
Abstract
UDP-glucuronosyltransferases (UGTs) are involved in phase II conjugation reactions of xenobiotics and differences in their isoform activities result in interindividual kinetic differences of UGT probe substrates. Here, extensive literature searches were performed to identify probe substrates (14) for various UGT isoforms (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7 and UGT2B15) and frequencies of human polymorphisms. Chemical-specific pharmacokinetic data were collected in a database to quantify interindividual differences in markers of acute (Cmax) and chronic (area under the curve, clearance) exposure. Using this database, UGT-related uncertainty factors were derived and compared to the default factor (i.e. 3.16) allowing for interindividual differences in kinetics. Overall, results show that pharmacokinetic data are predominantly available for Caucasian populations and scarce for other populations of different geographical ancestry. Furthermore, the relationships between UGT polymorphisms and pharmacokinetic parameters are rarely addressed in the included studies. The data show that UGT-related uncertainty factors were mostly below the default toxicokinetic uncertainty factor of 3.16, with the exception of five probe substrates (1-OH-midazolam, ezetimibe, raltegravir, SN38 and trifluoperazine), with three of these substrates being metabolised by the polymorphic isoform 1A1. Data gaps and future work to integrate UGT-related variability distributions with in vitro data to develop quantitative in vitro–in vivo extrapolations in chemical risk assessment are discussed.
Article Highlights
-
Extensive literature search of human kinetic parameters for UGT probe substrates.
-
Bayesian meta-analysis quantifying human variability in acute and chronic kinetic parameters.
-
UGT isoform-related uncertainty factors were below the 3.16 kinetic default uncertainty factor for most probe substrates.
-
Quantifying human variability in UGT polymorphisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucuronidation is an enzymatic reaction catalysed by UDP-glucuronosyltransferase (UGT) isoforms which involves the conjugation of endogenous substrates (e.g. bilirubin) and xenobiotics [e.g. pharmaceuticals (morphine), dietary chemicals (flavonoids), and environmental contaminants (mycotoxins)] with glucuronic acid (Dong et al. 2012; Lv et al. 2019). In humans, glucuronide conjugates are water soluble and readily excreted in the urine or the faeces resulting in increased elimination and most often inactivation of the compound, thereby contributing to xenobiotic detoxification (Fisher et al. 2001). Multiple UGT isoforms are often involved in xenobiotic metabolism, which, from a toxicological viewpoint, is advantageous as dysfunctionality of an isoform does not necessarily result in the impaired elimination and thus detoxification of chemicals (Lv et al. 2019). Since UGTs are ubiquitous in pharmacokinetic and toxicokinetic processes [absorption, distribution, metabolism and excretion (ADME)], their involvement in human metabolic variability is important.
The superfamily of UGT isoforms has a nomenclature which is based on similar features to that described for the cytochrome P450 (CYP) superfamily (Meech et al. 2019; Rowland et al. 2013). The subfamilies of UGT1A and UGT2B are predominantly expressed in the liver as well as in the intestine and kidney, where they mediate intestinal and hepatic first-pass glucuronidation of many phenolic compounds, including pharmaceuticals and natural phenols (Dong et al. 2012, Fig. 1). The most clinically relevant hepatic UGTs include UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7 and 2B15 (Rowland et al. 2013; Stingl et al. 2014). Other UGTs from the 2B subfamily are mainly responsible for the metabolism of endogenous compounds rather than xenobiotics, such as steroids (2B4, 2B15 and 2B17) and bile acids (2B4) (Fisher et al. 2001).

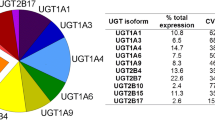
Average distribution of the major UDP-glucuronosyltransferase isoforms in human liver (a), intestine (b) and kidney (c) (Lv et al. 2019)
UGT isoforms are known to be highly polymorphic with more than a hundred variants described (Stingl et al. 2014). In most cases, these polymorphic variants result in lower expression levels and/or lower activity, and in some instances even complete loss of activity (Sim et al. 2013). Because of such changes in expression and/or activity, polymorphic UGT variants may cause higher plasma concentrations of (toxic) metabolites or parent compounds, resulting in chemical-induced toxicity. For example, UGT1A1 polymorphism is associated with irinotecan toxicity, while UGT2B7 polymorphism can affect plasma concentrations of valproic acid (Tsunedomi et al. 2017; Wang et al. 2018b). For other isoforms, comparable impact of UGT polymorphisms on internal drug concentrations has been observed (Stingl et al. 2014).
For the last 70 years, a 100-fold default uncertainty factor (UF) has been applied to derive chronic safe levels of exposure for non-cancer effects of chemicals. This default factor allows for interspecies differences (tenfold) and human variability (tenfold) to chemical exposure. In the 1990s, the tenfold factor allowing for human variability has been refined to a composite value of two factors of 3.16 (100.5), accounting, respectively, for interindividual differences in toxicokinetics and toxicodynamics (Renwick and Lazarus 1998). However, interindividual differences between healthy adults and potentially sensitive subgroups including neonates, elderly and poor metabolisers expressing polymorphic UGT genes may not be covered by the default kinetic factor (Dorne et al. 2001b; Renwick and Lazarus 1998). Under such circumstances, pathway-specific UFs or chemical-specific adjustment factors (CSAFs) have been proposed and can provide an option to replace such default UFs. Pathway-related UFs to account for variability have been described for CYP3A4 as well as efflux and influx transporters (Darney et al. 2019, 2020; Dorne et al. 2001b). Human variability in glucuronidation processes in relation to UFs has been described earlier by Dorne et al. (2001a); however, at the time, information on isoform specificity and genetic polymorphisms was very limited.
The manuscript aims to investigate human variability in UGT activity through (1) identifying isoform-specific UGT probe substrates and collecting pharmacokinetic data for intravenous and oral markers of acute (maximum concentration (Cmax)) and chronic exposure (clearance, area under the curve (AUC)) by means of extensive literature searches and meta-analyses, (2) quantifying interindividual differences in pharmacokinetics by means of hierarchical Bayesian meta-analyses to derive UGT-related variability distributions and UGT-related UFs. Such UGT-related UFs are relevant to refine toxicokinetic UFs for risk assessment of toxicants, nutrients and environmental xenobiotics that are metabolised by UGTs, and (3) unravelling the frequencies and pharmacokinetic consequences of UGT polymorphisms in human populations. A graphical abstract is depicted in Fig. 2.

Human variability in the pharmacokinetics of isoform-specific UGT probe substrates, genetic polymorphisms and UGT-related uncertainty factors
Materials and methods
Extensive literature searches (ELS) and data collection
UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7 and 2B15 were identified as the most clinically relevant UGT isoforms for xenobiotic metabolism (Rowland et al. 2013; Stingl et al. 2014). Probe substrates for these UGT isoforms were identified from the in vitro and in vivo literature as compounds metabolised by extensive glucuronidation (> 60% of the dose excreted in the urine) (Lv et al. 2019; Rowland et al. 2013; Stingl et al. 2014; Yang et al. 2017).
ELS were performed using two main databases (i.e. Scopus and PubMed) to identify human pharmacokinetic (PK) studies in non-phenotyped adults for isoform-specific UGT probe substrates in adults of different geographical ancestry or ethnic background. A PK database was then computed, including intravenous and oral markers of acute (Cmax) and chronic (clearance and AUC) exposure. Search queries for the probe substrate deferiprone are illustrated in Table 1 and queries for all other substrates are provided in Supplementary Material 1. Data reporting frequencies of UGT polymorphisms distribution and the associated PK parameters in phenotyped individuals were collected using a horizontal literature search in Google Scholar.
A two-step process was conducted to screen the retrieved studies from literature as described previously (Darney et al. 2019). This process was used to assess whether reported PK values were suitable for inclusion in the database. After removing duplicates, the following exclusion criteria were applied: 1. species other than humans, 2. in vitro studies, 3. development of analytical methods, 4. modelling studies, 5. pharmacodynamics investigations only, 6. substrates other than those identified as relevant and/or mixtures of substrates. Articles meeting the exclusion criteria were not considered for further evaluation. Furthermore, articles that were written in any other language than English or did not contain original research data (e.g. reviews) were excluded from analysis. Overall, data on non-phenotyped healthy individuals were collected and included in the meta-analysis (see “Data standardisation and meta-analyses”). The specific selection of subgroups is described in Supplementary Material 1. In a second step, the full text of the included papers was checked for PK parameter values after single-dose exposure. Repeated dosing studies and studies for which multiple formulas were administered to the same group of volunteers were excluded. However, for ethinylestradiol, data were included for both single dose and repeated dosing for 21 days, the standard regimen for anticonception drugs.
Data standardisation and meta-analyses
Meta-analyses of PK parameter values were performed in non-phenotyped subjects for each probe substrate to derive UGT-related variability distributions and UGT-related UFs. For this purpose, each PK parameter was normalised in a harmonised manner (Cmax expressed in ng/mL; AUC in ng*h/mL and clearance in L/h/kg bw) while applying body weight correction to the applied doses (mg/kg bw). If available, the reported (mean) body weight was used, or continent specific body weights were used to normalise the dose if body weight data were not available (Walpole et al. 2012). For SN38, the dose was normalised to body surface area instead of body weight, as this is the standard measure for this compound. If body surface area data were not available, a default value of 1.79 m2 was used (Sacco et al. 2010). Data from these studies were extracted mostly as arithmetic mean (AM) and standard deviation (SD), but sometimes geometric means (GM) and geometric standard deviations (GSD) were reported. Generally, PK data are recognised to follow a lognormal distribution (Dorne et al. 2001b; Naumann et al. 1997; Renwick and Lazarus 1998). Since GM and GSD are more appropriate to summarise a lognormal distribution, all pharmacokinetic data were described as GM and GSD using the following equations:
where X is the arithmetic mean and CVN is the coefficient of variation for normally distributed data:
In some studies, SD was not reported and was estimated from the standard error (SE, SEM) or CV using equations described previously (Darney et al. 2019).
The objective of the meta-analyses is to provide accurate information regarding interindividual differences in non-phenotyped adults of the PK parameters for a substrate expressed as distributions. Variability related to interstudy, intersubstrate and interindividual differences was analysed for each substrate and parameter and for each UGT isoform, through a decomposition of the PK parameter variance (clearance, AUC or Cmax) using a previously described hierarchical Bayesian model (Darney et al. 2019; Wiecek et al. 2019). For the meta-analysis, non-informative prior data were selected for most compounds, except for zidovudine and oxazepam for which kinetic variability was previously meta-analysed (Dorne et al. 2001a).
