
Abstract
The review presents metabolic properties of Ivermectin (IVM) as substrate and inhibitor of human P450 (P450, CYP) enzymes and drug transporters. IVM is metabolized, both in vivo and in vitro, by C-hydroxylation and O-demethylation reactions catalyzed by P450 3A4 as the major enzyme, with a contribution of P450 3A5 and 2C9. In samples from both in vitro and in vivo metabolism, a number of metabolites were detected and as major identified metabolites were 3″-O-demethylated, C4-methyl hydroxylated, C25 isobutyl-/isopropyl-hydroxylated, and products of oxidation reactions. Ivermectin inhibited P450 2C9, 2C19, 2D6, and CYP3A4 with IC50 values ranging from 5.3 μM to no inhibition suggesting that it is no or weak inhibitor of the enzymes. It is suggested that P-gp (MDR1) transporter participate in IVM efflux at low drug concentration with a slow transport rate. At the higher, micromolar concentration range, which saturates MDR1 (P-gp), MRP1, and to a lesser extent, MRP2 and MRP3 participate in IVM transport across physiological barriers. IVM exerts a potent inhibition of P-gp (ABCB1), MRP1 (ABCC1), MRP2 (ABCC2), and BCRP1 (ABCG2), and medium to weak inhibition of OATP1B1 (SLC21A6) and OATP1B3 (SLCOB3) transport activity. The metabolic and transport properties of IVM indicate that when IVM is co-administered with other drugs/chemicals that are potent inhibitors/inducers P4503A4 enzyme and of MDR1 (P-gp), BCRP or MRP transporters, or when polymorphisms of the drug transporters and P450 3A4 exist, drug–drug or drug–toxic chemical interactions might result in suboptimal response to the therapy or to toxic effects.
Similar content being viewed by others
Introduction
Avermectin compounds are a class of 16-membered macrolide lactone compounds produced by the fungus Streptomyces avermitilis, which synthesizes a complex of eight individual avermectin compounds (Burg et al. 1979). Ivermectin (IVM), as one of the avermectin derivative, is a semisynthetic derivative of avermectin B1 and consists of an 80:20 mixture of the equipotent homologous 22,23-dehydro B1a and B1b (Fig. 1). IVM is a widely used FDA-approved broad-spectrum antiparasitic drug used also to treat pest insects and was found to be especially effective to decrease P. vivax transmission (Hotson 2020; Campbell et al. 1983; Pinilla et al. 2018). As an anthelminthic drug, IVM is used to treat various parasitic infestations in veterinary and human medicine. Applications include river blindness (onchocerciasis) (African region Onchocerciasis Control Programme of West Africa, 1975–2002, (Mectizan) by Merck & Co. Inc., in 1988), head lice, scabies, lymphatic filariasis, ascariasis, enterobiasis, strongyloidiasis, and trichuriasis (Lindley 1987; Boatin and Richards 2006; Moncayo et al. 2008; Anselmi et al. 2015; Thomsen et al. 2016; King et al. 2018; Sharun et al. 2019).

Ivermectin B1a (IVM-B1a) and B1b (IVM-B1b)
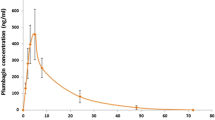
In the treatment of onchocerciasis, the optimal dose of IVM is 150 μg/kg once or three times yearly (González Canga et al. 2008). The drug is effective in most patients with scabies after a single oral dose of 0.2 mg/kg, which is the maximal FDA-approved dose, but often the regimen involves two or three repeated doses, separated by intervals of 1 or 2 weeks (Dourmishev et al. 2005). Pharmacokinetic data of IVM in humans following single oral dose (6 and 12 mg) showed that no unchanged or conjugated Ivermectin could be detected in urine, AUC and Cmax were dosing proportional and increased linearly with dose with tmax approximately 4 h and t1/2 elimination, depending on the doses applied, was up to 91 h. Due to its high lipid solubility, IVM is widely distributed within the body and strongly binds to plasma proteins (93.2 ± 4.4% (SD) (Klotz et al. 1990; Krishna and Klotz 1993; Guzzo et al. 2002; Muñoz et al. 2018).
In addition to its antiparasitic action, IVM was also reported to inhibit the growth of some human cancer cells. IVM has powerful antitumor effects, including the inhibition of proliferation, metastasis, and angiogenic activity in different cancer cells (Korystov et al. 2004; Melotti et al. 2014; Kwon et al. 2015; Dou et al. 2016; Zhu et al. 2017; Nambara et al. 2017; Juarez et al. 2018; Jiang et al. 2019; Liu et al. 2020; Tang et al. 2021).
