
Abstract
Alternaria molds can produce a variety of different mycotoxins, often resulting in food contamination with chemical mixtures, posing a challenge for risk assessment. Some of these metabolites possess estrogenic properties, an effect whose toxicological relevance is questioned in the light of the strong genotoxic and cytotoxic properties of co-occurring toxins. Thus, we tested a complex extract from A. alternata for estrogenic properties in Ishikawa cells. By assessing alkaline phosphatase activity, we did not observe estrogen receptor (ER) activation at non-cytotoxic concentrations (≤ 10 µg/ml). Furthermore, an extract stripped of highly genotoxic perylene quinones also did not mediate estrogenic effects, despite diminished genotoxic properties in the comet assay (≥ 10 µg/ml). Interestingly, both extracts impaired the estrogenicity of 17β-estradiol (E2) at non-cytotoxic concentrations (5–10 µg/ml), indicating anti-estrogenic effects which could not be explained by the presence of known mycoestrogens. A mechanism for this unexpected result might be the activation of the aryl hydrocarbon receptor (AhR) by Alternaria metabolites, as indicated by the induction of CYP1A1 transcription. While a direct influence on the metabolism of E2 could not be confirmed by LC–MS/MS, literature describing a direct interplay of the AhR with estrogenic pathways points to a corresponding mode of action. Taken together, the present study indicates AhR-mediated anti-estrogenic effects as a novel mechanism of naturally co-occurring Alternaria toxin mixtures. Furthermore, our results confirm their genotoxic activity and raise questions about the contribution of still undiscovered metabolites to toxicological properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Black molds of the genus Alternaria are ubiquitous fungi with a still unclear relevance as food contaminants. The infestation of crops designated for human consumption may result in the occurrence of a variety of toxic metabolites—the so-called Alternaria toxins—in food products. Particularly, tomatoes, vegetable oils, wheat flour and berries have been reported as potential sources of exposure (Lee et al. 2015; Puntscher et al. 2018a; Tournas and Katsoudas 2005).
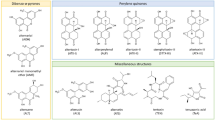
Alternaria spp. produce a cocktail of secondary metabolites, whereby the composition depends on the fungal strain and the respective growth conditions (Zwickel et al. 2016, 2018). The metabolite of highest prevalence in food samples is usually tenuazonic acid (TeA), a tetramic acid derivative (Fig. 1a). It exerts mild acute toxic properties, and is of little concern for human health according to regulative authorities (EFSA 2016).

Chemical structures of selected Alternaria toxins, grouped as a tenuazonic acid, b dibenzo-α-pyrones, c perylene quinones and d miscellaneous structures
Alternariol (AOH) and alternariol monomethyl ether (AME) represent the so far most-studied Alternaria metabolites. Together with other compounds including altenuene (ALT), iso-altenuene (iso-ALT) or altenuisol (ATL), they form the dibenzo-α-pyrone group of Alternaria toxins (Fig. 1b). Both AOH and AME have been found to act cytotoxic in different human cell lines in two-digit micromolar concentrations (Fehr et al. 2010). Also, they have been described to exert genotoxicity in colon carcinoma cells, an effect which is mainly caused ascribed to their ability to poison topoisomerase enzymes, which are crucial for maintaining DNA integrity by avoiding torsion stress during replication and transcription (Fehr et al. 2009). Moreover, the induction of oxidative stress might contribute to their DNA-damaging properties (Aichinger et al. 2017; Tiessen et al. 2013a). Recently, AOH and AME were also reported to cause cytotoxicity and genotoxicity in liver cell models, while metabolic activity contributes to cellular detoxification (Hessel-Pras et al. 2019). Thus, the latter metabolites are today considered as the main toxicological concern associated with Alternaria contaminations (EFSA 2016). However, AOH and several of its phase I and II metabolites were recently reported to act as mimetics of 17β-estradiol (E2), thus being able to bind to and activate estrogen receptors (ER), indicating the endocrine disruptive potential of these compounds (Dellafiora et al. 2018; Lehmann et al. 2006). Additionally, AOH was demonstrated to impact the endocrine system by increasing E2 and progesterone production and by up-regulating progesterone receptor expression (Frizzell et al. 2013). Furthermore, AOH was shown to potentiate the effects of other xenoestrogens, even at low concentrations where it does not exert estrogenic effects itself (Vejdovszky et al. 2017).
Another group of Alternaria toxins is constituted by the perylene quinones alterperylenol, altertoxins (ATX) I–III and stemphyltoxins (STE) I–III (Fig. 1c). Until now, these compounds were found, if at all, in comparably low concentrations in food (Puntscher et al. 2018a). Nevertheless, the analogues bearing an epoxide moiety like ATX-II possess potent genotoxic and mutagenic properties in vitro, which is suspected to result from the formation of DNA adducts (Tiessen et al. 2013b). In line with this hypothesis, ATX-II was shown to greatly exceed the DNA-breaking properties of AOH and AME, and to be one of the main genotoxic constituents in a complex extract of Alternaria alternata cultured on rice (Schwarz et al. 2012b). Another perylene quinone epoxide, STE-III, was recently described to impair DNA integrity to a comparable extent as ATX-II (Fleck et al. 2016). However, these toxins are expected to be rather unstable under physiologic conditions (Aichinger et al. 2018b) and during food processing (Puntscher et al. 2018b). Thus, their toxicological relevance in vivo remains to be clarified.