Overall, the meta-analyses provided variability and uncertainty distributions describing interindividual differences for each PK parameter using median values and 95% confidence intervals. The coefficient of variation (CV) was also estimated as follows:
where \({\uptau }_{j}\) is the interindividual difference of the activity for a substrate ‘j’.
UGT isoform-related UFs were calculated as the ratio between the percentile of choice and the median of the distribution for each PK parameter for 95th and 97.5th centiles.
Software
All statistical analyses were performed in R (version 3.5) and the Bayesian modelling was implemented in Jags (4.2.0) (Plummer 2003). Data processing and graphical display were performed in R (dplyr and ggplot2 packages) (R Development Core Team 2018; Wickham 2016; Wickham et al. 2020). References of the studies used to compile the database were stored and sorted in EndNote X8.
Results and discussion
Extensive literature searches and data collection
UGT isoforms can conjugate a wide variety of substrates and show a broad overlapping substrate specificity. This is advantageous when detoxifying chemicals; however, because of such overlap, identifying selective probe substrates for each isoform remains a challenge. Moreover, UGTs are also present in the gastrointestinal tract and pre-systemic conjugation occurs readily for a range of compounds. Here, to quantify isoform-specific variability in UGTs, selective probe substrates with available PK data for each isoform were selected. Moreover, differences in variability between oral and intravenous data were compared to investigate the contribution of bioavailability and pre-systemic conjugation after oral intake. A total of 14 isoform-specific UGT probe substrates covering both the UGT1A and UGT2B subfamilies were identified, namely 1-OH-midazolam (UGT1A4), codeine (UGT2B7), deferiprone (UGT1A6), entacapone (UGT1A9), ethinylestradiol (UGT1A1), ezetimibe (UGT1A1/UGT1A3), mycophenolic Acid (UGT1A9), oxazepam (UGT2B15/UGT1A9), propofol (UGT1A9), raltegravir (UGT1A1), SN38 (UGT1A1), telmisartan (UGT1A3), trifluoperazine (UGT1A4) and zidovudine (UGT2B7).
From the ELS, a total of 7173 papers were assessed from Scopus and PubMed (up to August 2019) and an extra 11 papers were retrieved from Google Scholar, for the 13 UGT isoform probe substrates and for zidovudine, 10 studies were included from a previous database (shared by Dr. N. Quinot, collated for EFSA/SCER/2014/06 project). PRISMA flow diagrams for the individual compounds are provided in Supplementary Material 2. Figure 3 provides a summary PRISMA diagram for all papers collected in the ELS (Moher et al. 2009). Overall, a total of 210 peer-reviewed publications were selected from the ELS and included in the database. Supplementary Material 1 provides the search queries for both Scopus and PubMed for the individual compounds. Table 2 illustrates the selected probe substrates, the structure of the compounds, bioavailability, percentage of glucuronidation and their site of glucuronidation.

PRISMA diagram illustrating the extensive literature searches performed for the 13 isoform-specific UGT probe substrates (UGT1A and UGT2B subfamilies) and human pharmacokinetic studies
Interindividual differences in the kinetics of isoform-specific UGT probe substrates and related uncertainty factors in non-phenotyped adults
Results of the meta-analyses are expressed as geometric means (normalised to dose and body weight) for the 14 isoform-specific UGT probe substrates and are illustrated for markers of acute (Cmax) and chronic (AUC/clearance) exposure after oral and intravenous administration in Fig. 4. The full dataset of extracted information used can be accessed on EFSA knowledge junction (Kasteel et al. 2020). Data availability was variable for each UGT probe substrate and interstudy differences are reported for each compound. For SN38, only patient data were available and no data on healthy adult individuals were reported in the literature. For deferiprone, no clear distinction could be made between healthy adult data and patient data for all three parameters, suggesting that the condition of the individuals did not influence the pharmacokinetics of this compound. In Fig. 5, isoform-specific interindividual differences in AUC are illustrated for world populations from different geographical ancestry or country of origin (one probe substrate per UGT isoform). These plots indicate that no clear differences in chronic exposure (AUC) can be demonstrated across world populations from different geographical ancestry and, therefore, there is indication of significant interethnic differences for these probe substrates. The same conclusion holds for other PK parameters and other probe substrates, which are illustrated in Supplementary Material 3.

Results of the meta-analyses reporting interindividual differences in non-phenotyped healthy adults for 14 isoform-specific UGT probe substrates. Data are expressed as log geometric means (GM) for markers of acute (Cmax) and chronic (AUC, clearance) exposure. AUC (normalised to dose, a), clearance (normalised to body weight, b), and Cmax (normalised to dose, c). Open squares: oral exposure; solid circles: IV exposure. Red data points: healthy volunteers; blue data points: patients. 21d: repeated dose for 21 days

Inter-individual differences in markers of chronic exposure (area under the concentration–time curve (AUC)) for isoform-specific UGT probe substrates across world populations of different geographical ancestry. For each UGT isoform included in the study, one probe substrate is shown. Graphs for other probe substrates and other PK parameters are accessible in Supplementary Material 3
Interindividual differences were higher compared to an earlier study (Dorne et al. 2001a), which included 11 probe substrates compared to 14 here; with an overlap of only two probe substrates (zidovudine (AZT) and oxazepam). In addition, Dorne et al. (2001a) mostly considered UGT2B7 probe substrates while UGT1A1-specific probe substrates were not included since they were not available at that time. Polymorphisms have the highest impact on the PK of UGT1A1 probe substrates, which may explain the larger interindividual differences in this study. Finally, the 2001 study investigated PK data for potentially sensitive subgroups including neonates, infants, children, and the elderly, but little-to-no data for these subgroups were available for the included probe substrates here (Dorne et al. 2001a). It is worth noting that UGT metabolism in neonates is impaired and that they show a low glucuronidation activity (Allegaert et al. 2009). Data for such PK differences in markers of acute and chronic exposure are still very limited for these subgroups, but can reach two- to threefold in comparison with healthy adults so that the default kinetic factor may be inadequate and an extra UF may be required to cover these subgroups (Dorne et al. 2001b, 2005).
UGT1A1
For UGT1A1, ethinylestradiol, ezetimibe, raltegravir and SN38 were identified as probe substrates. Besides pharmaceuticals, UGT1A1 is involved in the glucuronidation of several compounds important in (food) toxicology, including the naturally occurring food components resveratrol and several hydroxyflavones, the heterocyclic amine 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and the phytochemical ferulic acid (Brill et al. 2006; Li et al. 2011; Malfatti and Felton 2004; Tang et al. 2010). For ethinylestradiol, only single-dose studies were used to quantify the UGT1A1 variability. PK data were available for Europeans, East Asians, South Asians, Southeast Asians, North Americans, South Americans, North Africans and Middle Eastern adults with the majority of the datasets from North American and European studies. Data gaps were identified for specific groups like Central Americans and Africans. Chemical-specific CVs ranged from 35 to 72%, while isoform-related CVs ranged from 46 to 51% (Table 3). Overall, the UGT1A1 related UFs were most often below or close to the default TK UF of 3.16 for at least 97.5% of the healthy adults when considering the median value. However, our analysis by the Bayesian model takes into account the uncertainty around the estimation of the UF and this shows that, given the available data (number of studies and number of individuals per study), the default factor may not cover all possible cases. Indeed, the upper bound of the confidence interval is higher than 3.16. The chemical-specific data show that SN38, ezetimibe and raltegravir all have an UF97.5 higher than 3.16, ranging from 3.2 to 3.6 (Table 3). Ethinylestradiol is the only UGT1A1 probe substrate studied here that did not exceed the default kinetic UF for any of the parameters.
UGT1A3
UGT1A3 is a UGT isoform involved in the glucuronidation of the flavonoid icaritin and several other flavonoids (Chen et al. 2008; Wang et al. 2018a). In this study, two probe substrates were included for UGT1A3, namely telmisartan and ezetimibe. Isoform-related CVs varied from 37–62%. Highest variability was observed for telmisartan and ezetimibe clearance with CV values of 59 and 66%, respectively. It has been demonstrated previously in the literature that telmisartan shows high variability in PK parameters (Chen et al. 2013; Deppe et al. 2010; Kang et al. 2018; Stangier et al. 2000b). Overall, UGT1A3-related UFs were below the default TK UF of 3.16 (Table 4).
UGT1A4
The ginsenoside 20(S)-protopanaxadiol is one of the naturally occurring probe substrates of the UGT1A4 isoform (Li et al. 2016). UGT1A4 probe substrates selected here included trifluoperazine and 1-OH-midazolam. It is important to note that 1-OH-midazolam is a metabolite of midazolam which is formed by CYP3A4 and then conjugated by UGT1A4. Variability for trifluoperazine was quite extensive, although only a limited number of publications were available, and studies were all from Canada. Large interindividual differences in PK parameters has previously been demonstrated for trifluoperazine, independent of ethnicity (Midha et al. 1988). After oral administration, 1A4 shows the highest variability regarding acute exposure (Cmax) out of all isoforms with a CV of 62%. However, least variability was found for UGT1A4 in mRNA expression levels when compared with mRNA expression levels of UGT1A1, UGT1A3, UGT1A6 and UGT1A9 (Aueviriyavit et al. 2007). Despite this low variability in mRNA expression levels, an exceedance of the default TK UF is observed for the 97.5th percentile for 1-OH-midazolam (UF97.5: 3.3, Table 5).
UGT1A6
Of the seven UGT isoforms investigated in this study, UGT1A6 has been recognised as one of the minor isoforms for glucuronidation and drug metabolism (Stingl et al. 2014). However, it is involved in the glucuronidation of several pharmaceuticals, including acetaminophen and aspirin, and the remarkable sensitivity of cats to these analgesics is due to the lack of UGT1A6 expression in the feline liver (Shrestha et al. 2011). The natural occurring compound protocatechuic aldehyde is also metabolised by this UGT isoform (Liu et al. 2008). In this study, deferiprone was included as probe substrate for UGT1A6. Only data after oral administration were available and for all PK parameters, the CVs ranged from 36 to 48% Table 6) with UGT1A6-related UFs all below the default TK UF.
UGT1A9
For the UGT1A9 isoform, several relevant substrates include resveratrol, several flavonols and the natural flavouring agent estragole (Brill et al. 2006; Iyer et al. 2003; Wu et al. 2011). Probe substrates for this isoform included entacapone, mycophenolic acid, oxazepam, and propofol. Overall, isoform-related CVs varied between 23 and 41%. For oxazepam, variability in PK parameters was described previously (Dorne et al. 2001a). Compared to our results, variability in Cmax and AUC was comparable, while the calculated variability was lower for clearance in our study (33% against 51%). UGT1A9-related UFs did not exceed the UF of 3.16 (Table 7).
