
Abstract
The aim of the present study was to develop a generic rat physiologically based kinetic (PBK) model that includes a novel testing strategy where active biliary excretion is incorporated using estradiol-17β glucuronide (E217βG) as the model substance. A major challenge was the definition of the scaling factor for the in vitro to in vivo conversion of the PBK-model parameter Vmax. In vitro values for the Vmax and Km for transport of E217βG were found in the literature in four different studies based on experiments with primary rat hepatocytes. The required scaling factor was defined based on fitting the PBK model-based predicted values to reported experimental data on E217βG blood levels and cumulative biliary E217βG excretion. This resulted in a scaling factor of 129 mg protein/g liver. With this scaling factor the PBK model predicted the in vivo data for blood and cumulative biliary E217βG levels with on average of less than 1.8-fold deviation. The study provides a proof of principle on how biliary excretion can be included in a generic PBK model using primary hepatocytes to define the kinetic parameters that describe the biliary excretion.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The kinetic profile of a substance is of importance when considering human safety assessment and drug development. Prediction of these kinetic profiles based on absorption, distribution, metabolism and excretion (ADME) using physiologically based kinetic (PBK) modelling has been shown of value not only to predict blood levels of drugs following defined dose levels in forward dosimetry, but also in so-called reverse dosimetry for quantitative in vitro—in vivo extrapolation (QIVIVE) (Louisse et al. 2017; Rietjens et al. 2011). QIVIVE aims to contribute to the 3Rs of Russel and Burch (reduction, replacement and refinement) of animal experiments (Flecknell 2002; Louisse et al. 2015; Zhang et al. 2018). Up to now, proofs of principle for human safety assessment using PBK model-facilitated QIVIVE have been mainly provided for chemicals that do not depend on active excretion via either kidneys or liver (Fabian et al. 2019; Louisse et al. 2010; Moxon et al. 2020; Ning et al. 2019; Punt et al. 2019; Strikwold et al. 2017).
In drug development, there already have been studies on QIVIVE using PBK-modelling focussing on especially excretion of certain drugs via active transport from the liver to the bile (Chapy et al. 2015; Jamei et al. 2014; Jones et al. 2012). In addition, both endogenous and exogenous glucuronide conjugates have also been shown to be actively excreted via this route (Cronholm et al. 1971; Ge et al. 2016; Hjelle and Klaassen 1984). The common properties of the compounds for which this active transport from liver into bile was shown important are that they have a low membrane permeability, a molecular weight cut off of 475 Da (400 Da for rats), remain mostly unchanged and therefore are excreted via active uptake and efflux in and from the liver cells into bile (Yang et al. 2009).
Given these structural characteristics it can be foreseen that biliary excretion may not only be relevant for drugs but also for other chemicals, including new chemicals (Choi et al. 2019). For these substances, active excretion should be included in the PBK-models to obtain an adequate description of their kinetics and subsequent prediction of in vivo effective dose levels. However, due to the lack of well-established and validated in vitro assays to quantify kinetics for active excretion there is a lack of PBK-models including active biliary and/or renal excretion. When not including active excretion in QIVIVE, the prediction of the time-dependent blood concentration can deviate from the in vivo situation resulting in incorrect determination of points of departure for human safety assessment when based on PBK model-facilitated reverse dosimetry (Louisse et al. 2017).
To be of value for novel non-animal based testing strategies, parameters required for the PBK-models should preferably be defined using in silico and in vitro approaches. When using in vitro models for biliary excretion, important challenges relate to the type of in vitro model to be used and the subsequent translation of the in vitro data to the in vivo situation using adequate scaling factors (Choi et al. 2019). When using for example a cell model with overexpression of a transporter of interest, proper scaling depends on the expression level of the transporter in the cell model compared to its expression level in the organ in vivo. To solve this matter a relative expression factor (REF) can be used (Chan et al. 2019; Jamei et al. 2014). Moreover, because the activity of the transporter in the transfected cell can differ from its in vivo activity often a relative activity factor (RAF) is used as well (Izumi et al. 2018; Poirier et al. 2009). Other factors may further complicate this in vitro to in vivo scaling, such as the fact that the cell of origin used to generate the transfected cell model [e.g. human embryonic kidney cells, Chinese hamster ovaries, Xenopus Laevis oocytes, (Cattori et al. 2001; Eckhardt et al. 1999; van de Steeg et al. 2013)] may not fully represent the cell type of the designated organ making the in vitro to in vivo translation more complex. This implies that hepatocytes may provide an alternative model to define the kinetic parameters for biliary excretion (Cantrill and Houston 2017; Chu et al. 2013; Yabe et al. 2011). Therefore, the aim of the present study was to develop a generic rat PBK model incorporating a novel testing strategy of including active biliary excretion, focussing on hepatocytes as the in vitro cell model and the scaling factor to be used to obtain adequate in vitro to in vivo translation. This was done using estradiol-17β glucuronide (E217βG) (Fig. 1) as the model substance (molecular weight 448.5 g/mole). E217βG is an anionic endogenous oestrogen glucuronide conjugate well known for its high affinity for active transport in the liver where it is taken up from the blood (sinusoidal side) via the (rat) organic anion transporting polypeptides (Oatps) into the hepatocytes, to be subsequently excreted into the bile (Kanai et al. 1996). This uptake of E217βG into the hepatocytes via the Oatps is the rate-limiting step for its elimination (Varma et al. 2012).

