
Abstract
Drug-induced liver injury (DILI) is a serious health burden. It has diverse clinical presentations that can escalate to acute liver failure. The worldwide increase in the use of psychotropic drugs, their long-term use on a daily basis, common comorbidities of psychiatric and metabolic disorders, and polypharmacy in psychiatric patients increase the incidence of psychotropics-induced DILI. During the last 2 decades, hepatotoxicity of various antidepressants (ADs) and antipsychotics (APs) received much attention. Comprehensive review and discussion of accumulated literature data concerning this issue are performed in this study, as hepatotoxic effects of most commonly prescribed ADs and APs are classified, described, and discussed. The review focuses on ADs and APs characterized by the risk of causing liver damage and highlights the ones found to cause life-threatening or severe DILI cases. In parallel, an overview of hepatic oxidative stress, inflammation, and steatosis underlying DILI is provided, followed by extensive review and discussion of the pathophysiology of AD- and AP-induced DILI revealed in case reports, and animal and in vitro studies. The consequences of some ADs and APs ability to affect drug-metabolizing enzymes and therefore provoke drug–drug interactions are also addressed. Continuous collecting of data on drugs, mechanisms, and risk factors for DILI, as well as critical data reviewing, is crucial for easier DILI diagnosis and more efficient risk assessment of AD- and AP-induced DILI. Higher awareness of ADs and APs hepatotoxicity is the prerequisite for their safe use and optimal dosing.
Similar content being viewed by others


Introduction
Drug-induced liver injury (DILI) is an adverse reaction to drugs and/or their metabolites that may result in permanent loss of liver function and death. DILI covers a broad spectrum of clinical manifestations, from asymptomatic abnormalities in the liver function tests (prothrombin time, albumin, direct and indirect bilirubin, the level of aspartate, and alanine transaminase in serum) to symptomatic acute liver disease, prolonged jaundice, and disability, or overt acute or subacute liver failure (Chalasani et al. 2008). DILI is commonly divided into two types: intrinsic and idiosyncratic. Intrinsic DILI is dose-dependent, typically has a short latency period and predictable disease course, and is reproducible in animal models. Idiosyncratic DILI is a severe liver injury occurring very rarely and usually is not dose-dependent. It is characterized by unpredictable disease course, more varied manifestations, and is not reproducible in animal models (Ye et al. 2018). Drugs that cause idiosyncratic DILI typically also cause a mild and asymptomatic liver injury—transient, asymptomatic elevations in serum alanine aminotransferase levels (Mosedale and Watkins 2017). The lack of specific serological markers makes DILI hard to diagnose. The clinical symptoms are very diverse and may include tiredness, lack of appetite, nausea, vomiting, fever, joint and muscle pain, rash, and jaundice, while some patients remain asymptomatic. DILI is challenging not only concerning diagnosis but for the management as well. In most cases, the only treatment is to stop the drug administration and provide general supportive care (Navarro and Senior 2006).
The liver is the primary site for the metabolism of drugs, including antidepressants (ADs) and antipsychotics (APs). Thus, it is of great importance to understand how a specific drug and its metabolites affect the structure and function of this organ. Hepatotoxicity may result in hepatic steatosis (accumulation of fat in the liver), steatohepatitis (steatosis with inflammation), fibrosis (excessive accumulation of extracellular matrix proteins), and cirrhosis (formation of bridging fibrous septa and disruption of the standard architecture of the liver) (Aithal et al. 2011). ADs and APs, used in treating different psychiatric disorders such as depression, schizophrenia, and anxiety, may display hepatotoxicity, even at therapeutic doses (reviewed in detail in the following sections). More than 160 psychotropic drugs have been shown to produce hepatic side-effects (Dumortier et al. 2002). A more recent study reported that psychotropic drugs were responsible for 7.6% of DILI cases in a cohort of 185 patients (Licata et al. 2017). Thus, the strategy behind the choice of a psychotropic drug must consider its hepatotoxicity, especially in patients with other risk factors, such as alcoholism, drug abuse, polymedication, obesity, and diabetes.
DILI represents a burden that has been heavily underestimated due to the limitations of clinical trials to identify such rare events. However, it is among the leading causes of late-stage drug development interruption and post-marketing drug withdrawal (Raschi and de Ponti 2015). A systematic review which included 462 medical products withdrawn in the period 1953–2013 revealed that most products were withdrawn from the marketplace due to their hepatotoxicity (18%), followed by immune-related reactions (17%), neurotoxicity (16%), and cardiotoxicity (14%) (Onakpoya et al. 2016). DILI is the most common cause of acute liver failure (50%), ahead of viral infection or autoimmune hepatitis. In developing countries, people are at higher risk of acute liver failure due to viral infections, while in Western Europe and the United States, most cases arise from DILI (Bernal et al. 2010). Epidemiology data regarding DILI are limited, given that the incidence and prevalence of this disorder are underestimated. In China, the incidence of DILI has been rising year by year and now is recognized as a major public health concern (Yu et al. 2017; Shen et al. 2019). According to different national studies, summarized by Ahmad and Odin (2017), DILI incidence varies from 2 to 19 cases per 100,000 per year. However, despite low incidence, DILI should always be considered in acute liver injury cases when other possible aetiologies have been excluded. DILI frequency varies according to age, gender, and socio-economic status. As for the age, the elderly are more affected, probably due to multiple drug use and low tolerability of the therapy. Also, women and wealthier classes are more affected than men and developing societies, respectively (Licata et al. 2017).
Given a large number of new cases of psychiatric patients each year, the need and use of psychotropic drugs are on the constant rise. In the case of depression, the total estimated number of affected people increased by 18.4% between 2005 and 2015 (WHO 2017). Also, it is expected that the current worldwide health emergency caused by COVID-19 adversely affects mental health in many ways. It has already been established that COVID-19 survivors and frontline healthcare engaged with COVID-19 patients presented a high risk of developing symptoms of depression, anxiety, insomnia, fatigue, and post-traumatic stress disorder (Lai et al. 2020; Mazza et al. 2020; Rogers et al. 2020). In addition, fear of infection, as well as unemployment, working from home, home-schooling of children, and lack of contact with family members and friends are risk factors for psychiatric disorders in the general population (WHO 2020). Thus, the current COVID-19 pandemic is expected to increase the use of psychotropic drugs further in the near future. With exploding rates of mental disorders and the growing use of psychotropic drugs, more and more evidences emerge on their hepatotoxic effects.
This review aims to: (1) provide a brief insight into the hepatic metabolism of psychotropic drugs; (2) evaluate the hepatotoxic effects of commonly prescribed ADs and APs; (3) summarize the roles of oxidative stress, inflammation, and lipid accumulation in the pathophysiology of DILI; and (4) review and discuss these pathophysiological mechanisms in the course of AD- and AP-induced hepatotoxicity.
The most important means of assessing a drug’s risk for hepatotoxicity is to review the published case reports, animal studies, and cell-based in vitro studies. We conducted a comprehensive literature search for case reports, original articles, and reviews published from 1960 to 2020 using the search terms “hepatotoxicity,” “DILI,” “liver injury,” “oxidative stress,” “hepatitis,” “steatosis,” and “fatty liver,” cross-referenced with “antidepressant” and “antipsychotic.” This time interval was chosen, because the first ADs and APs were introduced in practice in the 1950s, while the first publications considering their hepatotoxic effects emerged in the 1960s. This approach cannot be used to compare the risks carried by different drugs, since many were introduced at different times and have been prescribed in different doses and rates. It should also be borne in mind that the extent to which the results obtained by animal studies can reasonably be generalised to humans remains open to question. Given that even humans differ in their response to drugs, it is reasonable to suspect if animal models are good predictors of human response (Shanks et al. 2009; Bailey et al. 2014). Thus, relevant in vitro studies with liver microsomes, hepatocytes, liver slices, and recombinant enzymes are very valuable and usually the only source of information reflecting the situation in humans (Martignoni et al. 2006). Taking all this into consideration, we made an effort to highlight the drugs associated with severe DILI cases, explain the potential molecular mechanisms underlying the phenomenon, and point out the ones which are less likely to cause DILI, reviewing relevant case studies, as well as in vivo and in vitro studies.
Hepatic metabolism of psychotropic drugs
Metabolism of psychotropic drugs occurs mainly in the liver, with the cytochrome P450-dependent monooxygenase (CYP) family of isoforms being the most important drug-metabolizing enzymes. CYP450 is a superfamily of heme-containing monooxidases that metabolize xenobiotics, steroids, fatty acids, and vitamins (Furge and Guengerich 2006). The highest expressed isoforms in the liver are CYPs 3A4, 2C9, 2C8, 2E1, and 1A2, while 2A6, 2D6, 2B6, 2C19, and 3A5 are less abundant but still more highly expressed in the liver than in any other organ (Zanger and Schwab 2013). It has been observed that drugs that are substrates of CYP450 enzymes have a higher likelihood of causing DILI in a dose-independent manner, while drugs that are CYP450 inhibitors have a higher likelihood of generating DILI only when they are administered at high daily doses (Yu et al. 2014). The majority of ADs and APs are substrates of CYP450 superfamily isoenzymes, whereby some of them were shown to be their inhibitors. CYP450 expression is influenced by genetic polymorphisms, xenobiotics, cytokines, and hormones, as well as sex and age. CYP3A4 is abundantly expressed in the liver in the majority of individuals and is responsible for the metabolism of > 50% of all clinically used drugs (Zanger and Schwab 2013).
Enzymes CYPs 3A4, 1A2, 2C9, 2C19, and 2D6 account for the metabolism of the majority of ADs and APs. The lipophilic property, which enables ADs to pass the cell membranes, might be one of the reasons why these drugs undergo extensive metabolism in the liver and show the first-pass effect, leading to variable bioavailability ranging from 30 to 80%. The linear relationship between dose and plasma concentrations exists for most ADs, except for paroxetine and fluvoxamine, whereby the time until peak plasma concentration is reached varies between 1 and 12 h (Mauri et al. 2014). Concerning APs, the relationship between dose and effect of several drugs vary considerably between patients, mainly due to pharmacokinetic differences influenced by age, changes in the first-pass effect, and the induction or inhibition of the metabolic system (Mauri et al. 2018). Most of the APs are also lipophilic and move freely across the lipoidal membranes. When administered orally, APs are well absorbed and undergo substantial pre-systemic elimination (bioavailability: 10–70%). They are highly bound to plasma proteins (75–99%) and tissues, and are extensively distributed (Javaid 1994). However, co-medication can influence pharmacokinetics. The patients with multiple comorbidities, which take many medications, are exposed to the risk of drug–drug interactions. Drug interactions are classified as either pharmacodynamic or pharmacokinetic. Pharmacodynamic drug interactions suggest that mechanisms of actions or adverse effects of two drugs used concomitantly are altered in nature, magnitude, or duration. Pharmacokinetic drug interactions occur where one drug alters the absorption, distribution, metabolism, or elimination of another (Preskorn and Werder 2006). Significant pharmacokinetic interactions with ADs and APs predominantly involve drug-induced changes in hepatic metabolism. Pharmacokinetic of these drugs can be altered by drugs that inhibit or induce metabolizing routes of appropriate CYP enzymes. Besides, some ADs and APs are inhibitors of the certain CYP enzymes themselves, so they can increase levels of other medications (Bleakley 2016). Psychotropic drugs that act as CYP450 inhibitors may cause adverse drug–drug interaction if co-administered with another drug which is CYP450 substrate.
Even though drug metabolism usually yields an inactive metabolite (detoxication), in some cases, metabolites can be more reactive than the parent drug (bioactivation) (Park et al. 2011). This is the case with some tricyclic antidepressants (TCAs) such as amitriptyline and imipramine, and APs like chlorpromazine and risperidone (Telles-Correia et al. 2017). Reactive metabolites may covalently bind cellular proteins, lipids, and nucleic acids, causing cell structure and function disruption. Binding to mitochondrial proteins may impair mitochondrial respiration and cause the leakage of superoxide radical anion (O2·−), which is one of the respiration mediators. Apart from producing reactive metabolites, CYP-mediated reactions during drug metabolism may also generate reactive oxygen species (ROS), including O2·−, hydrogen peroxide (H2O2), hydroxyl radical (.OH), hydroperoxyl radical (HOO.), and singlet oxygen (1O2) (Hrycay and Bandiera 2015).
Hepatotoxicity of ADs
ADs are drugs that relieve symptoms of depression, but are also used in treatments of anxiety disorders, panic, and obsessive–compulsive disorders. Monoamine oxidase inhibitors (MAOIs) and TCAs, the pioneering classes of ADs, were introduced in the clinical practice in the 1950s. MAOIs and TCAs were often related to liver toxicity (Table 1), but since their current use is infrequent, most of these cases were reported during the 1980s and ’90s. In the late 1990s, MAOIs and TCAs were largely replaced by safer and better tolerated new generations of ADs: selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), and serotonin antagonist and reuptake inhibitors (SARIs). MAOIs raised safety concerns due to their dietary and drug interactions (serotonin syndrome/toxicity, sympathomimetic drug interactions, and dietary tyramine interactions) (Grady and Stahl 2012), while both MAOIs and TCAs were associated with higher cardiac side-effects comparing to other ADs (Spindelegger et al. 2014). A major concern regarding TCAs is the extensive side effect profile, including anticholinergic symptoms, orthostatic hypotension, cardiac dysrhythmia, and neurological side-effects (including sedation and seizures) (Mullish et al. 2014). Thus, they are reserved primarily for depressed patients unresponsive to more commonly prescribed ADs such as the SSRIs or SNRIs. Fluoxetine, paroxetine, sertraline, citalopram, and fluvoxamine are SSRIs mostly linked with hepatotoxicity (Table 1). SNRIs venlafaxine and duloxetine, as well as SARI trazodone, are also strongly associated with hepatotoxic side-effects. All these drugs mostly demonstrate an idiosyncratic, unpredictable, and reversible hepatic injury. The injury may onset as early as after several days or after up to 6 months after drug administration and generally ends after the drug is withdrawn. Voican et al. (2014) reviewed relevant clinical data related to AD-induced liver injury published from 1965 onward and concluded that 0.5–3% of patients taking AD may develop asymptomatic mild elevation of serum aminotransferase levels.
