
Abstract
Despite the frequent infection of agricultural crops by Alternaria spp., their toxic secondary metabolites and potential food contaminants lack comprehensive metabolic characterization. In this study, we investigated their bioavailability, metabolism, and excretion in vivo. A complex Alternaria culture extract (50 mg/kg body weight) containing 11 known toxins and the isolated lead toxin altertoxin II (0.7 mg/kg body weight) were administered per gavage to groups of 14 Sprague Dawley rats each. After 3 h and 24 h, plasma, urine and feces were collected to determine toxin recoveries. For reliable quantitation, an LC–MS/MS method for the simultaneous detection of 20 Alternaria toxins and metabolites was developed and optimized for either biological matrix. The obtained results demonstrated efficient excretion of alternariol (AOH) and its monomethyl ether (AME) via feces (> 89%) and urine (> 2.6%) after 24 h, while the majority of tenuazonic acid was recovered in urine (20 and 87% after 3 and 24 h, respectively). Moreover, modified forms of AOH and AME were identified in urine and fecal samples confirming both, mammalian phase-I (4-hydroxy-AOH) and phase-II (sulfates) biotransformation in vivo. Despite the comparably high doses, perylene quinones were recovered only at very low levels (altertoxin I, alterperylenol, < 0.06% in urine and plasma, < 5% in feces) or not at all (highly genotoxic, epoxide-holding altertoxin II, stemphyltoxin III). Interestingly, altertoxin I was detected in all matrices of rats receiving altertoxin II and suggests enzymatic de-epoxidation in vivo. In conclusion, the present study contributes valuable information to advance our understanding of the emerging Alternaria mycotoxins and their relevance on food safety.
Similar content being viewed by others


Introduction
Alternaria mycotoxins can contaminate a broad variety of agricultural crops intended for human consumption. These include cereals like wheat, barley, and oats, as well as tomatoes, peppers, sunflower seeds, and fruits (Ostry 2008; Scott 2001; Solfrizzo 2017). Commonly known as prevalent plant pathogens, fungal species belonging to the genus Alternaria are producing these toxins as secondary metabolites. Despite their considerable adverse effects demonstrated in vitro, no legally binding guidelines or maximum levels for food or feed exist in the European Union to date (EFSA 2016). The lack of both, comprehensive occurrence data and toxicological characterization of Alternaria toxins, is a reason for incomplete exposure and risk assessment. Particularly data providing thorough toxicological in vivo characterization is scarce. Alternaria spp. are ubiquitously found in the environment and capable of spreading and proliferating even at low temperatures. Hence, plant or crop infestation can occur pre- and post-harvest, i.e. in the orchards or during transport, storage as well as after food processing. In fact, crop spoilage results in considerable economic losses worldwide.
Due to their highly diverse chemical structures, Alternaria toxins show a wide range of biological effects comprising cytotoxicity, genotoxicity, mutagenicity, and estrogenicity. However, most studies published to date, focus on single or a low number of these toxic molecules. Even the most recent dietary exposure assessment performed by the European Food Safety Authority (EFSA (2016)) included only the four key toxins alternariol (AOH), alternariol monomethyl ether (AME), tenuazonic acid (TeA), and tentoxin (TEN). Available occurrence data were evaluated using the TTC approach (thresholds of toxicological concern) indicating that human exposure to AOH and AME may exceed the TTC of 2.5 ng/kg body weight, while this was not the case for TEN and TeA (TTC of 1500 ng/kg body weight). The comparatively low TTC value for AOH and AME derives from their genotoxic and mutagenic effects (Brugger et al. 2006) related to topoisomerase I and II poisoning (Fehr et al. 2009). Furthermore, these compounds demonstrated estrogenic activity (Dellafiora et al. 2018; Lehmann et al. 2006). No genotoxic effects were described for TEN and TeA. TeA is reported to be acutely toxic to mice, rats and dogs (Griffin and Chu 1983; Pero et al. 1973), while TEN is a phytotoxin inhibiting photophosphorylation in chloroplasts (Arntzen 1972). The group of perylene quinones including altertoxin I (ATX-I), II (ATX-II), and III (ATX-III), alterperylenol (ALP), and stemphyltoxin III (STTX-III), gained increasing interest because of their high genotoxic potential. Especially ATX-II exhibited more potent genotoxic effects compared to AOH (Fleck et al. 2012; Schwarz et al. 2012).
Only limited data describing biotransformation of Alternaria toxins in plants and mammals is available in the literature. In general, biologically modified toxins often remain undetected by targeted analysis, while still contributing to the total contamination burden (Berthiller et al. 2013). Modified mycotoxins may still have toxicological impact and certain modifications can be reverted by hydrolysis during mammalian digestion. In phase-I metabolism, AOH and AME are hydroxylated at various positions as observed in microsomal incubations of rat, human, and porcine liver (Pfeiffer et al. 2007), in incubation with recombinant cytochrome P450 isoforms (Pfeiffer et al. 2008), and in rat liver slices (Burkhardt et al. 2011). In vivo, hydroxylated AOH metabolites have been identified in blood and feces of mice (Schuchardt et al. 2014). This study reported generally low systemic absorption and effective fecal (85–91%) and urinary (up to 9%) excretion rates of the orally administered radiolabeled AOH after 24 h. Similar results of poor absorption were reported for 14C-AME in Sprague Dawley rats (Pollock et al. 1982). Moreover, Sarkanj et al. (2018) reported AOH (up to 0.2 ng/mL) for the first time in human urine samples as new biomarkers of exposure. Catechols formed by hydroxylation of AOH and AME may be further methylated by catechol-O-methyltransferase (Pfeiffer et al. 2007; Tiessen et al. 2017), while AME was also reported to be demethylated by microsomes of rat, human, and porcine liver (Pfeiffer et al. 2007). However, AOH and AME are thermally and chemically rather stable in food matrices and therefore persistent during food processing (Estiarte et al. 2018; Siegel et al. 2010). Other dibenzopyrones are altenuene (ALT) and its isoform isoaltenuene (isoALT). Hydroxylation and methylation of catechols were confirmed also for these compounds in vitro (Pfeiffer et al. 2009a). However, no metabolism was observed for AOH, AME and ALT by human fecal microbiota (Lemke et al. 2016). Importantly, several conjugation products (phase-II metabolism) were reported for AOH and AME. Glucuronidation was confirmed for rat, human and porcine metabolism in vitro (Pfeiffer et al. 2009b), in Caco-2 cells (Burkhardt et al. 2009), in rat liver slices (Burkhardt et al. 2011), and mouse hepatoma cell lines (Burkhardt et al. 2012). Sulfation was reported for Caco-2 cells, rat liver cytosol (Burkhardt et al. 2009), and rat liver slices (Burkhardt et al. 2011), but also the fungus Alternaria alternata itself is capable of synthetizing this metabolite (Soukup et al. 2016). With respect to plant metabolism, glycosylations including β-d-glycopyranosides and 6′-malonyl-glucopyranosides were identified in tobacco BY-2 suspension cell culture (Hildebrand et al. 2015) and glucosylations of the sulfate conjugates in tomato tissue (Soukup et al. 2016). Rapid and nearly complete urinary excretion was confirmed for the comparatively polar tenuazonic acid (TeA) (Asam et al. 2013).