UGT2B7
UGT2B7 is a UGT isoform which conjugates natural compounds such as emodin, a Chinese traditional medicine, the natural sweetener stevioside and natural compounds from herbs such as andrographolide and estragole (Iyer et al. 2003; Tian et al. 2015; Wang et al. 2014; Wu et al. 2018). Selective pharmaceutical probe substrates included in this study were codeine and zidovudine and isoform-related CVs varied between 26 and 37% (Table 8). The PK database mainly consisted of Caucasians (North America and Europe) for both compounds. For codeine, five studies from the USA and four studies from Europe were available, and the remaining studies were from Asia or Australia. For zidovudine, six studies were available from North America, and three from South America and Europe. The variability as indicated by the CV was 26% for clearance, 28% for AUC, and 43% for the Cmax for zidovudine. While the calculated variability for clearance and Cmax is comparable to Dorne et al. (2001a), the AUC showed less variability (28%, 12 studies against 56%, 2 studies). UGT2B7-related UFs did not exceed the TK default UF.
UGT2B15
UGT2B15 is mostly responsible for the metabolism of endogenous compounds such as steroids (e.g. dihydrotestosterone and 17β-diol) (Chen et al. 1993). Environmental contaminants that are metabolised by UGT2B15 include bisphenol A (Hanioka et al. 2008). Major xenobiotic substrates for UGT2B15 include the pharmaceuticals lorazepam and S-oxazepam, although lorazepam is not recommended as a probe substrate for the isoform because of the involvement of several other UGT isoforms in its glucuronidation (Lv et al. 2019; Rowland et al. 2013). Oxazepam is a benzodiazepine which is administered as a racemic mixture, with R-oxazepam being glucuronidated by UGT1A9 and S-oxazepam being glucuronidated by UGT2B15. Variability in the ratio between the R-glucuronide and the S-glucuronide has been characterized particularly for the formation of the S-glucuronide (Patel et al. 1995). Table 7 shows that variability in oxazepam is 33% and 44% for markers of chronic exposure and 26% for markers of acute exposure and all calculated UGT2B15-related UFs are below the default TK UF. As oxazepam is the only substrate included for UGT2B15, calculated CVs and UFs for oxazepam are considered the overall isoform-specific CVs and UFs for UGT2B15.
Frequencies of UGT isoform polymorphisms in world populations and impact on the pharmacokinetics of probe substrates in non-phenotyped subjects
Frequencies of single nucleotide polymorphisms (SNPs) of UGT isoforms, namely UGT1A1*28, UGT1A3, UGT1A4*2 (C70A), UGT1A4*3 (T142G), UGT1A6*2, UGT1A9*22, UGT2B7 C802T and UGT2B15*2 are presented in Fig. 6 for world populations of different geographical ancestry. Data investigating the impact of UGT polymorphisms on in vivo PK parameters are limited and summarised in Table 9 for the probe substrates included in this study. Overall, the limited data show that such an impact still needs to be fully characterised for endogenous substrates and xenobiotics in world populations.

Frequencies of single nucleotide polymorphisms (SNPs) genotypes for UGT1A1*28 (a), UGT1A3 (b), UGT1A4*2 (C70A) (c) UGT1A4*3 (T142G) (d), UGT1A6*2 (e), UGT1A9*22 (f), UGT2B7 (C802T) (g), and UGT2B15*2 (h) in human populations of different geographical ancestries. C(E) central, N north, S south, E east, SE southeast
Understanding the functional role of UGT SNPs is a key aspect to quantify the relationship between their frequency distributions (Fig. 6) and the pharmacokinetic consequences on UGT conjugation across world populations. Table 9 provides an account of such pharmacokinetic consequences; however, available studies from the literature are still limited. The consequences can be two-sided: an increased UGT activity would result in detoxification and a decreased UGT activity would result in an increase in the concentration of the toxic form (parent compound). Well-known exceptions to this rule include carboxylic acid-containing drugs that are metabolised by UGTs and form acyl glucuronides, like mycophenolic acid and telmisartan. These acyl glucuronides can cause idiosyncratic drug toxicity by binding covalently to proteins (Iwamura et al. 2017). For mycophenolic acid, indeed protein adducts have been found in vivo and these can result in several adverse effects (Shipkova et al. 2002).
UGT1A1
UGT1A1 in humans is one of the most important UGT isoforms in terms of glucuronidation and is known to have multiple clinically relevant polymorphisms that can contribute to variability in PK parameters (Mehboob et al. 2017; Miners et al. 2002). Polymorphisms in UGT1A1 are extensively studied and alteration in its activity can result in Gilbert’s syndrome, one of the most common syndromes in humans (Burchell and Hume 1999; Stingl et al. 2014). Gilbert’s syndrome results in hyperbilirubinaemia, as UGT1A1 is responsible for the metabolism of bilirubin. The frequency and type of polymorphisms differ between individuals from different geographical ancestry or ethnic backgrounds (Weber 1999) and this is also apparent from the frequencies of mutations in UGT1A1 that are responsible for Gilbert’s syndrome. A dinucleotide polymorphism in the TATA box promoter (UGT1A1*28) resulting in reduced UGT1A1 expression and Gilbert’s syndrome is detected in only 3% of Asians and ~ 15% in Europeans, while it can be up to 36% in Africans (Beutler et al. 1998). When looking at the frequency distribution of this SNP, clear interindividual differences are indeed detected across populations of different geographical ancestry (Fig. 6a, for references see Supplementary Material 4). As expected, Asian populations (especially East and Southeast Asians), as well as Oceanians, more frequently express the wild-type genotype, whereas other ethnicities predominantly express the heterozygous genotype. In Europe and Middle East, mixed frequencies in wild-type and heterozygous genotypes are observed. Another SNP in this isoform, UGT1A1*6, results in an amino acid substitution at position 71 (G71R). This mutation also causes Gilbert’s syndrome and is more frequently detected in Asians (Burchell and Hume 1999).
As mentioned earlier, data gaps for pharmacokinetics of UGT1A1 probe substrates have been identified especially for Africans and Central Americans. Distribution of genotypes for UGT1A1*28 differs for these populations compared to Europeans and this highlights that PK data in phenotyped individuals from different geographical ancestries are needed to characterise isoform-specific UGT variability and related UFs. Besides, the high variability observed for SN38 may also be rationalised by the fact that only patient data were available and included in the meta-analysis, which may bias the analysis. Moreover, internal concentrations of SN38 can be influenced by the UGT1A1*28 mutation and some PK studies included only patients with the wild-type genotype, which also results in a bias in the calculation of the variability and the UF (Ri et al. 2018).
It is striking that variability in PK for ethinylestradiol is low (35–42%; Table 3) when compared to variability in PK for raltegravir and SN38 (up to 72%, Table 3). One possible explanation is the inclusion of only females as it is used as a contraceptive, and this may reduce variability. Indeed, genetic sex differences are known to have an important effect on interindividual differences in UGT enzymes as well and this aspect is further elaborated further down in the discussion. Another possible rationale may be that the identified SNPs have a larger impact on raltegravir and SN38 metabolism than on ethinylestradiol metabolism, which could be due to differences in docking resulting in different affinities and kinetics. Unfortunately, no studies that investigated the effect of UGT1A1 polymorphisms directly on ethinylestradiol PK in vivo were available. However, both ethinylestradiol and SN38 show significantly lower in vitro rates of metabolism with UGT1A1*28 polymorphic human liver microsomes (Zhang et al. 2007).
For the UGT1A1*28 polymorphism, significantly higher values for the AUCs were reported for SN38 which corresponds with a decrease in glucuronidation capacity (Table 9). For the UGT1A1*6 polymorphism, impact on PK parameters is less clear with only one study showing a significant increase in AUC for SN38. No in vivo PK data were available for the other UGT1A1 probe substrates and the effect of either UGT1A1 polymorphism on their PK parameters.
UGT1A3
For UGT1A3 polymorphisms, the frequency distribution is rather uniform across populations of different geographical ancestry. However, data were only available for three populations (North America, East-Asia and Europe) and the heterozygous genotype was the most represented one in all three populations (Fig. 6b). A contrasting exception was the observation of slightly higher frequencies for the wild type in East Asians compared to the other two populations.
UGT1A3 polymorphisms are associated with an increase in glucuronidation rates for a range of compounds. UGT1A3*2 (nucleotide changes T31C, G81A and T140C) polymorphism is correlated with an increase in glucuronidation of atorvastatin (Cho et al. 2012). Moreover, polymorphisms in UGT1A3 have been associated with polymorphisms in UGT1A1, which is due to a linkage disequilibrium within the UGT1A locus (Cho et al. 2012; Riedmaier et al. 2010; Saeki et al. 2006).
A study on telmisartan PK reported a significant influence of the *2a and *4a variants of the UGT1A3 polymorphisms, associated with a decrease and an increase in AUC, respectively (Ieiri et al. 2011, Table 9). Furthermore, a number of studies showed an impact of UGT1A1 and UGT2B7 polymorphisms on PK parameters of telmisartan, indicating that multiple UGT isoforms may be responsible for its glucuronidation and that multiple polymorphisms can, therefore, influence its PK parameters (Ieiri et al. 2011; Miura et al. 2009; Yamada et al. 2011).
UGT1A4
For UGT1A4, the *2 and *3 mutations are the two most common SNPs. UGT1A4*2 is a mutation at codon 24, resulting in an amino acid change from proline to threonine (P24T) because of a C70A SNP. UGT1A4*3 is a T142G SNP, resulting in an amino acid change at codon 48, from a leucine to a valine (L48V). In the frequency distribution data, no differences in C70A and T142G genotypes between populations from different geographical ancestries were observed (Fig. 6c/d). Compared to the mutation and the heterozygous genotype, the wild-type genotype is predominantly detected (C70A: > 80%, T142G: > 55%).
Studies on these SNPs show contradictory results. Neither polymorphism is significantly associated with trifluoperazine glucuronidation activity in vitro (Benoit-Biancamano et al. 2009b). However, decreased activities have been reported for benzidine, β-naphthylamine, steroids and tigogenin, but increased glucuronidation has been reported for clozapine and olanzapine with UGT1A4*3 (Ehmer et al. 2004; Ghotbi et al. 2010; Mori et al. 2005). This suggests that the impact of UGT1A4 mutations on PK parameters is substrate dependent, but the associated mechanism remains to be elucidated. The UGT1A4*3 has also been associated with decreased serum levels of lamotrigine which correspond to an increase in glucuronidation rates (Gulcebi et al. 2011; Reimers et al. 2016). For UGT1A4, no studies were encountered that studied effects of polymorphisms in this UGT isoform on in vivo PK parameters of the probe substrates.