Chemical structure of estradiol-17β glucuronide
Materials and methods
Physiologically based kinetic (PBK) modelling
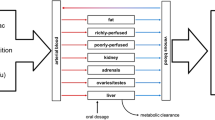
A conceptual PBK model was developed for E217βG in rat (Fig. 2). The model contained separate compartments for blood, fat and liver. All other organ tissues were divided in either a rapidly perfused tissue compartment (brain, heart, lungs, kidneys) or a slowly perfused tissue compartment (bone, skin, muscle). The gastro-intestinal tract was not included as a separate compartment since administration of E217βG was intravenously. Physiological and anatomical parameters such as tissue blood flow and tissue weight were obtained from Brown et al. (1997). Tissue:blood partition coefficients were determined by a mathematical method described by DeJongh et al. (1997) based on the log Kow of E217βG (2.05 ALOGPS). Table 1 presents a detailed overview of the parameters.

Schematic overview of the PBK model for E217βG including biliary excretion
The transport of E217βG from liver to bile was described by Michaelis–Menten kinetics using the following formula:
where AL is the change in the amount of E217βG in the liver over time (µmol/h), QL the blood flow to the liver (L/h), CB the concentration of E217βG in arterial blood (µmol/L), CVL the venous concentration of E217βG in the liver (µmol/L), VmaxE217bG and KmE217bG the maximum rate (µmol/h) and Michaelis–Menten constant (µmol/L) for the transport of E217βG. The model equations were encoded and solved using Berkeley Madonna 8.3.18 (UC Berkeley, CA, USA).
Although it is known that E217βG undergoes enterohepatic circulation, this was not taken into account in the model because for all the in vivo data included in this study the bile duct of the rats was cannulated, therefore, enterohepatic circulation could not take place.
In vitro kinetic data
The in vitro kinetic data for transport of E217βG into hepatocytes were obtained from the literature. Four studies, (Brock and Vore 1984; Brouwer et al. 1987; Ishizuka et al. 1998; Kouzuki et al. 1999) reported Vmax (pmol/min/mg protein) and Km (µM) values for the transport of E217βG into hepatocytes. Briefly, all studies used freshly isolated primary hepatocytes from Sprague–Dawley rats and radio-labelled E217βG. Three studies used a hepatocyte suspension system where concentration dependent uptake of [3H]-E217βG under linear conditions with respect to time was determined. At the end of each incubation, a fraction of hepatocyte suspension was removed from the incubations. This fraction was added to a tube containing silicon oil and 3 M KOH. The tube was centrifuged and cut at the silicone oil layer. The hepatocyte pellet was placed in a scintillation vial and radioactivity was measured. The other study cultured the freshly isolated hepatocytes on collagen-coated dishes. After washing the dishes three times with either a Krebs–Henseleit buffer or choline buffer, the uptake was initiated by adding [3H]-E217βG. The uptake was stopped and cells were washed three times using ice-cold Krebs–Henseleit buffer for both procedures. The cells were solubilised in 1 N NaOH, distilled water was added and [3H]-E217βG was measured with liquid scintillation. Protein content in all studies was determined using either the method of Lowry (Lowry et al. 1951) or the method of Bradford (Bradford 1976) with bovine serum albumin as a standard. The Vmax and Km values were determined by plotting the rate of uptake (pmol/min/mg protein) of [3H]-E217βG by the hepatocytes against the [3H]-E217βG concentration (µM) fitting the curve using the Michaelis–Menten equation.
Scaling factor
In the present study the scaling factor was determined by fitting the PBK model-based predicted values for blood concentrations and the cumulative biliary excretion of E217βG to the data reported for these endpoints in rat experiments found in literature. Table 2 presents an overview of available in vivo studies. They included in vivo rat kinetic data for the time-dependent plasma concentration of E217βG upon an intravenous dose level of 81 ng/kg bw (Gotoh et al. 2002; Morikawa et al. 2000) and 23 ng/kg bw (Slikker et al. 1983) and the time-dependent cumulative biliary excretion of E217βG upon an intravenous dose level 81 ng/kg bw (Gotoh et al. 2002; Morikawa et al. 2000) and 48 ng/kg bw (Takikawa et al. 1996). Three of these studies used male Sprague–Dawley rats, one study used female Sprague–Dawley rats (Slikker et al. 1983). All studies quantified [3H]-E217βG by liquid scintillation counting. Since the PBK model predicts whole blood concentrations and the in vivo data present time-dependent plasma concentrations, the plasma concentrations were converted to whole blood concentrations using the following formula:
where Cblood is the concentration of E217βG in whole blood (µmol/L), Cplasma the concentration of E217βG in plasma (µmol/L) and Hct the rat haematocrit, which was set at 40%, the average of the range published (Probst et al. 2006). (See figure S1 in supplementary material A for original plasma concentration data).
Comparison of the time-dependent plasma concentration reported by Morikawa et al. (2000) and Gotoh et al. (2002) using a similar dose of 81 ng/kg bw, indicated the data point at t = 1 h reported by Gotoh et al. (2002) to be an outlier (figure S1 supplementary material A), and this data point was aligned with that from Morikawa et al. (2000) before further use of the Gotoh et al. (2002) data. In addition, because the time-dependent plasma concentrations reported by Slikker et al. (1983) at a dose of 23 ng/kg bw completely overlapped with both in vivo data sets at a dose of 81 ng/kg bw (figure S2 supplementary material A), these data were corrected to bring them in line with the rest of the data by correcting the blood concentrations by a factor of 0.284 (23/81).