IVM also possess broad-spectrum antiviral property in vitro against RNA and deoxyribonucleic acid (DNA) viruses, including human immunodeficiency virus-1 (HIV-1), dengue virus (DENV), influenza, Venezuelan equine encephalitis virus (VEEV), a flavivirus, and Zika virus (Wagstaff et al. 2012; Mastrangelo et al. 2012; Pandey et al. 2020; Yang et al. 2020). A single dose of IVM effectively removed SARS-CoV-2 viral RNA in cell culture by 48 h. It was suggested that IVM might be used for the treatment of COVID-19 patients in the early phase with mild to moderate symptoms, PCR positive for SARS-CoV-2 virus, and those without comorbidities such as chronic obstructive pulmonary disease, diabetes, hypertension, obesity, acute or chronic renal failure, and coronary diseases. IVM suggested anti-CoV-2 effects are based on in vitro studies, showing that this compound inhibits the importin alpha/beta-1 nuclear transporter, and thus, in cell cultures, reduces the replication of various viruses including SARS-CoV-2 (Bray et al. 2020; Caly et al. 2020; Rizzo 2020; Sharun et al. 2020; Heidary and Gharebaghi 2020; Dixit et al. 2020; Chaccour et al. 2020a, b; Formiga et al.2020). In addition, in a silico-based analysis of Ivermectin’s molecular interaction indicates positive interaction of Ivermectin with viral protein targets, which is leading for SARS-CoV 2 N-protein NTD (nucleocapsid protein N-terminal domain) (Kaur et al. 2021). Avermectins have also antifungal activity and are used for the treatment of several metabolic disorders. Ivermectin plasma concentrations required to reach an in vitro antiviral efficacy (about 2–10 μM) might be very toxic as very high oral doses would be needed for antiviral use and overdose or uncontrolled administration of IVM can lead to the development of severe toxicity (El-Saber Batiha et al. 2020). Ivermectin has not been approved for the treatment of viral diseases, including SARS-CoV-2, and in April 2020, FDA issued a warning not to use Ivermectin to treat COVID-19 in humans (Bray et al. 2020).
The pharmacokinetics and hepatic metabolism of IVM was investigated in a subpopulation of the CF-1 mouse stock naturally deficient in mdr1a P-glycoprotein (multidrug-resistance protein). Intestinal excretion of (3H)IVM enhances in P-gp (+/+) animals. The absence of P-gp resulted in higher blood concentrations of IVM after oral dosing, suggesting enhanced absorption of IVM in (−/−) mice. Concentrations of (3H)IVM were always greater in the brains of (−/−) mice compared with (+/+) mice after either i.v. or oral administration. In contrast, liver concentration was not different between (+/+) and (−/−) animals after an i.v. dose. These results show the P-gp (−/−) and (+/+) subpopulations of CF-1 mice are useful for studying the role of P-gp in systemic exposure and tissue disposition of P-gp substrates (Umbenhauer et al. 1997; Kwei et al. 1999). A mechanism of antiparasitic action of IVM is inhibition of a glutamate-gated chloride channel and a GABA receptor, specific for some invertebrates. In mammals, IVM may also inhibit GABAergic neurotransmission by promoting the release of GABA and acting as a GABA receptor agonist exerting neurotoxic activity (Yang 2012; Ménez et al. 2012). Potential neurotoxic activity in humans depends on the absorption-extrusion activity of the drug from the gastrointestinal tract/blood–brain barrier which is regulated by P-gp, MRPs, ABCB1, and other ABC transporters. In the case of IVM, it was suggested that the severe central nervous system side effects seen in various vertebrates following IVM treatment may be due to an absence of, or functional deficiency in P-glycoprotein (Edwards 2003). Due to the wide range of its clinical applications and its possible co-administration with other drugs when used to treat various illnesses, there is a possibility that toxic and/or clinically significant drug–drug interactions might occur. Consequences of toxic drug–chemical and/or drug–drug interactions which might occur during co-treatment of IVM with other drugs/chemicals will depend on metabolic and pharmacokinetic properties of IVM and co-administered drugs, i.e., their interactions with drug-metabolizing enzymes (cytochrome P450s) and drug transporters involved in IVM metabolic pathways. The interactions might thus result in changes of activity and/or expression of drug metabolism enzymes and/or transporters with consequences such as changed responses to drug treatment or significant adverse/toxic effects.
In the earlier paper discussed was metabolism and interactions of chloroquine and hydroxychloroquine with human cytochrome with P450 enzymes and drug transporters. These drugs are since years used in the treatment and prevention of malaria and chronic inflammatory diseases but also proposed to be used in treatment of COVID-19 (alone or in combination with other drugs). The data on their metabolism showed that they are metabolized by white range of P450 enzymes in vitro (1A2, 2C8, 2C19, 2D6, and 3A4/5) and by P450s 2C8 and 3A4/5 in vivo. In addition, chloroquine effectively inhibited P450 2D6 in vitro and in vivo in particular in persons with limited P450 2D6 activity. Concluded was that by co-administration of the drugs with other drugs, substrates of P450 2D6 enzyme, attention should be given to substrate-specific metabolism by P450 2D6 alleles present in persons taking the drugs for possible drug–drug interactions (Rendic and Guengerich 2020).
The present paper summarizes data published on the metabolism of IVM by human cytochrome P450 enzymes, interactions with drug transporters, and the potential to cause drug–drug/drug–chemical interactions and toxicity which might occur in a wide range of its clinical applications.