Lastly, Alternaria spp. produce further secondary metabolites with miscellaneous structures, whose toxic properties are not yet elucidated sufficiently (Fig. 1d).
With risk assessment strategies strongly relying on exposure and toxicity data of single compounds, the general co-occurrence of mycotoxins in chemical mixtures poses a potential problem. Cumulative or even synergistic combinatory effects might exceed the effects of single compounds, and it is unclear which bioactive mechanism is of significant impact for adverse outcomes, and which are overlaid by other, potentially dominating, pathways. This particularly applies to the estrogenicity of dibenzo-α-pyrones, as growth-stimulating effects triggered by endocrine disruption are obviously only of potential relevance in sub-cytotoxic and sub-genotoxic concentrations.
Hence, in the present study, we addressed the question whether Alternaria toxins exert estrogenic effects also in complex mixtures or whether the cellular effects are dominated by genotoxic constituents. Therefore, a naturally occurring mixture of Alternaria toxins was generated as an extract from Alternaria alternata strain DSM 62010 cultured on rice. In addition, to allow conclusions on the role of genotoxic perylene chinones, a “reduced extract” (RE) was produced, that was stripped of ATX-II and STE-III by fractionation. Furthermore, as compared to the complete extract, the composition of the RE correlates better with occurrence data of Alternaria toxins in food, where epoxide-carrying perylene quinones are usually not reported. In Ishikawa cells, an estrogen-sensitive cell model expressing both isoforms of the estrogen receptor (ER), we investigated and compared estrogenic versus genotoxic properties of these extracts.
Materials and methods
Chemicals and assay kits
Cell culture media and supplements were obtained from GIBCO Invitrogen (Karlsruhe, Germany), cell culture flasks and dishes from Sarstedt (Nürnbrecht, Germany). 17β-Estradiol, alternariol, alternariol monomethyl ether, ethidium bromide, neutral red, triton X-100, benzo[a]pyrene and 4-nitrophenyl phosphate were purchased from Sigma-Aldrich (Schnelldorf, Germany). Maxwell 16 LEV simplyRNA kit was obtained from Promega, QuantiTect® Reverse Transcription Kit, QuantiTect® SYBR® Green PCR Kit and primers for CYP1A1, ESR1, ESR2, ALPP, GADPH and ACTB from Qiagen (Hilden, Germany). Normal and low melting Agarose were purchased from Bio-Rad, formamidopyrimidine-DNA glycosylase from New England Biolabs (Frankfurt, Germany).
Generation and characterization of Alternaria extracts
A complete extract (CE) of rice infected with the Alternaria alternata strain DSM 62010 was generated as recently described (Puntscher et al. 2019). To generate a “reduced extract” (RE), stripped of the highly genotoxic perylene quinones altertoxin-II (ATX-II) and stemphyltoxin-III (STE-III), aliquots of the CE were dissolved in acetonitrile/water (50/50, v/v) and injected into a HPLC–DAD system using a gradient of water and acetonitrile, a constant flow rate of 1 ml/min and a Phenomenex Luna 5u C18 column for separation of the contained metabolites. Fractions were collected manually. The fractions from min 2 to 14 and from min 19 to 36 were rejoined to form the RE, whereas the fraction containing ATX-II and STE-III (min 14–min 19) was sampled separately. Both extracts were characterized using a LC–MS/MS multi-method as recently described (Puntscher et al. 2018a).
Cell culture
Ishikawa cells were purchased from ECACC (Wiltshire, UK). Produced cells stocks were kept in liquid nitrogen and routinely checked for the absence of mycoplasma contamination. Roughly 2 weeks before starting experiments, cells were taken into culture and grown in minimum essential medium (MEM), supplemented with 5% (v/v) heat-inactivated fetal bovine serum (FBS), 1% l-glutamine and 1% penicillin/streptomycin (P/S). For all assays, cells were seeded in “assay medium”, a phenol red-free Dulbecco’s Modified Eagle Medium/F-12 nutrient mixture (DMEM/F12) supplemented with 5% charcoal–dextran stripped (CD-) FBS and 1% P/S.
Sulforhodamine B (SRB) assay
10,000 Ishikawa cells/well were seeded to 96-well plates in assay medium, allowed to grow for 48 h, and then incubated with the test mixtures and the respective controls for another 48 h. Subsequently, cells were fixed by addition of trichloroacetic acid. After 1 h at 4 °C, plates were washed four times with water and allowed to dry overnight. Afterwards, they were incubated with a solution of 0.4% (w/v) SRB for 1 h at room temperature, washed twice with water, twice with 1% (v/v) acetic acid and again dried overnight. Then, a TRIS buffer with a pH of 9 was added to resolve the dye, and the absorbance at 570 nm was determined with a PerkinElmer Victor3V plate reader. All samples were measured in triplicate per plate and their mean values, a measure for the protein content of the wells, were related to the solvent control (1% v/v DMSO).
Neutral red (NR) assay
Cell seeding and incubation were carried out as described for the SRB assay. Neutral red medium (4 µg/ml neutral red in DMEM/F-12 supplemented with 5% FCS and 1% P/S) was prepared 1 d before its use, incubated overnight at 37 °C, centrifuged for 10 min at 600 rcf and filtered to remove formed crystals. After 48 h incubation, cells were incubated with neutral red medium for 3 h at 37 °C. Subsequently, the medium was removed and cells were washed with Dulbecco’s PBS. After adding a destaining solution (1% acetic acid in a 1:1 mixture of water and ethanol), the plates were rapidly shaken and then, the absorption at 540 nm was measured with a PerkinElmer Victor3V plate reader. All samples were measured in triplicate per plate and their mean values, a measure for the ability of viable cells to incorporate the dye into their lysosomes, were related to the solvent control (1% v/v DMSO).