Very little is known about metabolic pathways of perylene quinones. The highly reactive epoxide groups of ATX-II and STTX-III has been suggested to cause DNA adducts leading to their severe genotoxicity (Fleck et al. 2016; Schwarz et al. 2012). Chemical reactions with thiols such as glutathione have been reported and support this hypothesis (Fleck et al. 2014b; Jarolim et al. 2017). However, the precise mechanism of action has not been elucidated so far. In human cell lines (and tomato tissue in case of ATX-II), both compounds were found to be reduced to their alcohol analogues, ATX-I and ALP, respectively (Fleck et al. 2014a, b; Puntscher et al. 2019c). Moreover, significant degradation or chemical reactivity of ATX-II was determined in the presence of the natural food component and the flavonoid delphinidin (Aichinger et al. 2018) and in tomato matrix (Puntscher et al. 2019c).
The aim of the present study was to elucidate Alternaria toxin in vivo metabolism by mimicking oral exposure of rats. Biological effects induced by a complex Alternaria culture extract comprising 11 quantified Alternaria toxins were compared to the effects of respective amounts of the isolated altertoxin II. Consequently, a major focus of this work was the development of an LC–MS/MS method for the reliable quantitation of multiple Alternaria toxins in different biological matrices comprising rat plasma, urine and feces. The application of this assay to the samples obtained from this experiment revealed intriguing information about the biological absorption, potential systemic availability, and the excretion rates of Alternaria toxins and metabolites.
Materials and methods
Chemicals and reagents
LC–MS grade acetonitrile (ACN), methanol (MeOH), and water were purchased from Honeywell (Seelze, Germany), ammonia solution (25% in water), ammonium acetate and formate from Sigma Aldrich. For rice extraction and preparative HPLC, Milli-Q water, MeOH (HPLC grade) and acetic and formic acid (p.a.) from Sigma Aldrich (Steinheim, Germany) and n-hexane (p.a.) from Carl Roth GmbH (Karlsruhe, Germany) were used. Ethyl acetate (anhydrous, 99.8%) was purchased from Sigma Aldrich (Steinheim, Germany). Alternaria toxin reference materials were acquired as follows: TeA and TEN were purchased from Sigma Aldrich (Steinheim, Germany), AOH and AME from Toronto Research Chemicals (Ontario, Canada) and ALS from Eubio (Vienna, Austria). ALT, isoALT and AA-III were kindly provided by Prof. Joachim Podlech (Institute of Organic Chemistry, Karlsruhe Institute of Technology, Germany) (Altemöller et al. 2006; Nemecek et al. 2013), AOH-3-Glc, AOH-9-Glc, AOH-3-Sulf, AME-3-Glc, and AME-3-Sulf were a gift from Dr. Hannes Mikula (Institute of Applied Synthetic Chemistry, Vienna University of Technology (TU Wien), Vienna, Austria) (Mikula et al. 2013). ATX-I, ATX-II, STTX-III and ALP were isolated from in-house-cultivated fungal cultures grown on rice by an optimized protocol based on Puntscher et al. (2019b) and Schwarz et al. (2012).
Stock solutions were prepared in MeOH at final concentrations of 10–500 µg/mL. Dibenzo-α-pyrone conjugates AOH-3-Glc, AOH-9-Glc, AOH-3-Sulf, AME-3-Glc, AME-3-Sulf were dissolved in water/ACN (20/80, v/v). By combining the individual stock solutions, multi-toxin working solutions containing all analytes were prepared freshly every 4 weeks resulting in concentrations of 1–60 µg/mL. All solid reference materials and solutions were stored at − 20 °C.
Alternaria culture extract
Long grain rice was artificially inoculated with the A. alternata strain DSM 62010 (DSMZ, Braunschweig, Germany) resulting in a heavily infested black culture after 21 days as reported by Puntscher et al. (2019b). After adding cold potassium chloride solution (0.15 mM, pH 3), the rice culture was extracted with ethyl acetate. The solvent of the combined organic phases was evaporated and the residual oily extract was stored at − 20 °C for further experiments.
Altertoxin-II isolation
The Alternaria toxin ATX-II was isolated form an Alternaria culture extract utilizing a semi-preparative HPLC-system including a Knauer Smartline 1000 (LPG) pump, a Knauer Manager 5000 controller, a manual injector, and a diode array detector (Agilent G1315D, with preparative flow cell). The instrument was operated with ClarityChrom Software, while the chromatographic data were acquired with ChemStation analysis software. The LC system included a Phenomenex Luna column (5u, 250 mm × 21.2 mm), equipped with a Phenomenex SecurityGuard column. A binary gradient program, using water (pH 3, adjusted with formic acid) as eluent A and methanol as eluent B, with a constant flow rate of 14 mL/min was used: After 1 min with 55% B, eluent B was increased to 58% within the following 45 min. Subsequently, the system was purged for 20 min with 100% B, before it was re-equilibrated at the starting conditions (55% B) for 20 min. Injection volumes were between 1 and 7 mL. The UV detector recorded chromatograms for the following wavelengths: 254 nm and wavelengths typically absorbed by perylene quinones: 262 nm, and 280 nm (Schwarz et al. 2012). The chromatographic peak at a retention time of 35–40 min was collected and identified as ATX-II using LC–MS/MS and HPLC–UV. After removing the methanol from the combined ATX-II containing eluent fractions by the use of a rotary evaporator, the remaining aqueous phase was extracted twice with cold chloroform using a separation funnel. Subsequently, the organic phase was again evaporated resulting in a yellowish residue, which was finally reconstituted in EtOH and immediately put to − 20 °C until further experiments. The chemical identity and purity (> 90%) of ATX-II was confirmed by LC–UV, LC–MS and 1H-NMR.
