1Dα and 5-HT1Dβ receptor subtypes. Eur J Pharmacol. 1995;291(1):9–15.
Bertaccini EJ. The molecular mechanisms of anesthetic action: updates and cutting edge developments from the field of molecular modeling. Pharmaceuticals. 2010;3(7):2178–96.
Ueda K, Kamaya H. Kinetic and thermodynamic aspects of the mechanism of general anaesthesia in a model system of firefly luminescence in vitro. Anesthesiology. 1973;38(5):425–36.
Göthert M. Die Sekretionsleistung des Nebennierenmarks unter dem Einfluß von Narkotica und Muskelrelaxantien. Berlin, Heidelberg, New York: Springer; 1972. ISBN 3-540-06058-8.
Göthert M, Dorn W, Loewenstein I. Inhibition of catecholamine release from the adrenal medulla by halothane. Naunyn-Schmiedeberg’s Arch Pharmacol. 1976;294(3):239–49.
Göthert M. Effects of halothane on the sympathetic nerve terminals of the rabbit heart. Differences in membrane actions of halothane and tetracaine. Naunyn-Schmiedeberg’s Arch Pharmacol. 1974;286(2):125–43.
Göthert M. Life in movement as a scientist. In: Geschichte und Wirken der pharmakologischen, klinisch-pharmakologischen und toxikologischen Institute im deutschsprachigen Raum (A Philippu ed). Wattens: Berenkamp; 2014, pp 185–210, ISBN 978-3-85093-325-4.
Starke K. Selectivity of ethanol on ligand-gated ion channels. Trends Pharmacol Sci. 1991;12(5):182.
Göthert M, Thielecke G. Inhibition by ethanol of noradrenaline output from peripheral sympathetic nerves: possible interaction of ethanol with neuronal receptors. Eur J Pharmacol. 1976;37(2):321–8.
Göthert M, Dührsen U, Rieckesmann JM. Ethanol, anaesthetics and other lipophilic drugs preferentially inhibit 5-hydroxytryptamine- and acetylcholine-induced noradrenaline release from sympathetic nerves. Arch int Pharmacodyn. 1979;242(2):196–209.
Göthert M, Fink K. Inhibition of N-methyl-d-aspartate (NMDA)- and glutamate-induced noradrenaline and acetylcholine release in the rat brain by ethanol. Naunyn-Schmiedeberg’s Arch Pharmacol. 1989;340(5):516–21.
Fink K, Göthert M. Inhibition of N-methyl-d-aspartate-induced noradrenaline release by alcohols is related to their hydrophobicity. Eur J Pharmacol. 1990;191(2):225–9.
Fink K, Schultheiß Göthert M. Inhibition of N-methyl-d-aspartate- and kainate-evoked noradrenaline release in human cerebral cortex slices by ethanol. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992;345(6):700–3.
Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–4.
Fink K, Göthert M, Molderings G, Schlicker E. N-methyl-d-aspartate (NMDA) receptor-mediated stimulation of noradrenaline release, but not release of other transmitters, in the rat brain cortex: location, characterization and desensitization. Naunyn-Schmiedeberg’s Arch Pharmacol. 1989;339(5):514–21.
Fink K, Göthert M, Schlicker E. Veratridine and other depolarizing agents counteract the inhibitory effect of Mg2+ ions on N-methyl-d-aspartate (NMDA)-induced noradrenaline release in vitro. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992;342(1):53–60.
Fink K, Schmitz V, Böing C, Göthert M. Stimulation of serotonin release in the rat brain cortex by activation of ionotropic glutamate receptors and its modulation via α2-heteroreceptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 1995;352(4):394–401.
Malinowska B, Göthert M, Godlewski G, Wrobel B, Bönisch H, Buczko W. Inhibitory effect of ethanol on the 5-hydroxytryptamine-induced Bezold–Jarisch reflex—involvement of peripheral 5-HT3 receptors. Eur J Pharmacol. 1995;293(1):71–6.
Malinowska B, Godlewski G, Buczko W, Göthert M. Facilitation by substance P and inhibition by (+)-tubocurarine of the 5-HT3 receptor-mediated Bezold-Jarisch reflex in rats. Eur J Pharmacol. 1996;315(2):159–64.
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54(2):161–202.
Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62(4):588–631.
Szabo B, Schlicker E. Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol. 2005;168:327–65.
Fan P. Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurons. J Neurophysiol. 1995;73(2):907–10.