Results and discussion
Metabolism of Ivermectin (IVM) by P450 enzymes
IVM is extensively metabolized by cytochrome P450 enzymes (P450s, CYP) both in vivo and in vitro. In vivo IVM metabolites are predominately excreted in feces with only 1% of the doses in the urine. Using human liver microsomes, human recombinant enzymes, hepatocytes and blood samples up to 13 metabolites were identified. The major metabolites of IVM-B1a in humans occurs by the following metabolic recitations: methyl-hydroxylation, O-demethylation, and oxidation reactions occurring at different positions of the molecule. The structure of the major five metabolites of IVM identified in vitro indicated that they occur by 3″-O-demethylation, C4-hydroxylation, C25 isobutyl-hydroxylation, and 3″-O-demethyl-4-hydroxymethyl metabolite formation (Fig. 2). The major contribution to the metabolism of IMV in vitro is attributed to P450 3A4. In addition, 3″-O-demethyl metabolite was also produced to a small extent by P450 3A5 and is considered as a minor metabolite. The other hydroxylated metabolite occurs also by P4502C8 (Table 1 and references therein). Of the major metabolites of IVM formed in vitro, three of them were identified also in human plasma and occur by 3″-O-demethylation, C4-hydroxylation, and the metabolite formed by both 3′-O-demethylation and C4-hydroxylation reactions. These reactions are catalyzed in vitro by P450 3A4 and 3″-O-demethylation was also catalyzed in minor extent by P450 3A5 (Zeng et al. 1998; Tipthara et al. 2021). The data showed that IVM is predominately substrate of P450 3A4 and that co-administration of drugs that are potent inhibitors or inducers of the enzyme activity, or polymorphism of the enzyme might provoke pronounced drug–drug interactions. Potent inhibitors of P450 3A4 activity are, for example, ketoconazole, econazole, troleandomycin, azamulin, cyclosporine, while potent inducers of P4503A4 activity are, for example, carbamazepine, cortisol, hydrocortisone, rifampicin, amprenavir, and clotrimazole (Rendic 2002).

Ivermectin (IVM) metabolites formed by cytochrome P450 enzymes
Inhibition of P450 enzymes by Ivermectin (IVM)
IVM was demonstrated as a weak, no, or medium inhibitor of the reactions catalyzed by P450 enzymes (Table 2 and references therein). The potency of inhibition for IC50 and % of inhibition values were rated according to the values and ranges as presented in Table 3. Taking into account these data and the data on the inhibition of P450 3A4 IVM was assigned as a medium to week inhibitor of P450 3A4. The IC50 values of inhibition of P450 2C9, 2D6, and 2C19 enzymes ranged from 5.3 to 13.8 μM, or no inhibition (Table 2 and references therein). Thus, the data reported show that they are at least somewhat dependent on the experimental model used, that the potential for drug–drug interactions due to inhibition of P450 enzymes is not likely, and that it will depend on the dose applied. As blood plasma concentrations of IVM in humans were reported to be rarely higher than 100 ng/ml (about 0.1 μM) at a standard oral dose, it might be concluded that IVM, when applied under clinically recommended dose, does not have the potential to cause drug–drug interaction potential on the level of inhibition of P450 enzymes. In addition, it was reported that co-administration of IVM and tribendimidine in rats did not significantly alter the pharmacokinetic behavior of each other and no clinically relevant drug–drug interactions between tribendimidine and Ivermectin on the level of P450 metabolism were observed (Neodo et al. 2018) .
Interaction of Ivermectin (IVM) with drug transporters
In the present chapter discussed is the role of transporters that belong to the ATP-binding cassette (ABC) transporter family and solute carrier family (SLC) transporters in the transport of IVM (Table 4 and references cited). ABC transporters are membrane-bound proteins that are present in all prokaryotes, as well as plants, fungi, yeast, and animals. In humans, the ABC transporters take part in the transport of a variety of compounds (drugs, environmental and physiological compounds) using the energy of ATP and have important role in drug disposition and effects/toxicities by affecting the absorption, distribution and excretion of drugs (Liu 2019).
The multiple drug-resistance transporter (MDR, P-glycoprotein, P-gp) is an export transporter discovered for its role in imparting resistance to drugs in cancer cells by pumping them out (Bradley et al. 1988). The transporter was in the course of research assigned in the literature under different names (Table 4). Its role as a drug export protein was realized in the late 1990s and with time, the number of proteins involved in drug transport processes was grown. P-gp is present in intestinal epithelial cells, liver, kidney, and brain. It was also realized that drug’s transport in or out of the cells, as well as passive diffusion, is dependent on the transporter’s activity, having important role in many cases of drug transport, and can have major effects on tissue selectivity of drugs. Another point is that P-gp export transporter can also lower the level of a drug in a cell and so slow the rate of its metabolism and influence pharmacokinetics and therapeutic activity of drugs, including anticancer drugs (Lin and Yamazaki 2003; Jiang et al. 2019). Similarly, MRP1, 2, and 3 are efflux transporters that are present in hepatocytes with wide substrate transport specificity, including important drugs. The main roles of the MRP transporter proteins are the efflux of xenobiotics (drugs and food components) and endogenous metabolites, transport of inflammatory mediators (e.g., LTC4), and defense against oxidative stress. In addition, P-gp and MRP1 play a role in the development of drug resistance of various types of cancer and contribute to inflammatory responses (Cole and Deeley 1998; Cole 2014; Nakanishi and Tamai 2015; Guengerich and Rendic 2010).