Experimental procedure
The animal study was approved by the Austrian Federal Ministry of Education, Science and Research (Austria, GZ: BMWFW-66.009/0120-WF/II/3b/2014) according to the animal welfare act 2012, BGBI. I Nr. 114/2012 (TVG 2012). Forty-two male Sprague Dawley rats, obtained from the Institute of Laboratory Animal Science and Genetics (Medical University of Vienna, Austria), were acclimated at the Center for Biochemical Research (Medical University of Vienna, Austria) for at least 14 days before the study was conducted. An overview of the animal study design is represented in Fig. S1. The rats were randomly assigned to one of the three test groups (each consisting of 14 rats) and received a single administration by gavage (Table 1). (1) The prepared Alternaria culture extract was administered to the “extract group” (50 mg/kg body weight, a dose determined as appropriate in a recently published pilot study (Puntscher et al. 2019b)). (2) Isolated ATX-II was administered to the ATX-II group (0.7 mg/kg body weight, corresponding to the amount quantified in the extract). (3) A solvent control not containing any Alternaria toxins was administered to the “control group”. For administration, extract and ATX-II doses were dissolved in a small amount of EtOH and sunflower seed oil resulting in a total administration volume of approximately 1 mL/300 g rat. All animals received the same amount of EtOH (90 µL/300 g body weight) and oil (900 µL/300 g body weight). Animals belonging to the control group received only the respective amount of the solvent EtOH in sunflower seed oil. The absence of Alternaria toxins in the respective the sunflower seed oil was confirmed by a validated LC–MS/MS method (sample #O7 in Puntscher et al. 2019a). Directly after administration, the animals were placed into metabolic cages of the type no. 3700M071 (diameter 23 cm, height 18 cm; from Techniplast, Varese, Italy). Both, feed and water were provided ad libitum. Collection funnels under the stainless steel grid floor of the metabolic cages allowed for the separated sampling of urine and feces. Body weight and temperature, as well as the consumption of water and feed during the experiment were documented for each rat (Table 1). Per day, six animals were treated: two from each administration group, of which one was euthanized after 3 and one after 24 h (see Fig. S1 for the study design). In total, seven animals of each group were killed after 3 h and seven after 24 h. Before euthanasia by cardiac puncture and cervical fracture, the animals were anesthetized by intravenous injection of ketamine (0.1 mL/100 g rat) and xylazine (0.1 mL/rat). Blood was collected directly from the heart using EDTA rinsed syringes. Aliquots were prepared for immediate hematology (1 mL in Vacuette, measured by an automated analyzer ADVIA 2120i, Siemens AG) and for separating plasma by centrifugation (4 °C, 2500 rcf, 10 min, by a Rotana 46 RC centrifuge, Hettich AG, Germany). All samples were directly put on dry ice for transport until storage at − 80 °C.
Sample preparation
Before extraction, fecal samples were lyophilized (for 24 h) to allow for quantitative analysis normalized to dry matter. The resulting dry fecal pellets were homogenized using a FastPrep-24 5G™ High Speed Homogenizer (MP Biomedicals Life Sciences, USA). Subsequently, fine powder aliquots of 20 mg were suspended in water (4 °C, 200 µL). Urine and plasma samples (200 µL) were also aliquoted on ice.
Urine, plasma, and fecal suspension (4 °C, 200 µL) were extracted by the addition of precooled extraction solvent (800 µL, ACN/MeOH/HAc, 49.5/49.5/1, v/v/v, − 20 °C). After thorough homogenization by vortexing and sonication (15 min in ice bath), the samples were stored at − 20 °C overnight to allow for precipitation of proteins. Subsequently, the samples were centrifuged (4 °C, 14,000 rpm, 10 min), before evaporating the supernatants (850 µL) to dryness, using a vacuum concentrator (Labconco, Missouri, USA) set to 4 °C. The resulting residues were reconstituted in 170 µL solvent (ACN/water (1:1, v/v, precooled to 4 °C) by careful vortexing and sonication (15 min in ice bath). After a final centrifugation step (4 °C, 20,000 rcf, 15 min for urine and fecal samples, 30 min for plasma samples), the supernatants were transferred to amber HPLC vials and stored at 4 °C in the LC autosampler for analysis. Thereafter, all samples were stored at − 80 °C. A fully 13C-labeled yeast extract was added to the plasma samples before extraction as internal standard for additional high-resolution mass spectroscopy experiments (Hermann et al. 2018).
If concentration results exceeded the provided calibration range, respective samples were diluted using ACN/water (50/50, v/v) and re-measured. This was the case for AOH, AME, TeA, and AST levels in fecal samples obtained from the extract group (dilution by a factor 10). Analogously, urine samples required a dilution by a factor of 2500 to quantify TeA. Solvent-matched calibration standards were applied for toxin quantitation in these cases. While matrix effects in highly diluted urine were neglected, they were taken into account for the less diluted fecal matrix. Hereby, the obtained results were corrected considering the signal ratio of solvent-matched and diluted fecal-matrix-matched (dilution 1–10) calibration standards.
LC–MS/MS analysis
Alternaria toxin and metabolite identification and quantitation was performed on a high-performance liquid chromatography triple quadrupole mass spectrometer system (HPLC UltiMate 3000, TSQ Vantage) coupled by a heated electrospray ionization interface (all Thermo Scientific). Chromatographic separation was achieved by the UHPLC column Acquity UPLC HSS T3 (1.8 µm, 2.1 × 100 mm, Waters, Austria) equipped with a VanGuard pre-column (1.8 µm, 2.1 mm, Waters, Austria) and a binary elution at a flow of 0.4 mL/min. The most favorable chromatographic performance was obtained by the use of an aqueous NH4F solution (0.3 mM) as eluent A and methanol as eluent B. The instrument’s autosampler and column oven were set to a temperature of 4 °C and 35 °C, respectively. For analysis, a sample volume of 5 µL was injected. To reduce matrix effects and to allow for the baseline separation of isomeric target analytes an optimized multi-step gradient was applied as follows: during the first minute, eluent B was kept at 10%, before increasing it to 30% within half a minute. Eluent B was further increased linearly to 55% until 12 min and to 100% until 16 min. After 2.5 min at 100% of eluent B, the column was re-equilibrated with the initial conditions (10% B) between 18.6 and 20 min, resulting in a total run time of 20 min. To reduce the contamination of the ion source and the MS inlet by the most polar sample fractions, a divert valve was directing the LC flow to waste between 0.5 and 1.5 min.