Barann M, Molderings G, Brüss M, Bönisch H, Urban BW, Göthert M. Direct inhibition by cannabinoids of human 5-HT3A receptors: probable involvement of an allosteric modulatory site. Br J Pharmacol. 2002;137(5):589–96.
Oz M, Zhang L, Morales M. Endogenous cannabinoid, anandamide, acts as a noncompetitive inhibitor on 5-HT3 receptor-mediated responses in Xenopus oocytes. Synapse. 2002;46(3):150–6.
Walstab J, Combrink S, Brüss M, Göthert M, Niesler B, Bönisch H. Aequorin luminescence-based assay for 5-hydroxytryptamine (serotonin) type 3 receptor characterization. Anal Biochem. 2007;368(2):185–92.
Godlewski G, Göthert M, Malinowska B. Cannabinoid receptor-independent inhibition by cannabinoid agonists of the peripheral 5-HT3 receptor-mediated von Bezold-Jarisch reflex. Br J Pharmacol. 2003;138(5):767–74.
Przegaliński E, Göthert M, Frankowska M, Filip M. WIN 55,212-2-induced reduction of cocaine hyperlocomotion: possible inhibition of 5-HT3 receptor function. Eur J Pharmacol. 2005;517(1–2):68–73.
Rácz I, Bilkei-Gorzo A, Markert A, Stamer F, Göthert M, Zimmer A. Anandamide effects on 5-HT3 receptors in vivo. Eur J Pharmacol. 2008;596(1–3):98–101.
Zeitz KP, Guy N, Malmberg AB, Dirajlal S, Martin WJ, Sun L, Bonhaus DW, Stucky CL, Julius D, Basbaum AI. The 5-HT3 subtype of serotonin receptor contributes to nociceptive processing via a novel subset of myelinated and unmyelinated nociceptors. J Neurosci. 2002;22(3):1010–9.
Bowker RM, Westlund KN, Sullivan MC, Wilber JF, Coulter JD. Descending serotonergic, peptidergic and cholinergic pathways from the raphe nuclei: a multiple transmitter complex. Brain Res. 1983;288(1–2):33–48.
McCleane GJ, Suzuki R, Dickenson AH. Does a single intravenous injection of the 5HT3 receptor antagonist ondansetron have an analgesic effect in neuropathic pain? A double-blinded, placebo-controlled cross-over study. Anesth Analg. 2003;97(5):1474–8.
Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27(6):329–36.
Oz M, Zhang L, Ravindran A, Morales M, Lupica CR. Differential effects of endogenous and synthetic cannabinoids on alpha7-nicotinic acetylcholine receptor-mediated responses in Xenopus oocytes. J Pharmacol Exp Ther. 2004;310(3):1152–60.
Baranowska U, Göthert M, Rudz R, Malinowska B. Methanandamide allosterically inhibits in vivo the function of peripheral nicotinic acetylcholine receptors containing the alpha 7-subunit. J Pharmacol Exp Ther. 2008;326(3):912–9.
Molderings GJ, Brüss M, Göthert M. Functional and molecular identification of 5-hydroxytryptamine receptors in rabbit pulmonary artery: involvement in complex regulation of noradrenaline release. Pharmacol Rep. 2006;58(2):188–99.
Molderings GJ, Göthert M, Fink K, Roth E, Schlicker E. Inhibition of noradrenaline release in the pig coronary artery via a novel serotonin receptor. Eur J Pharmacol. 1989;164(2):213–22.
Fink K, Zentner J, Göthert M. Subclassification of presynaptic 5-HT autoreceptors in the human cerebral cortex as 5-HT1Dβ receptors. Naunyn-Schmiedeberg’s Arch Pharma-col. 1995;352(4):451–4.
Acknowledgements
Open Access funding provided by Projekt DEAL. The authors are grateful to Barbara Hantschel for typing the manuscript and to Jatayu Holznagel, Marek Toczek and Anna Pędzińska-Betiuk for generating the figures.
Funding
The experimental work by Manfred Göthert cited in this review was supported by the Deutsche Forschungsgemeinschaft (German Research Council; DFG) and Collaborative Research Centres funded by the European Community.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
This review is dedicated to Polish-German cooperation in the spirit of confident and peaceful neighbourhood with best wishes for ongoing success.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Göthert, M., Bönisch, H., Malinowska, B. et al. Serotonin discovery and stepwise disclosure of 5-HT receptor complexity over four decades. Part II. Some contributions of Manfred Göthert. Pharmacol. Rep 72, 271–284 (2020). https://doi.org/10.1007/s43440-019-00047-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-019-00047-4