Single-cell gel electrophoresis (“comet assay”)
The comet assay was performed according to the guidelines of Tice et al. (Tice et al. 2000) with some modifications. Briefly, 200,000 cells were seeded to petri dishes (dm = 3.5 cm) using assay medium and allowed to attach for 24 h. Then, cells were incubated with the test mixtures or the solvent control for 1 h. After washing them thrice with PBS, trypsin was added to harvest singularized cells. An aliquot of the cells was stained with trypan blue for counting and assessing the viability. In case the latter was below 80%, the assay was not continued. 2× 30,000 cells per sample were embedded in agarose on object slides. Those were put in a lysis buffer (1% lauroyl sarcosilate, EDTA, 1% Triton-X100) overnight. Subsequently, slides were treated with formamidopyrimidine-DNA glycosylase (FPG) or FPG buffer for 30 min at 37 °C. After equilibrating for 20 min, electrophoresis (25 V, 300 mA) was carried out for 20 min under strong alkaline conditions (pH 13). Afterwards, the slides were neutralized and stained with ethidium bromide. Microscopic analysis was carried out on a Zeiss Axioskop, and the “Comet Assay IV” software (Perceptive Instruments, UK) was used to score 100 cells per slide, whose mean tail intensity was calculated as a measure for DNA damage.
Alkaline phosphatase (AlP) assay
The activity of AlP after 48 h of incubation was determined as a measure for ER activation as recently described (Aichinger et al. 2018a). In brief, we used assay medium to seed 10,000 cells/well into 96-well plates. After allowing to attach for 48 h, cells were incubated with E2, the extracts or combinations of the latter for another 48 h. Afterwards, cells were washed thrice with PBS and lyzed by quick freezing at − 80 °C for at least 20 min. Plates were allowed to thaw at room temperature for 5 min before the detection buffer (5 mM 4-nitrophenylphosphate, 1 M diethanolamine, 0.24 mM MgCl2, pH 9.8) was added. Subsequently, the increase of absorption at 405 nm was measured for 1 h every 3 min. The slope of the linear range of the obtained curve was calculated as a measure for the activity of the enzyme. Every incubation was measured in triplicate, and means of these technical replicates were used for further analysis.
Quantitative real-time (qRT-) PCR
100,000 Ishikawa cells/well were seeded in 12-well plates and grown for 72 h, using assay medium. Then, the cells were incubated with the solvent control, two concentrations of CE, 1 nM E2, a combination of CE and E2 or 5 µM of benzo[a]pyrene (B[a]P), which served as a positive control, for 24 h. RNA extraction was performed using a Maxwell™ 16 LEV simplyRNA Cells kit (Promega) according to the manufacturer’s manual. Briefly, the medium was removed, cells were washed with PBS, and a homogenization solution was added to the wells. Cells were harvested, singularized and lyzed. The lysates were transferred to cartridges and RNA extraction was carried out with the Maxwell™ 16 instrument. After centrifugation, the RNA content of the resulting supernatant was determined with a Nanodrop 2000/2000c spectral photometer.
A QuantiTect® Reverse Transcription Kit was used to convert mRNA to cDNA according to the manufacturer’s protocol. In brief, 1 µg RNA of each sample was mixed with RNAse-free water plus a gDNA wipeout solution, and was incubated for 2 min at 42 °C. The RT-MasterMix was added and the tubes were incubated for another 15 min at 42 °C and for 2 min at 95 °C. The resulting solutions of cDNA were stored at − 20 °C until further processing.
qRT-PCR was conducted with primers for CYP1A1, ESR1, ESR2 and ALPP genes, with GAPDH and ACTB serving as housekeeping genes. cDNA was mixed with a SYBR-green kit and the respective primer, and PCR was performed with a StepOne Plus™ thermocycler. 2−ΔΔCT values were calculated for each gene for a relative quantification of gene transcription as suggested by Schmittgen and Livak (2008).
LC–MS/MS
LC–MS/MS measurements to determine E2 metabolism were performed on a Dionex Ultimate 3000 UHPLC system coupled to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Scientific). An Acquity UPLC® HSS T3 column (1.8 µm, 2.1 × 100 mm; Waters) equipped with a VanGuard pre-column (1.8 µm) served to achieve chromatographic separation. As mobile phases, water with 0.3 mM ammonium fluoride as additive (A) and acetonitrile (B) were used at a flow rate of 0.4 mL/min. Measurements were performed in the negative electrospray ionization mode.
Results
Extract composition
Both the complete and the reduced extract were analyzed for a number of important Alternaria toxins with a LC–MS multi-method as described previously in detail (Puntscher et al. 2018a). Representing more than 50% of the total extract weight, tenuazonic acid (TeA) was found to be the dominant compound of the CE, followed by the perylene quinones STE-III, ATX-II, alterperylenol (AP) and ATX-I with concentrations around 1–2% (Table 1). AOH and AME were much less abundant, with a cumulative content of 0.16%. The aim in generating the RE was to strip the extract of the highly genotoxic ATX-II and STE-III. By removing the corresponding fractions, both compounds were reduced to levels below the LOD. Furthermore, small parts of AOH, AME, tentoxin (TEN), ATX-I and alterperylenol were lost during the work-up. Of note, altenusin (ALS) was the only metabolite that was found to be more concentrated in the RE. Additionally, the method included iso-altenuene, altenuic acid and several glycosides and sulfates of AOH and AME, all of which were not found in the extracts.