Combining the four in vitro kinetic data sets for the Vmax and Km of E217βG transport in rat hepatocytes with the six in vivo data sets on plasma E217βG and on cumulative biliary excretion of E217βG provided 24 fitted scaling factors, which were combined to generate a mean value for further PBK model-based predictions. All predicted curves and reported in vivo data were graphically illustrated using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA).
To define the scaling factor as described above in vitro Vmax values expressed in pmol/min/mg protein have to be converted to an in vivo Vmax value expressed in µmol/h/whole liver. This was done using the following formula:
where the factor 1,000,000 is used to convert pmol to µmol, 60 to convert minutes to hours, SF is the scaling factor expressed in mg protein/g liver, multiplied by the volume of the liver expressed in kilograms (Table 1), which is multiplied by 1000 to convert kilograms to grams.
The full model code is presented in Supplementary material B.
Sensitivity analysis
To assess the influence of the scaling factor on the model predictions and to assess the model parameters that can influence the model output most, a sensitivity analysis was performed for both the predicted concentration of E217βG in blood and its predicted cumulative biliary excretion. To carry out the sensitivity analysis, a dose level of one of the available rat studies was used, 81 ng/kg bw. The analysis was performed using the values for Km and Vmax obtained from the rat hepatocyte study with the lowest catalytic efficiency (CE) (calculated as Vmax/Km) (Brouwer et al. 1987) and the highest catalytic efficiency (Kouzuki et al. 1999). Based on the method reported by Evans and Andersen (2000) the sensitivity coefficients (SCs) for the model parameters were calculated as follows:
where C indicates the initial value of the model output, C′ indicates the modified value of the model output resulting from an increase in the parameter value. P indicates the initial parameter value and P′ indicates the modified parameter value after a 5% increase of its value, keeping all other parameters at their original value.
Results
In vitro kinetic data
Data from the four studies reporting values for the kinetic parameters Vmax and Km for E217βG transport by hepatocytes are listed in Table 3. For both the Km and Vmax the maximum fold difference between the values from the different data sets was not higher than 11. The resulting catalytic efficiencies (CE) vary less than 3.1-fold.
Predicted versus observed time-dependent blood levels of E217βG
Using the Vmax and Km values presented in Table 3, PBK model-based predictions for E217βG levels in blood were calculated optimising the scaling factor to obtain the best fit between predicted and actually reported experimental data for the three available in vivo data sets (Table 2). To this end an iterative process varying the scaling factor within the range of 5–500 mg protein/g liver was performed to find the best fit. For each in vivo data set four optimisations were carried out, one for each of the data sets for Vmax and Km derived from rat hepatocytes (Table 3). Since the four fitted predictions thus obtained for each of the three experimentally observed time-dependent blood concentration of intravenously administered E217βG in rats were similar, only the predictions for one out of the four data sets are displayed in Fig. 3 (all twelve predictions can be found in supplementary material A fig. S3). The scaling factors in mg protein/g liver for conversion of in vitro hepatocyte protein levels to in vivo liver protein levels optimised to obtain the predicted curves are presented in Table 4.

Predicted and observed blood concentrations (corrected from reported plasma concentrations) of E217βG in rats upon intravenous administration. Symbols represent rat in vivo data obtained at a dose of (a) 81 ng/kg bw (squares) (Morikawa et al. 2000), (b) 81 ng/kg bw (triangles) (Gotoh et al. 2002) and (c) 23 ng/kg bw (dots) (Slikker et al. 1983). Data represent the mean (and the SD where available). Predictions (lines) are based on the Vmax and Km values for hepatocyte transport of E217βG from rat hepatocyte system 1 obtained from literature and presented in Table 3. The predictions for all four Vmax and Km data sets are presented in supplementary material A figure S3. The details of the in vivo data sets are presented in Table 2
The data thus obtained reveal that with all kinetic hepatocyte data adequate fits can be obtained with scaling factors that differ only to a limited extent for the four kinetic hepatocyte data sets. The scaling factors for the fits of the 3 different in vivo data sets for blood levels of E217βG varied here amounting to at most a sixfold difference when comparing results within a hepatocyte data set.
Predicted versus observed time-dependent cumulative biliary excretion of E217βG
Next to the time-dependent blood concentration, the cumulative biliary excretion was predicted using the same approach (Fig. 4). Since also for these three data sets on the time-dependent biliary excretion the predictions obtained using the four Vmax and Km data sets were similar, only the predictions for one out of the four data sets are displayed in Fig. 4. All twelve predictions can be found in supplementary material A fig. S4. The values for the scaling factor in mg protein/g liver for conversion of in vitro hepatocyte protein levels to in vivo liver protein levels optimised to obtain the predicted curves are presented in Table 4.