AD-induced DILI is generally of hepatocellular pattern, and less frequently of the cholestatic or mixed pattern. Regarding the pathophysiology, it is usually immune-allergic or metabolic (Voican et al. 2014). The observational study of Friedrich et al. (2016), based on data from a multicentre drug surveillance program conducted from 1993 to 2011 in German-speaking countries, reported that ADs caused DILI in 0.08% of patients. Their findings also suggested that SSRIs are less likely to contribute to DILI development compared to other classes of ADs. However, a recent study of Billioti de Gage et al. (2018) is not in line with this conclusion. These authors conducted a cohort study using data from the French National Health Insurance Database which included almost 5 million patients initiated on an AD from 2010 to 2015. They identified 382 severe DILI cases, but did not find evidence that SNRIs have a higher risk of serious liver injury than SSRIs. Interestingly, their analysis showed that duloxetine displays lower hepatotoxicity than SSRIs. However, one should bear in mind that duloxetine has previously been identified, in pre-marketing clinical trials and post-marketing monitoring, as a causative agent of liver injury (DeSanty and Amabile 2007) and thereby it had not been prescribed to patients believed to present higher susceptibility to DILI (e.g., elderly, obese patients, individuals with diabetes, chronic renal failure, etc.). Taking both studies into consideration, it is safe to assume that SSRIs are less likely to precipitate DILI comparing to TCAs and MAOIs, but not comparing to SNRIs. With regard to SARI, the incidence of severe liver failure leading to death or liver transplantation due to nefazodone is 1 per 250,000–300,000 patient-years of treatment. This drug carries a Food and Drug Administration (FDA) “black box” warning for hepatotoxicity and is withdrawn from use in many countries, so it will not be further discussed in this review. Fulminant hepatic failure leading to liver transplantation or death has also been reported for another SARI trazodone, but to a lesser extent comparing to nefazodone. Trazodone is still in wide use and will be discussed (Voican et al. 2014).
It is also important to note that SSRIs have the potential to cause drug–drug interactions through inhibition of CYP isoforms, which can contribute to DILI (Crewe et al. 1992). Fluoxetine and paroxetine are potent CYP2D6 inhibitors, whereas norfluoxetine, the main metabolite of fluoxetine, has a moderate inhibitory effect on CYP3A4 isoenzyme. Sertraline is a moderate inhibitor of CYP2D6, while citalopram has little effect on the major CYP isoforms (Hemeryck and Belpaire 2002). Thus, these SSRIs can substantially elevate the levels of co-prescribed drugs, which are dependent on these specific CYP enzymes. Consequent high exposure to the co-prescribed drug may result in both hepatic and non-hepatic complications (Preskorn and Werder 2006). Given the long half-life of both fluoxetine (2–6 days) and norfluoxetine (7–15 days), a special attention should be paid to fluoxetine administration, as its inhibitory effects on CYP- isoenzymes can persist for weeks after drug discontinuation (Bergstrom et al. 1993). Also, co-treatments with fluvoxamine and clozapine resulted in elevated plasma levels of clozapine, most likely due to drug–drug interaction. Of the SSRIs, fluvoxamine is one of the most potent inhibitors of the CYP1A2, isoenzyme predominantly involved in clozapine metabolism (Heeringa et al. 1999). Fluvoxamine also inhibits CYP3A4 and CYP2D6 (van Harten 1995). Co-administration of aripiprazole with fluvoxamine was shown to cause hepatic damage mediated by CYP3A4 inhibition and aripiprazole accumulation (Shastry et al. 2013). Therefore, co-administration of fluvoxamine with clozapine or aripiprazole should be avoided if possible or co-prescribed with caution.
Growing evidence suggests that obesity and diabetes mellitus are both risk factors for DILI. It is important to point out that a reciprocal link between depression and obesity has been noted. The meta-analysis on the longitudinal relationship between depression, overweight, and obesity, which included 15 studies, confirmed that the risk of depression is increased in obese people, as well as vice versa; depression is predictive for developing obesity (Luppino et al. 2010). This, together with the fact that obesity increases the risk of DILI, highlights the need to recognize the hepatotoxic potential of ADs.
The available data show that all listed ADs are associated with the risk of hepatotoxicity. Life-threatening or severe DILI has been reported for MAOI phenelzine, TCA imipramine, SSRI sertraline, SNRIs venlafaxine and duloxetine, and SARI trazodone, while SSRIs citalopram and fluvoxamine are characterized by lower risk.
Hepatotoxicity of APs
APs are drugs primarily prescribed to control schizophrenia and bipolar disorders, but are also used in the treatment of dementia and major depression. Typical, classical, or first-generation antipsychotics (FGA) were introduced more than 60 years ago. Those drugs, strong dopamine D2 receptor antagonists, are effective against hallucinations and delusions, but cause heavy extra-pyramidal side-effects (Miyamoto et al. 2005). In addition, commonly prescribed FGA, such as chlorpromazine and haloperidol, frequently resulted in elevated serum levels of the liver enzymes but also with severe liver damage (Table 2).
Atypical, second-generation antipsychotics (SGA), including clozapine, risperidone, olanzapine, and quetiapine, are available since the 1990s. These drugs antagonize not only D2 receptors but serotonin 5-HT2A/5-HT2C, histaminergic H1, and muscarinic M3 receptors as well (Meltzer 2013). Besides managing hallucinations and delusions, they are effective against anhedonia and apathy. Therefore, they are used in the treatment of depressive disorders alone or in combination with ADs (Wang and Si 2013). Unlike FGAs, SGAs produce minimal extra-pyramidal side-effects. On the other hand, they are strongly associated with adverse metabolic effects like weight gain, obesity, hyperlipidaemia, insulin resistance, hyperglycaemia, and diabetes (De Hert et al. 2012).
Clozapine is mostly associated with hepatotoxicity among SGAs (Telles-Correia et al. 2017). The asymptomatic rise in serum transaminase level is noticed in up to 60% of patients treated with clozapine, whereby 15–30% experienced twofold or threefold elevation (Wu Chou et al. 2014). The increase in serum levels of the liver enzymes is most often transient and asymptomatic. Still, many cases reported clozapine-induced liver damage and even fulminant liver failure accompanying moderate doses of clozapine (Table 2). A case report of hepatotoxicity from a low dose of clozapine (125 mg per day) was also reported (Wu Chou et al. 2014). However, in most cases, patients would recover from clozapine-induced DILI after stopping the treatment. Although knowledge regarding side-effects mostly comes from case studies, the relationship between clozapine and the hepatic injury was also unequivocally demonstrated in animal studies (Table 2). Both case and animal studies provide solid evidence on risperidone hepatotoxicity, as well (Table 2). Risperidone has been associated with clinically non-significant abnormalities in liver function tests, mostly with serum alkaline phosphatase increase, in 52.5% of cases. Marked elevation of serum levels of the liver enzymes was reported in 0.8% of cases, in a patient cohort encompassing 120 children and adolescents (Erdogan et al. 2008). Also, several case studies reported olanzapine-induced hepatotoxicity. In line with that, a number of animal studies demonstrated various hepatic biochemical and histological changes due to olanzapine (Table 2, discussed in more detail in further text). Quetiapine, another SGA, has been associated with cholestasis and rare fulminant hepatic failure (Table 2). Ilgin et al. (2018) demonstrated dose-dependent hepatotoxic effects of this drug in rats. Histopathology of the animal liver revealed necrotic regions accompanied by sinusoidal dilatation.
Most APs are hepatically metabolized by CYP2D6 and CYP3A4, although clozapine and olanzapine are predominantly metabolized via CYP1A2. Clozapine was shown to inhibit CYPs 2C19, 2D6, and 3A4, and therefore has the potential to cause pharmacokinetic drug–drug interactions (Urichuk et al. 2008). This is of particular importance for schizophrenia patients who are likely to take other drugs concomitantly.
The presented data show that both FGAs and SGAs are associated with a risk of hepatotoxicity. Life-threatening or severe DILI has been reported for FGA chlorpromazine and SGAs clozapine and quetiapine. Aripiprazole bears lower risk compared to other APs listed in Table 2. However, it should be borne in mind that this SGA was last introduced into practice, so it has been available for a shorter time for pharmacovigilance (pharmacological science related to the collection, detection, assessment, monitoring, and prevention of adverse effects of pharmaceutical products) to detect post-marketing signals of DILI.
Major pathophysiological pathways in DILI
Drugs may induce damage in the liver through multiple molecular mechanisms, including direct hepatic toxicity, as well as innate and adaptive immune response. Drugs and/or their metabolites can cause direct hepatic toxicity through oxidative stress by accumulating ROS and inducing mitochondrial dysfunction. They can also trigger inflammation and lipid accumulation, causing impairments in hepatic structure and function (Ye et al. 2018). Oxidative stress, inflammation, and lipid accumulation, as major pathophysiological pathways in DILI, will be discussed in more detail further in the text. Overview of molecular mechanisms will be followed by review and discussion of knowledge regarding AD- and AP-related case reports, animal, and in vitro studies. Figures summarizing oxidative, inflammatory, and, finally, steatogenic pathways demonstrated for listed ADs (Table 1) and APs (Table 2) will be presented at the end of each section dedicated to respective phenomena.
Oxidative stress in DILI
Oxidative stress is strongly associated with DILI. Various drugs induce oxidative stress in the liver by promoting ROS production and lipid peroxidation, as well as by reducing antioxidants levels (Li et al. 2015). The main sources of ROS in the liver are mitochondria and CYP450 enzymes in hepatocytes. Besides, significant sources of ROS generation are immune cells, primarily Kupffer cells (resident hepatic macrophages), and neutrophils (Cesaratto et al. 2004). ROS introduce carbonyl groups into amino acid side chains, affecting protein structure and function (Stadtman and Levine 2000). Furthermore, ROS may oxidize polyunsaturated fatty acids in the process of lipid peroxidation. Lipid peroxidation within the membrane bilayer increases membrane permeability and violates the integrity of the cells and organelles (Wong-Ekkabut et al. 2007). ROS may also cause different types of mutation of the DNA and oxidative histone modifications, and affect DNA methylation and DNA-associated proteins, interfering with gene expression profiles (Kreuz and Fischle 2016).
Hepatocytes are provided with an antioxidant system consisting of several enzymatic (superoxide dismutase—SOD, catalase—CAT, and glutathione peroxidase—GPx) and non-enzymatic (glutathione—GSH, vitamin E, vitamin C) antioxidant molecules. GSH, the most abundant thiol in cells, is capable of reacting directly with ROS and prevent damage to macromolecules. Besides, GSH is involved in peroxide reduction and drug detoxification in reactions that are catalysed by GPx and GST, respectively (Gupta et al. 2005). This is why, the deficit in GSH is the main indicator of a pro-oxidant state and is commonly associated with liver injury (Han et al. 2006). Antioxidant enzymes in the liver are influenced by various factors, including stress (Filipović et al. 2010), hormones (Pajović and Saicić 2008), as well as xenobiotics (Durašević et al. 2010; Djurasevic et al. 2019).
Pro-oxidant effects of ADs in the liver
Oxidative stress in the brain plays a major role in the pathophysiology of depression, and the antioxidant effects of ADs may result in the remission of depression (Xu et al. 2014). However, ADs seem to have antioxidant or pro-oxidant effects, depending on the tissue and the dose applied. Thus, in the case of SSRIs, the majority of data highlight an antioxidant effect in brain tissues, while in the liver, these drugs mainly show a pro-oxidant effect (Ștefan et al. 2020). The influence of ADs on hepatic mitochondria homeostasis and redox balance, in general, was mostly studied in the case of SSRI fluoxetine. Souza et al. (1994) showed that fluoxetine, as well as its main metabolite norfluoxetine, disturbed oxidative phosphorylation and increased pro-oxidant production in isolated hepatic mitochondria, while our results showed that fluoxetine cause oxidative damage of lipids and proteins in rat liver (Zlatković et al. 2014). In line with that, increased total oxidant status paralleled with a decrease in total antioxidant capacity were reported in the liver of fluoxetine-treated rats (Yilmaz et al. 2016). Accordingly, Elgebaly et al. (2018) reported a decreased level of GSH and activity of SOD, CAT, and GPx, accompanied by increased lipid peroxidation and NO production in rat liver due to fluoxetine. The same study revealed the up-regulation of pro-apoptotic Bax and caspase-3 and down-regulation of anti-apoptotic Bcl-2, indicating pro-apoptotic events in hepatocytes (Fig. 1). The authors speculated that an imbalance between Bax and Bcl-2 was a consequence of oxidative stress. It is known that ROS down-regulates Bcl-2 and originates the pro-apoptotic mitochondrial signalling (Herrera et al. 2001). Besides, fluoxetine easily crosses the mitochondrial membrane and interferes with membrane-bound proteins causing pro-apoptotic events (de Oliveira 2016). Hepatic oxidative stress due to fluoxetine is also, at least in part, a result of increased O2•− production coming from intensive CYP-mediated metabolism (Bondy and Naderi 1994). It is known that CYP-derived ROS can cause oxidative damage of macromolecules, impair mitochondrial function, and trigger signalling resulting in cell death (Begriche et al. 2011; Du et al. 2016).