Impact of extracts on ER stimulation
Estrogenicity of extracts was determined via assessing the activity of alkaline phosphatase (AlP), an enzyme whose transcription is regulated by ER activity. The impact on the expression of AlP was measured after 48 h incubation of Ishikawa cells as conversion of 4-nitrophenylphosphate to 4-nitrophenol, which was determined photometrically. No induction of estrogenic stimuli could be observed for concentrations up to 10 µg/ml for both extracts (Fig. 2a). At even higher concentrations, the signal significantly declined, probably due to cytotoxicity.
However, when the extracts were incubated in combination with 1 nM E2, a significant decrease in 4-NP formation was observed at concentrations ≥ 5 µg/ml (Fig. 2b), thus indicating anti-estrogenic properties of both extracts, independent from the content of ATX-II and STE-III.

Estrogenic impact of Alternaria extracts, a alone or b on the activity of E2, as measured by the AlP assay. All values are depicted as means + SD of at least four independent experiments and in relation to the stimulative effect of 1 nM E2. Significant differences to the respective solvent control (a) or the lowest applied concentration of the respective extract (b) were calculated using one-way ANOVA, followed by Fisher’s LSD post hoc testing, and are indicated with “***” (p < 0.001)
Contribution of AOH and AME
To assess whether at least a part of the observed anti-estrogenic effects was caused by the previously described mycoestrogens AOH and AME, the two compounds were purchased, mixed in the ratio at which they were found in the CE, and used to perform AlP assays in Ishikawa cells. Those measurements revealed no estrogenic effects for up to a cumulative concentration of 550 nM, and no diminishing effect on the estrogenicity of 1 nM E2 at any applied concentration (Fig. 3). AlP activity was significantly increased only at the highest measured dose (2.5 µM AOH/2 µM AME), which would correspond to a complete extract concentration of approximately 830 µg/ml, a dose high above any applied concentration and clearly beyond the cytotoxic/genotoxic threshold range.

The possible contribution of AOH and AME to the estrogenic impact of extracts, as measured by the AlP assay. Values are depicted as means + SD of at least three independent experiments and in relation to the positive control (1 nM E2). Significant differences to the solvent control were calculated using one-way ANOVA, followed by Fisher’s LSD post hoc testing, and are indicated with “***” (p < 0.001). No significant differences were found between the combinations of AOH, AME and E2 with both E2 alone or the combination with the lowest applied mycotoxin concentration
Cytotoxicity
The influence of Alternaria extracts on cell viability and growth of Ishikawa cells was assessed at the level of protein content (SRB assay) after 48 h incubation. For the CE, the onset of cytotoxicity was observed at concentrations ≥ 10 µg/ml (Fig. 4a). Of note, the removal of ATX-II and STE-III failed to diminish cytotoxicity. The RE also reduced the cellular protein at concentrations ≥ 10 µg/ml, but at 5 µg/ml, already a non-significant reduction was apparent. Cell viability was significantly lower after treatment with the RE as compared to the CE at both 5 µg/ml and 10 µg/ml. At 50 µg/ml, both extracts were highly toxic and reduced cell viability below 40%, regardless of a combination with E2. The incubation with 1 nM E2 alone did not result in an increase of cellular protein.
To exclude false-negative results, data for the CE were reconfirmed by assessing lysosomal activity by neutral red staining (Fig. 4b). Herewith, no significant cytotoxic effect of the CE could be observed for up to 10 µg/ml. Of note, 1 nM E2 lead to a significant (one-sided Student’s t test, p < 0.05) increase of absorption, but the combination with 10 µg/ml CE reduced the lysosomal activity to 91.5%, which could point at a reduction of estrogen-induced cell growth.

Cytotoxicity of A. alternata extracts, as determined by a SRB or b neutral red assay. Values are displayed as means + SD of at least four independent experiments and in relation to the solvent control (1% v/v DMSO), which is indicated as a dotted line. Significant differences between extracts and solvent control were calculated using one-way ANOVA, followed by Fisher’s LSD post hoc testing, and are indicated with “a” (p < 0.05). Significant differences of extract/E2 to 1 nM E2 were calculated using one-way ANOVA, followed by Fisher’s LSD post hoc testing, and are indicated with “b” (p < 0.05). Differences between the two extracts at the same concentration were analyzed by Student’s t test and are indicated by “*” (p < 0.05)
Genotoxicity
The induction of DNA strand breaks was measured by comet assay after 1 h incubation of Ishikawa cells with the extracts. Herewith, we found both extracts to significantly damage the DNA at concentrations ≥ 5 µg/ml (Fig. 5). The genotoxicity of the CE was significantly higher as compared to the RE at 5 and 10 µg/ml. Additionally, we observed both extracts to induce FPG-sensitive sites at different concentrations, thus indicating an oxidative damage of DNA bases at doses as low as 1 µg/ml.