Predicted and observed cumulative biliary excretion of E217βG in rats upon intravenous administration expressed as percentage of the dose (%). Symbols represent rat in vivo data obtained at a dose of (a) 81 ng/kg bw (squares) (Morikawa et al. 2000), (b) 81 ng/kg bw (triangles) (Gotoh et al. 2002) and (c) 48 ng/kg bw (diamonds) (Takikawa et al. 1996). Data represent the mean (and the SD where available). Predictions (lines) are based on the Vmax and Km values for hepatocyte transport of E217βG from rat hepatocyte system 1 obtained from literature and presented in Table 3. The predictions for all four Vmax and Km data sets are presented in supplementary material A figure S4. The details of the in vivo data sets are presented in Table 2
The data obtained reveal that also for prediction of the cumulative biliary excretion all kinetic hepatocyte data can provide adequate fits with scaling factors that differ only to a limited extent for the four kinetic hepatocyte data sets. The scaling factors for the fits of the 3 different in vivo data sets for blood E217βG varied somewhat more compared with the scaling factors obtained from the time-dependent blood concentration amounting from a sixfold to a ninefold difference when comparing results within a hepatocyte data set. Taking all 24 scaling factors together resulted in an average scaling factor of 129 ± 24 mg protein/g liver (mean ± SEM).
Evaluation of the scaling factor
To further evaluate the scaling factor, the in vivo kinetic data on time-dependent E217βG blood concentration and cumulative biliary excretion were predicted using this average value and compared to the experimental data in Figs. 5, 6. The results obtained reveal that with this average scaling factor all experimental data for blood E217βG levels were on average predicted with a less than 1.8-fold deviation, while for the data for the cumulative biliary excretion of E217βG the averaged deviation was less than 1.4-fold. The largest deviations were observed for the in vivo data on blood E217βG levels of Slikker et al. (1983) where the model somewhat overpredicted the clearance of E217βG from the blood (Fig. 5c, f, i, l) ranging from a 4 to 12- fold difference and for the in vivo data on cumulative biliary excretion reported by Gotoh et al. (2002) that were too, somewhat overpredicted with a maximum of sixfold deviation (Fig. 6b, e, h, k).

Predictions of the blood concentration in time of E217βG in rats upon intravenous administration using the average value for the scaling factor (129 mg protein/g liver) compared with the observed blood concentrations. Symbols represent rat in vivo kinetic data obtained at a dose of (a, d, g, j) 81 ng/kg bw (squares) (Morikawa et al. 2000), (b, e, h, k) 81 ng/kg bw (triangles) (Gotoh et al. 2002) and (c, f, i, l) 23 ng/kg bw (dots) (Slikker et al. 1983). Data represent the mean (and the SD where available). Predictions (lines) are based on the Vmax and Km values for hepatocyte transport of E217βG obtained from literature and presented in Table 3

Predictions of the cumulative biliary excretion in time of E217βG in rats upon intravenous administration expressed as percentage of the dose (%) using the average value for the scaling factor (129 mg protein/g liver) compared with the observed cumulative biliary excretion. Symbols represent rat in vivo kinetic data obtained at a dose of (a, d, g, j) 81 ng/kg bw (squares) (Morikawa et al. 2000), (b, e, h, k) 81 ng/kg bw (triangles) (Gotoh et al. 2002) and (c, f, i, l) 48 ng/kg bw (diamonds) (Takikawa et al. 1996). Data represent the mean (and the SD where available). Predictions (lines) are based on the Vmax and Km values for hepatocyte transport of E217βG obtained from literature and presented in Table 3
Sensitivity analysis
To further evaluate the influence of the scaling factor on the model predictions and also elucidate which PBK-model parameters influence the predictions most a sensitivity analysis was performed. The predicted blood concentration and cumulative biliary excretion levels of E217βG at a time point of 0.5 h were used as the basis for this analysis. Only parameters with a normalized sensitivity coefficient > 0.1 are presented. The results reveal that for prediction of both the blood E217βG concentration (Fig. 7a) and the cumulative biliary excretion (Fig. 7b) the blood flow to slowly perfused tissue (QSc) and the blood flow to rapidly perfused tissue (QRc) are most influential parameters while for prediction of the cumulative biliary excretion also the blood flow to the liver (QLc) is influential. At different catalytic efficiencies (CE) for the hepatocyte transport the normalized sensitivity coefficients are somewhat different, but the parameters that have the largest influence remain the same. The influence of the scaling factor on both model outcomes shows not to be substantial with a sensitivity coefficient at the highest and lowest CE of 0.2 and -0.4 for the blood concentration and 0.04 and 0.13 for the cumulative biliary excretion.