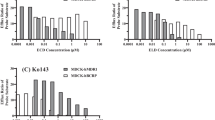
IVM is a potent inhibitor of the MDR1 (P-gp) (Pouliot et all 1997; Didier and Loor 2015) and MRP transporting proteins (Table 4 and referenced cited). For instance, RH123 accumulation into LS180V cells was used as an assay for P-gp inhibition in the screening of 17 clinically used drugs. The results showed that IVM was one of the most potent P-gp inhibitors, causing a ninefold increase in RH123 accumulation compared to control cells (Perloff et al. 2003). The data presented in Table 4 with IC50 ranging from 0.1 to 2.5 µM classify IVM to potent inhibitors of the P-gp mediated transport (Table 3). The data also show that MRP1, and to a lesser extent MRP2 and MRP3 take part also to IVM flux across physiological barriers (Schwab et al. 2003) and that IVM is also a substrate of P-gp. Thus, the MDR1 and MRP binding properties and its large molecular size (Fig. 1) make IVM potential competing inhibitor when co-administrated with other drugs/chemicals which are P-gp and MRPs substrates. IVM also inhibited the P-gp, MRP1, 2, and 3 ATPase activity after stimulation by their respective activators (Table 4 and references cited).
Organic-anion transporting polypeptides (OATP transporters, OATPs) are influx transporters having important role of drug transport in intestine and transport across blood–brain barrier (OATP1A2, OATP2B1) and hepatocytes (OATP1B1, OATP1B3). Thus, OATPs might have impact on drug disposition and efficacy by facilitating oral absorption and drug access to the liver and brain (Nakanishi and Tamai 2015; Varma et al. 2010; Shitara et al. 2013). IVM, when interacting with organic anion transporter polypeptide family, showed to be weak to a medium inhibitor of the activity (Table 3 and Table 4 and references cited). Thus, drug–drug interactions on the level of OATPs by IVM might be possible with application of high doses of IVM.
BCRP (ABCG2) is a member of the ATP-binding cassette (ABC) superfamily of transmembrane transporters and localized to the apical plasma membrane of cells. It actively transports xenobiotic and endogenous substrates out of the cell with energy derived from the hydrolysis of ATP. BCRP was discovered in cancer cells but present also in normal cells preventing the accumulation of chemicals and their toxicity. It has a broad substrate specificity and important role by modulating efflux and having an impact on the pharmacokinetics of drugs, nutrients, and toxicokinetic of toxicants, and to have an important role in transporter-mediated drug–drug interactions (Bricsak and Aleksunes 2015; Safar et al. 2019). IVM inhibits BCRP (Table 4 and references cited) and according to the reported IC50 values (1–3.1 µM) IVM is classified as a potent to medium potency inhibitor (Table 3). Consequently, this interaction may also have implications in the metabolism and distribution of IVM and give rise to drug–drug interactions (Jani et al. 2011). The properties of IVM as a potent multidrug-resistance reversal agent, when combined with antitumor agents, led to its application as enhancer of antitumor activity. IVM drastically reversed the resistance of the tumor cells to the chemotherapeutic drugs both in vitro and in vivo. IVM reversed the resistance by inhibiting the proliferation of several tumor cells by regulating multiple signaling pathways and not by directly inhibiting P-gp activity. The inhibition of the transcriptional factor NF-κB led to the reduced P-gp transcription in tumor cells leading to reversal of the resistance of the tumor cells (Jiang et al. 2019; Tang et al. 2021).
On the level of P450 enzymes, drug–drug interaction might be provoked by co-administration with drugs which are potent inhibitors of P450 3A enzymes by lowering its extrusion from the body. Alternatively, potent inducers of P450 3A activity might affect IVM systemic exposure lowering its therapeutic effect. On the level of drug transport, by inhibition P-gp or MRPs, unexpected high plasma concentration and potential toxic effects of IVM might be elicited. On another side, induction of transporter’s activity in the host, as observed for induction of ABC transporters by rifampicin (Chaccour et al. 2017), or its synthesis by co-administered drugs, might result in subtherapeutic concentrations at the site of action, or cause the development of strains resistant to the drug (James and Davey 2009). Drug–drug interactions at the level of transporters may affect systemic exposure blocking influx transporters in the intestine, but increase it by modulating influx and efflux transporters in the liver and efflux transporters in the small intestine (Nakanishi and Tamai 2015). In addition to co-administered drugs, also polymorphism of drug transporters and P450 enzymes as well as foods might influence the metabolism and pharmacokinetics of IVM in humans. The plasma concentration of IVM in healthy volunteers was found as much higher in beer drinkers, but orange juice decreased AUC values of IVM. The latter effect was ascribed to the inhibition of drug transporters (González Canga et al. 2008). Thus, the results and discussion presented suggest that co-administration of IVM with other drugs, polymorphism of the drug-metabolizing enzymes and transporters or even kinds of food, might affect biotransformation and transport of IVM affecting its pharmacokinetic properties and provoke drug–drug interactions or toxic effects.
Concluding remarks
In clinical practice, IVM is often co-administered with other drugs when different diseases are treated (parasitic, cancers, and COVID-19). Therefore, the effect of IVM and co-administered drugs on P450 enzymes and ABC-mediated transport have to be considered to provide the most effective treatments for patients. IVM is metabolized, both in vivo and in vitro, by C-hydroxylation and O-demethylation reactions catalyzed by P450 3A4, and with a contribution of P450 3A5 and 2C9 enzymes.