UGT1A6
For UGT1A6, the most prominent mutation is UGT1A6*2, which is the result of two substitutions: T181A and R184S (Ciotti et al. 1997). The linkage disequilibrium between these polymorphisms is very high, as they are only 11 nucleotides apart (nucleotides 541 and 552) (McGreavey et al. 2005). A linkage disequilibrium between UGT1A6*2 and UGT1A1*28 is also observed (Lampe et al. 1999). No differences are seen across world populations in the frequency distribution of this polymorphism (Fig. 6e).
No impact on deferiprone PK was found in vivo for UGT1A6*2 (Limenta et al. 2008). However, an in vitro study showed that the UGT1A6*2 variant could lead to either a decrease or an increase in glucuronidation capacity for various phenolic compounds (Ciotti et al. 1997; Nagar et al. 2004). Lampe et al. (1999) showed that genetic sex had more influence on the PK parameters of deferiprone whereas polymorphism had no impact. This may be due to both the variation in UGT1A6 content and activity between males and females. Indeed, glucuronidation capacity has been shown to be higher in males with a 50% higher UGT1A6 protein content in males compared to that in females (Bock et al. 1994; Court et al. 2001).
UGT1A9
For UGT1A9, SNPs have been associated with a range of impacts on the PK of xenobiotics. T98C (UGT1A9*3) may result in a decrease in glucuronidation activity, although the reported results are contradictory (Girard et al. 2004; Jiao et al. 2008; Villeneuve et al. 2003). The T-275A SNP, which is located in the promotor region, is associated with an increase in glucuronidation rates, while in another study, the glucuronidation rate of mycophenolic acid remained unaffected (Girard et al. 2004; Jiao et al. 2008; Kuypers et al. 2005; Mazidi et al. 2013). Multiple linkage disequilibria are known for polymorphisms in UGT1A9 since SNPs in UGT1A9 are linked to SNPs in UGT1A7 and UGT1A6 (Saeki et al. 2006). The frequency distributions of these genotypes across several populations are described in Supplementary Material 5.
The SNP with the most apparent differences in frequencies between populations is a ‘T’ deleted at position-118 in the promotor region of the gene, UGT1A9*22 (Cecchin et al. 2009). Japanese individuals show a different distribution compared to that in other populations including other Asian populations (Fig. 6f). In other world populations, the heterozygous genotype is the most occurring, while in Japan most prominent frequencies are a mix between the heterozygous genotype and the homozygous mutation. It is shown that combinations of haplotypes differ between Caucasians and Asians and this might explain the large differences in frequencies observed here (Saeki et al. 2006).
The effect of UGT1A9*22 on PK parameters remains unclear since an increased transcriptional activity has been reported, but it was not associated with an impact on mycophenolic acid PK parameters (Jiao et al. 2008; Yamanaka et al. 2004; Zhang et al. 2008). A significant decrease is demonstrated in AUC for SN38 with this mutation, although SN38 is mainly metabolised by UGT1A1 (Han et al. 2006).
UGT2B7
For UGT2B7, the frequencies of the C802T mutation are quite comparable for the three world regions (Europe, North America, South America, Fig. 6g) represented in the PK database and indeed, not much variability is observed in the PK parameters of zidovudine. The SNP C802T in UGT2B7 results in an amino acid substitution at residue 268, from histidine to tyrosine (H268Y, UGT2B7*2) at the N-terminal substrate binding site of the enzyme (Yuan et al. 2015). It is demonstrated that this variant form has the same localisation as the wild type. Moreover, it is demonstrated that UGT2B7*2 can form both homodimers and heterodimers with wild-type and other polymorphic enzymes, albeit with a decrease in affinity (Yuan et al. 2015). Coffman et al. (1998) showed that the 268Y form of the UGT was ten times more efficient in the glucuronidation of buprenorphine than the 268H form. However, no differences were detected for some other opioids, like morphine and codeine. In another study of Coffman et al. (2003), it was demonstrated that opioids bind to amino acids 84–118 of the UGT, which implies that mutations at other places are less likely to influence the binding of opioids to UGT. However, also polymorphisms outside the substrate-binding pocket can still influence the dynamics of substrate binding by, for example, altering the packing of the enzyme and thereby influencing the active site (Rutherford et al. 2008).
It is demonstrated that UGT2B7*2 in a hetero-dimer with the wild-type enzyme has an impaired glucuronidation activity for zidovudine (Yuan et al. 2015). For other chemicals including valproic acid, tamoxifen, and lamotrigine, UGT2B7 polymorphism has been shown to affect plasma concentrations (Blevins-Primeau et al. 2009; Du et al. 2016; Petrenaite et al. 2018; Sun et al. 2015; Wang et al. 2018b). Molecular docking would provide an insight into the binding of substrates to UGT2B7 and other UGTs and the effect of polymorphisms hereon. However, a complete crystal structure is not available yet for human UGTs (Dong et al. 2012). The partial crystal structure of UGT2B7 that is available does not include the N-terminal substrate-binding domain and consequently does not provide insight into substrate binding (Miley et al. 2007). No in vivo data exploring the relationship between UGT2B7 polymorphisms and PK parameters of zidovudine or codeine were available. Only two studies investigated the impact of UGT2B7*2 on telmisartan PK in Japanese adults. In both studies, no significant differences in AUC were found (Miura et al. 2009; Yamada et al. 2011).
UGT2B15
For UGT2B15, the most common polymorphism is known as UGT2B15*2 and this mutation results in the substitution of an aspartic acid with a tyrosine at position 85 (D85Y). The frequency distribution of this polymorphism is comparable for different populations (Fig. 6h). In one study, different ethnicities (African-American, Hispanic-American, Chinese-American, Japanese-American and Caucasian-American) in North-America were compared and all different ethnicities showed approximately the same distribution, with the heterozygous genotype being the predominant genotype (Riedy et al. 2000).
For this polymorphism, no differences were found in relation to the metabolic and PK profile of tamoxifen (Romero-Lorca et al. 2015; Sutiman et al. 2016). However, acetaminophen total clearance was significantly influenced by this polymorphism (Court et al. 2017). Moreover, in vitro data show lower median activities for S-oxazepam glucuronidation with microsomes containing the UGT2B15*2 polymorphism and a lower intrinsic clearance of bisphenol A with this SNP (Court et al. 2004; Hanioka et al. 2011). Finally, lower systemic clearance of lorazepam is reported in Asian individuals homozygous for UGT2B15*2 and the authors suggested that this polymorphism is a major contributor to interindividual differences in lorazepam PK (Chung et al. 2005). A significant increase in AUC has been observed for UGT2B15*2 for individuals with at least one polymorphic gene. According to the study of He et al. (2009), the polymorphism accounts for 34% of the interindividual differences in oxazepam oral clearance (Table 9).
Conclusions and future perspectives
This manuscript aimed to quantify interindividual differences in UGT isoform-specific metabolism for probe substrates. Hierarchical Bayesian meta-analyses for pharmacokinetic markers of acute (Cmax) and chronic exposure (AUC/clearance) were performed for a total of 14 probe substrates of the seven clinically most relevant UGT isoforms (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7 and UGT2B15). The resulting variability distributions and the UGT-related UFs showed that the default factor of 3.16 would not be exceeded for at least 97.5% of non-phenotyped healthy adults when considering the median value, with a few exceptions (1-OH-midazolam, ezetimibe, raltegravir, SN38 and trifluoperazine).
Overall, interindividual differences in kinetics for intravenous- and oral routes of administration were comparable. A possible explanation for such similarities lies in the fact that UGTs are more abundant in the liver compared to the intestine, so that the impact of first-pass metabolism for the included probe substrates is low (Lv et al. 2019). In contrast, similar analysis performed for CYP3A4 probe substrates revealed larger interindividual differences for markers of oral chronic exposure compared to their IV counterparts (Darney et al. 2019). Several UGT isoforms are also expressed in the kidney, including UGT1A6, UGT1A9 and UGT2B7 (Ohno and Nakajin 2009). This would have no influence on the first-pass metabolism, but variability estimates are likely to reflect hepatic and renal UGT metabolism for the probe substrates metabolised by these isoforms.
Overall, data gaps have been identified from this human UGT PK database for a range of non-phenotyped and phentoyped populations of different geographical ancestries as well as sensitive subgroups of the population, including neonates, children and the elderly. A typical example is the lack of PK data for the African population which shows broad genetic diversity in the frequency of UGT polymorphisms. Such PK data are needed to integrate genotype frequencies in different populations and to generate distributions to address interphenotypic differences which then allow the derivation of UGT-related UFs as well as chemical-specific adjustment factors (Campbell and Tishkoff 2008; Gaibar et al. 2018; Novillo et al. 2018).
Indeed, different UGT polymorphisms can have (substrate-dependent) impact on interphenotypic differences in PK parameters, particularly for the UGT1A1 isoform while new polymorphisms are still being characterised (Liu et al. 2019). In this light, it is recommended to investigate interphenotypic differences in relation to UGT polymorphisms rather than geographical ancestry since polymorphisms are better predictors of altered PK compared to ethnicity alone (Darney et al. 2019; Wu et al. 2018).
Although isoform-specific variability was investigated here using specific probe substrates, most often several UGT isoforms are involved in the glucuronidation of xenobiotics in a concentration-dependent manner. For example, acetaminophen glucuronidation by human liver microsomes can be mediated by multiple UGTs. Three isoforms are most active, UGT1A1 is the main contributor at toxic concentrations and UGT1A6 is the most active at low concentrations (Court et al. 2001). Besides the contribution of several isoforms to the glucuronidation of one compound, other factors could also contribute to interindividual differences in metabolism by UGTs. For example, correlations have been established between UGT abundances and their activity, and variability in glucuronidation is comparable to variability in UGT protein abundance (Achour et al. 2017). In addition to interphenotypic differences, age differences have been described to impact UGT expression and activities, particularly in neonates and young infants, leading to slower kinetics and elimination through a reduction of PK parameters by several folds compared to that in healthy adults (Bhatt et al. 2019; Court 2010; Dorne et al. 2001b).
UGTs are also involved in the metabolism of large numbers of xenobiotics, other than pharmaceuticals, like environmental contaminants and naturally occurring compounds. However, for these compounds multiple UGT isoforms are often involved in their conjugation. For example, isoflavones are conjugated by multiple UGT isoforms in human liver microsomes (Tang et al. 2009). Besides the involvement of several UGT isoforms in conjugation, human kinetic data for most environmental contaminants and food-relevant chemicals are still scarce in the literature.