Sensitivity coefficients of the PBK-model parameters for prediction of a the E217βG blood concentration and b the cumulative active biliary excretion of E217βG at an intravenous dose of 81 ng/kw bw, t = 0.5 h with the Vmax and Km that result in the lowest catalytic efficiency (CE) (black bars) (Brouwer et al. 1987) and the Vmax and Km that result in the highest catalytic efficiency (grey bars) (Kouzuki et al. 1999) (see Table 3). VFc = volume fat tissue, VLc = volume liver tissue, VRc = volume of rapidly perfused tissues, VSc = volume of slowly perfused tissue, QFc = blood flow to fat, QLc = blood flow to liver, QRc = blood flow to rapidly perfused tissue, QSc = blood flow to slowly perfused tissue, PF = partition coefficient of fat, PR = partition coefficient of rapidly perfused tissue, PS = partition coefficient of slowly perfused tissue, Vmax = maximum rate of E217βG transport in hepatocytes, Km = Michaelis–Menten constant of E217βG transport in hepatocytes, SF = scaling factor
Discussion
The aim of the present study was to provide a proof of principle for including biliary excretion into a generic rat PBK model with a major focus on determining the in vitro to in vivo scaling factor to be used when defining the kinetics of biliary transport using primary rat hepatocytes. A scaling factor is required to convert a kinetic value obtained in the in vitro cellular model, to a value that reflects the same biological function for the relevant whole organ. In this study, the scaling factor was needed to translate the in vitro Vmax value for E217βG transport in hepatocytes expressed in pmol/min/mg protein to an in vivo rate for E217βG transport by the liver expressed in µmol/h. The scaling factor expressed in mg protein/g liver enabled use of the in vitro hepatocyte Vmax value in the PBK model to enable prediction of in vivo blood and cumulative biliary excretion levels of intravenously administered E217βG. We used E217βG as a model substance because the substance is excreted as such, with its active excretion into bile via the activity of the Oatp transporters in the sinusoidal membrane of hepatocytes being the rate-limiting step for its elimination (Varma et al. 2012). Using the obtained value for the scaling factor, 129 mg protein/g liver, resulted in PBK model-based predictions of the blood and cumulative biliary excretion levels of E217βG that were on average within a 1.8-fold deviation from reported in vivo data, thus showing the value of the use of primary hepatocytes as an in vitro system to determine kinetic parameters for describing biliary excretion.
It is of importance to note that main deviations between predicted and observed in vivo data could be related to especially differences between reported experimental data at the same dose level. Even though the in vivo time-dependent blood concentration and the corresponding cumulative biliary excretion reported by Gotoh et al. (2002) and Morikawa et al. (2000) were obtained from the same laboratory, the outcomes were different resulting in different values for the individual scaling factors obtained when fitting the data. This difference between the experimental data sets might be explained by the two year time-lap between the two studies and the fact that animal experiments are depending on an array of guideline protocols and conditions that might have been slightly changed over the two years influencing the (expected) outcome (Council 2011). This deviation in reported in vivo data becomes even more apparent when looking at the in vivo data from Slikker et al. (1983) where at one third of the dose level the data for E217βG blood levels were overlapping with the data of the two studies using a threefold higher dose level. This discrepancy could be related to the sex of the rats used (female not male) in the Slikker et al. (1983) study. However, Gotoh et al. (2002) reported in vivo data on blood levels of E217βG in female Sprague–Dawley rats, and showed that these levels were comparable to the levels found in the male Sprague–Dawley rats. Other factors that could play a role in the differences in in vivo data are the vehicle used for the IV administration, which was distilled water: polyethylene glycol: ethanol (10:4:1 v/v) in the studies reported by Gotoh et al. (2002) and Morikawa et al. (2000) but saline: propylene glycol: ethanol (10:4:1 v/v) in the study of Slikker et al. (1983). Use of a different vehicle may influence the bioavailability, which may have been higher in the Slikker et al. (1983) study. Furthermore, the type and rate of IV administration, which for one study was an IV infusion (Takikawa et al. 1996) while for the other three studies a bolus IV administration was utilized.
In the present study the apparent deviation shown in the data reported by Slikker et al. (1983) was corrected for before using the data to define the scaling factor. Without this correction scaling factors obtained for the Slikker et al. (1983) data set appeared to amount to a maximum 10 mg protein/g liver, representing values that are not substantially different from the ones reported in Table 4. This observation corroborates the results of the sensitivity analysis that revealed that the scaling factor was not an influential parameter for the PBK model-based predictions.
Until now, not much has been published about the scaling factor expressed in mg protein/g liver to convert in vitro hepatocyte protein levels to in vivo liver data in rats. A study by Sohlenius-Sternbeck (2006) reported a protein concentration in rat liver homogenate of 112 mg/g liver, but did not define how that value relates to primary hepatocytes, the model system of the present study. However, to further support the scaling factor now obtained the following theoretical approach could be applied to approximate the scaling factor by the use of a hepatocellularity number. There are a few studies that determined and reported this number ranging from 117 to 135 × 106 cells (hepatocytes)/g liver (Bayliss et al. 1999; Houston 1994, Sohlenius-Sternbeck 2006). Sohlenius-Sternbeck (2006) also reported on the protein concentration in a hepatocyte suspension (0.985 mg/106 cells). Together with the weight of the liver (Table 1) these numbers would result in a scaling factor ranging from of 115 to 132 mg protein/g liver. Our modelled and averaged scaling factor based on 24 fitted predictions using in vitro input data from rat hepatocytes on active uptake of E217βG and in vivo kinetic data is fully in line with this theoretical estimate.
The predictions made using the defined scaling factor revealed (1) that the value 129 mg protein/g liver is suitable to translate the in vitro Vmax from hepatocytes to an in vivo Vmax in liver obtaining predictions that are in line with the in vivo kinetic data for blood and cumulative biliary excretion levels of E217βG and also (2) that hepatocytes provide an adequate in vitro model to obtain kinetic parameters Vmax and Km for active uptake of an Oatp substrate, E217βG in our study. Given the uncertainty in scaling factors that would be required when using transfected cell models, use of hepatocytes to describe biliary excretion may be preferred over the use of in vitro systems with an overexpression of individual transporters. This is also because E217βG has affinity for more than one Oatp which will all be taken into account when using hepatocytes known to contain multiple Oatps (e.g. 1a1, 1a5, 361 1b2, 2b1) thus better mimicking the overall transport in the organ of interest (Hagenbuch and Meier 2003; Richert et al. 2006). Taking all together it is concluded that freshly isolated hepatocytes provide an adequate in vitro system to investigate uptake via active transport resulting in kinetic parameters that are able to include biliary excretion into PBK-models (Harris et al. 2004; Sahi et al. 2010). Use of this model also eliminates the need for the use of REFs and RAFs to scale expression and activity of transporters from in vitro systems to a full organ, diminishing uncertainties.