P-gp (MDR1) participates in IVM efflux at low drug concentration with slow transport rate. In the higher, micromolar concentration range, which saturates MDR1 (P-gp), MRP1, and to a lesser extent MRP2 and MRP3 take part in IVM flux across physiological barriers. IVM exert a potent inhibition of P-gp (ABCB1), MRP1 (ABCC1), MRP2 (ABCC2), and BCRP1 (ABCG2), and medium to weak inhibition of OATP1B1 (SLC21A6) and OATP1B3 (SLCOB3) transport activity. The data reported emphasize the potentially unsafe/toxic effects when IVM is applied at higher doses, in the subjects with an impaired P-gp transporter function (Ménez et al. 2012). It might be well expected that in the clinic, following application of higher doses of IVM, the pharmacokinetics of co-administered drugs might be critically altered as result to drug–drug interactions and that toxic effect might be provoked as a result of the inhibition of P-gp and/or MRP transporters. In addition, polymorphism of the enzymes and transporters which take part in IVM metabolism might additionally contribute to the effects.
The presented data show complex interactions of IVM being a substrate of P450 3A enzymes, and substrate and inhibitor of P-gp and MRP, and an inhibitor of BCRP transporter. Besides its FDA-approved antiparasitic activity, it exerted potent anticancer and antiviral effects. Therefore, co-administration of IVM with other drugs in complex treatments should take particular attention to develop the most effective treatment strategies for patients receiving multiple drug regimens to avoid unwonted and toxic effects.
Availability of data and materials
All the data are available in the text and tables of the review.
References
Anselmi M, Buonfrate D, Guevara Espinoza A et al (2015) Mass Administration of ivermectin for the elimination of onchocerciasis significantly reduced and maintained low the prevalence of Strongyloides stercoralis in esmeraldas. Ecuador. PLoS Negl Trop Dis 9(11):e0004150. https://doi.org/10.1371/journal.pntd.0004150 (Published 2015 Nov 5)
Bapiro TE, Egnell AC, Hasler JA, Masimirembwa CM (2001) Application of higher throughput screening (HTS) inhibition assays to evaluate the interaction of antiparasitic drugs with cytochrome P450s. Drug Metab Dispos 29(1):30–35
Bircsak KM, Aleksunes LM (2015) Interaction of isoflavones with the BCRP/ABCG2 drug transporter. Curr Drug Metab 16(2):124–140. https://doi.org/10.2174/138920021602150713114921
Boatin BA, Richards FO Jr (2006) Control of onchocerciasis. Adv Parasitol 61:349–394. https://doi.org/10.1016/S0065-308X(05)61009-3
Bradley G, Juranka PF, Ling V (1988) Mechanism of multidrug resistance. Biochim Biophys Acta 948(1):87–128. https://doi.org/10.1016/0304-419x(88)90006-6
Bray M, Rayner C, Noël F, Jans D, Wagstaff K (2020) Ivermectin and COVID-19: A report in Antiviral Research, widespread interest, an FDA warning, two letters to the editor and the authors’ responses. Antivir Res 178:104805. https://doi.org/10.1016/j.antiviral.2020.104805
Burg RW, Miller BM, Baker EE et al (1979) Avermectins, new family of potent anthelmintic agents: producing organism and fermentation. Antimicrob Agents Chemother 15(3):361–367. https://doi.org/10.1128/aac.15.3.361
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM (2020) The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res 178:104787. https://doi.org/10.1016/j.antiviral.2020.104787
Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA (1983) Ivermectin: a potent new antiparasitic agent. Science 221(4613):823–828. https://doi.org/10.1126/science.6308762
Chaccour CJ, Hammann F, Alustiza M et al (2017) Cytochrome P450/ABC transporter inhibition simultaneously enhances ivermectin pharmacokinetics in the mammal host and pharmacodynamics in Anopheles gambiae. Sci Rep 7(1):8535. https://doi.org/10.1038/s41598-017-08 (Published 2017 Aug 17)
Chaccour C, Hammann F, Ramón-García S, Rabinovich NR (2020a) Ivermectin and COVID-19: keeping rigor in times of urgency. Am J Trop Med Hyg 102(6):1156–1157. https://doi.org/10.4269/ajtmh.20-0271
Chaccour C, Ruiz-Castillo P, Richardson MA et al (2020b) The SARS-CoV-2 Ivermectin Navarra-ISGlobal Trial (SAINT) to Evaluate the Potential of Ivermectin to Reduce COVID-19 Transmission in low risk, non-severe COVID-19 patients in the first 48 hours after symptoms onset: a structured summary of a study protocol for a randomized control pilot trial. Trials 21(1):498. https://doi.org/10.1186/s13063-020-04421-z (Published 2020 Jun 8)