DNA strand breaks after 1 h incubation with reduced extract (RE) and complete extract (CE), as measured with the comet assay. UV radiation served as a positive control (PC). Data are presented as mean ± SD of at least four independent experiments. Significant differences (p < 0.05) to the respective solvent control (SC, 1% v/v DMSO) were calculated by one-way ANOVA, followed by Fisher´s LSD post hoc test and are indicated with “+” (p < 0.05). Significant differences (p < 0.05) between the same concentrations of RE and CE were calculated by Student’s t test are indicated by “#”. “*”Indicate significant differences (p < 0.05) between FPG treated and untreated samples, which were analyzed by Student’s t test
Gene transcription
A possible influence of a 24 h incubation of Ishikawa cells with 5 µM or 10 µM of the complete Alternaria extract on the transcription of several genes was analyzed by measuring mRNA transcript levels with qRT-PCR in comparison to the solvent control (1% DMSO). For both concentrations, and also for 1 nM E2, we did not observe any significant change of ESR1 (ERα) or ESR2 (ERβ) transcription (Fig. 6a) as compared to the solvent control.
The transcription of ALPP (AlP) was 18-fold induced by 1 nM E2, but not by the extract (Fig. 6b). Of note, the combined incubation of E2 with 10 µg/ml CE resulted in significantly lower ALPP transcript levels (RQ = 3.9), thus confirming the antagonistic interaction observed in ALP assays.

Transcript levels of aESR, bALPP and cCYP1A1 genes in Ishikawa cells after a 24 h incubation with CE and/or E2, as measured by qRT-PCR. mRNA quantities in relation to the solvent control (RQ), normalized to β-actin and GADPH (2−ΔΔCT) are shown. Data are presented as mean + SD of three independent experiments. Significant differences (p < 0.05) were calculated by Student’s t test and are indicated with “*” (compared to the solvent control) or “#” (compared to other samples)
Likewise, we measured mRNA levels of CYP1A1, a gene induced by the aryl hydrocarbon receptor (AhR) activation, after a 24 h incubation of Ishikawa cells with the CE (Fig. 6c). Here, a dose-dependent increase of gene transcription was observed at 5 µg/ml (RQ = 3.9) and 10 µg/ml (RQ = 11.0). Furthermore, 5 µM of the known AhR activator B[a]P, which served as a positive control, potentiated gene transcription by a factor of 16.8. As expected, E2 did not induce CYP1A1 transcription by itself or alter the impact of the CE in this regard.
E2 metabolism
To assess whether the observed induction of CYP1A1 transcription might enhance the metabolic degradation of E2, we incubated Ishikawa cells for 24 h with the hormone both in and without the presence of 10 µg/ml CE. After incubation, E2 levels were measured in supernatant media by LC–MS/MS. E2 was quite stable in cell culture medium, declining only marginally from 0.83 nM to 0.68 nM after 48 h of incubation at 37 °C. In the presence of cells, E2 levels were reduced to 0.170 ± 0.026 nM (mean ± SD) over 24 h of incubation. The depletion was not significantly different in the presence of the CE (0.143 ± 0.025 nM). Of note, considerable amounts of a metabolite with m/z 351 were observed only in the samples incubated with both the CE and E2. Both the mass and the fragmentation pattern were identical with estradiol-3-sulfate, which was included in our method, but the retention time differed. Hence, the exact identity of this compound could not be revealed. A shift in retention time of estradiol-3-sulfate due to effects of the matrix or the appearance of another estradiol sulfate isomer seems possible.
Discussion
In the past, significant research has been conducted to investigate the effects of Alternaria toxins as individual compounds. However, these fungi produce a large and diverse pattern of toxins, which might lead to an overlay of distinct bioactivities depending on the qualitative and quantitative compositions of the exposure. In particular, it is up to question whether estrogenic effects of dibenzo-α-pyrone derivatives are of relevance for risk assessment considering the co-occurrence with highly genotoxic compounds.
To address these points, we focused on determining estrogenic and genotoxic effects of Alternaria toxins in a realistic mixture, an extract of the Alternaria alternata strain DSM 62010 cultured on rice.
The characterization of the full extract by LC–MS revealed TeA, a rather weak toxin, to be the most abundant constituent. However, the most outstanding result of the extract analysis arises from comparison of the toxin pattern to the metabolome of other Alternaria strains (Zwickel et al. 2018). Under the applied culture conditions, DSM 62010 apparently produces much higher amounts of the perylene quinones ATX-I, ATX-II, AP and STE-III (Table 1). Thus, it was speculated that the well-described cytotoxic and genotoxic effects of ATX-II and STE-III (Fleck et al. 2016; Schwarz et al. 2012b) might overlay any possible impact of co-occurring dibenzo-α-pyrones on ER stimulation. However, we recently demonstrated that the stability of ATX-II appears to be limited under physiological conditions, presumably due to the reactivity of the epoxide moiety (Aichinger et al. 2018b). This might contribute to the fact that—on the contrary to AOH and AME—ATX-II and STTX-III were not detectable in the urine or feces of rats after administration of our extract (Puntscher et al. 2019). Therefore, it cannot be excluded that ATX-II and STE-III might be of relevance in the gastrointestinal tract, but the present data argue about a potential loss of these genotoxic perylene chinones, so they are likely insignificant for the evaluation of systemic toxic effects like endocrine disruption. For that reason, we produced a reduced extract (RE) which consisted of the complete extract (CE) stripped of those two metabolites and compared the two extracts regarding their toxicological impact on the estrogen-sensitive human adenocarcinoma Ishikawa cell line.
Interestingly, even as the absence of genotoxic perylene quinones was confirmed by LC–MS/MS (Table 1), the RE exerted similar cytotoxic properties as the CE after 48 h of cell incubation (Fig. 4). It seems that other compounds are mainly responsible for those effects.