Validation experiments
As specified in the Commission Decision (EC) No. 657/2002 (EC 2002) and the Eurachem Laboratory Guide for the validation of analytical methods (Magnusson 2014), the method’s performance was evaluated by the determination of the following validation parameters: selectivity, linearity, matrix effects, extraction recovery, limit of detection (LOD), limit of quantification (LOQ), repeatability (intraday precision, RSDr), and intermediate precision (interday precision, RSDR). Target analytes were quantified based on the chromatographic peak area of two respective SRM transitions with the most favorable signal-to-noise ratio for each matrix (quantifier and qualifier transitions for the quantitation and identification confirmation, respectively). Moreover, the ion ratios between these signals and their chromatographic retention time of according reference standards were considered as further identification criteria.
Due to a lack of certified reference materials, spiking experiments of representative blank matrices were conducted at two concentration levels (“low” and “high”) to evaluate the method’s performance. Therefore, the biological samples obtained by control group animals were pooled. In case of plasma, samples of 14 control rats (7 killed after 3 h and 7 after 24 h) were combined. For blank urine and lyophilized blank feces mixtures were derived from seven control rats. Urine and fecal samples collected after 24 h were considered as more representative. Moreover, only very limited amounts of these were obtained after 3 h.
To confirm the selectivity of the selected SRM transitions, the absence of matrix signals interfering or co-eluting with target analytes has been investigated by the comparison of respective blank and spiked samples. The calibration curves (5–7 concentration levels) in neat solvent and matched with matrix were evaluated for sufficient linearity by the respective regression coefficients (R2). Furthermore, the ratio between the slopes of the regression lines (matrix-matched vs. solvent calibration) was used to determine matrix effects potentially causing signal suppression or enhancement (SSE). The extraction recovery (RE) of each analyte was calculated as the ratio of the concentration in spiked blank matrix samples quantified by matrix-matched calibration and the according known spiking level. It was determined at two concentration levels: the lower spiking concentration (“low”) was chosen in the range of 2–5 times the limits of quantification of the respective analyte. The second concentration level (“high”) was set higher by a factor of five. The multi-toxin working solution (see “Chemicals and reagents”) was further diluted with methanol to provide for two individual spiking solutions, of which a volume of 25 µL was used for spiking either low or high spiking levels. Blank urine and plasma aliquots (4 °C, 200 µL) were directly spiked, while lyophilized fecal samples (4 °C, 20 mg) were spiked before adding water (4 °C, 200 µL, see “Sample preparation”). Before proceeding with the sample preparation, the spiked samples were kept on ice for 15 min to allow the analytes to interact with the according matrix. The whole sample preparation was carried out at 4 °C as we intentionally developed the protocol to allow for untargeted metabolomics in exactly the same set of samples. The extraction of blank matrix and spiked samples were performed in triplicate for each matrix. During a period of 3 months, this procedure was repeated three times to complete a robust in-house validation giving three independent sample extraction batches and LC–MS/MS measurements. Moreover, the repeatability (intraday precision, RSDr), and intermediate precision (interday precision, RSDR) were calculated from the standard deviations of the analyte concentrations quantified in the spiked samples. Based on the analysis of blank matrices spiked at the low concentration level as well as the lowest matrix-matched calibration levels, the LOD and LOQ values were manually estimated based on a signal-to-noise ratio of three and six, respectively.
Results and discussion
Physiological observations
No visually apparent health impairments of the animals were noted before the experiment was started. Their average body weight was 309 ± 27 g and the average body temperature was 38.2 ± 0.6 °C. An overview of parameters recorded during the experiment is given in Table 1. This includes details on administration, the animals’ consumption of feed and water, excretions, and changes in body temperature and weight gain. Statistical analysis was carried out applying unpaired heteroscedastic Student’s t tests. It was noted that rats of all groups lost weight 3 h post-administration (5.7 ± 2.6 g), and gained weight after 24 h. Usually, rats consume more feed and water during night hours. However, all animals which received the complex Alternaria extract gained significantly less weight after 24 h (weight gain of 1.0 ± 2.7 g, p = 0.043) than animals from the ATX-II group (2.1 ± 3.4 g) and the control group (5.2 ± 3.6 g). These results are in line with those reported in our pilot study (Puntscher et al. 2019b). The animals’ body temperature of all groups decreased by 0.7 ± 0.6 °C and 0.8 ± 0.7 °C after 3 and 24 h, respectively. This effect is assumed to derive from the animal handling before the administration and may not be directly associated to the administration itself. However, body temperatures of rats that received the extract appeared slightly lower compared to the control group while not reaching significance (p = 0.051). Moreover, rats of the extract group consumed more water (p = 0.195) and significantly more feed (p = 0.013) compared to the animals of the other two groups after 3 h. They also excreted significantly more urine and feces (calculations based on the fecal dry mass). The latter was of soft and mucous consistence and partly of intensive malodor. By examining the content of the gastrointestinal tract, rats of the extract group (after 3 h) appeared to suffer from partly severe digestion problems indicated by anomalous, mucous intestinal contents. One of these rats was suffering from diarrhea, which may be considered a result of the treatment. Most animals of the extract group (24 h) also demonstrated visibly enlarged Peyer’s patches in the small intestine. Peyer’s patches (PPs) are considered as key players of the mucosal immune host response toward antigens and bacteria of the gut (Jung et al. 2010). Albeit there are large variations in size, shape and distribution of PPs between individuals, the observation of enlarged PPs in animals treated with Alternaria extract might be not neglected as this could be considered a potential sign of immune response.
A commercially available creatinine assay (Abcam, Cambridge, UK) was used for colorimetric determination of urinary creatinine levels. No significant differences were observed for urinary creatinine concentrations between animals of the different groups and time points (57 ± 12 mg/dL and 54 ± 10 mg/dL for 3 h and 24 h, respectively). This may suggest no apparent impairment of the renal function. Toxin concentrations in urine were not corrected for creatinine concentrations in the present study, because the aim was to determine the absolute amounts of toxins and metabolites excreted. As expected, higher relative standard deviations were observed for quantified excretions and consumptions after 3 h compared to after 24 h. For single animals, no urine or fecal samples were obtained after the short period and some rats did not consume feed or water.
Hematology
The parameters analyzed in the differential blood test (routine WBC differential) were generally within the normal ranges and included white blood cells (WBC), neutrophils (Neut), lymphocytes (Lymph), monocytes (Mono), eosinophils (Eos), basophils (Baso), large unstained cells (LUC), lobularity index (LI), mean peroxidase index (MPXI), white blood cell peroxidase (WBCP). No significant differences were observed between the test groups for both time points. A decrease of Neut levels was observed for all groups from 24.4 ± 9.5% after 3 h to 9.7 ± 1.9% after 24 h. This was suggested to be an effect of the animal handling and may not be related to the toxin administration.