Taken all together, investigation of isoform-specific UGT-related age and interphenotypic differences in world populations will allow the characterisation and publication of full variability distributions for human populations in an open source format (as illustrated here with the relatively limited data available). For food-relevant compounds, it is foreseen that such distributions can then be combined with in vitro data characterising the kinetics of UGT isoform-specific metabolism for a whole host of compounds including flavourings, food additives, pesticides, mycotoxins and other contaminants to develop quantitative in vitro–in vivo extrapolation (QIVIVE) models. It is important to note that the isoform-specific distributions and uncertainty factors generated in this study have been drawn from pharmaceutical data and can be applied to a large number of UGT substrates with short half-lives. As variability between the UGT isoforms has been shown to be relatively similar, chemical-specific variability can also be derived, even for compounds conjugated by multiple UGT isoforms. A major data gap is the lack of human in vivo PK data and mechanistically validated in vitro assays in human intestinal, liver, and kidney cells. Further research and validation efforts in these areas would allow to characterise either direct isoform-specific UGT metabolism, cytochrome P450 and/or influx or efflux transport with subsequent UGT conjugation as well as differential renal or bile excretion to further develop such QIVIVE models and gain experience and confidence in their use in daily chemical risk assessment.
References
Achour B et al (2017) Quantitative characterization of major hepatic UDP-glucuronosyltransferase enzymes in human liver microsomes: comparison of two proteomic methods and correlation with catalytic activity. Drug Metab Dispos Biol Fate Chem 45:1102–1112. https://doi.org/10.1124/dmd.117.076703
Allegaert K, Vanhaesebrouck S, Verbesselt R, van den Anker JN (2009) In vivo glucuronidation activity of drugs in neonates: extensive interindividual variability despite their young age. Ther Drug Monit 31:411–415. https://doi.org/10.1097/FTD.0b013e3181a8cc0a
Ando Y, Saka H, Asai G, Sugiura S, Shimokata K, Kamataki T (1998) UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol 9:845–847. https://doi.org/10.1023/a:1008438109725
Armstrong VW, Tenderich G, Shipkova M, Parsa A, Koerfer R, Schroder H, Oellerich M (2005) Pharmacokinetics and bioavailability of mycophenolic acid after intravenous administration and oral administration of mycophenolate mofetil to heart transplant recipients. Ther Drug Monit 27:315–321. https://doi.org/10.1097/01.ftd.0000163949.40686.0f
Aueviriyavit S, Furihata T, Morimoto K, Kobayashi K, Chiba K (2007) Hepatocyte nuclear factor 1 alpha and 4 alpha are factors involved in interindividual variability in the expression of UGT1A6 and UGT1A9 but not UGT1A1, UGT1A3 and UGT1A4 mRNA in human livers. Drug Metab Pharmacokinet 22:391–398
Bae J-W, Choi C-I, Lee J-H, Jang C-G, Chung M-W, Lee S-Y (2011) Effects of UDP-glucuronosyltransferase polymorphisms on the pharmacokinetics of ezetimibe in healthy subjects. Eur J Clin Pharmacol 67:39–45. https://doi.org/10.1007/s00228-010-0899-x
Barbier O, Turgeon D, Girard C, Green MD, Tephly TR, Hum DW, Bélanger A (2000) 3′-azido-3′-deoxythimidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7). Drug Metab Dispos 28:497–502
Benoit-Biancamano M-O, Connelly J, Villeneuve L, Caron P, Guillemette C (2009a) Deferiprone glucuronidation by human tissues and recombinant UDP glucuronosyltransferase 1A6: an in vitro investigation of genetic and splice variants. Drug Metab Dispos 37:322–329. https://doi.org/10.1124/dmd.108.023101
Benoit-Biancamano MO, Adam JP, Bernard O, Court MH, Leblanc MH, Caron P, Guillemette C (2009b) A pharmacogenetics study of the human glucuronosyltransferase UGT1A4. Pharmacogenet Genom 19:945–954. https://doi.org/10.1097/FPC.0b013e3283331637
Beutler E, Gelbart T, Demina A (1998) Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci 95:8170–8174. https://doi.org/10.1073/pnas.95.14.8170
Bhatt DK et al (2019) Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther 105:131–141. https://doi.org/10.1002/cpt.1109
Blevins-Primeau AS, Sun D, Chen G, Sharma AK, Gallagher CJ, Amin S, Lazarus P (2009) Functional significance of UDP-glucuronosyltransferase variants in the metabolism of active tamoxifen metabolites. Cancer Res 69:1892–1900. https://doi.org/10.1158/0008-5472.can-08-3708
Bock KW, Schrenk D, Forster A, Griese E-U, Mörike K, Brockmeier D, Eichelbaum M (1994) The influence of environmental and genetic factors on CYP2D6, CYP1A2 and UDP-glucuronosyltransferases in man using sparteine, caffeine, and paracetamol as probes. Pharmacogenet Genom 4:209–218. https://doi.org/10.1097/00008571-199408000-00005
Bomgaars LR et al (2007) Phase II trial of irinotecan in children with refractory solid tumors: a children's oncology group study. J Clin Oncol 25:4622–4627. https://doi.org/10.1200/JCO.2007.11.6103
Brainard DM, Wenning LA, Stone JA, Wagner JA, Iwamoto M (2011) Clinical pharmacology profile of raltegravir, an HIV-1 integrase strand transfer inhibitor. J Clin Pharmacol 51:1376–1402. https://doi.org/10.1177/0091270010387428
Brill SS et al (2006) Glucuronidation of trans-resveratrol by human liver and intestinal microsomes and UGT isoforms. J Pharm Pharmacol 58:469–479. https://doi.org/10.1211/jpp.58.4.0006
Bullingham R, Monroe S, Nicholls A, Hale M (1996) Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single-dose oral and intravenous administration. J Clin Pharmacol 36:315–324. https://doi.org/10.1002/j.1552-4604.1996.tb04207.x
Burchell B, Hume R (1999) Molecular genetic basis of Gilbert's syndrome. J Gastroenterol Hepatol 14:960–966. https://doi.org/10.1046/j.1440-1746.1999.01984.x
Campbell MC, Tishkoff SA (2008) African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genom Hum Genet 9:403–433. https://doi.org/10.1146/annurev.genom.9.081307.164258
Cecchin E et al (2009) Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol Off J Am Soc Clin Oncol 27:2457–2465. https://doi.org/10.1200/jco.2008.19.0314
Chen F, Ritter JK, Wang MG, McBride OW, Lubet RA, Owens IS (1993) Characterization of a cloned human dihydrotestosterone/androstanediol UDP-glucuronosyltransferase and its comparison to other steroid isoforms. Biochemistry 32:10648–10657. https://doi.org/10.1021/bi00091a015
Chen Y, Xie S, Chen S, Zeng S (2008) Glucuronidation of flavonoids by recombinant UGT1A3 and UGT1A9. Biochem Pharmacol 76:416–425. https://doi.org/10.1016/j.bcp.2008.05.007
Chen WQ et al (2013) Polymorphism of ORM1 is associated with the pharmacokinetics of telmisartan. PLoS One 8:e70341. https://doi.org/10.1371/journal.pone.0070341
Cho SK, Oh ES, Park K, Park MS, Chung JY (2012) The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharmacogenet Genom 22:598–605. https://doi.org/10.1097/FPC.0b013e3283544085
Chung J-Y et al (2005) Effect of the UGT2B15 genotype on the pharmacokinetics, pharmacodynamics, and drug interactions of intravenous lorazepam in healthy volunteers. Clin Pharmacol Ther 77:486–494. https://doi.org/10.1016/j.clpt.2005.02.006
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2014 Dec 23—Identifier NCT01989455, A Blinded, Placebo-controlled study of the safety and pharmacokinetics of single doses of intravenous deferiprone in healthy volunteers; 2013 Nov 21 [cited 2020 01 23]. https://clinicaltrials.gov/ct2/show/results/NCT01989455?view=results
Ciotti M, Marrone A, Potter C, Owens IS (1997) Genetic polymorphism in the human UGT1A6 (planar phenol) UDP-glucuronosyltransferase: pharmacological implications. Pharmacogenet Genom 7:485–495. https://doi.org/10.1097/00008571-199712000-00007
Coffman BL, Rios GR, King CD, Tephly TR (1997) Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos 25:1–4
Coffman BL, King CD, Rios GR, Tephly TR (1998) The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268). Drug Metab Dispos 26:73–77
Coffman BL, Kearney WR, Goldsmith S, Knosp BM, Tephly TR (2003) Opioids bind to the amino acids 84 to 118 of UDP-glucuronosyltransferase UGT2B7. Mol Pharmacol 63:283–288. https://doi.org/10.1124/mol.63.2.283
Court MH (2010) Interindividual variability in hepatic drug glucuronidation: studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab Rev 42:209–224. https://doi.org/10.3109/03602530903209288
Court MH, Duan SX, von Moltke LL, Greenblatt DJ, Patten CJ, Miners JO, Mackenzie PI (2001) Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther 299:998–1006
Court MH, Duan SX, Guillemette C, Journault K, Krishnaswamy S, von Moltke LL, Greenblatt DJ (2002) Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTs): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab Dispos 30:1257–1265. https://doi.org/10.1124/dmd.30.11.1257
Court MH, Hao Q, Krishnaswamy S, Bekaii-Saab T, Al-Rohaimi A, von Moltke LL, Greenblatt DJ (2004) UDP-glucuronosyltransferase (UGT) 2B15 pharmacogenetics: UGT2B15 D85Y genotype and gender are major determinants of oxazepam glucuronidation by human liver. J Pharmacol Exp Ther 310:656–665. https://doi.org/10.1124/jpet.104.067660
Court MH, Zhu Z, Masse G, Duan SX, James LP, Harmatz JS, Greenblatt DJ (2017) Race, gender, and genetic polymorphism contribute to variability in acetaminophen pharmacokinetics, metabolism, and protein-adduct concentrations in healthy African-American and European-American Volunteers. J Pharmacol Exp Ther 362:431–440. https://doi.org/10.1124/jpet.117.242107