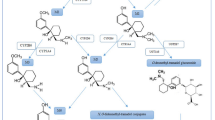
Oxidative stress in AD- and AP-induced DILI. Summarizing hepatic oxidative stress pathways demonstrated for listed antidepressants (ADs) and antipsychotics (APs). a Drug bioactivation by CYP450 enzymes into reactive metabolite that can bind macromolecules covalently and impair cell structure and function: amitriptyline, imipramine, tianeptine, citalopram, duloxetine (ADs); chlorpromazine, risperidone, clozapine, olanzapine (APs); b CYP-mediated generation of reactive oxygen species (ROS) and oxidative damage of macromolecules: fluoxetine, sertraline, citalopram (ADs); chlorpromazine, haloperidol, clozapine (APs); c Covalent binding of reactive drug metabolites to mitochondrial proteins, compromising mitochondrial respiration and causing O2·− hyperproduction: fluoxetine, sertraline (ADs); d Initiating apoptosis by down-regulation of anti-apoptotic Bcl-2, up-regulation of pro-apoptotic Bax, cytochrome C release from mitochondria and caspase-3 activation: fluoxetine (AD); e Compromising antioxidant defence by decreasing glutathione (GSH) level and activity of glutathione-S transferase (GST), glutathione peroxidase (GPx) and catalase (CAT): fluoxetine, citalopram (ADs); chlorpromazine, haloperidol, clozapine (APs)
Regarding SSRIs other than fluoxetine, sertraline and citalopram have been associated with oxidative stress as well. Sertraline was demonstrated to inhibit mitochondrial respiration complexes by uncoupling oxidative phosphorylation in isolated hepatic mitochondria (Li et al. 2012) (Fig. 1). In addition, 10-day sertraline treatment was found to increase lipid peroxidation and NO production in the mice liver (Abdel-Salam et al. 2013). It has been shown recently that DNA damage plays an important role in sertraline-induced hepatic cytotoxicity. The same study has shown that the parental form of sertraline is likely the main cause of liver toxicity, while CYP2D6, 2C19, 2B6, and 2C9-mediated metabolism is responsible for its detoxification (Chen et al. 2020). Citalopram has also been associated with ROS overproduction and antioxidant defence depletion in rat liver. Ahmadian et al. (2017) indicate that metabolic activation plays a crucial role in citalopram-induced hepatotoxicity. TCAs imipramine (Duda et al. 2016) and amitriptyline (Bautista-Ferrufino et al. 2011) were also shown to increase ROS production and lipid oxidation in rat and mice liver.
Concerning SNRIs, venlafaxine and duloxetine were demonstrated to induce oxidative stress in hepatocytes. Mitochondria and lysosomes are proposed as the primary targets in venlafaxine-mediated cell damage (Ahmadian et al. 2016). Mitochondrial dysfunction may be triggered by the drug itself or its reactive drug metabolite, generated through the activity of CYP450 enzymes. In the case of duloxetine, hepatotoxicity might come from oxidative stress triggered by hydroxylated and epoxide metabolites, which can release free radicals or ROS (Mishra et al. 2016). This is corroborated by our findings that duloxetine increase GPx and GST activity in the rat liver (Đorđević et al. 2016). SARI trazodone also demonstrated pro-oxidant action by inducing oxidative stress in isolated rat liver mitochondria (Dykens et al. 2008).
Pro-oxidant effects of APs in the liver
There is a large body of data concerning the roles of oxidative stress in psychiatric disorders and the redox status in patients treated with FGA and SGA, which is reviewed elsewhere (Padurariu et al. 2010; Hendouei et al. 2018; Lin and Lane 2019). However, far less data are available on how APs influence redox balance in the liver, as most studies were conducted on brain tissue and blood serum.
Chlorpromazine was shown to impair the antioxidant system by decreasing SOD and CAT activity and reducing GSH-dependent defence, as well as to cause lipid peroxidation and nitrosative damage in the rat liver (Dejanovic et al. 2017). Chlorpromazine-induced hepatotoxicity is the result of its bioactivation in hepatocytes, which can be done by (1) CYP-catalysed pathway, which yields highly reactive quinone imine metabolite; or (2) peroxidase-catalysed oxidation, which generates toxic radicals (MacAllister et al. 2013). Haloperidol, another FGA, was also shown to decrease GSH level and produce lipid peroxidation in rat and mice liver (Andreazza et al. 2015; El-Awdan et al. 2015; Abdel-Salam et al. 2018).
Regarding SGAs, clozapine was shown to cause severe adverse effects, including metabolic syndrome (Pillinger et al. 2020) and agranulocytosis (Mijovic 2020), as well as hepatotoxicity (Table 2). Both metabolic syndrome and agranulocytosis were previously linked to oxidative stress (Fischer et al. 1991; Ando and Fujita 2009). Oxidative bioactivation of clozapine by CYP450 generates highly reactive nitrenium ion, clozapine-N-oxide. This reactive metabolite causes ROS formation, imposes oxidative stress, and requires GSH-mediated neutralization (Dragovic et al. 2013) (Fig. 1). In line with that, we found that clozapine increased GST activity, decreased GSH level, and caused oxidative damage of lipids and proteins in the rat liver (Zlatković et al. 2014). Due to noted severe adverse effects caused by highly reactive nitrenium metabolite, the use of clozapine is restricted. In an effort to modify the clozapine structure to minimize the nitrenium ion formation, another SGA was designed: quetiapine. This AP has not been associated with hepatic oxidative stress so far. On the other hand, risperidone-induced hepatotoxicity, associated with oxidative stress, mitochondria, and lysosome damage, was reported in rats (Eftekhari et al. 2016). Our results also revealed that olanzapine, like clozapine, increases hepatic GST activity, probably due to the elimination of reactive olanzapine metabolites (Todorović et al. 2016). Four major oxidative metabolites of olanzapine, 4′-N-desmethyl olanzapine, 7-OH olanzapine, 2-OH olanzapine, and N–O olanzapine were formed in vitro by human liver microsomes (Wrighton and Ring 1999).
Liver inflammation in DILI
Activation of the immune response in DILI involves activation of hepatic resident immune cells such as Kupffer cells, natural killer, and natural killer T cells, as well as recruitment of peripheral immune cells like lymphocytes and neutrophils (Barnes et al. 2013).
Reactive drug metabolites may trigger apoptosis and necrosis and initiate an inflammatory response through the release of damage-associated molecular patterns (DAMP) from damaged cells. Kupffer cells express a plethora of receptors necessary for the detection of DAMP (e.g., Toll-like receptors), and therefore mediate the initial response to acute cell injury, which may include the release of inflammatory cytokines and chemokines (Woolbright and Jaeschke 2018). Toll-like receptor family activates nuclear factor-kappa b (NF-κB), a pro-inflammatory transcription factor that promotes production not only of cytokines but ROS as well (Morgan and Liu 2011). Thus, activated Kupffer cells, a major source of inflammatory mediators, including cytokines, O2·−, NO, eicosanoids, chemokines, as well as lysosomal and proteolytic enzymes, demonstrate increased cytotoxicity and chemotaxis (Kolios et al. 2006).
DILI is often associated with immune cell infiltrates, and the nature and size of these infiltrates may serve as a prognosis of the progress and severity of tissue damage. The liver is a site where lymphocytes undergo apoptosis during the resolution phase of the peripheral immune response. However, infiltrating activated B and T lymphocytes have been associated with hepatic histopathology (Ikeda and Yoshikawa 2003). Cytotoxic (CD8 +) T cells were found to be dominant lymphocytes present in liver biopsies of subjects with DILI (Foureau et al. 2014). Besides direct toxicity, reactive drug metabolite can act as a hapten which covalently binds to protein and form an immunogenic hapten–carrier adduct. These adduct molecules activate antigen-presenting cells, which further activate T cells that may be cytotoxic to other cells, including hepatocytes. Also, covalent modification of self-proteins by reactive drug haptens may trigger the loss of self-recognition. Consequently, immune cells whose primary role is to fight against non-self-cells, such as bacteria, would then attack self-cells, leading to autoimmune-mediated hepatitis (Pohl et al. 1989; Woolbright and Jaeschke 2018).
Liver eosinophilia has often been associated with DILI. It was reported in 40% of 570 case reports reviewed by Bjornsson et al. (2007). Eosinophils have been associated with detrimental effects on one hand, but also with remodelling and repairing ones on the other. Although the role played by eosinophils in DILI is ambiguous, in the clinical context, the presence of eosinophilia, in most cases, indicates a good prognosis (Bjornsson et al. 2007).
Finally, neutrophils in an infiltrate are common in the early response to tissue damage (Jaeschke and Tadashi 2006). Neutrophils are essential mediators of drug hepatotoxicity, since they are a significant source of ROS, mainly O2·− produced by NADPH oxidase 2. Inside neutrophils O2·− dismutates to H2O2, which may diffuse into hepatocytes and provoke intracellular oxidative stress, or be used by myeloperoxidase to generate hypochlorous acid, a potent oxidant and chlorinating agent (Jaeschke and Tadashi 2006). Thus, neutrophil-derived ROS mediate further damage to liver tissue (Fig. 2).

Inflammation in AD- and AP-induced DILI. Summarizing hepatic inflammatory pathways demonstrated for listed antidepressants (ADs) and antipsychotics (APs). a Necrosis and apoptosis caused by reactive drug metabolite and/or CYP-derived reactive oxygen species (ROS) and release of damage-associated molecular patterns (DAMP): amitryptiline, fluoxetine, duloxetine (ADs); haloperidol, clozapine, olanzapine, quetiapine (APs); b activation of Kupffer cells through DAMP-mediated toll-like receptors (TLR) activation, and initiation of inflammatory response—activation of nuclear factor-kappa B (NF-κB); increase in production and release of cytokines (TNF-α and IL-1β), ROS, NO and chemokines: fluoxetine, duloxetine, sertraline (ADs); haloperidol (AP); c infiltration of inflammatory cells, mainly lymphocytes and eosinophils, triggered by chemokines/cytokines leakage: amitriptyline, fluoxetine, paroxetine, sertraline, citalopram, venlafaxine, duloxetine (ADs); haloperidol, clozapine, risperidone, olanzapine, quetiapine (APs); d Neutrophil infiltration and activation; increased production and release of ROS; ROS diffusion to hepatocytes and reinforcement of oxidative damage: fluoxetine (AD); clozapine, olanzapine (APs)
Pro-inflammatory effects of ADs in the liver
AD-associated DILI is generally immuno-allergic or metabolic, regarding pathophysiology. Hepatitis, inflammatory liver disease, was reported in patients treated with various TCA drugs. A case of disturbed hepatic architecture due to massive liver necrosis with portal areas containing lymphocytes and a significant amount of eosinophils was described in an imipramine-treated patient (Ilan et al. 1996). Amitriptyline-induced hepatitis with infiltration of mononuclear cells, neutrophils, and eosinophils (Larrey et al. 1988), as well as with massive necrosis and an intense inflammatory reaction (Danan et al. 1984) were also demonstrated. Recently, another acute hepatitis case due to amitriptyline was reported (Chen et al. 2019). In addition, acute hepatitis was described in a patient 8 weeks after beginning tianeptine administration (Bricquir et al. 1994).
SSRI-induced hepatitis is rare, but can be serious. Chronic hepatitis has been rarely attributed to fluoxetine therapy, although acute hepatitis was reported in several cases (Table 1). A patient with moderately expanded portal tracts, infiltrated by lymphocytes and plasma cells, was described in a case of hepatitis associated with paroxetine (Colakoglu et al. 2005) (Fig. 2). Curiously, fatal hepatitis was reported in a patient treated with sertraline (Fartoux-Heymann et al. 2001), a drug that was considered less dangerous for the liver, comparing to other SSRIs. The authors speculated that sertraline-induced hepatitis was due to immunization against self-proteins covalently modified by a reactive metabolite generated in situ in the liver. However, the reactive metabolites have not been identified yet, although it is known that sertraline is extensively metabolized by hepatic CYP450 (Fartoux-Heymann et al. 2001). In addition, lobular inflammation and a mild prominence of eosinophils were reported in another patient treated with sertraline (Suen et al. 2013) (Fig. 2).
The other group of the new generation ADs, SNRIs, also rarely causes hepatitis. Venlafaxine-induced hepatitis was reported only in a few cases. Surprisingly, the first case was described in a patient who received venlafaxine in a low dose (75 mg per day) (Phillips et al. 2006). However, fulminant hepatic failure was reported in a patient receiving venlafaxine combined with trazodone at normal therapeutic doses (Detry et al. 2009). Cholestatic hepatitis involving mixed portal inflammatory infiltrates along with eosinophils was also described in a patient taking venlafaxine (Stadlmann et al. 2012). The post-marketing reports of hepatitis and cholestatic jaundice in patients receiving duloxetine emerged in late 2005. Fulminant hepatic failure in a duloxetine-treated patient reported by Hanje et al. (2006) was associated with a mixed inflammatory infiltrate within the portal and lobular areas composed mainly of neutrophils with occasional eosinophils and lymphocytes. Neutrophils are important mediators of drug hepatotoxicity, since they are a significant source of ROS, which further damage liver tissue (Jaeschke and Tadashi 2006). Vuppalanchi et al. (2010) described in detail a case of duloxetine-associated hepatitis. The histological analysis showed a pronounced portal, and sinusoidal infiltrates consisted of activated lymphocytes. Besides, Kupffer cells appeared activated, and rare foci of necrosis were detected, while endothelitis was present in central and portal venules. SARI trazodone, combined with diazepam, was demonstrated to cause hepatocellular liver injury with confluent necrosis and modest inflammation (Carvalhana et al. 2016).