A similar picture was revealed at the level of genotoxicity, which was measured by Comet assay after a 1 h incubation of Ishikawa cells with both extracts. In this setting, the CE was found to impair DNA integrity at concentrations ≥ 5 µg/ml, and at 1 µg/ml, significant oxidative damage of DNA bases was detectable (Fig. 5). But also the RE caused DNA damage, albeit at slightly higher concentrations (10 µg/mL), which suggests that other compounds are contributing significantly to the genotoxicity of the applied extracts. However, it seems highly unlikely that one of the other Alternaria toxins whose concentration in the extracts was assessed by LC–MS plays an important role in that regard. AOH and AME were previously reported to act genotoxic in two-digit micromolar concentrations in vitro (Fehr et al. 2009; Schwarz et al. 2012a), but their cumulative concentration in the highest applied extract dose was calculated to be 54 nM and is thus approximately a factor 1000 lower than needed to impair DNA integrity. The toxicity of ATX-I is not very well described in literature, but the compound was found to be more than 10 times less genotoxic as compared to ATX-II in several cell lines (Fleck et al. 2014), while it was less concentrated in the CE than both ATX-II and STE-III. For AP, no data on its impact on the DNA exists so far. However, the compound is structurally similar to ATX-I and particularly also lacks the epoxide group that is suspected to cause strong genotoxicity of other perylene quinones. Thus, one can assume that we missed one or several major contributing genotoxicants. One suspect compound could be ATX-III, a perylene quinone carrying two epoxide groups, which is usually not included in analytical methods due to low prevalence, but is suspected to be of even higher mutagenicity and genotoxicity than ATX-II (Stack and Prival 1986).
Due to the genotoxic and cytotoxic properties of both extracts, their potential estrogenicity could not be observed in AlP assays (Fig. 2a). But interestingly, when combined with 1 nM E2, both extracts were able to diminish the estrogenic stimulus of the endogenous hormone to trigger AlP enzyme synthesis (Fig. 2b), an effect that was also confirmed at the level of gene transcription (Fig. 6b). Of note, this effect was observed at non- and low-cytotoxic concentrations (≤ 10 µg/ml, Fig. 4), thus proving a direct interplay with ER activation or signaling. This finding could be of toxicological relevance, as the interference with endogenous estrogenic stimuli is considered a toxic mode of action, potentially causing problems with fertility and adverse pregnancy outcomes (Kaplan-Kraicer et al. 1996; Zhang et al. 2017).
It is, however, not entirely clear which of the present Alternaria toxins are responsible for this anti-estrogenic effects. For AOH and AME, two compounds previously reported to interact with the ER (Dellafiora et al. 2018; Lehmann et al. 2006), we tested concentrations corresponding to the amount of the substances in the CE by AlP assay, both with and without adding E2. At the doses present in 10 µg/ml CE (25 nM AOH, 20 nM AME), we did not observe any effects as compared to the controls (Fig. 3). Only at a 100-fold higher concentration, this binary mixture induced ER-related enzymatic activity, while a suppressing effect on E2 estrogenicity could not be found at any concentration. This suggests that other compounds are responsible for the anti-estrogenic activity of the extracts.
Regarding the underlying mechanism for the latter effect, we could rule out a possible influence of the extracts on ER gene transcription by qRT-PCR (Fig. 6a). One conceivable mode of action is an interplay of AhR/ARNT signaling with ER-related responses. This receptor is activated by the presence of molecules with a planar hydrocarbon structure and subsequently triggers the transcription and expression of phase I metabolic enzymes, in particular CYP1A1 (Hu et al. 2007; Pahlke et al. 2016). Interestingly, CYP1A1 is also involved in the metabolic inactivation of E2, and thus an activation of the AhR/ARNT pathway was previously reported to increase the metabolic degradation of E2 (Suchar et al. 1996).
Additionally, the AhR was described to interact with estrogen-mediated processes by increasing the ubiquitinylation and proteasomal degradation of the ER (Ohtake et al. 2009; Wormke et al. 2003) as well as by competing with ERs for nuclear co-regulatory proteins (Beischlag et al. 2002; Nguyen et al. 1999).
Recently, both AOH and AME were found to activate the AhR, resulting in enhanced transcription of CYP1A1, an enzyme of phase I metabolism with both compounds representing substrates as well (Pahlke et al. 2016; Schreck et al. 2012). But ATX-II was also found to induce CYP1A1 transcription as a marker for AhR activation in human esophagal cells, even at very low concentrations (Pahlke et al. 2016), giving reason to speculations that due to their planar structure, AhR/ARNT signaling is a common molecular mechanism for perylene quinones in general.
Thus, we assessed the ability of the CE to induce AhR/ARNT signaling via determining a possible induction of CYP1A1 transcription, and found the extract to act potently in that regard (Fig. 6c). This lead us to the question whether an incubation of cells with the extract would lead to an enhanced degradation of E2, which we assessed by LC–MS measurements. Therewith, E2 levels were found to be slightly and non-significantly lower in co-incubated samples (see LC–MS results section). Of note, we observed the occurrence of an additional metabolite, presumably an estradiol sulfate not included in our analytical method. However, one has to note that the impact of the CE on E2 metabolism is—if present at all—much too low to explain the strong anti-estrogenic effects. As explained above, AhR activation is expected to interfere with ERs by additional modes of actions, which might be responsible for causing the intriguing activities of the investigated extracts.