Darney K et al (2019) Inter-ethnic differences in CYP3A4 metabolism: a Bayesian meta-analysis for the refinement of uncertainty factors in chemical risk assessment. Comput Toxicol 12:100092. https://doi.org/10.1016/j.comtox.2019.100092
Darney K et al (2020) Human variability in influx and efflux transporters in relation to uncertainty factors for chemical risk assessment. Food Chem Toxicol 140:111305
Denlinger CS et al (2009) Pharmacokinetic analysis of irinotecan plus bevacizumab in patients with advanced solid tumors. Cancer Chemother Pharmacol 65:97–105. https://doi.org/10.1007/s00280-009-1008-7
Deppe S, Böger RH, Weiss J, Benndorf RA (2010) Telmisartan: a review of its pharmacodynamic and pharmacokinetic properties. Expert Opin Drug Metab Toxicol 6:863–871. https://doi.org/10.1517/17425255.2010.494597
Dong D, Ako R, Hu M, Wu B (2012) Understanding substrate selectivity of human UDP-glucuronosyltransferases through QSAR modeling and analysis of homologous enzymes. Xenobiotica Fate Foreign Compd Biol Syst 42:808–820. https://doi.org/10.3109/00498254.2012.663515
Dorne JLCM, Walton K, Renwick AG (2001a) Human variability in glucuronidation in relation to uncertainty factors for risk assessment. Food Chem Toxicol 39:1153–1173. https://doi.org/10.1016/S0278-6915(01)00087-4
Dorne JLCM, Walton K, Renwick AG (2001b) Uncertainty factors for chemical risk assessment: human variability in the pharmacokinetics of CYP1A2 probe substrates. Food and Chem Toxicol 39:681–696. https://doi.org/10.1016/S0278-6915(01)00005-9
Dorne JLCM, Walton K, Renwick AG (2005) Human variability in xenobiotic metabolism and pathway-related uncertainty factors for chemical risk assessment: a review. Food Chem Toxicol 43:203–216. https://doi.org/10.1016/j.fct.2004.05.011
Du Z, Jiao Y, Shi L (2016) Association of UGT2B7 and UGT1A4 polymorphisms with serum concentration of antiepileptic drugs in children. Med Sci Monit 22:4107–4113. https://doi.org/10.12659/MSM.897626
Ebner T, Remmel RP, Burchell B (1993) Human bilirubin UDP-glucuronosyltransferase catalyzes the glucuronidation of ethinylestradiol. Mol Pharmacol 43:649–654
Ehmer U, Vogel A, Schütte JK, Krone B, Manns MP, Strassburg CP (2004) Variation of hepatic glucuronidation: novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology 39:970–977. https://doi.org/10.1002/hep.20131
Favetta P, Degoute CS, Perdrix JP, Dufresne C, Boulieu R, Guitton J (2002) Propofol metabolites in man following propofol induction and maintenance. Br J Anaesth 88:653–658. https://doi.org/10.1093/bja/88.5.653
Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA (2001) The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev 33:273–297. https://doi.org/10.1081/DMR-120000653
Fotherby K (1996) Bioavailability of orally administered sex steroids used in oral contraception and hormone replacement therapy. Contraception 54:59–69. https://doi.org/10.1016/0010-7824(96)00136-9
Fukuda M et al (2018) Relationship between UGT1A1*27 and UGT1A1*7 polymorphisms and irinotecan-related toxicities in patients with lung cancer. Thorac Cancer 9:51–58. https://doi.org/10.1111/1759-7714.12535
Furman WL et al (2009) Tyrosine kinase inhibitor enhances the bioavailability of oral irinotecan in pediatric patients with refractory solid tumors. J Clin Oncol Off J Am Soc Clin Oncol 27:4599–4604. https://doi.org/10.1200/JCO.2008.19.6642
Gaibar M, Novillo A, Romero-Lorca A, Esteban ME, Fernández-Santander A (2018) Pharmacogenetics of ugt genes in North African populations. Pharmacogenom J 18:609–612. https://doi.org/10.1038/s41397-018-0034-4
Gallicano KD et al (1999) Induction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patients. Br J Clin Pharmacol 48:168–179. https://doi.org/10.1046/j.1365-2125.1999.00987.x
Ghosal A et al (2004) Identification of human UDP-Glucuronosyltransferase enzyme(s) responsible for the glucuronidation of ezetimibe (Zetia). Drug Metab Dispos 32:314–320. https://doi.org/10.1124/dmd.32.3.314
Ghotbi R, Mannheimer B, Aklillu E, Suda A, Bertilsson L, Eliasson E, Ösby U (2010) Carriers of the UGT1A4 142T%3eG gene variant are predisposed to reduced olanzapine exposure—an impact similar to male gender or smoking in schizophrenic patients. Eur J Clin Pharmacol 66:465–474. https://doi.org/10.1007/s00228-009-0783-8
Girard H et al (2004) Identification of common polymorphisms in the promoter of the UGT1A9 gene: evidence that UGT1A9 protein and activity levels are strongly genetically controlled in the liver. Pharmacogenetics 14:501–515. https://doi.org/10.1097/01.fpc.0000114754.08559.27
Gulcebi MI, Ozkaynakcı A, Goren MZ, Aker RG, Ozkara C, Onat FY (2011) The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res 95:1–8. https://doi.org/10.1016/j.eplepsyres.2011.01.016
Han J-Y et al (2006) Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol 24:2237–2244. https://doi.org/10.1200/JCO.2005.03.0239
Han J-Y, Lim H-S, Park YH, Lee SY, Lee JS (2009) Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer 63:115–120. https://doi.org/10.1016/j.lungcan.2007.12.003
Hanioka N, Ozawa S, Jinno H, Ando M, Saito Y, Sawada J (2001) Human liver UDP-glucuronosyltransferase isoforms involved in the glucuronidation of 7-ethyl-10-hydroxycamptothecin. Xenobiotica 31:687–699. https://doi.org/10.1080/00498250110057341
Hanioka N, Naito T, Narimatsu S (2008) Human UDP-glucuronosyltransferase isoforms involved in bisphenol A glucuronidation. Chemosphere 74:33–36. https://doi.org/10.1016/j.chemosphere.2008.09.053
Hanioka N, Oka H, Nagaoka K, Ikushiro S, Narimatsu S (2011) Effect of UDP-glucuronosyltransferase 2B15 polymorphism on bisphenol A glucuronidation. Arch Toxicol 85:1373–1381. https://doi.org/10.1007/s00204-011-0690-5
Hazama S et al (2010) Phase I study of irinotecan and doxifluridine for metastatic colorectal cancer focusing on the UGT1A1*28 polymorphism. Cancer Sci 101:722–727. https://doi.org/10.1111/j.1349-7006.2009.01428.x
He X, Hesse LM, Hazarika S, Masse G, Harmatz JS, Greenblatt DJ, Court MH (2009) Evidence for oxazepam as an in vivo probe of UGT2B15: oxazepam clearance is reduced by UGT2B15 D85Y polymorphism but unaffected by UGT2B17 deletion. Br J Clin Pharmacol 68:721–730. https://doi.org/10.1111/j.1365-2125.2009.03519.x
Heikkinen H, Saraheimo M, Antila S, Ottoila P, Pentikäinen PJ (2001) Pharmacokinetics of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, in man. Eur J Clin Pharmacol 56:821–826. https://doi.org/10.1007/s002280000244
Heizmann P, Eckert M, Ziegler W (1983) Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharmacol 16:43S–49S. https://doi.org/10.1111/j.1365-2125.1983.tb02270.x
Hyland R, Osborne T, Payne A, Kempshall S, Logan YR, Ezzeddine K, Jones B (2009) In vitro and in vivo glucuronidation of midazolam in humans. Br J Clin Pharmacol 67:445–454. https://doi.org/10.1111/j.1365-2125.2009.03386.x
Ieiri I et al (2011) Pharmacokinetic and pharmacogenomic profiles of telmisartan after the oral microdose and therapeutic dose. Pharmacogenet Genom 21:495–505. https://doi.org/10.1097/FPC.0b013e3283489ce2
Innocenti F et al (2004) Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol Off J Am Soc Clin Oncol 22:1382–1388. https://doi.org/10.1200/jco.2004.07.173
Iwamura A, Nakajima M, Oda S, Yokoi T (2017) Toxicological potential of acyl glucuronides and its assessment. Drug Metab Pharmacokinet 32:2–11. https://doi.org/10.1016/j.dmpk.2016.11.002
Iyer L et al (2002) UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenom J 2:43–47. https://doi.org/10.1038/sj.tpj.6500072
Iyer LV, Ho MN, Shinn WM, Bradford WW, Tanga MJ, Nath SS, Green CE (2003) Glucuronidation of 1′-hydroxyestragole (1′-HE) by human UDP-glucuronosyltransferases UGT2B7 and UGT1A9. Toxicol Sci 73:36–43. https://doi.org/10.1093/toxsci/kfg066
Jaeckle KA et al (2010) Phase II NCCTG trial of RT + irinotecan and adjuvant BCNU plus irinotecan for newly diagnosed GBM. J Neurooncol 99:73–80. https://doi.org/10.1007/s11060-009-0103-2
Jiao Z et al (2008) Population pharmacokinetic modelling for enterohepatic circulation of mycophenolic acid in healthy Chinese and the influence of polymorphisms in UGT1A9. Br J Clin Pharmacol 65:893–907. https://doi.org/10.1111/j.1365-2125.2008.03109.x
Kang WY et al (2018) Pharmacokinetic and bioequivalence study of a telmisartan/S-amlodipine fixed-dose combination (CKD-828) formulation and coadministered telmisartan and S-amlodipine in healthy subjects. Drug Des Dev Ther 12:545–553. https://doi.org/10.2147/dddt.S156492
Kassahun K et al (2007) Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos 35:1657–1663. https://doi.org/10.1124/dmd.107.016196
Kasteel EEJ, Darney K, Kramer NI, Dorne JLCM, Lautz LS (2020) Pharmacokinetic data for UGT probe substrates in adults. https://doi.org/10.5281/zenodo.3631235
Klecker RW Jr, Collins JM, Yarchoan R, Thomas R, Jenkins JF, Broder S, Myers CE (1987) Plasma and cerebrospinal fluid pharmacokinetics of 3′-azido-3′-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther 41:407–412. https://doi.org/10.1038/clpt.1987.49
Kosoglou T, Statkevich P, Johnson-Levonas A, Paolini JF, Bergman AJ, Alton KB (2005) Ezetimibe. Clin Pharmacokinet 44:467–494. https://doi.org/10.2165/00003088-200544050-00002
Kuypers DR, Naesens M, Vermeire S, Vanrenterghem Y (2005) The impact of uridine diphosphate-glucuronosyltransferase 1A9 (UGT1A9) gene promoter region single-nucleotide polymorphisms T-275A and C-2152T on early mycophenolic acid dose-interval exposure in de novo renal allograft recipients. Clin Pharmacol Ther 78:351–361. https://doi.org/10.1016/j.clpt.2005.06.007