Regarding animal studies, there are little data about inflammatory changes in the liver under AD treatment. Most studies concerning the immuno-modulatory effects of ADs were conducted in blood and brain tissues. However, few studies described the pro-inflammatory effects of fluoxetine in rat liver. Özden et al. (2005) described lobular inflammation, focal necrosis, apoptosis, portal area inflammation, and Kupffer cell hyperplasia. In line with that, we found the infiltrates of neutrophils, lymphocytes, and macrophages in portal triads; Kupffer and inflammatory cells in sinusoids, and focal necrosis in the liver of fluoxetine-treated rats (Zlatković et al. 2014) (Fig. 2). Like fluoxetine, sertraline was found to induce hepatic inflammation. Analysing the liver of rabbits exposed to sertraline, Almansour et al. (2018) noted sinusoidal dilatation; lobular and periportal hepatic spaces infiltrated mostly by lymphocytes and plasma cells; portal vessel congestion, as well as Kupffer cell increased number and hyperplasia. Similarly, a mild inflammatory cell infiltrate, composed mainly of lymphocytes, was detected in citalopram-treated rats (Ahmadian et al. 2017). SSRIs were also reported to increase the production of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β in an in vitro whole blood assay (Munzer et al. 2013). Thus, scarce data regarding liver inflammatory changes under ADs are mostly related to SSRI drugs. However, it is noteworthy that liver inflammation, including infiltration of inflammatory cells and dilated sinusoids, was observed in rats treated with an SNRI venlafaxine, as well (Paulis et al. 2018). Here is also important to address the question concerning the applicability of rodent models for understanding hepatic pro-inflammatory effects of drugs. A recent study of Ölander et al. (2020) showed that human and murine hepatocytes were fairly similar, whereas non-parenchymal cells, including Kupffer cells, tended towards larger species differences. Namely, they found substantial proteomic differences between human and murine non-parenchymal cell types. This interspecies variability in protein expression contributes to the debatable predictive value of animal models in toxicity of drugs, including their hepatic pro-inflammatory effects.
Pro-inflammatory effects of APs in the liver
Hepatitis has been associated with antipsychotic medication, since it was reported in patients treated with both FGAs and SGAs, including haloperidol, clozapine, risperidone, olanzapine, quetiapine, and aripiprazole (Table 2). Severe cases of hepatitis, even fatal in a small minority, have been observed. Massive infiltration of inflammatory cells, predominantly eosinophils, in the vicinity of portal bile ducts was noticed in a patient with haloperidol-induced hepatitis (Fuller et al. 1977) (Fig. 2). With regard to SGA, acute necrotic hepatitis, as well as cholestatic hepatitis with focal necrosis and eosinophil infiltration were reported after clozapine treatment (Slim et al. 2016). Also, immune-allergic hepatitis with eosinophilia and high levels of anti-smooth-muscle antibodies was described in a patient taking risperidone (Esposito et al. 2005). Hepatocellular damage, centrilobular necrosis, and infiltrates in the portal areas containing mononuclear granulocytes, lymphocytes, and eosinophils were observed in some patients treated with olanzapine (Slim et al. 2016) (Fig. 2). Extensive hepatocyte necrosis with nonspecific inflammatory infiltrates was found after quetiapine treatment (Shpaner et al. 2008; Slim et al. 2016). Finally, portal inflammatory cellular reaction with eosinophilia was reported in a patient using aripiprazole (Kornischka and Cordes 2016).
Animal studies have also shown hepatic inflammatory changes in response to AP treatment. Haloperidol-treated rats manifested centrilobular necrosis and inflammatory cell infiltration, especially in the periportal area, as well as high TNF-α immunoreactivity in the liver (Abdel-Salam et al. 2018). Our study reported infiltrates of neutrophils, lymphocytes, and macrophages in portal triads; Kupffer and inflammatory cells in sinusoids, and focal necrosis in the liver of clozapine-treated rats (Zlatković et al. 2014) (Fig. 2). We also described portal inflammation and focal hepatocyte necrosis in olanzapine-treated rats (Todorović et al. 2016). In line with that, Elbakary (2017) detected various histological changes, including dilatation and congestion of central veins, inflammatory cellular infiltration in the portal areas, cytoplasmic vacuolation of hepatocytes, mitochondrial degeneration, bile ducts dilatation, and excessive deposition of lipid droplets in olanzapine-treated rats. Noted cellular infiltration was described as the consequence of ROS, cytokines, and chemokines released from activated Kupffer cells. Besides, Mahmoud and El-deek (2019) (Fig. 2) recently reported lobular hepatitis, marked dilation and congestion of central vein and blood sinusoids, spotty necrosis, and portal tract expansion by inflammatory cells in rat liver due to olanzapine.
Fatty liver changes in DILI
Hepatic steatosis, or fatty liver, is a reversible condition in which fat accumulates in hepatocytes due to an imbalance between lipid deposition and removal. An increase in fatty acid influx from the diet and adipose tissue lipolysis, or de novo lipogenesis, and a decrease in fatty acid removal through β-oxidation or very-low-density lipoprotein (VLDL) secretion, can all lead to the intracellular lipid droplets accumulation and hepatic steatosis (Marchesini et al. 2016). All of the noted mechanisms can be affected by different ADs and APs.
Steatosis occurs in two primary forms, macrovesicular and microvesicular. Macrovesicular steatosis or macrosteatosis is characterized by a single, large fat vacuole per hepatocyte that fills up most of the cytoplasm and displaces the nucleus to the periphery of the cell. Macrosteatosis has a good long-term prognosis with rare progression to fibrosis or cirrhosis (Bessone et al. 2018). On the other hand, diffuse, microvesicular steatosis, or microsteatosis consists of enlarged hepatocytes with multiple small droplets in the cytoplasm, while the nucleus holds a central location. It is often associated with chronic lipid peroxidation, development of steatohepatitis, liver insufficiency, and encephalopathy, and can be life-threatening when extensive or long-lasting (Amacher and Chalasani 2014).
Steatogenic effects of ADs in the liver
Hepatic steatogenic effects of different AD classes were shown in patients, animal models, as well as in in vitro studies. Regarding case studies, moderate steatosis due to imipramine treatment was reported (Horst et al. 1980). Moderate macro- and microvesicular fat accumulation, involving 40% of hepatocytes, was detected in a patient who developed acute hepatitis after taking tianeptine for 8 weeks (Bricquir et al. 1994). Recently, a case of fluoxetine-induced hepatomegaly (enlarged liver) with steatosis was described (Agrawal et al. 2019). In the case of SNRI, moderate-to-marked steatosis was detected in patients who received venlafaxine (Levine et al. 1996), as well as duloxetine (Hanje et al. 2006; Vuppalanchi et al. 2010).
In vitro and animal studies provided substantial evidence that various ADs have the steatogenic effect. TCAs amitriptyline and imipramine were shown to promote the activation of sterol regulatory element-binding proteins (SREBP), the most important regulators of cellular cholesterol and free fatty acids’ (FFAs) biosynthesis (Fig. 3). This effect was observed in cultured human liver cells (Raeder et al. 2006). Besides, the steatogenic effect of amitriptyline was shown in vivo in rat liver (Sahini et al. 2014; Kampa et al. 2020). Interestingly, the other two TCAs, amineptine and tianeptine, were shown to cause mild hepatic steatosis due to heptanoic chain attached to a tricyclic moiety. This heptanoic chain undergoes mitochondrial β-oxidation, which shortens it to the 5-carbon and 3-carbon derivatives. Therefore, hepatic mitochondria of the patients receiving amineptine or tianeptine are exposed to C7, C5, and C3 analogues of natural FFAs, which reversibly inhibit β-oxidation of medium- and short-chain fatty acids (Fromenty et al. 1989; Pessayre et al. 2012). As a result, FFAs increasingly undergo esterification into triglycerides that accumulate in hepatocytes as benign large lipid droplets (Bessone et al. 2018). As opposed to the steatogenic effects of ADs described so far, a recent study demonstrated that MAOI phenelzine reduced hepatic lipid content in mice fed with a high-fat diet (Mercader et al. 2019).

Mechanisms of fat accumulation in hepatocytes due to AD- and AP-induced DILI. Summarizing hepatic steatogenic pathways demonstrated for listed antidepressants (ADs) and antipsychotics (APs). a Activation of sterol regulatory element-binding proteins (SREBR) and up-regulation of lipogenic enzymes resulting in increased triglycerides’ (TG) synthesis and accumulation in fat vacuole: amitryptiline, imipramine (ADs); clozapine (AP); b down-regulation of lipolytic enzymes and consequential accumulation of TG in fat vacuole: fluoxetine (AD); clozapine (AP); c reduced β-oxidation of free fatty acids (FFAs) in mitochondria: amineptine, tianeptine (ADs); clozapine (AP); d increased de novo lipogenesis induced by hyperglycaemia and hyperinsulinemia: fluoxetine (AD); clozapine, olanzapine (APs). e Reduced transport of long-chain fatty acids into the mitochondria due to decreased levels of available carrier l-carnitine: clozapine, olanzapine (APs); f decreased extrahepatic transport of TG due to down-regulation of very-low-density lipoprotein (VLDL) secretion-related proteins apoB and apoE: olanzapine (AP)
The long-term use of SSRIs is associated with obesity/weight gain, diabetes mellitus, and dyslipidemia in children and adults (Jerrell 2010). Also, prenatal exposure to SSRIs, the most common ADs used in pregnancy, increases the risk of childhood overweight (Grzeskowiak et al. 2013). The rodent model demonstrated that fetal and neonatal exposure to fluoxetine cause adiposity, fatty liver, and insulin resistance (De Long et al. 2015). Indeed, the relationship between obesity, hepatic lipid accumulation, insulin resistance, and hepatic inflammation was described in both clinical and animal studies (Gruben et al. 2014). Obesity and fatty liver changes have been associated with increased production of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β (Ma et al. 2008). As mentioned in the section “Pro-inflammatory effects of ADs in the liver,” SSRIs were shown to increase the production of these pro-inflammatory mediators (Munzer et al. 2013). In vitro studies have found that fluoxetine regulates lipogenic and lipolytic genes to promote hepatic lipid accumulation (Fig. 3). Specifically, fluoxetine was reported to induce hepatic lipid accumulation in primary mouse hepatocytes via both promotion of de novo lipogenesis and reduction of lipolysis (Feng et al. 2012; Xiong et al. 2014). These findings are supported by the results of a recent in vivo study of Pan et al. (2018), who showed that fluoxetine increased hepatic triglyceride level by increasing the expression of lipogenic and decreasing the expression of lipolytic enzymes in the mice liver (Fig. 3). In line with that, we noticed macrovesicular fatty changes in the liver of rats treated with fluoxetine (Zlatković et al. 2014). Chronic exposure to sertraline was shown to down-regulate expression of drug- and arachidonic acid-metabolizing enzymes in hepatic tissue, indicating that patients using this drug may be at risk of hepatotoxicity with reduced capacity to metabolize drugs and fatty acids (Almansour et al. 2018). SSRI fluvoxamine was also demonstrated to cause fat accumulation in the liver of mice by shifting the metabolism towards fatty acid synthesis (Rozenblit-Susan et al. 2016).
Steatogenic effects of APs in the liver
Many AP drugs, particularly SGAs, are strongly associated with adverse metabolic effects, including steatosis. Even though some FGAs demonstrated steatogenic effects, like chlorpromazine did in the rat liver (Yang et al. 2015), steatosis is primarily associated with SGAs. Fatty liver was identified in two cases of pediatric patients treated with risperidone. Both cases were accompanied by obesity and abnormal serum levels of the liver enzymes (Kumra et al. 1997). Risperidone-associated steatohepatitis, accompanied by excessive weight gain, was also described (Holtmann et al. 2003). A study of Morlán-Coarasa et al. (2016) aimed to investigate the development of fatty liver during the first 3 years of SGA treatment (aripiprazole, risperidone, quetiapine, and ziprasidone) in 191 non-affective psychosis patients. After 3 years, 25.1% of individuals scored ≥ 60 on a fatty liver index, representing a strong prediction of steatosis. The fatty liver index predicts fatty liver disease based on the body mass index, waist circumference, and blood levels of triglyceride and ɣ-glutamyltransferase, an enzyme involved in drug detoxification.
SGAs, primarily clozapine and olanzapine and, to a lesser extent, quetiapine, risperidone, and aripiprazole can trigger body weight gain, insulin resistance, and metabolic syndrome. All these metabolic disorders promote liver lipogenesis and can subsequently lead to fatty liver and steatohepatitis (Slim et al. 2016). Insulin resistance exposes hepatocytes to an overload of glucose and insulin due to decreased glucose uptake by peripheral tissues and compensatory hyperinsulinemia, respectively. The resulting hyperglycaemia and hyperinsulinemia can promote de novo lipogenesis (Matsuzaka and Shimano 2011). Besides, insulin represses the synthesis of apolipoprotein B (apoB), the main VLDL protein cofactor, reducing the exportation of hepatic lipids via VLDL (Charlton et al. 2002). Thus, high insulin levels further increase hepatic triglyceride levels by blocking VLDL-mediated lipid secretion. Although AP-induced steatosis may be the indirect result of drug-induced obesity and insulin resistance, an experimental study on primary rat hepatocytes showed that clozapine and olanzapine can directly increase de novo lipogenesis, as well (Lauressergues et al. 2010) (Fig. 3). A later study on the human hepatocyte cell culture demonstrated that other APs like haloperidol, olanzapine, and risperidone activated SREBP and increased expression of target genes involved in FFAs biosynthesis, resulting in intracellular lipids accumulation (Lauressergues et al. 2012). These results corroborated the findings of Raeder et al. (2006) that clozapine activates SREBP-mediated lipogenic gene expression in cultured human liver cells. These findings were confirmed in our in vivo study, where clozapine was shown to induce rat liver steatosis (Zlatković et al. 2014). Liver steatosis, as well as an increase in plasma triglycerides and FFAs, due to clozapine treatment, was confirmed by later studies (Li et al. 2017; Liu et al. 2017). Moreover, a single intraperitoneal injection of clozapine (50 mg/kg) was demonstrated to down-regulate genes involved in FFA β-oxidation and lipolysis, leading to lipid accumulation in rat liver (Fernø et al. 2009) (Fig. 3). It was found recently that clozapine can contribute to the fatty liver by reducing renal reabsorption of l-carnitine, an amino acid that acts as a carrier for transporting long-chain fatty acids into the mitochondria for β-oxidation (Wang et al. 2019). Similar to clozapine, olanzapine is strongly associated with fatty liver changes. This AP dysregulated glucose and lipid metabolism, caused mild glucose intolerance, increased body weight, and caused hepatic steatosis in the mouse model after only 4 weeks. When combined with a high-fat diet, olanzapine caused an even greater increase in weight gain, dysregulation of glucose and lipid metabolism, and more pronounced liver damage (Isaacson et al. 2020). These results indicate that obesity may aggravate olanzapine-induced side-effects. Soliman et al. (2013) reported that olanzapine- and aripiprazole-treated rats developed steatosis in the liver, whereby more prominent changes were caused by olanzapine. Hepatic abnormalities of lipid and glucose metabolism due to olanzapine have been intensively studied lately. All these studies accumulated evidence that olanzapine has multifaceted effects on hepatic lipid metabolism, including (1) increasing de novo lipogenesis; (2) decreasing lipoprotein internalization and cholesterol clearance; and (3) reducing VLDL secretion through down-regulation of VLDL secretion-related proteins, including apoB and apoE; which all together result with lipid accumulation in the liver (Chen et al. 2018; Liu et al. 2019; Mahmoud and El-deek 2019; Ren et al. 2019a, b) (Fig. 3). In addition, a recent study demonstrated that olanzapine, like clozapine, decreases the level of available l-carnitine and, therefore, negatively affects β-oxidation of FFAs in mice liver (Jiang et al. 2019). Thus, l-carnitine supplementation could be a promising strategy to prevent or ameliorate olanzapine- and clozapine-induced steatosis. Further on, visceral adiposity associated with hepatic steatosis in mice after long-term risperidone treatment was demonstrated, as well (Auger et al. 2014). Risperidone was also shown to significantly increase gene expression of major lipogenic factors, including fatty acid synthase, in the mouse and rat liver (Auger et al. 2018; Azirak et al. 2019) (Fig. 3).