Development of the analytical method
The aim of the present study was to develop a generic sample preparation suitable for both, targeted (quantitation of Alternaria toxins) and untargeted (metabolomics) analysis (data not included in this work). Therefore, biological samples were stored at − 80 °C until sample preparation and all further processing was carried out on ice.
To establish an adequate method allowing for the base-line separation of toxin isomers, a sufficient reduction of the challenging matrix effects, and a satisfying ionization of the target analytes by electrospray ionization, ten binary eluent systems were tested. Therefore, five aqueous eluents (A) including LC–MS grade water with 5 mM NH4Ac (adjusted pH to 8 using NH4OH; 0.1% HAc; 0.1% HAc, 1% HAc, 0.1% formic acid) and 0.3 mM NH4F have been combined with both ACN or MeOH (as eluents B). In all combinations, MeOH demonstrated better chromatographic results than ACN. For method development, blank matrix extracts (urine, plasma, fecal samples) were spiked with low concentrations of a multi-Alternaria-toxin working solution (see “Chemicals and reagents”). In general, matrix effects were significantly different in each matrix. The most challenging matrix for most eluent combinations appeared to be plasma with signal suppressions of up to 100% and partly shifting and tailing peaks. The polar analytes TeA, AA-III and AOH- and AME-sulfates, as well as the included perylene quinones, demonstrated better peak shapes with basic eluents. However, the overall signal-to-noise ratio was most favorable with 0.3 mM NH4F as eluent A (Preindl et al. 2019; Yanes et al. 2011). Despite the still interfering matrix suppression in fecal samples, the overall performance was most suitable using this eluent A and MeOH as eluent B. The elution gradient was further optimized to improve chromatographic separation of isomers.
Ionization and MS parameters were tuned by infusion of individual analytes in neat solvent into a constant LC flow. Up to five SRM transitions were evaluated for each analyte and matrix to determine the most favorable matrix-specific signal-to-noise ratios (Table S1). To increase the required number of chromatographic data points, a scheduled SRM acquisition of four segments was applied.
Method validation
The performance of the developed method was evaluated based on the analysis of spiked blank matrix samples at concentration levels in the low ng/mL or ng/g range. Respective parameters and results are reported in Table 2. As expected, the strict regulations outlined for the analytically less challenging food matrixes according to Commission Decision (EC) No. 657/2002 (EC 2002) were not met for all analytes. While the method performance demonstrated reproducible performance, extraction recoveries were limited by the sample preparation at low temperatures (below 5 °C). This protocol was optimized for a dual analysis enabling broad coverage of endogenous metabolites as well for untargeted metabolomics analysis. Therefore, these conditions were considered as an acceptable compromise for this elegant two-in-one approach that has not been applied in mycotoxin research so far to the best of our knowledge. The method’s specificity was confirmed by the measurement and comparison of respective blank matrix and spiked blank matrix samples. To assure the signal identity of more challenging analytes, additional SRM transitions were taken into account. This was the case for AOH-3-Sulf, ATX-I and ATX-II in urine and feces. Moreover, AOH, AME, TeA, and AST were identified in supposedly blank urine and fecal samples (AST just in feces) obtained from the control group of these experiments. Naturally contaminated rat feed was confirmed to contribute additional exposure (see Table S2). However, the low amounts (< LOQ, < 1%) were considered as neglectable with respect to validation. Matrix effects (SSE, e.g.m the signal suppression and enhancement) were determined to be more severe in fecal matrix, followed by urine. The developed sample preparation utilized fourfold higher concentrations of fecal extracts compared to our previous study (Puntscher et al. 2019b). Urine extracts were analyzed in the volume of the original sample (no dilution). For the toxins ATX-II and AST, the analysis in feces was significantly affected by matrix effects and can therefore only be regarded as semi-quantitative. High noise levels resulted in high LOD levels for AST and ATX-II in fecal matrix. However, the determined low matrix effects for all analytes in plasma would allow for quantitation using solvent-matched calibration.
The majority of the analytes demonstrated reproducible and almost identical RTs for the different matrixes. Even the polar compounds TeA, AA-III, ALS, AOH-3-Sulf and AME-3-Sulf demonstrated stable RTs within single batches, even though their chromatographic behavior was affected by matrix-related differences and minor pH changes of the eluent in different measurement sequences (reported in Puntscher et al. 2018). The linearity of the matrix- and solvent-matched calibration curves (covering at least three orders of magnitude) was verified by adequate regression coefficients (R2) of 0.97–0.99. The achieved LOD and LOQ values were significantly lower compared to the analytical tools initially used for biomonitoring (Puntscher et al. 2019b). Even LOD values in the pg/mL range were achieved for certain toxins and demonstrate the high sensitivity. The method’s performance proved to generate reproducible data and to be fit-for-purpose.
Determination of Alternaria toxins in rat biospecimen
Overall, 13 different Alternaria toxins and metabolites were identified in the analyzed rat samples, namely the dibenzopyrones AOH, AME, their respective 4-hydroxyl- and 3-sulfate-metabolites, and ALT, the perylene quinones ATX-I and ALP, as well as TEN, TeA, ALS, and AST. The quantified toxin concentrations in the respective biological matrices are reported in Table 3. Furthermore, percentual ratios are stated, representing the toxins’ biological recoveries in the respective samples considering the administered dose. For the calculation of the toxin recovery in urine and feces, the volumes of collected urine and the dry masses of fecal samples were taken into account. The estimation of the animals’ total plasma volume was based on the individual body weights at the time of blood sampling. According to Bogdanske et al. (2010), the plasma volume of laboratory rats can be approximated as 4.15 mL/100 g body weight. Biological recovery of toxins determined below their respective LOQ values were calculated by assuming a toxin concentration of LOQ/2.