Lampe JW, Bigler J, Horner NK, Potter JD (1999) UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenet Genom 9:341–350. https://doi.org/10.1097/00008571-199906000-00009
Lautala P, Ethell BT, Taskinen J, Burchell B (2000) The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP-glucuronosyltransferases. Drug Metabol Dispos 28:1385–1389
Li X et al (2011) Identification of the human UDP-glucuronosyltransferase isoforms involved in the glucuronidation of the phytochemical ferulic acid. Drug Metab Pharmacokinet 26:341–350. https://doi.org/10.2133/dmpk.dmpk-10-rg-125
Li J, He C, Fang L, Yang L, Wang Z (2016) Identification of human UDP-glucuronosyltransferase 1A4 as the major isozyme responsible for the glucuronidation of 20(S)-protopanaxadiol in human liver microsomes. Int J Mol Sci 17:205–205. https://doi.org/10.3390/ijms17030205
Limenta LMG et al (2008) UGT1A6 genotype-related pharmacokinetics of deferiprone (L1) in healthy volunteers. Br J Clin Pharmacol 65:908–916. https://doi.org/10.1111/j.1365-2125.2008.03103.x
Liu H-X, Liu Y, Zhang J-W, Li W, Liu H-T, Yang L (2008) UDP-glucuronosyltransferase 1A6 is the major isozyme responsible for protocatechuic aldehyde glucuronidation in human liver microsomes. Drug Metab Dispos 36:1562–1569. https://doi.org/10.1124/dmd.108.020560
Liu D, Zhang L, Duan L, Wu J, Hu M, Liu Z, Wang C (2019) Potential of herb–drug/herb interactions between substrates and inhibitors of UGTs derived from herbal medicines. Pharmacol Res 150:104510. https://doi.org/10.1016/j.phrs.2019.104510
Lv X, Zhang J-B, Hou J, Dou T-Y, Ge G-B, Hu W-Z, Yang L (2019) Chemical probes for human UDP-glucuronosyltransferases: a comprehensive review. Biotechnol J 14:1800002. https://doi.org/10.1002/biot.201800002
Malfatti MA, Felton JS (2004) Human UDP-glucuronosyltransferase 1A1 is the primary enzyme responsible for the N-glucuronidation of N-hydroxy-PhIP in vitro. Chem Res Toxicol 17:1137–1144. https://doi.org/10.1021/tx049898m
Mazidi T et al (2013) Impact of UGT1A9 polymorphism on mycophenolic acid pharmacokinetic parameters in stable renal transplant patients. Iran J Pharm Res 12:547–556. https://doi.org/10.22037/IJPR.2013.1334
McGreavey LE et al (2005) No evidence that polymorphisms in CYP2C8, CYP2C9, UGT1A6, PPARδ and PPARγ act as modifiers of the protective effect of regular NSAID use on the risk of colorectal carcinoma. Pharmacogenet Genom 15:713–721. https://doi.org/10.1097/01.fpc.0000174786.85238.63
Meech R et al (2019) The UDP-glycosyltransferase (UGT) superfamily: new members new functions, and novel paradigms. Physiol Rev 99:1153–1222. https://doi.org/10.1152/physrev.00058.2017
Mehboob H, Tahir IM, Iqbal T, Akhter N, Munir N, Riaz M (2017) Genetic Polymorphism of UDP-Glucuronosyltransferase. In: Genetic Polymorphisms. IntechOpen, Narasimha Reddy Parine, p 159 (ISBN: 978-953-51-3516-6). https://doi.org/10.5772/intechopen.69206
Midha KK, Korchinski ED, Roscoe RMH, Hawes EM, Cooper JK, McKay G (1984) Relative bioavailability of a commercial trifluoperazine tablet formulation using a radioimmunoassay technique. J Pharm Sci 73:261–263. https://doi.org/10.1002/jps.2600730231
Midha KK, Hawes EM, Hubbard JW, Korchinski ED, McKay G (1988) A pharmacokinetic study of trifluoperazine in two ethnic populations. Psychopharmacology 95:333–338. https://doi.org/10.1007/BF00181943
Miley MJ, Zielinska AK, Keenan JE, Bratton SM, Radominska-Pandya A, Redinbo MR (2007) Crystal structure of the cofactor-binding domain of the human phase II drug-metabolism enzyme UDP-glucuronosyltransferase 2B7. J Mol Biol 369:498–511. https://doi.org/10.1016/j.jmb.2007.03.066
Miners JO, McKinnon RA, Mackenzie PI (2002) Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology 181–182:453–456. https://doi.org/10.1016/S0300-483X(02)00449-3
Miura M, Satoh S, Inoue K, Saito M, Habuchi T, Suzuki T (2009) Telmisartan pharmacokinetics in Japanese renal transplant recipients. Clin Chim Acta 399:83–87. https://doi.org/10.1016/j.cca.2008.09.020
Moher D, Liberati A, Tetzlaff J, Altman DG, The PG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6:e1000097. https://doi.org/10.1371/journal.pmed.1000097
Mori A, Maruo Y, Iwai M, Sato H, Takeuchi Y (2005) UDP-glucuronosyltransferase 1a4 polymorphisms in a japanese population and kinetics of clozapine glucuronidation. Drug Metab Dispos 33:672–675. https://doi.org/10.1124/dmd.104.002576
Nagar S, Zalatoris JJ, Blanchard RL (2004) Human UGT1A6 pharmacogenetics: identification of a novel SNP, characterization of allele frequencies and functional analysis of recombinant allozymes in human liver tissue and in cultured cells. Pharmacogenetics 14:487–499. https://doi.org/10.1097/01.fpc.0000114771.78957.cb
Naumann BD, Weideman PA, Dixit R, Grossman SJ, Shen CF, Sargent EV (1997) Use of toxicokinetic and toxicodynamic data to reduce uncertainties when setting occupational exposure limits for pharmaceuticals. Hum Ecol Risk Assess Int J 3:555–565. https://doi.org/10.1080/10807039709383711
Novillo A et al (2018) UDP-glucuronosyltransferase genetic variation in North African populations: a comparison with African and European data. Ann Hum Biol 45:516–523. https://doi.org/10.1080/03014460.2018.1559354
Ohno S, Nakajin S (2009) Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 37:32–40. https://doi.org/10.1124/dmd.108.023598
Paoluzzi L et al (2004) Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38. J Clin Pharmacol 44:854–860. https://doi.org/10.1177/0091270004267159
Patel M, Tang BK, Grant DM, Kalow W (1995) Interindividual variability in the glucuronidation of (S) oxazepam contrasted with that of (R) oxazepam. Pharmacogenetics 5:287–297
Petrenaite V, Öhman I, Ekström L, Sæbye D, Hansen TF, Tomson T, Sabers A (2018) UGT polymorphisms and lamotrigine clearance during pregnancy. Epilepsy Res 140:199–208. https://doi.org/10.1016/j.eplepsyres.2018.01.011
Picard N, Ratanasavanh D, Prémaud A, Le Meur Y, Marquet P (2005) Identification of the UDP-glucuronosyltransferase isoforms involved in mycophenolic acid phase II metabolism. Drug Metab Dispos 33:139–146. https://doi.org/10.1124/dmd.104.001651
Plummer M (2003) JAGS: a program for analysis of bayesian graphical models using gibbs sampling. In: Hornik K, Leisch F, Zeileis A (eds) Proceedings of the 3rd international workshop on distributed statistical computing. Vienna, Austria, pp 1–10
R Development Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Reimers A, Sjursen W, Helde G, Brodtkorb E (2016) Frequencies of UGT1A4*2 (P24T) and *3 (L48V) and their effects on serum concentrations of lamotrigine. Eur J Drug Metab Pharmacokinet 41:149–155. https://doi.org/10.1007/s13318-014-0247-0
Renwick AG, Lazarus NR (1998) Human variability and noncancer risk assessment—an analysis of the default uncertainty factor. Regul Toxicol Pharmacol 27:3–20. https://doi.org/10.1006/rtph.1997.1195
Ri M et al (2018) A phase I/II study for dose-finding, and to investigate the safety, pharmacokinetics and preliminary efficacy of NK012, an SN-38-incorporating macromolecular polymeric micelle, in patients with multiple myeloma. Intern Med (Tokyo, Japan) 57:939–946. https://doi.org/10.2169/internalmedicine.9567-17
Riedmaier S et al (2010) UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin Pharmacol Ther 87:65–73. https://doi.org/10.1038/clpt.2009.181
Riedy M, Wang J-Y, Miller AP, Buckler A, Hall J, Guida M (2000) Genomic organization of the UGT2b gene cluster on human chromosome 4q13. Pharmacogenet Genom 10:251–260
Rodrat S, Yamanont P, Tankanitlert J, Chantraraksri U, Fucharoen S, Morales NP (2012) Comparison of pharmacokinetics and urinary iron excretion of two single doses of deferiprone in β-thalassemia/hemoglobin E patients. Pharmacology 90:88–94. https://doi.org/10.1159/000339658
Rogers JF, Findlay JWA, Hull JH, Butz RF, Jones EC, Bustrack JA, Welch RM (1982) Codeine disposition in smokers and nonsmokers. Clin Pharmacol Ther 32:218–227. https://doi.org/10.1038/clpt.1982.151
Romero-Lorca A, Novillo A, Gaibar M, Bandres F, Fernandez-Santander A (2015) Impacts of the glucuronidase genotypes UGT1A4, UGT2B7, UGT2B15 and UGT2B17 on tamoxifen metabolism in breast cancer patients. PLoS One 10:e0132269. https://doi.org/10.1371/journal.pone.0132269
Rowland A, Miners JO, Mackenzie PI (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 45:1121–1132. https://doi.org/10.1016/j.biocel.2013.02.019
Rutherford K, Parson WW, Daggett V (2008) The histamine N-methyltransferase T105I polymorphism affects active site structure and dynamics. Biochemistry 47:893–901. https://doi.org/10.1021/bi701737f
Sacco JJ, Botten J, Macbeth F, Bagust A, Clark P (2010) The average body surface area of adult cancer patients in the UK: a multicentre retrospective study. PLoS One 5:e8933. https://doi.org/10.1371/journal.pone.0008933
Saeki M et al (2006) Haplotype structures of the UGT1A gene complex in a Japanese population. Pharmacogenom J 6:63–75. https://doi.org/10.1038/sj.tpj.6500335
Satoh T et al (2011) Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci 102:1868–1873. https://doi.org/10.1111/j.1349-7006.2011.02030.x
Seo K-A, Bae SK, Choi Y-K, Choi CS, Liu K-H, Shin J-G (2010) Metabolism of 1′- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab Dispos 38:2007–2013. https://doi.org/10.1124/dmd.110.035295
Seo K-A, Kim H-J, Jeong ES, Abdalla N, Choi C-S, Kim D-H, Shin J-G (2014) In vitro assay of six UDP-glucuronosyltransferase isoforms in human liver microsomes, using cocktails of probe substrates and liquid chromatography–tandem mass spectrometry. Drug Metab Dispos 42:1803–1810. https://doi.org/10.1124/dmd.114.058818