Conclusions
All ADs and APs listed in this review have the potential to cause hepatotoxicity, even at therapeutic doses. AD- and AP-induced DILI can be mediated through multiple molecular mechanisms, including unwanted side reactions and products of drug metabolism, hepatic oxidative stress, inflammation, and steatosis. Although fulminant and irreversible liver injury due to ADs or APs is a rare event, hepatotoxicity of these drugs should be recognized and carefully considered. These drugs carry a potential risk for liver damage, particularly in patients with polypharmacy or reduced drug metabolism. Besides, patients suffering from disorders that share pathogenic mechanisms with DILI, such as diabetes and metabolic syndrome, bear a higher risk of developing psychotropics-induced hepatotoxicity. In this respect, therapy must be carefully planned, tailored, and monitored, not just in terms of the effect on a psychiatric disorder of patient, but hepatic homeostasis, as well. For example, patients prone to DILI may benefit from lower doses and slower upwards titration to avoid the hepatotoxic effects. When using ADs and APs associated with a greater risk of hepatotoxicity (nefazodone, phenelzine, imipramine, duloxetine, and trazodone as well as chlorpromazine and clozapine, respectively), the serum levels of the liver enzymes should be monitored regularly. Also, these drugs should not be initiated in individuals with pre-existing liver disease, as well as in elderly patients, and alcohol and illegal drug users. Even though there is no clear evidence that pre-existing chronic liver disease increases the likelihood of developing DILI, baseline abnormalities can complicate patient monitoring. Also, a special attention should be paid when implementing therapy combining SGA and AD, which is often the case in treatment-resistant depressive patients. Even though SGA combined with an SSRI can ameliorate depressive symptoms, one should always bear in mind that drug–drug interactions may have serious, even life-threatening consequences. Aripiprazole, quetiapine, and olanzapine have US FDA approval as an augmentation therapy to ADs, but one should always consider their association with metabolic disorders, which may promote liver lipogenesis. For example, patients could benefit from monitoring for signs of the fatty liver when taking these drugs together with fluoxetine; SSRI also related to adiposity and steatosis. Besides, SSRI fluvoxamine co-administration with SGAs clozapine or aripiprazole should be co-prescribed with special caution due to drug–drug interactions described in section “Hepatotoxicity of ADs.” Thus, clinicians should carefully assess the potential risk-to-benefit ratio when planning to implement adjunctive pharmacotherapy. Personalized medicine should be the preferred approach whenever possible and mandatory for risk groups of patients. Finally, patients should be informed that ADs and APs can be associated with liver damage, even liver failure, and encouraged to refer to a primary care physician or psychiatrist if any DILI-related symptoms emerge. In addition, consumption of alcohol, illegal drugs, and over-the-counter medications during ADs and APs treatment may increase the risk of severe liver abnormalities, so it should be strongly discouraged.
References:
Abdel-Salam OME, Youness ER, Khadrawy YA, Sleem AA (2013) Brain and liver oxidative stress after sertraline and haloperidol treatment in mice. J Basic Clin Physiol Pharmacol 24:115–123. https://doi.org/10.1515/jbcpp-2012-0022
Abdel-Salam OME, Sleem AA, Youness ER et al (2018) Bone marrow-derived stem cells protect against haloperidol-induced brain and liver damage in mice. Biomed Pharmacol J 11:11–22. https://doi.org/10.13005/bpj/1343
Abdullah AA, Ibrahim AS, Ahmed HF (2015) Sertraline Induced Acute Hepatitis: A Case Report. Health (Irvine Calif) 07:476–480. https://doi.org/10.4236/health.2015.74056
Agrawal R, Almoghrabi A, Attar BM, Gandhi S (2019) Fluoxetine-induced Stevens-Johnson syndrome and liver injury. J Clin Pharm Ther 44:115–118. https://doi.org/10.1111/jcpt.12760
Ahmad J, Odin JA (2017) Epidemiology and genetic risk factors of drug hepatotoxicity. Clin Liver Dis 21:55–72. https://doi.org/10.1016/j.cld.2016.08.004
Ahmadian E, Babaei H, Nayebi AM et al (2016) Venlafaxine-induced cytotoxicity towards isolated rat hepatocytes involves oxidative stress and mitochondrial/lysosomal dysfunction. Adv Pharm Bull 6:521–530. https://doi.org/10.15171/apb.2016.066
Ahmadian E, Eftekhari A, Fard JK et al (2017) In vitro and in vivo evaluation of the mechanisms of citalopram induced hepatotoxicity. Arch Pharm Res 40:1296–1313. https://doi.org/10.1007/s12272-016-0766-0
Aithal GP, Watkins PB, Andrade RJ et al (2011) Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther 89:806–815. https://doi.org/10.1038/clpt.2011.58
Almansour MI, Jarrar YB, Jarrar BM (2018) In vivo investigation on the chronic hepatotoxicity induced by sertraline. Environ Toxicol Pharmacol 61:107–115. https://doi.org/10.1016/j.etap.2018.05.021
Amacher DE, Chalasani N (2014) Drug-induced hepatic steatosis. Semin Liver Dis 34:205–214. https://doi.org/10.1055/s-0034-1375960
Ando K, Fujita T (2009) Metabolic syndrome and oxidative stress. Free Radic Biol Med 47:213–218. https://doi.org/10.1016/j.freeradbiomed.2009.04.030
Andreazza AC, Barakauskas VE, Fazeli S et al (2015) Effects of haloperidol and clozapine administration on oxidative stress in rat brain, liver and serum. Neurosci Lett 591:36–40. https://doi.org/10.1016/j.neulet.2015.02.028
Auger F, Duriez P, Martin-Nizard F et al (2014) Long-TErm risperidone treatment induces visceral adiposity associated with hepatic steatosis in mice: a magnetic resonance approach. Schizophr Res Treatment 2014:1–11. https://doi.org/10.1155/2014/429291
Auger F, Martin F, Pétrault O et al (2018) Risperidone-induced metabolic dysfunction is attenuated by Curcuma longa extract administration in mice. Metab Brain Dis 33:63–77. https://doi.org/10.1007/s11011-017-0133-y
Azaz-Livshits T, Hershko A, Ben-Chetrit E (2002) Paroxetine associated hepatotoxicity: A report of 3 cases and a review of the literature. Pharmacopsychiatry 35:112–115. https://doi.org/10.1055/s-2002-31515
Azirak S, Bilgic S, Korkmaz DT et al (2019) The protective effect of resveratrol against risperidone-induced liver damage through an action on FAS gene expression. Gen Physiol Biophys 38:215–225. https://doi.org/10.4149/gpb
Bailey J, Thew M, Balls M et al (2014) An analysis of the use of animal models in predicting human toxicology and drug safety. Altern Lab Anim 42:181–199. https://doi.org/10.1177/026119291404200306
Barnes M, Dixon LJ, Liu Z et al (2013) Macrophages and kupffer cells in drug-induced liver injury. In: Kaplowitz N, DeLeve L (eds) Drug-Induced liver disease, 3rd edn. Elsevier Inc., Amsterdam, pp 147–155
Bautista-Ferrufino MR, Cordero MD, Sánchez-Alcázar JA et al (2011) Amitriptyline induces coenzyme Q deficiency and oxidative damage in mouse lung and liver. Toxicol Lett 204:32–37. https://doi.org/10.1016/j.toxlet.2011.03.033
Begriche K, Massart J, Robin MA et al (2011) Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J Hepatol 54:773–794. https://doi.org/10.1016/j.jhep.2010.11.006
Bellmann R, Feistritzer C, Zoller H et al (2004) Treatment of intractable pruritus in drug induced cholestasis with albumin dialysis: A report of two cases. ASAIO J 50:387–391. https://doi.org/10.1097/01.MAT.0000132552.58214.00
Bergstrom R, Beasley C, Levy N et al (1993) The effects of renal and hepatic disease on the pharmacokinetics, renal tolerance, and risk-benefit profile of fluoxetine. Int Clin Psychopharmaco 8:261–266. https://doi.org/10.1097/00004850-199300840-00009
Bernal W, Auzinger G, Dhawan A, Wendon J (2010) Acute liver failure. Lancet 376:190–201. https://doi.org/10.1016/S0140-6736(10)60274-7
Bessone F, Dirchwolf M, Rodil MA et al (2018) Review article: drug-induced liver injury in the context of nonalcoholic fatty liver disease—a physiopathological and clinical integrated view. Aliment Pharmacol Ther 48:892–913. https://doi.org/10.1111/apt.14952
Billioti de Gage S, Collin C, Le-Tri T et al (2018) Antidepressants and hepatotoxicity: a cohort study among 5 Million Individuals Registered in the French National Health Insurance Database. CNS Drugs 32:673–684. https://doi.org/10.1007/s40263-018-0537-1
Bjornsson E, Kalaitzakis E, Olsson R (2007) The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug-induced liver injury. Aliment Pharmacol Ther 25:1411–1421. https://doi.org/10.1111/j.1365-2036.2007.03330.x
Bleakley S (2016) Antidepressant drug interactions: Evidence and clinical significance. Prog Neurol Psychiatry 20:21–27. https://doi.org/10.1002/pnp.429
Bondy SC, Naderi S (1994) Contribution of hepatic cytochrome P450 systems to the generation of reactive oxygen species. Biochem Pharmacol 48:155–159. https://doi.org/10.1016/0006-2952(94)90235-6
Bonkovsky HL, Blanchette PL, Schned AR (1986) Severe liver injury due to phenelzine with unique hepatic deposition of extracellular material. Am J Med 80:689–692. https://doi.org/10.1016/0002-9343(86)90826-0
Breuer RI (1965) Chlorpromazine hepatotoxicity manifested by a selective and sustained rise of serum alkaline phosphatase activity - Report of a case. Am J Dig Dis 10:727–731. https://doi.org/10.1007/BF02236073
Bricquir YL, Larrey D, Blanc P et al (1994) Tianeptine - an instance of drug-induced hepatotoxicity predicted by prospective experimental studies. J Hepatol 21:771–773. https://doi.org/10.1016/S0168-8278(94)80237-8
Brown C, Telio S, Warnock C, Wong A (2013) Clozapine Toxicity and Hepatitis. J Clin Psychopharmacol 33:570–571
Cai Q, Benson MA, Talbot TJ et al (1999) Acute hepatitis due to fluoxetine therapy. Mayo Clin Proc 74:692–694. https://doi.org/10.4065/74.7.692
Carvalhana S, Oliveira A, Ferreira P et al (2016) Acute liver failure due to trazodone and diazepam. GE Port J Gastroenterol 24:40–42. https://doi.org/10.1159/000450878
Castanheira L, Fernandes E, Levy P, Coentre R (2019) Aripiprazole-induced hepatitis: a case report. 17:551–555. https://doi.org/10.9758/cpn.2019.17.4.551
Cesaratto L, Vascotto C, Calligaris S, Tell G (2004) The importance of redox state in liver damage. Ann Hepatol Off J Mex Assoc Hepatol 3:86–92. https://doi.org/10.1016/s1665-2681(19)32099-x
Chalasani N, Fontana RJ, Bonkovsky HL et al (2008) Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 135:1924-1934.e4. https://doi.org/10.1053/j.gastro.2008.09.011
Chang A, Krygier DS, Chatur N, Yoshida EM (2009) Clozapine-induced fatal fulminant hepatic failure: a case report. Can J Gastroenterol 23:376–378. https://doi.org/10.1155/2009/503916
Charlton M, Sreekumar R, Rasmussen D et al (2002) Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 35:898–904. https://doi.org/10.1053/jhep.2002.32527
Chen CH, Shyue SK, Hsu CP, Lee TS (2018) Atypical antipsychotic drug olanzapine deregulates hepatic lipid metabolism and aortic inflammation and aggravates atherosclerosis. Cell Physiol Biochem 50:1216–1229. https://doi.org/10.1159/000494573
Chen T, Tzeng N, Tai Y (2019) Amitriptyline-Induced acute kidney injury and acute hepatitis : a case report. Am J Ther 2:1–2
Chen S, Wu Q, Li X et al (2020) The role of hepatic cytochrome P450s in the cytotoxicity of sertraline. Arch Toxicol 94:2401–2411. https://doi.org/10.1007/s00204-020-02753-y
Cheng YY, Min MY, Wei CC et al (2017) CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol Int 11:221–241. https://doi.org/10.1007/s12072-017-9793-2
Colakoglu O, Tankurt E, Unsal B et al (2005) Toxic hepatitis associated with paroxetine. Int J Clin Pract 59:861–862. https://doi.org/10.1111/j.1368-5031.2005.00572.x
Crewe H, Lennard M, Tucker G et al (1992) The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol 34:262–265. https://doi.org/10.1111/j.1365-2125.1992.tb04134.x