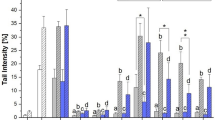
The analysis of samples obtained from the extract group revealed intriguing information regarding bioavailability, their prevalence in plasma, and the excretion efficiency after 3 and 24 h. Six Alternaria toxins were determined in plasma sampled after 3 h, namely TeA, AME-3-Sulf, ATX-I, ALP, ALS, and AST. Their concentrations declined after 24 h. In contrast to the identification of AOH and 4OH-AOH reported for mice plasma by Schuchardt et al. (2014), these compounds were not detected in any rat plasma sample in the present study. However, the mentioned mouse study conducted high oral administrations of 200–2000 µg/kg body weight. Moreover, it was reported that the total radioactivity in plasma deriving from labeled AOH did not exceed 0.06% of the administered dose. Thus, the significantly lower (and more realistic) dose of AOH and AME in the present study (32–39 µg/kg body weight) was not assumed to be detectable by means of the developed method. Interestingly, AME-3-Sulf (0.6 ± 0.5 ng/mL) was determined in plasma sampled after 3 h, accounting for 0.06% of the administered AME. For biological recovery calculations of modified toxins determined in the analyzed samples, the respective molecular masses were considered. AOH and AME were mostly excreted without biological modification via feces after 24 h (89 and 142%, respectively) and via urine (0.1% after 3 h and 2.6-2.8% after 24 h for both toxins, see Fig. 1). Employing an AME extraction recovery of 70% (determined for the higher spiking level) instead of the averaged 56%, the calculated excretion rate for AME was 113%. However, the resulting low bioavailabilities and high fecal excretion rates (> 85% after 24 h) are in line with previous in vivo studies investigating exclusively AOH or AME (Pollock et al. 1982; Schuchardt et al. 2014). Considering respective phase-II metabolites, low amounts of AME-3-Sulf were also present in urine. After 3 and 24 h, 0.01% and 0.6% AME equivalents were determined, which is in line with Puntscher et al. (2019b). In fecal samples, levels reached 2% after 24 h. Moreover, AOH-3-Sulf was identified for the first time in rat urine accounting for the excretion of 0.8% of the administered parent toxin after 24 h. In urine sampled after 3 h, AOH-3-Sulf was detected as well, with an averaged signal below the LOQ. Two rats demonstrated AOH-3-Sulf at 8.4–8.5 ng/mL. Considering these results, the systematic absorption and metabolic modification of AOH and AME was confirmed. Moreover, hydroxylated metabolites of AOH (and AME) were determined in fecal samples after 24 h. 4OH-AOH accounted for 1% of AOH intake, while the signals for the more challenging analyte 4OH-AME were close to the detection limit and therefore not as reliable. As expected, no glucose-conjugates which typically are formed by plant metabolism, were identified in any sample. Due to the lack of respective reference standards, glucuronides, typical mammalian metabolites, were not investigated in this study.

Biological recovery of Alternaria toxins in fecal and urinary excretions of rats sampled 24 h after the administration of a complex Alternaria culture extract
ALT, another dibenzopyrone present in the extract at similar levels as AOH and AME, was detected only in fecal samples after 24 h. The concentration accounted for 7% of the administrated dose and was not determined before in the reported pilot study (Puntscher et al. 2019b). Its isomer isoALT was neither determined in the administered extract, nor in any of the analyzed biological samples. Highest excretion rates of TEN were found in fecal samples after 24 h (45%). Moreover, 0.1 and 0.9% were detected in urine after 3 and 24 h. TeA, which was present in the extract at comparably high concentrations, was recovered in all matrixes. Plasma levels reached 0.2% after 3 h and declined to 0.02% after 24 h. Urinary excretion was confirmed as the main excretion strategy with 20 and 87% after 3 and 24 h, respectively (see Fig. 1). Rapid and efficient urinary excretion was also reported for rats (Puntscher et al. 2019b) and humans (Asam et al. 2013). Only low TeA concentrations were determined in fecal samples (0.6 and 2% after 3 and 24 h, respectively).
Perylene quinones, which were administered at comparatively high levels, were either recovered at very low levels (ATX-I and ALP) or not at all (epoxide-holding analogues ATX-II and STTX-III). ATX-I and ALP were recovered in plasma (up to 0.06% after 3 h), in urine (up to 0.05% after 24 h) and in fecal samples (4–5% after 24 h, see Fig. 1). Due to interfering matrix effects, reliable quantitation of more than half of the ALP signals was not possible. However, these results are in line with previous in vivo (Puntscher et al. 2019b) and in vitro studies demonstrating the highly reactive potential and low persistency of epoxide-holding perylene quinones (Aichinger et al. 2018; Fleck et al. 2014a, b; Puntscher et al. 2019c). Of note, ATX-I was also determined in plasma, urine and fecal samples of rats receiving the isolated ATX-II (ATX-II group). The respective concentrations were lower by a factor 5–10 compared to the samples of the extract group and partly below the LOQ (in plasma and in urine after 24 h). However, these results clearly suggest de-epoxidation of ATX-II in vivo resulting in the formation of ATX-I as suggested in in vitro models (Fleck et al. 2014a, b) and for plant metabolism (Puntscher et al. 2019c) before. In general, the enzymatic reduction of epoxides by mammalian epoxide hydrolases is known as a major detoxification mechanism (Decker et al. 2009).
Low amounts of the toxins ALS (up to 0.6%) and AST (up to 0.12%) were recovered in plasma from rats receiving the extract. In respective urinary and fecal samples, the quantitation of ALS was not possible due to highly varying matrix effects between the individual samples. However, AST was determined in urine (< LOQ) and fecal samples. The method’s performance was not adequate for quantitative analysis of AST in feces due to severe carryover effects and co-eluting matrix components. Despite these analytical challenges, the results indicate high fecal excretion rates of this barely studied fungal metabolite.
Moreover, low concentrations of single Alternaria toxins were determined in samples of the control and the ATX-II group. By LC–MS/MS analysis of sampled feed aliquots, this cereal-based product was identified as a contributing toxin source. In fact, AOH, AME, TeA, and AST (2.7, 1.4, 596, and 1.5 ng/g) were quantified in feed by standard addition (further details are reported in the supplementary material). This explains the contamination of rat feces sampled after 3 h, which was not expected due to gastrointestinal transit times of 15–24 h (Bove 2015).
Conclusions and outlook
In the present study, we report the investigation of the bioavailability, metabolism, and excretion of Alternaria toxins in Sprague Dawley rats (see Fig. 2). After the administration of a complex fungal culture extract comprising 11 determined and quantified Alternaria toxins (extract group) and the isolated highly genotoxic altertoxin II (ATX-II group), rat plasma, urine, and feces were sampled after 3 and 24 h and analyzed by a novel LC–MS/MS method. Extensive spiking experiments proofed the analysis to be reliable for these challenging biological matrices and therefore fit-for-purpose. Up to ten partly modified toxins were determined in rat plasma, urine and fecal samples. Toxin concentrations in plasma were generally below 1% of the administered dose after 3 h (0.01–0.6%) and even lower after 24 h. While the dibenzopyrones AOH and AME showed a low bioavailability and high excretion rates within 24 h mostly via feces (> 89%), TeA was predominantly excreted via urine (87%). Biotransformed metabolites of AOH and AME were identified including 4OH-AOH (putatively also 4OH-AME), AOH-3-Sulf, and AME-3-Sulf, which were determined in fecal samples, but partly also in urine. Epoxide-holding perylene quinones ATX-II and STTX-III were not recovered in any sample, while their analogues ATX-I and ALP were determined in all matrixes at very low levels confirming their potential bioavailability. Moreover, ATX-I was identified in samples deriving from rats, which just received the isolated ATX-II. This may suggest chemical reduction during digestion or even enzymatic de-epoxidation mechanisms in vivo as a potential detoxification pathway.