Cole SP, Deeley RG (1998) Multidrug resistance mediated by the ATP-binding cassette transporter protein MRP. BioEssays 20(11):931–940. https://doi.org/10.1002/(SICI)1521-1878(199811)20:11%3c931::AID-BIES8%3e3.0.CO;2-J
Cole SP (2014) Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem 289(45):30880–30888. https://doi.org/10.1074/jbc.R114.609248
Dalzell AM, Mistry P, Wright J, Williams FM, Brown CD (2015) Characterization of multidrug transporter-mediated efflux of avermectins in human and mouse neuroblastoma cell lines. Toxicol Lett 235(3):189–198. https://doi.org/10.1016/j.toxlet.2015.04.005
Didier A, Loor F (2015) The abamectin derivative ivermectin is a potent P-glycoprotein inhibitor. Anticancer Drugs 7(7):745–751. https://doi.org/10.1097/00001813-199609000-00005
Dixit A, Yadav R, Singh AV (2020) Ivermectin: potential role as repurposed drug for COVID-19. Malays J Med Sci 27(4):154–158. https://doi.org/10.21315/mjms2020.27.4.15
Dou Q, Chen HN, Wang K et al (2016) Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt axis in breast cancer. Cancer Res 76(15):4457–4469. https://doi.org/10.1158/0008-5472.CAN-15-2887
Dourmishev AL, Dourmishev LA, Schwartz RA (2005) Ivermectin: pharmacology and application in dermatology. Int J Dermatol 44(12):981–988. https://doi.org/10.1111/j.1365-4632.2004.02253.x
Edwards G (2003) Ivermectin: does P-glycoprotein play a role in neurotoxicity? Filaria J 2 Suppl 1(Suppl 1):S8. https://doi.org/10.1186/1475-2883-2-S1-S8
El-Saber Batiha G, Alqahtani A, Ilesanmi OB et al (2020) Avermectin derivatives, pharmacokinetics, therapeutic and toxic dosages, mechanism of action, and their biological effects. Pharmaceuticals (Basel). 13(8):196. https://doi.org/10.3390/ph13080196 (Published 2020 Aug 17)
Formiga FR, Leblanc R, de Souza RJ, Farias LP, de Oliveira RN, Pena L (2020) Ivermectin: an award-winning drug with expected antiviral activity against COVID-19. J Control Release S0168–3659(20):30580. https://doi.org/10.1016/j.jconrel.2020.10.009
González Canga A, Sahagún Prieto AM, Diez Liébana MJ, Fernández Martínez N, Sierra Vega M, García Vieitez JJ (2008) The pharmacokinetics and interactions of ivermectin in humans–a mini-review. AAPS J 10(1):42–46. https://doi.org/10.1208/s12248-007-9000-9
Griffin J, Fletcher N, Clemence R, Blanchflower S, Brayden DJ (2005) Selamectin is a potent substrate and inhibitor of human and canine P-glycoprotein. J Vet Pharmacol Ther 28(3):257–265. https://doi.org/10.1111/j.1365-2885.2005.00655.x
Guengerich FP, Rendic S (2010) Update information on drug metabolism systems–2009, part I. Curr Drug Metab 11(1):1–3. https://doi.org/10.2174/138920010791110908
Guzzo CA, Furtek CI, Porras AG et al (2002) Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol 42(10):1122–1133. https://doi.org/10.1177/009127002401382731
Heidary F, Gharebaghi R (2020) Ivermectin: a systematic review from antiviral effects to COVID-19 complementary regimen. J Antibiot (Tokyo) 73(9):593–602. https://doi.org/10.1038/s41429-020-0336-zR
Hotson IK (2020) The avermectins: a new family of antiparasitic agents. J S Afr Vet Assoc 53(2):87–90
James CE, Davey MW (2009) Increased expression of ABC transport proteins is associated with ivermectin resistance in the model nematode Caenorhabditis elegans. Int J Parasitol 39(2):213–220. https://doi.org/10.1016/j.ijpara.2008.06.009
Jani M, Makai I, Kis E et al (2011) Ivermectin interacts with human ABCG2. J Pharm Sci 100(1):94–97. https://doi.org/10.1002/jps.22262
Jiang L, Wang P, Sun YJ, Wu YJ (2019) Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-κB pathway. J Exp Clin Cancer Res 38(1):265. https://doi.org/10.1186/s13046-019-1251-7 (Published 2019 Jun 18)
Juarez M, Schcolnik-Cabrera A, Dueñas-Gonzalez A (2018) The multitargeted drug ivermectin: from an antiparasitic agent to a repositioned cancer drug. Am J Cancer Res 8(2):317–331
Karlgren M, Vildhede A, Norinder U et al (2012) Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem 55(10):4740–4763. https://doi.org/10.1021/jm300212s
Kaur H, Shekhar N, Sharma S, Sarma P, Prakash A, Medhi B (2021) Ivermectin as a potential drug for treatment of COVID-19: an in-sync review with clinical and computational attributes. Pharmacol Rep. https://doi.org/10.1007/s43440-020-00195-y (published online ahead of print, 2021 Jan 3)
King CL, Suamani J, Sanuku N et al (2018) A trial of a triple-drug treatment for lymphatic filariasis. N Engl J Med 379(19):1801–1810. https://doi.org/10.1056/NEJMoa1706854
Klotz U, Ogbuokiri JE, Okonkwo PO (1990) Ivermectin binds avidly to plasma proteins. Eur J Clin Pharmacol. 39(6):607–608. https://doi.org/10.1007/BF00316107