Danan G, Bernuau J, Moullot X et al (1984) Amitriptyline-induced fulminant. Hepatitis 30:179–184. https://doi.org/10.1159/000199103
Daneshmend TK, Scott GL, Bradfield JWB (1979) Angiosarcoma of liver associated with phenelzine. Br Med J 1:1679. https://doi.org/10.1136/bmj.1.6179.1679
Das A, Guarda L, Allen L (2017) Liver injury associated with quetiapine an illustrative case report. J Clin Psychopharmacol 37:623–625. https://doi.org/10.1097/JCP.0000000000000749
De Hert M, Detraux J, Van Winkel R et al (2012) Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol 8:114–126. https://doi.org/10.1038/nrendo.2011.156
De Long NE, Barry EJ, Pinelli C et al (2015) Antenatal exposure to the selective serotonin reuptake inhibitor fluoxetine leads to postnatal metabolic and endocrine changes associated with type 2 diabetes in Wistar rats. Toxicol Appl Pharmacol 285:32–40. https://doi.org/10.1016/j.taap.2015.03.006
de Oliveira MR (2016) Fluoxetine and the mitochondria: A review of the toxicological aspects. Toxicol Lett 258:185–191. https://doi.org/10.1016/j.toxlet.2016.07.001
Dejanovic B, Lavrnja I, Nikolic M et al (2017) Effects of agmatine on chlorpromazine toxicity in the liver of Wistar rats: the possible role of oxidant/antioxidant imbalance. Exp Anim 66:17–27. https://doi.org/10.1538/expanim.16-0010
DeSanty KP, Amabile CM (2007) Antidepressant-induced liver injury. Ann Pharmacother 41:1201–1211. https://doi.org/10.1345/aph.1K114
Detry O, Delwaide J, De Roover A et al (2009) Fulminant hepatic failure induced by venlafaxine and trazodone therapy: a case report. Transplant Proc 41:3435–3436. https://doi.org/10.1016/j.transproceed.2009.09.022
Djurasevic S, Todorovic Z, Pavlovic S, Pejic S (2019) Cadmium and fullerenes in liver diseases. In: Watson R, Preedy V (eds) Dietary interventions in liver disease: foods, nutrients, and dietary supplements. Elsevier Inc., Amsterdam, pp 333–344
Domínguez-Jiménez JL, Puente-Gutiérrez JJ, Pelado-García EM et al (2012) Liver toxicity due to olanzapine. Rev Esp Enfermedades Dig 104:617–618. https://doi.org/10.4321/S1130-01082012001100017
Đorđević N, Perić I, Stanisavljević A et al (2016) Duloxetine enhances hepatic GSH-dependent defense in rats. In: Čupić Ž, Anić S (eds) 13th International conference on fundamental and applied aspects of physical chemistry. Society of Physical Chemists of Serbia, Belgrade, pp 443–446
Dragovic S, Gunness P, Ingelman-Sundberg M et al (2013) Characterization of human cytochrome P450s involved in the bioactivation of clozapine. Drug Metab Dispos 41:651–658. https://doi.org/10.1124/dmd.112.050484
Du K, Ramachandran A, Jaeschke H (2016) Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol 10:148–156. https://doi.org/10.1016/j.redox.2016.10.001
Duda W, Curzytek K, Kubera M et al (2016) the effect of chronic mild stress and imipramine on the markers of oxidative stress and antioxidant system in rat liver. Neurotox Res 30:173–184. https://doi.org/10.1007/s12640-016-9614-8
Dumortier G, Cabaret W, Stamatiadis L et al (2002) Hepatic tolerance of atypical antipsychotic drugs. Encephale 28:542–551
Durašević SF, Dordević J, Jasnić N et al (2010) The influence of vitamin E supplementation on the oxidative status of rat liver. Arch Biol Sci 62:677–681. https://doi.org/10.2298/ABS1003677D
Dusi N (2019) Late-onset cholestatic liver injury during combination treatment with chlorpromazine and olanzapine a case report. J Clin Psychopharmacol 39:175–176
Dykens JA, Jamieson JD, Marroquin LD et al (2008) In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol Sci 103:335–345. https://doi.org/10.1093/toxsci/kfn056
Eftekhari A, Ahmadian E, Azarmi Y et al (2016) In vitro/vivo studies towards mechanisms of risperidone-induced oxidative stress and the protective role of coenzyme Q10 and N-acetylcysteine. Toxicol Mech Methods 26:520–528. https://doi.org/10.1080/15376516.2016.1204641
El Hajj I, Sharara AI, Rockey DC (2004) Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol 16:1415–1418. https://doi.org/10.1097/00042737-200412000-00029
El-Awdan SA, Abdel Jaleel GA, Saleh DO (2015) Alleviation of haloperidol induced oxidative stress in rats: Effects of sucrose vs grape seed extract. Bull Fac Pharm Cairo Univ 53:29–35. https://doi.org/10.1016/j.bfopcu.2015.02.004
Elbakary RH (2017) Histological study of the effects of olanzapine on the liver of adult male albino rat with and without Vitamin C. Egypt J Histol 40:1–11. https://doi.org/10.21608/EJH.2017.3692
Elgebaly HA, Mosa NM, Allach M et al (2018) Olive oil and leaf extract prevent fluoxetine-induced hepatotoxicity by attenuating oxidative stress, inflammation and apoptosis. Biomed Pharmacother 98:446–453. https://doi.org/10.1016/j.biopha.2017.12.101
Erdogan A, Atasoy N, Akkurt H et al (2008) Risperidone and liver function tests in children and adolescents: A short-term prospective study. Prog Neuro-Psychopharmacol Biol Psychiatry 32:849–857. https://doi.org/10.1016/j.pnpbp.2007.12.032
Esposito D, Brocvielle H, Becquemont L et al (2005) Risperidone-induced immunoallergic hepatitis. Am J Psychiatry 162:1984. https://doi.org/10.1176/appi.ajp.162.10.1984
Fartoux-Heymann L, Hézode C, Zafrani ES et al (2001) Acute fatal hepatitis related to sertraline. J Hepatol 35:683–684. https://doi.org/10.1016/S0168-8278(01)00159-3
Feng XM, Xiong J, Qin H et al (2012) Fluoxetine induces hepatic lipid accumulation via both promotion of the SREBP1c-related lipogenesis and reduction of lipolysis in primary mouse hepatocytes. CNS Neurosci Ther 18:974–980. https://doi.org/10.1111/cns.12014
Fernandes NF, Martin RR, Schenker S (2000) Case Reports. Trazodone-induced hepatotoxicity: a case report with comments on drug-induced hepatotoxicity. Am J Gastroenterol 95:532–535. https://doi.org/10.1111/j.1572-0241.2000.t01-1-01780.x
Fernø J, Vik-Mo AO, Jassim G et al (2009) Acute clozapine exposure in vivo induces lipid accumulation and marked sequential changes in the expression of SREBP, PPAR, and LXR target genes in rat liver. Psychopharmacology 203:73–84. https://doi.org/10.1007/s00213-008-1370-x
Filipović D, Mandić LM, Kanazir D, Pajović SB (2010) Acute and/or chronic stress models modulate CuZnSOD and MnSOD protein expression in rat liver. Mol Cell Biochem 338:167–174. https://doi.org/10.1007/s11010-009-0350-8
Fischer V, Haar J, Greiner L et al (1991) Possible role of free radical formation in clozapine (clozaril)-induced agranulocytosis. Mol Pharmacol 40:846–853
Foureau DM, Walling TL, Maddukuri V et al (2014) Comparative analysis of portal hepatic infiltrating leucocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Clin Exp Immunol 180:40–51. https://doi.org/10.1111/cei.12558
Friedenberg FK, Rothstein KD (1996) Hepatitis secondary to fluoxetine treatment. Am J Psychiatry 153:580. https://doi.org/10.1176/ajp.153.4.580a
Friedrich ME, Akimova E, Huf W et al (2016) Drug-induced liver injury during antidepressant treatment: results of AMSP, a drug surveillance program. Int J Neuropsychopharmacol 19:1–9. https://doi.org/10.1093/ijnp/pyv126
Fromenty B, Freneaux E, Labbe G et al (1989) Tianeptine, a new tricyclic antidepressant metabolized by β-oxidation of its heptanoic side chain, inhibits the mitochondrial oxidation of medium and short chain fatty acids in mice. Biochem Pharmacol 38:3743–3751. https://doi.org/10.1016/0006-2952(89)90580-7
Fuller CM, Yassinger S, Donlon P et al (1977) Haloperidol induced liver disease. West J Med 127:515–518
Furge LL, Guengerich FP (2006) Cytochrome P450 enzymes in drug metabolism and chemical toxicology: an introduction. Biochem Mol Biol Educ 34:66–74. https://doi.org/10.1002/bmb.2006.49403402066
Gabriel R, Wojtanowicz T, Farokhpay R, Bota R (2019) Acute transaminitis after initial days of starting haloperidol. Ment Illn 11:33–35. https://doi.org/10.4081/mi.2019.8113
Grady MM, Stahl SM (2012) Practical guide for prescribing MAOIs: debunking myths and removing barriers. CNS Spectr 17:2–10. https://doi.org/10.1017/S109285291200003X
Gruben N, Shiri-Sverdlov R, Koonen DPY, Hofker MH (2014) Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta Mol Basis Dis 1842:2329–2343. https://doi.org/10.1016/j.bbadis.2014.08.004
Grzeskowiak LE, Gilbert AL, Sørensen TIA et al (2013) Prenatal exposure to selective serotonin reuptake inhibitors and childhood overweight at 7 years of age. Ann Epidemiol 23:681–687. https://doi.org/10.1016/j.annepidem.2013.08.005
Gupta S, Pandey R, Katyal R et al (2005) Lipid peroxide levels and antioxidant status in alcoholic liver disease. Indian J Clin Biochem 20:67–71. https://doi.org/10.1007/BF02893045
Han D, Hanawa N, Saberi B, Kaplowitz N (2006) Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am J Physiol Gastrointest Liver Physiol 291:1–7. https://doi.org/10.1152/ajpgi.00001.2006
Hanje AJ, Pell LJ, Votolato NA et al (2006) Case report: fulminant hepatic failure involving duloxetine hydrochloride. Clin Gastroenterol Hepatol 4:912–917. https://doi.org/10.1016/j.cgh.2006.04.018
Heeringa M, Beurskens R, Schouten W, Verduijn MM (1999) Elevated plasma levels of clozapine after concomitant use of fluvoxamine. Pharm World Sci 21:243–244. https://doi.org/10.1023/A:1008748110692
Helmchen C, Boerner RJ, Meyendorf R, Hegerl U (1996) Reversible hepatotoxicity of paroxetine in a patient with major depression. Pharmacopsychiatry 29:223–226. https://doi.org/10.1055/s-2007-979576
Hemeryck A, Belpaire FM (2002) Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab 3:13–37. https://doi.org/10.2174/1389200023338017
Hendouei N, Farnia S, Mohseni F et al (2018) Alterations in oxidative stress markers and its correlation with clinical fi ndings in schizophrenic patients consuming perphenazine, clozapine and risperidone. Biomed Pharmacother 103:965–972. https://doi.org/10.1016/j.biopha.2018.04.109
Herrera B, Fernández M, Álvarez AM et al (2001) Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome c release in apoptosis induced by transforming growth factor β in rat fetal hepatocytes. Hepatology 34:548–556. https://doi.org/10.1053/jhep.2001.27447
Holtmann M, Kopf D, Mayer M et al (2003) Risperidone-associated steatohepatitis and excessive weight-gain. Pharmacopsychiatry 36:206–207
Horst DA, Grace ND, LeCompte PM (1980) Prolonged cholestasis and progressive hepatic fibrosis following imipramine therapy. Gastroenterology 79:550–554. https://doi.org/10.1016/0016-5085(80)90383-2
Hrycay EG, Bandiera SM (2015) Involvement of cytochrome P450 in reactive oxygen species formation and cancer. Adv Pharmacol 74:35–84. https://doi.org/10.1016/bs.apha.2015.03.003
Hummer M, Kurz M, Kurzthaler I et al (1997) Hepatotoxicity of clozapine. J Clin Psychopharmacol 17:314–317. https://doi.org/10.1097/00004714-199708000-00012
Ikeda M, Yoshikawa H (2003) Apoptosis-inducing protein derived from hepatocyte selectively induces apoptosis in lymphocytes. Immunology 108:116–122. https://doi.org/10.1046/j.1365-2567.2003.01542.x
Ilan Y, Samuel D, Reynes M, Tur-Kaspa R (1996) Hepatic failure associated with imipramine therapy. Pharmacopsychiatry 29:79–80. https://doi.org/10.1055/s-2007-979549
Ilgin S, Burukoglu D, Baysal M et al (2018) Assessment of hepatotoxic effects of quetiapine at repeated doses in rats. Anadolu Univ J Sci Technol C Life Sci Biotechnol 7:196–206. https://doi.org/10.18036/aubtdc.349864
Inkielewicz-Stêpniak I (2011) Impact of fluoxetine on liver damage in rats. Pharmacol Reports 63:441–447. https://doi.org/10.1016/S1734-1140(11)70510-2
Isaacson RH, Beier JI, Kh N et al (2020) ScienceDirect Olanzapine-induced liver injury in mice : aggravation by high-fat diet and protection with sulforaphane. J Nutr Biochem 81:108399. https://doi.org/10.1016/j.jnutbio.2020.108399
Jadallah K, Limauro D, Colatrella A (2003) Acute hepatocellular-cholestatic liver injury after olanzapine therapy. Ann Intern Med 138:357–358. https://doi.org/10.7326/0003-4819-138-4-200302180-00024
Jaeschke H, Tadashi H (2006) Role of neutrophils in acute inflammatory liver injury. Liver Int 26:912–919. https://doi.org/10.1111/j.1478-3231.2006.01327.x