With this study we demonstrated a proof of principle by introducing a novel testing strategy for including biliary excretion into a generic PBK model at the same time defining the scaling factor for the vitro system (primary rat hepatocytes) used to obtain adequate in vitro to in vivo translation. We confirmed based on the predictions using the averaged scaling factor that primary rat hepatocytes can be the gold standard for studies on biliary transport in addition to their use as the golden standard for many in vitro (human) hepatic endpoints (e.g. hepatic metabolism, hepatoxicity, induction/inhibition of cytochrome P450s) (Guguen-Guillouzo and Guillouzo 2010; Zeilinger et al. 2016). However, known downfalls of the use of primary hepatocytes are the inter individual differences and their non-high through put character. To what extent such bottlenecks would affect their use as a model for biliary transport remains to be elucidated. This also holds for their use to mimic biliary transport of other substrates for Oatps and substrates for other hepatic transporters (Oat2 and Oct1) involved in biliary excretion. To this end similar studies with other substrates for Oatps such as statins and ACE inhibitors (Izumi et al. 2018; Watanabe et al. 2010) or for other hepatic transporters such as antineoplastic drugs, antivirals. antidiuretics and some alkaloids (Burckhardt 2012; Lozano et al. 2013; Marada et al. 2015) may be of use. Nevertheless, the proof of principle described in the present paper provides a first important step towards including biliary excretion in PBK-models for reverse dosimetry based QIVIVE and alternative testing strategies.
References
Bayliss MK, Bell JA, Jenner WN, Park GR, Wilson K (1999) Utility of hepatocytes to model species differences in the metabolism of loxtidine and to predict pharmacokinetic parameters in rat, dog and man. Xenobiotica 29(3):253–268. https://doi.org/10.1080/004982599238650
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Brock WJ, Vore M (1984) Characterization of uptake of steroid glucuronides into isolated male and female rat hepatocytes. J Pharmacol Exp Ther 229(1):175–181
Brouwer KL, Durham S, Vore M (1987) Multiple carriers for uptake of [3H]estradiol-17 beta(beta-D-glucuronide) in isolated rat hepatocytes. Mol Pharmacol 32(4):519–523
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13(4):407–484. https://doi.org/10.1177/074823379701300401
Burckhardt G (2012) Drug transport by Organic Anion Transporters (OATs). Pharmacol Ther 136(1):106–130. https://doi.org/10.1016/j.pharmthera.2012.07.010
Cantrill CH, Houston JB (2017) Understanding the interplay between uptake and efflux transporters within in vitro systems in defining hepatocellular drug concentrations. J Pharm Sci 106(9):11. https://doi.org/10.1016/j.xphs.2017.04.056
Cattori V, van Montfoort JE, Stieger B et al (2001) Localization of organic anion transporting polypeptide 4 (Oatp4) in rat liver and comparison of its substrate specificity with Oatp1, Oatp2 and Oatp3. Pflugers Arch 443(2):188–195. https://doi.org/10.1007/s004240100697
Chan JCY, Tan SPF, Upton Z, Chan ECY (2019) Bottom-up physiologically-based biokinetic modelling as an alternative to animal testing. Altex 36(4):597–612. https://doi.org/10.14573/altex.1812051
Chapy H, Klieber S, Brun P, Gerbal-Chaloin S, Boulenc X, Nicolas O (2015) PBPK modelling of irbesartan: incorporation of hepatic uptake. Biopharm Drug Dispos 36(8):491–506. https://doi.org/10.1002/bdd.1961
Choi GW, Lee YB, Cho HY (2019) Interpretation of non-clinical data for prediction of human pharmacokinetic parameters: in vitro-in vivo extrapolation and allometric scaling. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11040168
Chu X, Korzekwa K, Elsby R et al (2013) Intracellular drug concentrations and transporters: measurement, modelling, and implications for the liver. Clin Pharmacol Ther 94(1):126–141. https://doi.org/10.1038/clpt.2013.78
Council NR (2011) Animal Research in a Global Environment: Meeting the Challenges. In: Proceedings of the November 2008 International Workshop. The National Academies Collection: Reports funded by National Institutes of Health, Washington (DC)
Cronholm T, Eriksson H, Gustafsson JA (1971) Excretion of endogenous steroids and metabolites of (4–14C)pregnenolone in bile of female rats. Eur J Biochem 19(3):424–432. https://doi.org/10.1111/j.1432-1033.1971.tb01332.x
DeJongh J, Verhaar HJ, Hermens JL (1997) A quantitative property-property relationship (QPPR) approach to estimate in vitro tissue-blood partition coefficients of organic chemicals in rats and humans. Arch Toxicol 72(1):17–25
Eckhardt U, Schroeder A, Stieger B et al (1999) Polyspecific substrate uptake by the hepatic organic anion transporter Oatp1 in stably transfected CHO cells. Am J Physiol 276(4):G1037–G1042. https://doi.org/10.1152/ajpgi.1999.276.4.G1037