Overview on the administration, absorption, metabolism, and excretion of Alternaria toxins in rats
This study contributes important insights in mammalian metabolism of Alternaria toxins and confirmed the relevance of their biologically modified forms. A putatively low systemic absorption does not exclude toxicological effects induced by acute or chronic exposure to these compounds. In this study, physiological aberrations such as digestive disturbance were observed for rats, which received the complex culture extract. With the identification of toxicologically relevant tissues and target organs, such as the gastrointestinal tract and the liver, it is crucial to investigate toxicological endpoints in follow-up studies. Moreover, the establishment of suitable biomarkers of exposure in animal and human bio-fluids (human biomonitoring, HBM) is required for future risk assessments and a more reliable understanding of the impact of Alternaria toxins on global food safety.
Abbreviations
- ADME:
-
Absorption, distribution, metabolism, excretion
- LC:
-
Liquid chromatography
- SRM:
-
Selected reaction monitoring
- MS:
-
Mass spectrometry
- AA-III:
-
Altenuic acid
- ALT:
-
Altenuene
- ALP:
-
Alterperylenol
- AME:
-
Alternariol monomethyl ether
- AME-3-Glc:
-
Alternariol monomethyl ether-3-glucoside
- AME-3-Sulf:
-
Alternariol monomethyl ether-3-sulfate
- AOH:
-
Alternariol
- AOH-3-Glc:
-
Alternariol-3-glucoside
- AOH-3-Sulf:
-
Alternariol-3-sulfate
- AOH-9-Glc:
-
Alternariol-9-glucoside
- ALS:
-
Altenusin
- AST:
-
Altersetin
- ATX-I:
-
Altertoxin I
- ATX-II:
-
Altertoxin II
- isoALT:
-
Isoaltenuene
- STTX-III:
-
Stemphyltoxin III
- TeA:
-
Tenuazonic acid
- TEN:
-
Tentoxin
- ACN:
-
Acetonitrile
- HAc:
-
Acetic acid
- MeOH:
-
Methanol
- NH4Ac:
-
Ammonium acetate
- NH4F:
-
Ammonium fluoride
References
Aichinger G, Puntscher H, Beisl J, Kütt M-L, Warth B, Marko D (2018) Delphinidin protects colon carcinoma cells against the genotoxic effects of the mycotoxin altertoxin II. Toxicol Lett 284:136–142
Altemöller M, Podlech J, Fenske D (2006) Total synthesis of altenuene and isoaltenuene. Eur J Org Chem 2006:1678–1684
Arntzen C (1972) Inhibition of photophosphorylation by tentoxin, a cyclic tetrapeptide. Biochim Biophys Acta (BBA) Bioenerg 283:539–542
Asam S, Habler K, Rychlik M (2013) Determination of tenuazonic acid in human urine by means of a stable isotope dilution assay. Anal Bioanal Chem 405:4149–4158
Berthiller F, Crews C, Dall’Asta C, Saeger SD, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, Speijers G, Stroka J (2013) Masked mycotoxins: a review. Mol Nutr Food Res 57:165–186
Bogdanske JJ, Hubbard-Van Stelle S, Riley MR, Schiffman BM (2010) Laboratory rat procedural techniques: manual and DVD. CRC Press, Boca Raton
Bove GM (2015) A non-invasive method to evaluate gastrointestinal transit behavior in rat. J Pharmacol Toxicol Methods 74:1–6
Brugger E-M, Wagner J, Schumacher DM, Koch K, Podlech J, Metzler M, Lehmann L (2006) Mutagenicity of the mycotoxin alternariol in cultured mammalian cells. Toxicol Lett 164:221–230
Burkhardt B, Pfeiffer E, Metzler M (2009) Absorption and metabolism of the mycotoxins alternariol and alternariol-9-methyl ether in Caco-2 cells in vitro. Mycotoxin Res 25:149
Burkhardt B, Wittenauer J, Pfeiffer E, Schauer U, Metzler M (2011) Oxidative metabolism of the mycotoxins alternariol and alternariol-9-methyl ether in precision-cut rat liver slices in vitro. Mol Nutr Food Res 55:1079–1086
Burkhardt B, Jung S, Pfeiffer E, Weiss C, Metzler M (2012) Mouse hepatoma cell lines differing in aryl hydrocarbon receptor-mediated signaling have different activities for glucuronidation. Arch Toxicol 86:643–649
Decker M, Arand M, Cronin A (2009) Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch Toxicol 83:297–318
Dellafiora L, Warth B, Schmidt V, Del Favero G, Mikula H, Fröhlich J, Marko D (2018) An integrated in silico/in vitro approach to assess the xenoestrogenic potential of Alternaria mycotoxins and metabolites. Food Chem 248:253–261
EC (2002) Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Union L221:8–36
EFSA (2016) Dietary exposure assessment to Alternaria toxins in the European population. EFSA J 14:e04654
Estiarte N, Crespo-Sempere A, Marín S, Ramos A, Worobo R (2018) Stability of alternariol and alternariol monomethyl ether during food processing of tomato products. Food Chem 245:951–957
Fehr M, Pahlke G, Fritz J, Christensen MO, Boege F, Altemöller M, Podlech J, Marko D (2009) Alternariol acts as a topoisomerase poison, preferentially affecting the IIα isoform. Mol Nutr Food Res 53:441–451
Fleck SC, Burkhardt B, Pfeiffer E, Metzler M (2012) Alternaria toxins: altertoxin II is a much stronger mutagen and DNA strand breaking mycotoxin than alternariol and its methyl ether in cultured mammalian cells. Toxicol Lett 214:27–32
Fleck SC, Pfeiffer E, Metzler M (2014a) Permeation and metabolism of Alternaria mycotoxins with perylene quinone structure in cultured Caco-2 cells. Mycotoxin Res 30:17–23
Fleck SC, Pfeiffer E, Podlech J, Metzler M (2014b) Epoxide reduction to an alcohol: a novel metabolic pathway for perylene quinone-type Alternaria mycotoxins in mammalian cells. Chem Res Toxicol 27:247–253
Fleck SC, Sauter F, Pfeiffer E, Metzler M, Hartwig A, Köberle B (2016) DNA damage and repair kinetics of the Alternaria mycotoxins alternariol, altertoxin II and stemphyltoxin III in cultured cells. Mutat Res Genet Toxicol Environ Mutagen 798:27–34