Javaid JI (1994) Clinical pharmacokinetics of antipsychotics. J Clin Pharmacol 34:286–295. https://doi.org/10.1002/j.1552-4604.1994.tb01995.x
Jerrell JM (2010) Neuroendocrine-related adverse events associated with antidepressant treatment in children and adolescents. CNS Neurosci Ther 16:83–90. https://doi.org/10.1111/j.1755-5949.2009.00106.x
Jiang T, Zhang Y, Bai M et al (2019) Up-regulation of hepatic fatty acid transporters and inhibition/down- regulation of hepatic OCTN2 contribute to olanzapine-induced liver steatosis. Toxicol Lett 316:183–193. https://doi.org/10.1016/j.toxlet.2019.08.013
Kampa JM, Sahin M, Slopianka M et al (2020) Mass spectrometry imaging reveals lipid upregulation and bile acid changes indicating amitriptyline induced steatosis in a rat model. Toxicol Lett 325:43–50. https://doi.org/10.1016/j.toxlet.2020.02.007
Kolios G, Valatas V, Kouroumalis E (2006) Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol 12:7413–7420. https://doi.org/10.3748/wjg.v12.i46.7413
Kornischka J, Cordes J (2016) Acute drug-induced hepatitis during aripiprazole monotherapy: a case report. J Pharmacovigil 04:2–4. https://doi.org/10.4172/2329-6887.1000201
Krebs S, Dormann H, Muth-Selbach U et al (2001) Risperidone-induced cholestatic hepatitis. Eur J Gastroenterol Hepatol 13:67–69. https://doi.org/10.1097/00042737-200101000-00013
Kreuz S, Fischle W (2016) Oxidative stress signaling to chromatin in health and disease. Epigenomics 8:843–862. https://doi.org/10.2217/epi-2016-0002
Kumra S, Herion D, Jacobsen LK et al (1997) Case study: Risperidone-induced hepatotoxicity in pediatric patients. J Am Acad Child Adolesc Psychiatry 36:701–705. https://doi.org/10.1097/00004583-199705000-00022
Lai J, Ma S, Wang Y et al (2020) Factors associated with mental health outcomes among health care workers exposed to coronavirus disease 2019. JAMA Netw open 3:e203976. https://doi.org/10.1001/jamanetworkopen.2020.3976
Larrey D, Amouyal G, Pessayre D et al (1988) Amitriptyline-induced cholestasis prolonged. Gastroenterology 94:200–203
Lauressergues E, Staels B, Valeille K et al (2010) Antipsychotic drug action on SREBPs-related lipogenesis and cholesterogenesis in primary rat hepatocytes. Naunyn Schmiedebergs Arch Pharmacol 381:427–439. https://doi.org/10.1007/s00210-010-0499-4
Lauressergues E, Martin F, Helleboid A et al (2011) Overweight induced by chronic risperidone exposure is correlated with overexpression of the SREBP-1c and FAS genes in mouse liver. Naunyn Schmiedebergs Arch Pharmacol 383:423–436. https://doi.org/10.1007/s00210-010-0597-3
Lauressergues E, Bert E, Duriez P et al (2012) Does endoplasmic reticulum stress participate in APD-induced hepatic metabolic dysregulation? Neuropharmacology 62:784–796. https://doi.org/10.1016/j.neuropharm.2011.08.048
Levine B, Jenkins AJ, Queen M et al (1996) Distribution of venlafaxine in three postmortem cases. J Anal Toxicol 20:502–505. https://doi.org/10.1093/jat/20.6.502
Li Y, Couch L, Higuchi M et al (2012) Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol Sci 127:582–591. https://doi.org/10.1093/toxsci/kfs100
Li S, Tan H-Y, Wang N et al (2015) The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci 16:26087–26124. https://doi.org/10.3390/ijms161125942
Li Y, Su R, Xu S et al (2017) Artesunate prevents rats from the clozapine-induced hepatic steatosis and elevation in plasma triglycerides. Neuropsychiatr Dis Treat 13:2477–2487. https://doi.org/10.2147/NDT.S145069
Licata A, Minissale MG, Calvaruso V, Craxì A (2017) DILI and epidemiology. Eur Rev Med Pharmacol Sci 21:112–121
Lin CH, Lane HY (2019) Early identification and intervention of schizophrenia: Insight from hypotheses of glutamate dysfunction and oxidative stress. Front Psychiatry 10:1–9. https://doi.org/10.3389/fpsyt.2019.00093
Liu Z, Cui C, Xu P et al (2017) Curcumin activates AMPK pathway and regulates lipid metabolism in rats following prolonged clozapine exposure. Front Neurosci. https://doi.org/10.3389/fnins.2017.00558
Liu X, Zhao X, Deng C et al (2019) Simvastatin improves olanzapine-induced dyslipidemia in rats through inhibiting hepatic mTOR signaling pathway. Acta Pharmacol Sin 40:1049–1057. https://doi.org/10.1038/s41401-019-0212-1
López-Torresa E, Süvegesa Á, Peñas-Lledó EM et al (2014) Liver enzyme abnormalities during antipsychotic treatment: A case report of risperidone-associated hepatotoxicity. Drug Metabol Drug Interact 29:123–126. https://doi.org/10.1515/dmdi-2013-0064
Lui SY, Tso S, Lam M, Cheung EFC (2009) Possible olanzapine-induced hepatotoxicity in a young Chinese patient. Hong Kong Med J 15:394–396
Luppino F, de Wit L, Bouvy P et al (2010) Overweight, obesity, and depression. Arch Gen Psychiatry 67:220–229
Ma KL, Ruan XZ, Powis SH et al (2008) Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 48:770–781. https://doi.org/10.1002/hep.22423
MacAllister SL, Young C, Guzdek A et al (2013) Molecular cytotoxic mechanisms of chlorpromazine in isolated rat hepatocytes. Can J Physiol Pharmacol 91:56–63. https://doi.org/10.1139/cjpp-2012-0223
Mahmoud GS, El-deek HEM (2019) Melatonin modulates inflammatory mediators and improves olanzapine-induced hepatic steatosis in rat model of schizophrenia. Int J Physiol Pathophysiol Pharmacol 11:64–75
Marchesini G, Petta S, Dalle Grave R (2016) Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 63:2032–2043. https://doi.org/10.1002/hep.28392
Martignoni M, Groothuis GMM, De KR (2006) Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2:875–894. https://doi.org/10.1517/17425255.2.6.875
Matsuzaka T, Shimano H (2011) Molecular mechanisms involved in hepatic steatosis and insulin resistance. J Diabetes Investig 2:170–175. https://doi.org/10.1111/j.2040-1124.2011.00111.x
Mauri MC, Fiorentini A, Paletta S, Altamura AC (2014) Pharmacokinetics of antidepressants in patients with hepatic impairment. Clin Pharmacokinet 53:1069–1081. https://doi.org/10.1007/s40262-014-0187-5
Mauri MC, Paletta S, Di Pace C et al (2018) Clinical pharmacokinetics of atypical antipsychotics: an update. Clin Pharmacokinet 57:1493–1528. https://doi.org/10.1007/s40262-018-0664-3
Mazza M, De Lorenzo R, Conte C et al (2020) Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav Immun 89:594–600. https://doi.org/10.1016/j.bbi.2020.07.037
McNamara RK, Magrisso IJ, Hofacer R et al (2012) Omega-3 fatty acid deficiency augments risperidone-induced hepatic steatosis in rats: positive association with stearoyl-CoA desaturase. Pharmacol Res 66:283–291. https://doi.org/10.1016/j.phrs.2012.06.010
Meltzer HY (2013) Update on typical and atypical antipsychotic drugs. Annu Rev Med 64:393–406. https://doi.org/10.1146/annurev-med-050911-161504
Mercader J, Sabater AG, Le Gonidec S et al (2019) Oral phenelzine treatment mitigates metabolic disturbances in mice fed a high-fat diet. J Pharmacol Exp Ther 371:555–566. https://doi.org/10.1124/jpet.119.259895
Mijovic A (2020) Clozapine-induced agranulocytosis. Ann Hematol 99:2477–2482. https://doi.org/10.1007/s00277-020-04215-y
Milkiewicz P, Chilton AP, Hubscher SG, Elias E (2003) Antidepressant induced cholestasis: hepatocellular redistribution of multidrug resistant protein (MRP2). Gut 52:300–303. https://doi.org/10.1136/gut.52.2.300
Mishra A, Keshari AK, Singh AK et al (2016) Oxidative stress-based hepatotoxicity of duloxetine in wistar rats. Int J Pharm Pharm Sci 8:28–32. https://doi.org/10.22159/ijpps.2016v8i11.8857
Miyamoto S, Duncan GE, Marx CE, Lieberman JA (2005) Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 10:79–104. https://doi.org/10.1038/sj.mp.4001556
Moradpolr D, Altorfer J, Flury R et al (1994) Chlorpromazine-induced vanishing bile duct syndrome leading to biliary cirrhosis. Hepatology 20:1437–1441. https://doi.org/10.1002/hep.1840200610
Morgan MJ, Liu Z (2011) Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 21:103–115. https://doi.org/10.1038/cr.2010.178
Morlán-Coarasa MJ, Arias-Loste MT, Ortiz-García de la Foz V et al (2016) Incidence of non-alcoholic fatty liver disease and metabolic dysfunction in first episode schizophrenia and related psychotic disorders: a 3-year prospective randomized interventional study. Psychopharmacology 233:3947–3952. https://doi.org/10.1007/s00213-016-4422-7
Mosedale M, Watkins PB (2017) Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 101:469–480. https://doi.org/10.1002/cpt.564
Moskovitz R, DeVane C, Harris R, Stewart R (1982) Toxic hepatitis and single daily dosage imipramine therapy. J Clin Psychiatry 43:165–166
Mullish BH, Kabir MS, Thursz MR, Dhar A (2014) Review article: depression and the use of antidepressants in patients with chronic liver disease or liver transplantation. Aliment Pharmacol Ther 40:880–892. https://doi.org/10.1111/apt.12925
Mullock BM, Hall DE, Shaw LJ, Hinton RH (1983) Immune responses to chlorpromazine in rats. Biochem Pharmacol 32:2733–2738. https://doi.org/10.1016/0006-2952(83)90084-9
Munzer A, Sack U, Mergl R et al (2013) Impact of antidepressants on cytokine production of depressed patients in vitro. Toxins (Basel) 5:2227–2240. https://doi.org/10.3390/toxins5112227
Mutairi FA, Dwivedi G, Al Ameel T (2012) Fulminant hepatic failure in association with quetiapine: a case report. J Med Case Rep 6:1. https://doi.org/10.1186/1752-1947-6-418
Navarro V, Senior J (2006) Drug-Related Hepatotoxicity. N Engl J Med 354:731–739. https://doi.org/10.1056/NEJMra052270
Neumann H, Csepregi A, Evert M, Malfertheiner P (2008) Drug-induced liver disease related to citalopram. J Clin Psychopharmacol 28:240–260
Ölander M, Wiśniewski JR, Artursson P (2020) Cell-type-resolved proteomic analysis of the human liver. Liver Int 40:1770–1780. https://doi.org/10.1111/liv.14452
Onakpoya IJ, Heneghan CJ, Aronson JK (2016) Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med 14:1–11. https://doi.org/10.1186/s12916-016-0553-2
Ouanes S, Damak R, Hajri M et al (2015) Haloperidol-induced Cytolytic Hepatitis. Eur Psychiatry 30:1597
Ozcanli T, Erdogan A, Ozdemir S et al (2006) Severe liver enzyme elevations after three years of olanzapine treatment: a case report and review of olanzapine associated hepatotoxicity. Prog Neuropsychopharmacol Biol Psychiatry 30:1163–1166. https://doi.org/10.1016/j.pnpbp.2006.03.014
Özden H, Bildirici K, Üstüner D et al (2005) Histopathologic examination of rat liver after experimental application of fluoxetine. Türk Ekopatol Derg 11:9–15
Padurariu M, Ciobica A, Dobrin I, Stefanescu C (2010) Evaluation of antioxidant enzymes activities and lipid peroxidation in schizophrenic patients treated with typical and atypical antipsychotics. Neurosci Lett 479:317–320. https://doi.org/10.1016/j.neulet.2010.05.088
Pajović SB, Saicić ZS (2008) Modulation of antioxidant enzyme activities by sexual steroid hormones. Physiol Res 57:801–811
Pan S-J, Tan Y-L, Yao S-W et al (2018) Fluoxetine induces lipid metabolism abnormalities by acting on the liver in patients and mice with depression. Acta Pharmacol Sin 39:1463–1472. https://doi.org/10.1038/aps.2017.207
Park YM, Lee BH, Lee HJ, Kang SG (2010) Cholestatic jaundice induced by duloxetine in a patient with major depressive disorder. Psychiatry Investig 7:228–230. https://doi.org/10.4306/pi.2010.7.3.228
Park BK, Laverty H, Srivastava A et al (2011) Drug bioactivation and protein adduct formation in the pathogenesis of drug-induced toxicity. Chem Biol Interact 192:30–36. https://doi.org/10.1016/j.cbi.2010.09.011
Paulis MG, Hafez EM, El-Tahawy NF, Aly MKM (2018) Toxicological assessment of venlafaxine: acute and subchronic toxicity study in rats. Int J Toxicol 37:327–334. https://doi.org/10.1177/1091581818777470
Pawelczyk T, Pawelczyk A, Rabe-Jablonska J (2014) Clinical case discussion olanzapine-induced triglyceride and aminotransferase elevations without weight gain or hyperglycemia normalized after switching to aripiprazole. J Psychiatr Pr 20:301–307. https://doi.org/10.1097/01.pra.0000452568.92449.3f