Evans MV, Andersen ME (2000) Sensitivity analysis of a physiological model for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): assessing the impact of specific model parameters on sequestration in liver and fat in the rat. Toxicol Sci 54(1):71–80. https://doi.org/10.1093/toxsci/54.1.71
Fabian E, Gomes C, Birk B et al (2019) In vitro-to-in vivo extrapolation (IVIVE) by PBTK modelling for animal-free risk assessment approaches of potential endocrine-disrupting compounds. Arch Toxicol 93(2):401–416. https://doi.org/10.1007/s00204-018-2372-z
Flecknell P (2002) Replacement, reduction and refinement. Altex 19(2):73–78
Ge S, Tu Y, Hu M (2016) Challenges and opportunities with predicting in vivo phase ii metabolism via glucuronidation from in vitro data. Curr Pharmacol Rep 2(6):326–338. https://doi.org/10.1007/s40495-016-0076-8
Gotoh Y, Kato Y, Stieger B, Meier PJ, Sugiyama Y (2002) Gender difference in the Oatp1-mediated tubular reabsorption of estradiol 17beta-D-glucuronide in rats. Am J Physiol Endocrinol Metab 282(6):E1245–E1254. https://doi.org/10.1152/ajpendo.00363.2001
Guguen-Guillouzo C, Guillouzo A (2010) General review on in vitro hepatocyte models and their applications. Methods Mol Biol 640:1–40. https://doi.org/10.1007/978-1-60761-688-7_1
Hagenbuch B, Meier PJ (2003) The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta 1609(1):1–18. https://doi.org/10.1016/s0005-2736(02)00633-8
Harris AJ, Shaddock JG, Delongchamp R, Dragan Y, Casciano DA (2004) Comparison of Basal gene expression in cultured primary rat hepatocytes and freshly isolated rat hepatocytes. Toxicol Mech Methods 14(5):257–270. https://doi.org/10.1080/15376520490434629
Hjelle JJ, Klaassen CD (1984) Glucuronidation and biliary excretion of acetaminophen in rats. J Pharmacol Exp Ther 228(2):407–413
Houston JB (1994) Relevance of in vitro kinetic parameters to in vivo metabolism of xenobiotics. Toxicol In Vitro 8(4):507–512. https://doi.org/10.1016/0887-2333(94)90002-7
Ishizuka H, Konno K, Naganuma H et al (1998) Transport of temocaprilat into rat hepatocytes: role of organic anion transporting polypeptide. J Pharmacol Exp Ther 287(1):37–42
Izumi S, Nozaki Y, Kusuhara H et al (2018) Relative activity factor (RAF)-based scaling of uptake clearance mediated by organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in human hepatocytes. Mol Pharm 15(6):2277–2288. https://doi.org/10.1021/acs.molpharmaceut.8b00138
Jamei M, Bajot F, Neuhoff S et al (2014) A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin Pharmacokinet 53(1):73–87. https://doi.org/10.1007/s40262-013-0097-y
Jones HM, Barton HA, Lai Y et al (2012) Mechanistic pharmacokinetic modelling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab Dispos 40(5):1007–1017. https://doi.org/10.1124/dmd.111.042994
Kanai N, Lu R, Bao Y, Wolkoff AW, Vore M, Schuster VL (1996) Estradiol 17 beta-D-glucuronide is a high-affinity substrate for oatp organic anion transporter. Am J Physiol 270(2 Pt 2):F326–F331. https://doi.org/10.1152/ajprenal.1996.270.2.F326
Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y (1999) Contribution of organic anion transporting polypeptide to uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther 288(2):627–634
Louisse J, de Jong E, van de Sandt JJ et al (2010) The use of in vitro toxicity data and physiologically based kinetic modelling to predict dose-response curves for in vivo developmental toxicity of glycol ethers in rat and man. Toxicol Sci 118(2):470–484. https://doi.org/10.1093/toxsci/kfq270
Louisse J, Bosgra S, Blaauboer BJ, Rietjens IM, Verwei M (2015) Prediction of in vivo developmental toxicity of all-trans-retinoic acid based on in vitro toxicity data and in silico physiologically based kinetic modelling. Arch Toxicol 89(7):1135–1148. https://doi.org/10.1007/s00204-014-1289-4
Louisse J, Beekmann K, Rietjens IM (2017) Use of physiologically based kinetic modelling-based reverse dosimetry to predict in vivo toxicity from in vitro data. Chem Res Toxicol 30(1):114–125. https://doi.org/10.1021/acs.chemrestox.6b00302
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Lozano E, Herraez E, Briz O et al (2013) Role of the plasma membrane transporter of organic cations OCT1 and its genetic variants in modern liver pharmacology. Biomed Res Int 2013:692071. https://doi.org/10.1155/2013/692071
Marada VV, Florl S, Kuhne A, Muller J, Burckhardt G, Hagos Y (2015) Interaction of human organic anion transporter 2 (OAT2) and sodium taurocholate cotransporting polypeptide (NTCP) with antineoplastic drugs. Pharmacol Res 91:78–87. https://doi.org/10.1016/j.phrs.2014.11.002