Korystov YN, Ermakova NV, Kublik LN et al (2004) Avermectins inhibit multidrug resistance of tumor cells. Eur J Pharmacol 493(1–3):57–64. https://doi.org/10.1016/j.ejphar.2004.03.067
Krishna DR, Klotz U (1990) Determination of ivermectin in human plasma by high-performance liquid chromatography. Arzneimittelforschung 43(5):609–611 (PMID: 8329009)
Kwei GY, Alvaro RF, Chen Q et al (1999) Disposition of ivermectin and cyclosporin A in CF-1 mice deficient in mdr1a P-glycoprotein. Drug Metab Dispos 27(5):581–587
Kwon YJ, Petrie K, Leibovitch BA et al (2015) Selective inhibition of SIN3 corepressor with avermectins as a novel therapeutic strategy in triple-negative breast cancer. Mol Cancer Ther 14(8):1824–1836. https://doi.org/10.1158/1535-7163.MCT-14-0980-T
Lespine A, Dupuy J, Orlowski S et al (2006) Interaction of ivermectin with multidrug resistance proteins (MRP1, 2 and 3). Chem Biol Interact 159(3):169–179. https://doi.org/10.1016/j.cbi.2005.11.002
Lespine A, Martin S, Dupuy J et al (2007) Interaction of macrocyclic lactones with P-glycoprotein: structure-affinity relationship. Eur J Pharm Sci 30(1):84–94. https://doi.org/10.1016/j.ejps.2006.10.004
Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42(1):59–98. https://doi.org/10.2165/00003088-200342010-00003
Lindley D (1987) Merck’s new drug free to WHO for river blindness programme. Nature 329(6142):752. https://doi.org/10.1038/329752a0
Liu X (2019) Transporter-mediated drug-drug interactions and their significance. Adv Exp Med Biol 1141:241–291. https://doi.org/10.1007/978-981-13-7647-4_5
Liu J, Zhang K, Cheng L, Zhu H, Xu T (2020) Progress in understanding the molecular mechanisms underlying the antitumour effects of ivermectin. Drug Des Devel Ther 14:285–296. https://doi.org/10.2147/DDDT.S237393 (Published 2020 Jan 21)
Mastrangelo E, Pezzullo M, De Burghgraeve T et al (2012) Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J Antimicrob Chemother 67(8):1884–1894. https://doi.org/10.1093/jac/dks147
Melotti A, Mas C, Kuciak M, Lorente-Trigos A, Borges I, Ruiz i Altaba A, (2014) The river blindness drug Ivermectin and related macrocyclic lactones inhibit WNT-TCF pathway responses in human cancer. EMBO Mol Med 6(10):1263–1278. https://doi.org/10.15252/emmm.201404084
Ménez C, Sutra JF, Prichard R, Lespine A (2012) Relative neurotoxicity of ivermectin and moxidectin in Mdr1ab (-/-) mice and effects on mammalian GABA(A) channel activity. PLoS Negl Trop Dis 6(11):e1883. https://doi.org/10.1371/journal.pntd.0001883
Moncayo AL, Vaca M, Amorim L et al (2008) Impact of long-term treatment with ivermectin on the prevalence and intensity of soil-transmitted helminth infections. PLoS Negl Trop Dis 2(9):e293. https://doi.org/10.1371/journal.pntd.0000293 (Published 2008 Sep 10)
Muñoz J, Ballester MR, Antonijoan RM et al (2018) Safety and pharmacokinetic profile of fixed-dose ivermectin with an innovative 18mg tablet in healthy adult volunteers. PLoS Negl Trop Dis 12(1):e0006020. https://doi.org/10.1371/journal.pntd.0006020 (Published 2018 Jan 18)
Nakanishi T, Tamai I (2015) Interaction of drug or food with drug transporters in intestine and liver. Curr Drug Metab 16(9):753–764. https://doi.org/10.2174/138920021609151201113537
Nambara S, Masuda T, Nishio M et al (2017) Antitumor effects of the antiparasitic agent ivermectin via inhibition of Yes-associated protein 1 expression in gastric cancer. Oncotarget 8(64):107666–107677. https://doi.org/10.18632/oncotarget.22587 (Published 2017 Nov 21)
Neodo A, Schulz JD, Huwyler J, Keiser J (2018) In vitro and in vivo drug-drug interaction study of the effects of ivermectin and oxantel pamoate on tribendimidine. Antimicrob Agents Chemother 63(1):e00762-e818. https://doi.org/10.1128/AAC.00762-18 (Published 2018 Dec 21)
Pandey S, Pathak SK, Pandey A et al (2020) Ivermectin in COVID-19: What do we know? Diabetes Metab Syndr 14(6):1921–1922. https://doi.org/10.1016/j.dsx.2020.09.027
Perloff MD, Störmer E, von Moltke LL, Greenblatt DJ (2003) Rapid assessment of P-glycoprotein inhibition and induction in vitro. Pharm Res 20(8):1177–1183. https://doi.org/10.1023/a:1025092829696
Pinilla YT, Lopes CPS, Sampaio SV et al (2018) Promising approach to reducing Malaria transmission by ivermectin: sporontocidal effect against Plasmodium vivax in the South American vectors Anopheles aquasalis and Anopheles darlingi. PLoS Negl Trop Dis 12(2):e0006221. https://doi.org/10.1371/journal.pntd.0006221 (Published 2018 Feb 14)