Taken together, we hereby report complex extracts of Alternaria alternata to act genotoxic and anti-estrogenic in Ishikawa cells. The known toxins ATX-II and STE-III contributed, as expected, to the overall genotoxicity of the extract, but additional genotoxic constituents remain to be identified. In sub-cytotoxic concentrations, the complex extract suppressed the estrogenic effectiveness of E2, arguing for an endocrine disruptive potential of Alternaria toxins. Concerning the underlying mechanisms, we found the complete extract to induce the AhR/ARNT pathway, which might at least in part contribute to the observed phenomena. With respect to the endocrine disruptive potential, the present study shows that naturally occurring complex mixtures of Alternaria toxins might not only contain weak estrogens like AOH, AME and respective metabolites, but seem to provide also anti-estrogenic activity, whereby the relevant compounds remain to be identified.
Abbreviations
- AhR:
-
Aryl hydrocarbon receptor
- AME:
-
Alternariol monomethyl ether
- AlP:
-
Alkaline phosphatase
- AOH:
-
Alternariol
- AP:
-
Alterperylenol
- ATX:
-
Altertoxin
- DMEM/F12:
-
Dulbecco’s modified Eagle medium/F-12 nutrient mixture
- DMSO:
-
Dimethyl sulfoxide
- E2:
-
17β-estradiol
- FBS:
-
Fetal bovine serum
- MEM:
-
Minimum essential medium
- NR:
-
Neutral red
- PBS:
-
Phosphate-buffered saline solution
- qRT-PCR:
-
Quantitative real-time PCR
- STE-III:
-
Stemphyltoxin-III
References
Aichinger G, Beisl J, Marko D (2017) Genistein and delphinidin antagonize the genotoxic effects of the mycotoxin alternariol in human colon carcinoma cells. Mol Nutr Food Res. https://doi.org/10.1002/mnfr.201600462
Aichinger G, Beisl J, Marko D (2018a) The hop polyphenols xanthohumol and 8-prenyl-naringenin antagonize the estrogenic effects of fusarium mycotoxins in human endometrial cancer cells. Front Nutr. https://doi.org/10.3389/fnut.2018.00085
Aichinger G, Puntscher H, Beisl J, Kutt ML, Warth B, Marko D (2018b) Delphinidin protects colon carcinoma cells against the genotoxic effects of the mycotoxin altertoxin II. Toxicol Lett 284:136–142. https://doi.org/10.1016/j.toxlet.2017.12.002
Beischlag TV, Wang S, Rose DW et al (2002) Recruitment of the NCoA/SRC-1/p160 family of transcriptional coactivators by the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator complex. Mol Cell Biol 22(12):4319–4333
Dellafiora L, Warth B, Schmidt V et al (2018) An integrated in silico/in vitro approach to assess the xenoestrogenic potential of Alternaria mycotoxins and metabolites. Food Chem 248:253–261. https://doi.org/10.1016/j.foodchem.2017.12.013
EFSA (2016) Dietary exposure assessment to Alternaria toxins in the European population. EFSA J 14(12):e04654-n/a. https://doi.org/10.2903/j.efsa.2016.4654
Fehr M, Pahlke G, Fritz J et al (2009) Alternariol acts as a topoisomerase poison, preferentially affecting the IIalpha isoform. Mol Nutr Food Res 53(4):441–451. https://doi.org/10.1002/mnfr.200700379
Fehr M, Baechler S, Kropat C et al (2010) Repair of DNA damage induced by the mycotoxin alternariol involves tyrosyl-DNA phosphodiesterase 1. Mycotoxin Res 26(4):247–256. https://doi.org/10.1007/s12550-010-0063-6
Fleck SC, Pfeiffer E, Podlech J, Metzler M (2014) Epoxide reduction to an alcohol: a novel metabolic pathway for perylene quinone-type Alternaria mycotoxins in mammalian cells. Chem Res Toxicol 27(2):247–253. https://doi.org/10.1021/tx400366w
Fleck SC, Sauter F, Pfeiffer E, Metzler M, Hartwig A, Koberle B (2016) DNA damage and repair kinetics of the Alternaria mycotoxins alternariol, altertoxin II and stemphyltoxin III in cultured cells. Mutat Res Genet Toxicol Environ Mutagen 798–799:27–34. https://doi.org/10.1016/j.mrgentox.2016.02.001
Frizzell C, Ndossi D, Kalayou S et al (2013) An in vitro investigation of endocrine disrupting effects of the mycotoxin alternariol. Toxicol Appl Pharmacol 271(1):64–71. https://doi.org/10.1016/j.taap.2013.05.002
Hessel-Pras S, Kieshauer J, Roenn G, Luckert C, Braeuning A, Lampen A (2019) In vitro characterization of hepatic toxicity of Alternaria toxins. Mycotoxin Research 35(2):157–168. https://doi.org/10.1007/s12550-018-0339-9
Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR (2007) Induction of CYP1A1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol 71(6):1475–1486. https://doi.org/10.1124/mol.106.032748
Kaplan-Kraicer R, Kostovetsky I, Bardin CW, Shalgi R (1996) Tamoxifen and RU39411 synergize with mifepristone to produce preimplantation pregnancy loss by increasing embryo transport (rat). Contraception 53(6):367–373. https://doi.org/10.1016/0010-7824(96)00087-X
Lee HB, Patriarca A, Magan N (2015) Alternaria in food: ecophysiology, mycotoxin production and toxicology. Mycobiology 43(2):93–106