Morikawa A, Goto Y, Suzuki H, Hirohashi T, Sugiyama Y (2000) Biliary excretion of 17beta-estradiol 17beta-D-glucuronide is predominantly mediated by cMOAT/MRP2. Pharm Res 17(5):546–552
Moxon TE, Li H, Lee MY et al (2020) Application of physiologically based kinetic (PBK) modelling in the next generation risk assessment of dermally applied consumer products. Toxicol In Vitro 63:104746. https://doi.org/10.1016/j.tiv.2019.104746
Ning J, Chen L, Strikwold M, Louisse J, Wesseling S, Rietjens I (2019) Use of an in vitro-in silico testing strategy to predict inter-species and inter-ethnic human differences in liver toxicity of the pyrrolizidine alkaloids lasiocarpine and riddelliine. Arch Toxicol 93(3):801–818. https://doi.org/10.1007/s00204-019-02397-7
Poirier A, Cascais AC, Funk C, Lave T (2009) Prediction of pharmacokinetic profile of valsartan in human based on in vitro uptake transport data. J Pharmacokinet Pharmacodyn 36(6):585–611. https://doi.org/10.1007/s10928-009-9139-3
Probst RJ, Lim JM, Bird DN, Pole GL, Sato AK, Claybaugh JR (2006) Gender differences in the blood volume of conscious Sprague-Dawley rats. J Am Assoc Lab Anim Sci 45(2):49–52
Punt A, Aartse A, Bovee TFH et al (2019) Quantitative in vitro-to-in vivo extrapolation (QIVIVE) of estrogenic and anti-androgenic potencies of BPA and BADGE analogues. Arch Toxicol 93(7):1941–1953. https://doi.org/10.1007/s00204-019-02479-6
Richert L, Liguori MJ, Abadie C et al (2006) Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab Dispos 34(5):870–879. https://doi.org/10.1124/dmd.105.007708
Rietjens IM, Louisse J, Punt A (2011) Tutorial on physiologically based kinetic modelling in molecular nutrition and food research. Mol Nutr Food Res 55(6):941–956. https://doi.org/10.1002/mnfr.201000655
Sahi J, Grepper S, Smith C (2010) Hepatocytes as a tool in drug metabolism, transport and safety evaluations in drug discovery. Curr Drug Discov Technol 7(3):188–198. https://doi.org/10.2174/157016310793180576
Slikker W Jr, Vore M, Bailey JR, Meyers M, Montgomery C (1983) Hepatotoxic effects of estradiol-17 beta-D-glucuronide in the rat and monkey. J Pharmacol Exp Ther 225(1):138–143
Sohlenius-Sternbeck AK (2006) Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro 20(8):1582–1586. https://doi.org/10.1016/j.tiv.2006.06.003
Strikwold M, Spenkelink B, de Haan LHJ, Woutersen RA, Punt A, Rietjens I (2017) Integrating in vitro data and physiologically based kinetic (PBK) modelling to assess the in vivo potential developmental toxicity of a series of phenols. Arch Toxicol 91(5):2119–2133. https://doi.org/10.1007/s00204-016-1881-x
Takikawa H, Yamazaki R, Sano N, Yamanaka M (1996) Biliary excretion of estradiol-17 beta-glucuronide in the rat. Hepatology 23(3):607–613. https://doi.org/10.1053/jhep.1996.v23.pm0008617443
van de Steeg E, Greupink R, Schreurs M et al (2013) Drug-drug interactions between rosuvastatin and oral antidiabetic drugs occurring at the level of OATP1B1. Drug Metab Dispos 41(3):592–601. https://doi.org/10.1124/dmd.112.049023
Varma MV, Chang G, Lai Y et al (2012) Physicochemical property space of hepatobiliary transport and computational models for predicting rat biliary excretion. Drug Metab Dispos 40(8):1527–1537. https://doi.org/10.1124/dmd.112.044628
Watanabe T, Kusuhara H, Maeda K et al (2010) Investigation of the rate-determining process in the hepatic elimination of HMG-CoA reductase inhibitors in rats and humans. Drug Metab Dispos 38(2):215–222. https://doi.org/10.1124/dmd.109.030254
Yabe Y, Galetin A, Houston JB (2011) Kinetic characterization of rat hepatic uptake of 16 actively transported drugs. Drug Metab Dispos 39(10):1808–1814. https://doi.org/10.1124/dmd.111.040477
Yang X, Gandhi YA, Duignan DB, Morris ME (2009) Prediction of biliary excretion in rats and humans using molecular weight and quantitative structure-pharmacokinetic relationships. AAPS J 11(3):511–525. https://doi.org/10.1208/s12248-009-9124-1
Zeilinger K, Freyer N, Damm G, Seehofer D, Knospel F (2016) Cell sources for in vitro human liver cell culture models. Exp Biol Med (Maywood) 241(15):1684–1698. https://doi.org/10.1177/1535370216657448
Zhang M, van Ravenzwaay B, Fabian E, Rietjens I, Louisse J (2018) Towards a generic physiologically based kinetic model to predict in vivo uterotrophic responses in rats by reverse dosimetry of in vitro estrogenicity data. Arch Toxicol 92(3):1075–1088. https://doi.org/10.1007/s00204-017-2140-5
Acknowledgements
This work was supported by BASF SE.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noorlander, A., Fabian, E., van Ravenzwaay, B. et al. Novel testing strategy for prediction of rat biliary excretion of intravenously administered estradiol-17β glucuronide. Arch Toxicol 95, 91–102 (2021). https://doi.org/10.1007/s00204-020-02908-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02908-x