Shipkova M et al (2002) Pharmacokinetics and protein adduct formation of the pharmacologically active acyl glucuronide metabolite of mycophenolic acid in pediatric renal transplant recipients. Ther Drug Monit 24:390–399
Shrestha B et al (2011) Evolution of a major drug metabolizing enzyme defect in the domestic cat and other felidae: phylogenetic timing and the role of hypercarnivory. PLoS One 6:e18046. https://doi.org/10.1371/journal.pone.0018046
Sim SC, Kacevska M, Ingelman-Sundberg M (2013) Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenom J 13:1–11. https://doi.org/10.1038/tpj.2012.45
Slatter JG et al (2000) Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos Biol Fate Chem 28:423–433
Sonne J et al (1988) Bioavailability and pharmacokinetics of oxazepam. Eur J Clin Pharmacol 35:385–389. https://doi.org/10.1007/BF00561369
Stangier J, Su C, Roth W (2000a) Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J Int Med Res 28:149–167. https://doi.org/10.1177/147323000002800401
Stangier J et al (2000b) Absorption, metabolism, and excretion of intravenously and orally administered [14C]telmisartan in healthy volunteers. J Clin Pharmacol 40:1312–1322
Stewart CF et al (2007) UGT1A1 promoter genotype correlates with SN-38 pharmacokinetics, but not severe toxicity in patients receiving low-dose irinotecan. J Clin Oncol 25:2594–2600. https://doi.org/10.1200/JCO.2006.10.2301
Stingl JC, Bartels H, Viviani R, Lehmann ML, Brockmöller J (2014) Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: a quantitative systematic review. Pharmacol Ther 141:92–116. https://doi.org/10.1016/j.pharmthera.2013.09.002
Sun Y-X et al (2015) The influence of UGT2B7 genotype on valproic acid pharmacokinetics in Chinese epilepsy patients. Epilepsy Res 114:78–80. https://doi.org/10.1016/j.eplepsyres.2015.04.015
Sutiman N et al (2016) Pharmacogenetics of UGT1A4, UGT2B7 and UGT2B15 and their influence on tamoxifen disposition in Asian breast cancer patients. Clin Pharmacokinet 55:1239–1250. https://doi.org/10.1007/s40262-016-0402-7
Tang L, Singh R, Liu Z, Hu M (2009) Structure and concentration changes affect characterization of UGT isoform-specific metabolism of isoflavones. Mol Pharm 6:1466–1482. https://doi.org/10.1021/mp8002557
Tang L et al (2010) Use of glucuronidation fingerprinting to describe and predict mono- and dihydroxyflavone metabolism by recombinant UGT isoforms and human intestinal and liver microsomes. Mol Pharm 7:664–679. https://doi.org/10.1021/mp900223c
Tian X et al (2015) Erratum to: regioselective glucuronidation of andrographolide and its major derivatives: metabolite identification isozyme contribution, and species differences. AAPS J 17:479–479. https://doi.org/10.1208/s12248-014-9683-7
Toffoli G et al (2006) The role of UGT1A1* 28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol 24:3061–3068. https://doi.org/10.1200/JCO.2005.05.5400
Tsunedomi R, Hazama S, Okayama N, Oka M, Nagano H (2017) Rapid and sensitive detection of UGT1A1 polymorphisms associated with irinotecan toxicity by a novel DNA microarray. Cancer Sci 108:1504–1509. https://doi.org/10.1111/cas.13272
Villeneuve L, Girard H, Fortier LC, Gagne JF, Guillemette C (2003) Novel functional polymorphisms in the UGT1A7 and UGT1A9 glucuronidating enzymes in Caucasian and African-American subjects and their impact on the metabolism of 7-ethyl-10-hydroxycamptothecin and flavopiridol anticancer drugs. J Pharmacol Exp Ther 307:117–128. https://doi.org/10.1124/jpet.103.054072
Walpole SC, Prieto-Merino D, Edwards P, Cleland J, Stevens G, Roberts I (2012) The weight of nations: an estimation of adult human biomass. BMC Public Health 12:439. https://doi.org/10.1186/1471-2458-12-439
Wang M, Lu J, Li J, Qi H, Wang Y, Zhang H (2014) Steviol glucuronidation and its potential interaction with UDP-glucuronosyltransferase 2B7 substrates. Food Chem Toxicol 64:135–143. https://doi.org/10.1016/j.fct.2013.11.028
Wang L et al (2018a) Glucuronidation of icaritin by human liver microsomes, human intestine microsomes and expressed UDP-glucuronosyltransferase enzymes: identification of UGT1A3, 1A9 and 2B7 as the main contributing enzymes. Xenobiotica Fate Foreign Compd Biol Syst 48:357–367. https://doi.org/10.1080/00498254.2017.1323139
Wang P, Lin X-Q, Cai W-K, Xu G-L, Zhou M-D, Yang M, He G-H (2018b) Effect of UGT2B7 genotypes on plasma concentration of valproic acid: a meta-analysis. Eur J Clin Pharmacol 74:433–442. https://doi.org/10.1007/s00228-017-2395-z
Weber WW (1999) Populations and genetic polymorphisms. Mol Diagn 4:299–307. https://doi.org/10.1016/S1084-8592(99)80006-X
Wenning LA et al (2009) Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin Pharmacol Ther 85:623–627. https://doi.org/10.1038/clpt.2009.12
Wickham H (2016) ggplot2: Elegant graphics for data analysis. Springer, New York
Wickham H, François R, Henry L, Müller K (2020) dplyr: A grammar of data manipulation. R Package Version 0.8.4. https://CRAN.R-project.org/package=dplyr
Wiecek W, Dorne J-L, Quignot N, Bechaux C, Amzal B (2019) A generic Bayesian hierarchical model for the meta-analysis of human population variability in kinetics and its applications in chemical risk assessment. Comput Toxicol 12:100106. https://doi.org/10.1016/j.comtox.2019.100106
Wienen W et al (2000) A review on telmisartan: a novel, long-acting angiotensin ii-receptor antagonist. Cardiovasc Drug Rev 18:127–154. https://doi.org/10.1111/j.1527-3466.2000.tb00039.x
Wikberg T, Vuorela A, Ottoila P, Taskinen J (1993) Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab Dispos 21:81–92
Williams MC, Goldzieher JW (1980) Chromatographic patterns of urinary ethynyl estrogen metabolites in various populations. Steroids 36:255–282. https://doi.org/10.1016/0039-128X(80)90001-X
Wu B, Xu B, Hu M (2011) Regioselective glucuronidation of flavonols by six human UGT1A isoforms. Pharm Res 28:1905–1918. https://doi.org/10.1007/s11095-011-0418-5
Wu L et al (2018) Gender differences in the hepatotoxicity and toxicokinetics of emodin: the potential mechanisms mediated by UGT2B7 and MRP2. Mol Pharm 15:3931–3945. https://doi.org/10.1021/acs.molpharmaceut.8b00387
Yamada A et al (2011) The impact of pharmacogenetics of metabolic enzymes and transporters on the pharmacokinetics of telmisartan in healthy volunteers. Pharmacogenet Genom 21:523–530. https://doi.org/10.1097/FPC.0b013e3283482502
Yamanaka H et al (2004) A novel polymorphism in the promoter region of human UGT1A9 gene (UGT1A9*22) and its effects on the transcriptional activity. Pharmacogenetics 14:329–332. https://doi.org/10.1097/00008571-200405000-00008
Yang N, Sun R, Liao X, Aa J, Wang G (2017) UDP-glucuronosyltransferases (UGTs) and their related metabolic cross-talk with internal homeostasis: a systematic review of UGT isoforms for precision medicine. Pharmacol Res 121:169–183. https://doi.org/10.1016/j.phrs.2017.05.001
Yuan L, Qian S, Xiao Y, Sun H, Zeng S (2015) Homo- and hetero-dimerization of human UDP-glucuronosyltransferase 2B7 (UGT2B7) wild type and its allelic variants affect zidovudine glucuronidation activity. Biochem Pharmacol 95:58–70. https://doi.org/10.1016/j.bcp.2015.03.002
Yue Q, Svensson J, Alm C, Sjoqvist F, Sawe J (1989) Interindividual and interethnic differences in the demethylation and glucuronidation of codeine. Br J Clin Pharmacol 28:629–637. https://doi.org/10.1111/j.1365-2125.1989.tb03555.x
Zhang D et al (2007) Characterization of the UDP glucuronosyltransferase activity of human liver microsomes genotyped for the UGT1A1*28 polymorphism. Drug Metab Dispos 35:2270–2280. https://doi.org/10.1124/dmd.107.017806
Zhang WX et al (2008) Influence of uridine diphosphate (UDP)-glucuronosyltransferases and ABCC2 genetic polymorphisms on the pharmacokinetics of mycophenolic acid and its metabolites in Chinese renal transplant recipients. Xenobiotica 38:1422–1436. https://doi.org/10.1080/00498250802488585
Acknowledgements
We thank Nadia Quinot for making the zidovudine data available (EFSA/SCER/2014/06) and Remco Westerink for helping review the manuscript.
Funding
This work was supported by the European Food Safety Authority (EFSA) [Contract number: GP/EFSA/SCER/2015/01].
Author information
Authors and Affiliations
Contributions
EK and LL carried out the data collection, analysis of the results and drafted the manuscript with equal contribution. KD performed the data analysis and assisted with data interpretation and drafting the manuscript. JLD and NK assisted in the design of the study and interpretation of the results, critical review and editing of the manuscript. All authors commented on previous versions of the manuscript and have read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Ms. Emma Kasteel, Mr. Keyvin Darney, Dr. Nynke Kramer and Ms. Leonie Lautz all report the grant GP/EFSA/SCER/2015/01 from the European Food Safety Authority under which this study was funded and conducted. No conflicts of interests were identified. Dr. Dorne works as a member of staff at European Food Safety Authority and the design of the study as part of the grant proposal. No conflicts of interests were identified.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kasteel, E.E.J., Darney, K., Kramer, N.I. et al. Human variability in isoform-specific UDP-glucuronosyltransferases: markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch Toxicol 94, 2637–2661 (2020). https://doi.org/10.1007/s00204-020-02765-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02765-8