Pouliot JF, L’Heureux F, Liu Z, Prichard RK, Georges E (1997) Reversal of P-glycoprotein-associated multidrug resistance by ivermectin. Biochem Pharmacol 53(1):17–25. https://doi.org/10.1016/s0006-2952(96)00656-9
Rendic S (2002) Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev 34(1–2):83–448. https://doi.org/10.1081/dmr-120001392
Rendic S, Guengerich FP (2020) Metabolism and interactions of chloroquine and hydroxychloroquine with human cytochrome P450 enzymes and drug transporters. Curr Drug Metab 21(14):1127–1135. https://doi.org/10.2174/1389200221999201208211537
Rizzo E (2020) Ivermectin, antiviral properties and COVID-19: a possible new mechanism of action. Naunyn Schmiedebergs Arch Pharmacol 393(7):1153–1156. https://doi.org/10.1007/s00210-020-01902-5
Safar Z, Kis E, Erdo F, Zolnerciks JK, Krajcsi P (2019) ABCG2/BCRP: variants, transporter interaction profile of substrates and inhibitors. Expert Opin Drug Metab Toxicol 15(4):313–328. https://doi.org/10.1080/17425255.2019.1591373
Schwab D, Fischer H, Tabatabaei A, Poli S, Huwyler J (2003) Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J Med Chem 46(9):1716–1725. https://doi.org/10.1021/jm021012t
Sharun K, Shyamkumar TS, Aneesha VA, Dhama K, Pawde AM, Pal A (2019) Current therapeutic applications and pharmacokinetic modulations of ivermectin. Vet World 12(8):1204–1211. https://doi.org/10.14202/vetworld.2019.1204-1211
Sharun K, Dhama K, Patel SK et al (2020) Ivermectin, a new candidate therapeutic against SARS-CoV-2/COVID-19. Ann Clin Microbiol Antimicrob 19(1):23. https://doi.org/10.1186/s12941-020-00368-w (Published 2020 May 30)
Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y (2013) Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 34(1):45–78. https://doi.org/10.1002/bdd.1823
Tang M, Hu X, Wang Y et al (2021) Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol Res 163:105207. https://doi.org/10.1016/j.phrs.2020.105207
Telbisz Á, Ambrus C, Mózner O et al (2021) Interactions of potential anti-covid-19 compounds with multispecific ABC and OATP drug transporters. Pharmaceutics 13(1):81. https://doi.org/10.3390/pharmaceutics13010081 (Published 2021 Jan 9)
Thomsen EK, Sanuku N, Baea M et al (2016) Efficacy, safety, and pharmacokinetics of coadministered diethylcarbamazine, albendazole, and ivermectin for treatment of Bancroftian filariasis. Clin Infect Dis 62(3):334–341. https://doi.org/10.1093/cid/civ882
Tipthara P, Kobylinski KC, Godejohann M et al (2021) Identification of the metabolites of ivermectin in humans. Pharmacol Res Perspect 9(1):e00712. https://doi.org/10.1002/prp2.712
Umbenhauer DR, Lankas GR, Pippert TR et al (1997) Identification of a P-glycoprotein-deficient subpopulation in the CF-1 mouse strain using a restriction fragment length polymorphism. Toxicol Appl Pharmacol 146(1):88–94. https://doi.org/10.1006/taap.1997.8225
Varma MV, Ambler CM, Ullah M et al (2010) Targeting intestinal transporters for optimizing oral drug absorption. Curr Drug Metab 11(9):730–742. https://doi.org/10.2174/138920010794328850
Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA (2012) Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J 443(3):851–856. https://doi.org/10.1042/BJ20120150
Yang CC (2012) Acute human toxicity of macrocyclic lactones. Curr Pharm Biotechnol 13(6):999–1003. https://doi.org/10.2174/138920112800399059
Yang SNY, Atkinson SC, Wang C et al (2020) The broad spectrum antiviral ivermectin targets the host nuclear transport importin α/β1 heterodimer. Antivir Res 177:104760. https://doi.org/10.1016/j.antiviral.2020.104760
Zeng Z, Andrew NW, Arison BH, Luffer-Atlas D, Wang RW (1998) Identification of cytochrome P4503A4 as the major enzyme responsible for the metabolism of ivermectin by human liver microsomes. Xenobiotica 28(3):313–321. https://doi.org/10.1080/004982598239597
Zhu M, Li Y, Zhou Z (2017) Antibiotic ivermectin preferentially targets renal cancer through inducing mitochondrial dysfunction and oxidative damage. Biochem Biophys Res Commun 492(3):373–378. https://doi.org/10.1016/j.bbrc.2017.08.097
Funding
No funding.
Author information
Authors and Affiliations
Ethics declarations
Conflict of interest
The author declare no conflict of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rendic, S.P. Metabolism and interactions of Ivermectin with human cytochrome P450 enzymes and drug transporters, possible adverse and toxic effects. Arch Toxicol 95, 1535–1546 (2021). https://doi.org/10.1007/s00204-021-03025-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-021-03025-z