Lehmann L, Wagner J, Metzler M (2006) Estrogenic and clastogenic potential of the mycotoxin alternariol in cultured mammalian cells. Food Chem Toxicol 44(3):398–408. https://doi.org/10.1016/j.fct.2005.08.013
Nguyen TA, Hoivik D, Lee JE, Safe S (1999) Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch Biochem Biophys 367(2):250–257. https://doi.org/10.1006/abbi.1999.1282
Ohtake F, Fujii-Kuriyama Y, Kato S (2009) AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem Pharmacol 77(4):474–484. https://doi.org/10.1016/j.bcp.2008.08.034
Pahlke G, Tiessen C, Domnanich K et al (2016) Impact of Alternaria toxins on CYP1A1 expression in different human tumor cells and relevance for genotoxicity. Toxicol Lett 240(1):93–104. https://doi.org/10.1016/j.toxlet.2015.10.003
Puntscher H, Kütt M-L, Skrinjar P et al (2018a) Tracking emerging mycotoxins in food: development of an LC–MS/MS method for free and modified Alternaria toxins. Anal Bioanal Chem 410(18):4481–4494. https://doi.org/10.1007/s00216-018-1105-8
Puntscher H, Marko D, Warth B (2018b) The fate of altertoxin ii during tomato food processing. Front Nutr. https://doi.org/10.26434/chemrxiv.7251686
Puntscher H, Hankele S, Tillmann K et al (2019) First insights into Alternaria multi-toxin in vivo metabolism. Toxicol Lett 301:168–178. https://doi.org/10.1016/j.toxlet.2018.10.006
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108
Schreck I, Deigendesch U, Burkhardt B, Marko D, Weiss C (2012) The Alternaria mycotoxins alternariol and alternariol methyl ether induce cytochrome P450 1A1 and apoptosis in murine hepatoma cells dependent on the aryl hydrocarbon receptor. Arch Toxicol 86(4):625–632. https://doi.org/10.1007/s00204-011-0781-3
Schwarz C, Kreutzer M, Marko D (2012a) Minor contribution of alternariol, alternariol monomethyl ether and tenuazonic acid to the genotoxic properties of extracts from Alternaria alternata infested rice. Toxicol Lett 214(1):46–52. https://doi.org/10.1016/j.toxlet.2012.08.002
Schwarz C, Tiessen C, Kreutzer M, Stark T, Hofmann T, Marko D (2012b) Characterization of a genotoxic impact compound in Alternaria alternata infested rice as altertoxin II. Arch Toxicol 86(12):1911–1925. https://doi.org/10.1007/s00204-012-0958-4
Stack ME, Prival MJ (1986) Mutagenicity of the Alternaria metabolites altertoxins I, II, and III. Appl Environ Microbiol 52(4):718–722
Suchar LA, Chang RL, Thomas PE, Rosen RT, Lech J, Conney AH (1996) Effects of phenobarbital, dexamethasone, and 3-methylcholanthrene administration on the metabolism of 17 beta-estradiol by liver microsomes from female rats. Endocrinology 137(2):663–676. https://doi.org/10.1210/endo.137.2.8593816
Tice RR, Agurell E, Anderson D et al (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35(3):206–221. https://doi.org/10.1002/(sici)1098-2280(2000)35:3%3c206:aid-em8%3e3.0.co;2-j
Tiessen C, Fehr M, Schwarz C et al (2013a) Modulation of the cellular redox status by the Alternaria toxins alternariol and alternariol monomethyl ether. Toxicol Lett 216(1):23–30. https://doi.org/10.1016/j.toxlet.2012.11.005
Tiessen C, Gehrke H, Kropat C et al (2013b) Role of topoisomerase inhibition and DNA repair mechanism in the genotoxicity of alternariol and altertoxin II. World Mycotoxin J 6(3):233–244
Tournas VH, Katsoudas E (2005) Mould and yeast flora in fresh berries, grapes and citrus fruits. Int J Food Microbiol 105(1):11–17. https://doi.org/10.1016/j.ijfoodmicro.2005.05.002
Vejdovszky K, Hahn K, Braun D, Warth B, Marko D (2017) Synergistic estrogenic effects of Fusarium and Alternaria mycotoxins in vitro. Arch Toxicol 91(3):1447–1460. https://doi.org/10.1007/s00204-016-1795-7
Wormke M, Stoner M, Saville B et al (2003) The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol Cell Biol 23(6):1843–1855
Zhang Z, Hu Y, Guo J, et al. (2017) Fluorene-9-bisphenol is anti-oestrogenic and may cause adverse pregnancy outcomes in mice. Nat Commun 8:14585. https://doi.org/10.1038/ncomms14585. https://www.nature.com/articles/ncomms14585#supplementary-information
Zwickel T, Kahl SM, Klaffke H, Rychlik M, Muller ME (2016) Spotlight on the underdogs-an analysis of underrepresented Alternaria mycotoxins formed depending on varying substrate, time and temperature conditions. Toxins. https://doi.org/10.3390/toxins8110344
Zwickel T, Kahl SM, Rychlik M, Müller MEH (2018) Chemotaxonomy of mycotoxigenic small-spored Alternaria fungi—do multitoxin mixtures act as an indicator for species differentiation? Front Microbiol 9:1368. https://doi.org/10.3389/fmicb.2018.01368
Acknowledgements
Open access funding provided by the University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aichinger, G., Krüger, F., Puntscher, H. et al. Naturally occurring mixtures of Alternaria toxins: anti-estrogenic and genotoxic effects in vitro. Arch Toxicol 93, 3021–3031 (2019). https://doi.org/10.1007/s00204-019-02545-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02545-z