Persky S, Reinus JF (2003) Sertraline hepatotoxicity: a case report and review of the literature on selective serotonin reuptake inhibitor hepatotoxicity. Dig Dis Sci 48:939–944. https://doi.org/10.1023/A:1023007831047
Pessayre D, Fromenty B, Berson A et al (2012) Central role of mitochondria in drug-induced liver injury. Drug Metab Rev 44:34–87. https://doi.org/10.3109/03602532.2011.604086
Phillips BB, Digmann RR, Beck MG (2006) Hepatitis associated with low-dose venlafaxine for postmenopausal vasomotor symptoms. Ann Pharmacother 40:323–327. https://doi.org/10.1345/aph.1G339
Pillinger T, Mccutcheon RA, Vano L et al (2020) Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: a systematic review and network meta-analysis. Lancet Psychiatry 7:64–77. https://doi.org/10.1016/S2215-0366(19)30416-X
Pohl LR, Kenna JG, Satoh H et al (1989) Neoantigens associated with halothane hepatitis. Drug Metab Rev 20:203–217. https://doi.org/10.3109/03602538909103537
Pompili M, Tittoto P, Mascianà R et al (2008) Acute hepatitis associated with use of paroxetine. Intern Emerg Med 3:275–277. https://doi.org/10.1007/s11739-008-0111-9
Preskorn S, Werder S (2006) Detrimental antidepressant drug-drug interactions: are they clinically relevant? Neuropsychopharmacology 31:1605–1612. https://doi.org/10.1038/sj.npp.1301070
Raeder MB, Fernø J, Vik-Mo AO, Steen VM (2006) SREBP activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol Cell Biochem 289:167–173. https://doi.org/10.1007/s11010-006-9160-4
Raschi E, de Ponti F (2015) Drug- and herb-induced liver injury: progress, current challenges and emerging signals of post-marketing risk. World J Hepatol 7:1761–1771. https://doi.org/10.4254/wjh.v7.i13.1761
Read AE, Harrison CV, Sherlock S (1961) Chronic chlorpromazine jaundice. Am J Med 31:249–258. https://doi.org/10.1016/0002-9343(61)90113-9
Ren L, Sun D, Zhou X et al (2019a) Chronic treatment with the modified Longdan Xiegan Tang attenuates olanzapine-induced fatty liver in rats by regulating hepatic de novo lipogenesis and fatty acid beta-oxidation-associated gene expression mediated by SREBP-1c, PPAR-alpha and AMPK-alpha. J Ethnopharmacol 232:176–187. https://doi.org/10.1016/j.jep.2018.12.034
Ren L, Zhou X, Huang X et al (2019b) The IRS/PI3K/Akt signaling pathway mediates olanzapine-induced hepatic insulin resistance in male rats. Life Sci 217:229–236. https://doi.org/10.1016/j.lfs.2018.12.015
Rettman KS, McClintock C (2001) Hepatotoxicity after short-term trazodone therapy. Ann Pharmacother 35:1559–1561. https://doi.org/10.1345/aph.10406
Rogers JP, Chesney E, Oliver D et al (2020) Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. The Lancet Psychiatry. https://doi.org/10.1016/S2215-0366(20)30203-0
Rozenblit-Susan S, Chapnik N, Froy O (2016) Metabolic effect of fluvoxamine in mouse peripheral tissues. Mol Cell Endocrinol 424:12–22. https://doi.org/10.1016/j.mce.2016.01.009
Sahini N, Selvaraj S, Borlak J (2014) Whole genome transcript profiling of drug induced steatosis in rats reveals a gene signature predictive of outcome. PLoS ONE 9:1–26. https://doi.org/10.1371/journal.pone.0114085
Shanks N, Greek R, Greek J (2009) Are animal models predictive for humans? Philos Ethics. Hum Med 20:1–20. https://doi.org/10.1186/1747-5341-4-2
Shastry CS, Shafeeque AA, Ashwathnarayana BJ (2013) Effect of combination of aripiprazole with carbamazepine and fluvoxamine on liver functions in experimental animals. Indian J Pharmacol 45:121–125. https://doi.org/10.4103/0253-7613.108280
Shen T, Liu Y, Shang J et al (2019) Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology 156:2230-2241.e11. https://doi.org/10.1053/j.gastro.2019.02.002
Shpaner A, Li W, Ankoma-Sey V, Botero RC (2008) Drug-induced liver injury: Hepatotoxicity of quetiapine revisited. Eur J Gastroenterol Hepatol 20:1106–1109. https://doi.org/10.1097/MEG.0b013e3282f8e3a0
Slim M, Medina-Caliz I, Gonzalez-Jimenez A et al (2016) Hepatic safety of atypical antipsychotics: current evidence and future directions. Drug Saf 39:925–943. https://doi.org/10.1007/s40264-016-0436-7
Soliman HM, Wagih HM, Algaidi SA, Hafiz AH (2013) Histological evaluation of the role of atypical antipsychotic drugs in inducing non-alcoholic fatty liver disease in adult male albino rats (light and electron microscopic study). Folia Biol 59:174–180
Solomons K, Gooch S, Wong A (2005) Toxicity with selective serotonin reuptake inhibitors. Am J Psychiatry 162:1225. https://doi.org/10.1176/appi.ajp.162.6.1225
Souza MEJ, Polizello ACM, Uyemura SA et al (1994) Effect of fluoxetine on rat liver mitochondria. Biochem Pharmacol 48:535–541. https://doi.org/10.1016/0006-2952(94)90283-6
Spindelegger CJ, Papageorgiou K, Grohmann R et al (2014) Cardiovascular adverse reactions during antidepressant treatment: A drug surveillance report of German-speaking countries between 1993 and 2010. Int J Neuropsychopharmacol 18:1–9. https://doi.org/10.1093/ijnp/pyu080
Stadlmann S, Portmann S, Tschopp S, Terracciano LM (2012) Venlafaxine-induced cholestatic hepatitis case report and review of literature. Am J Surg Pathol 36:1724–1728
Stadtman ER, Levine RL (2000) Protein Oxidation. Ann NY Acad Sci 899:191–208. https://doi.org/10.1111/j.1749-6632.2000.tb06187.x
Ștefan M, Kiss B, Gutleb A, Loghin F (2020) Redox metabolism modulation as a mechanism in SSRI toxicity and pharmacological effects. Arch Toxicol 94:1417–1441. https://doi.org/10.1007/s00204-020-02721-6
Suen C, Boyapati R, Simpson I, Dev A (2013) Acute liver injury secondary to sertraline. BMJ Case. https://doi.org/10.1136/bcr-2013-201022
Tabak F, Gunduz F, Tahan V et al (2009) Sertraline hepatotoxicity: report of a case and review of the literature. Dig Dis Sci 54:1589–1591. https://doi.org/10.1007/s10620-008-0524-3
Takács A, Thomas N, Pantelis C (2019) Clozapine rechallenge in a patient with clozapine- induced hepatitis. Australas Psychiatry 27:535. https://doi.org/10.1177/1039856219848824
Telles-Correia D, Barbosa A, Cortez-Pinto H et al (2017) Psychotropic drugs and liver disease: a critical review of pharmacokinetics and liver toxicity. World J Gastrointest Pharmacol Ther 8:26. https://doi.org/10.4292/wjgpt.v8.i1.26
Todorović N, Tomanović N, Gass P, Filipović D (2016) Olanzapine modulation of hepatic oxidative stress and inflammation in socially isolated rats. Eur J Pharm Sci 81:94–102. https://doi.org/10.1016/j.ejps.2015.10.010
Urichuk L, Prior TI, Dursun S, Baker G (2008) Metabolism of atypical antipsychotics: involvement of cytochrome p450 enzymes and relevance for drug-drug interactions. Curr Drug Metab 9:410–418
Van Harten J (1995) Overview of the pharmacokinetics of fluvoxamine. Clin Pharmacokinet 29:1–9. https://doi.org/10.2165/00003088-199500291-00003
Voican CS, Corruble E, Naveau S, Perlemuter G (2014) Antidepressant-induced liver injury: a review for clinicians. Am J Psychiatry 171:404–415. https://doi.org/10.1176/appi.ajp.2013.13050709
Vuppalanchi R, Hayashi PH, Chalasani N et al (2010) Duloxetine hepatotoxicity: a case-series from the drug-induced liver injury network. Aliment Pharmacol Ther 32:1174–1183. https://doi.org/10.1111/j.1365-2036.2010.04449.x
Wang P, Si T (2013) Use of antipsychotics in the treatment of depressive disorders. Shanghai Arch Psychiatry 25:134–140. https://doi.org/10.3969/j.issn.1002-0829.2013.03.002
Wang W, Bai M, Jiang T et al (2019) Clozapine-induced reduction of L-carnitine reabsorption via inhibition / down-regulation of renal carnitine / organic cation transporter 2 contributes to liver lipid metabolic disorder in mice. Toxicol Appl Pharmacol 363:47–56. https://doi.org/10.1016/j.taap.2018.11.007
WHO (2017) Depression and Other Common Mental Disorders: Global Health Estimates. Geneva: World Health Organization; 2017. Licence: CC BY-NC-SA 3.0 IGO
WHO (2020) Mental health & COVID-19. https://www.who.int/teams/mental-health-and-substance-use/covid-19. Accessed 10 Nov 2020
Wong-Ekkabut J, Xu Z, Triampo W et al (2007) Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys J 93:4225–4236. https://doi.org/10.1529/biophysj.107.112565
Woolbright BL, Jaeschke H (2018) Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr Pharmacol Rep 4:346–357. https://doi.org/10.1007/s40495-018-0147-0
Wright TM, Vandenberg AM (2007) Risperidone- and quetiapine-induced cholestasis. Ann Pharmacother 41:1518–1523. https://doi.org/10.1345/aph.1K145
Wrighton SA, Ring BJ (1999) Predicting drug interactions and pharmacokinetic variability with in vitro methods: The olanzapine experience. Drug Metab Rev 31:15–28. https://doi.org/10.1081/DMR-100101905
Wu Chou AI, Lu ML, Shen WW (2014) Hepatotoxicity induced by clozapine: A case report and review of literature. Neuropsychiatr Dis Treat 10:1585–1587. https://doi.org/10.2147/NDT.S67654
Xiong J, Yang H, Wu L et al (2014) Fluoxetine suppresses AMP-activated protein kinase signaling pathway to promote hepatic lipid accumulation in primary mouse hepatocytes. Int J Biochem Cell Biol 54:236–244. https://doi.org/10.1016/j.biocel.2014.07.019
Xu Y, Wang C, Klabnik JJ, Donnell JMO (2014) Novel therapeutic targets in depression and anxiety: antioxidants as a candidate treatment. Curr Neuropharmacol. https://doi.org/10.2174/1570159X11666131120231448
Yajima D, Motani H, Hayakawa M et al (2009) The relationship between cell membrane damage and lipid peroxidation under the condition of hypoxia-reoxygenation: analysis of the mechanism using antioxidants and electron transport inhibitors. Cell Biochem Funct 27:338–343. https://doi.org/10.1002/cbf.1578
Yang Q, Yang F, Tang X et al (2015) Chlorpromazine-induced perturbations of bile acids and free fatty acids in cholestatic liver injury prevented by the Chinese herbal compound Yin-Chen-Hao-Tang. BMC Complement Altern Med 15:1–12. https://doi.org/10.1186/s12906-015-0627-2
Ye H, Nelson L, Gómez Del Moral M et al (2018) Dissecting the molecular pathophysiology of drug-induced liver injury. World J Gastroenterol 24:1373–1385. https://doi.org/10.3748/wjg.v24.i13.1373
Yilmaz A, Elbey B, Yazgan ÜC et al (2016) Protective effects of caffeic acid phenethyl ester on fluoxetine-induced hepatotoxicity: an experimental study. Biomed Res Int. https://doi.org/10.1155/2016/1247191
Yu K, Geng X, Chen M et al (2014) High daily dose and being a substrate of cytochrome P450 enzymes are two important predictors of drug-induced liver injury. Drug Metab Dispos 42:744–750. https://doi.org/10.1124/dmd.113.056267
Yuan W, Williams B (2012) Acute hepatic failure involving duloxetine hydrochloride. J Neuropsychiatry Clin Neurosci 24:48–49. https://doi.org/10.1176/appi.neuropsych.11040083
Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. https://doi.org/10.1016/j.pharmthera.2012.12.007
Zlatković J, Todorović N, Tomanović N et al (2014) Chronic administration of fluoxetine or clozapine induces oxidative stress in rat liver: a histopathological study. Eur J Pharm Sci 59:20–30. https://doi.org/10.1016/j.ejps.2014.04.010
Acknowledgments
The research was funded by the Ministry of Education, Science and Technological Development of the Republic of Serbia, grant numbers: 451-03-68/2020-14/ 200017 and 451-03-68/2020-14/200178.
Funding
The research was funded by the Ministry of Education, Science, and Technological Development of the Republic of Serbia, grant numbers: 451–03-68/2020–14/ 200017 and 451–03-68/2020–14/200178.
Author information
Authors and Affiliations
Contributions
Specific author contributions: NTV had the idea for the article, conceptualised, searched and reviewed literature, and illustrated and drafted the manuscript. JĐ, SP, NĐ, and SBP revised it critically for important intellectual content.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Todorović Vukotić, N., Đorđević, J., Pejić, S. et al. Antidepressants- and antipsychotics-induced hepatotoxicity. Arch Toxicol 95, 767–789 (2021). https://doi.org/10.1007/s00204-020-02963-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02963-4