Griffin GF, Chu F (1983) Toxicity of the Alternaria metabolites alternariol, alternariol methyl ether, altenuene, and tenuazonic acid in the chicken embryo assay. Appl Environ Microbiol 46:1420–1422
Hermann G, Schwaiger M, Volejnik P, Koellensperger G (2018) 13C-labelled yeast as internal standard for LC–MS/MS and LC high resolution MS based amino acid quantification in human plasma. J Pharm Biomed Anal 155:329–334
Hildebrand AA, Kohn BN, Pfeiffer E, Wefers D, Metzler M, Bunzel M (2015) Conjugation of the mycotoxins alternariol and alternariol monomethyl ether in tobacco suspension cells. J Agric Food Chem 63:4728–4736
Jarolim K, Del Favero G, Pahlke G, Dostal V, Zimmermann K, Heiss E, Ellmer D, Stark TD, Hofmann T, Marko D (2017) Activation of the Nrf2-ARE pathway by the Alternaria alternata mycotoxins altertoxin I and II. Arch Toxicol 91:203–216
Jung C, Hugot J-P, Barreau F (2010) Peyer’s patches: the immune sensors of the intestine. Int J Inflamm 2010:823710
Lehmann L, Wagner J, Metzler M (2006) Estrogenic and clastogenic potential of the mycotoxin alternariol in cultured mammalian cells. Food Chem Toxicol 44:398–408
Lemke A, Burkhardt B, Bunzel D, Pfeiffer E, Metzler M, Huch M, Kulling S, Franz C (2016) Alternaria toxins of the alternariol type are not metabolised by human faecal microbiota. World Mycotoxin J 9:41–50
Magnusson B (2014) The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics. In: Eurachem, p 57
Mikula H, Skrinjar P, Sohr B, Ellmer D, Hametner C, Fröhlich J (2013) Total synthesis of masked Alternaria mycotoxins—sulfates and glucosides of alternariol (AOH) and alternariol-9-methyl ether (AME). Tetrahedron 69:10322–10330
Nemecek G, Thomas R, Goesmann H, Feldmann C, Podlech J (2013) Structure elucidation and total synthesis of altenuic acid III and studies towards the total synthesis of altenuic acid II. Eur J Org Chem 2013:6420–6432
Ostry V (2008) Alternaria mycotoxins: an overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J 1:175–188
Pero R, Posner H, Blois M, Harvan D, Spalding J (1973) Toxicity of metabolites produced by the “Alternaria”. Environ Health Perspect 4:87
Pfeiffer E, Schebb NH, Podlech J, Metzler M (2007) Novel oxidative in vitro metabolites of the mycotoxins alternariol and alternariol methyl ether. Mol Nutr Food Res 51:307–316
Pfeiffer E, Burkhardt B, Altemöller M, Podlech J, Metzler M (2008) Activities of human recombinant cytochrome P450 isoforms and human hepatic microsomes for the hydroxylation of Alternaria toxins. Mycotoxin Res 24:117
Pfeiffer E, Herrmann C, Altemöller M, Podlech J, Metzler M (2009a) Oxidative in vitro metabolism of the Alternaria toxins altenuene and isoaltenuene. Mol Nutr Food Res 53:452–459
Pfeiffer E, Schmit C, Burkhardt B, Altemöller M, Podlech J, Metzler M (2009b) Glucuronidation of the mycotoxins alternariol and alternariol-9-methyl ether in vitro: chemical structures of glucuronides and activities of human UDP-glucuronosyltransferase isoforms. Mycotoxin Res 25:3–10
Pollock G, DiSabatino C, Heimsch R, Coulombe R (1982) The distribution, elimination, and metabolism of 14C-alternariol monomethyl ether. J Environ Sci Health Part B 17:109–124
Preindl K, Braun D, Aichinger G, Sieri S, Fang M, Marko D, Warth B (2019) A generic liquid chromatography−tandem mass spectrometry exposome method for the determination of xenoestrogens in biological matrices. Anal Chem 91:11334–11342
Puntscher H, Kütt M-L, Skrinjar P, Mikula H, Podlech J, Fröhlich J, Marko D, Warth B (2018) Tracking emerging mycotoxins in food: development of an LC–MS/MS method for free and modified Alternaria toxins. Anal Bioanal Chem 410:4481–4494
Puntscher H, Cobankovic I, Marko D, Warth B (2019a) Quantitation of free and modified Alternaria mycotoxins in European food products by LC–MS/MS. Food Control 102:157–165
Puntscher H, Hankele S, Tillmann K, Attakpah E, Braun D, Kütt M-L, Del Favero G, Aichinger G, Pahlke G, Höger H, Marko D, Warth B (2019b) First insights into Alternaria multi-toxin in vivo metabolism. Toxicol Lett 301:168–178
Puntscher H, Marko D, Warth B (2019c) The fate of Altertoxin II during tomato processing steps at a laboratory scale. Front Nutr 6:92
Sarkanj B, Ezekiel CN, Turner PC, Abia WA, Rychlik M, Krska R, Sulyok M, Warth B (2018) Ultra-sensitive, stable isotope assisted quantification of multiple urinary mycotoxin exposure biomarkers. Anal Chim Acta 1019:84–92
Schuchardt S, Ziemann C, Hansen T (2014) Combined toxicokinetic and in vivo genotoxicity study on Alternaria toxins. EFSA Support Publ 11(11):679E
Schwarz C, Tiessen C, Kreutzer M, Stark T, Hofmann T, Marko D (2012) Characterization of a genotoxic impact compound in Alternaria alternata infested rice as altertoxin II. Arch Toxicol 86:1911–1925
Scott PM (2001) Analysis of agricultural commodities and foods for Alternaria mycotoxins. J AOAC Int 84:1809–1817
Siegel D, Feist M, Proske M, Koch M, Nehls I (2010) Degradation of the Alternaria mycotoxins alternariol, alternariol monomethyl ether, and altenuene upon bread baking. J Agric Food Chem 58:9622–9630
Solfrizzo M (2017) Recent advances on Alternaria mycotoxins. Curr Opin Food Sci 17:57–61
Soukup ST, Kohn BN, Pfeiffer E, Geisen R, Metzler M, Bunzel M, Kulling SE (2016) Sulfoglucosides as novel modified forms of the mycotoxins alternariol and alternariol monomethyl ether. J Agric Food Chem 64:8892–8901
Tiessen C, Ellmer D, Mikula H, Pahlke G, Warth B, Gehrke H, Zimmermann K, Heiss E, Fröhlich J, Marko D (2017) Impact of phase I metabolism on uptake, oxidative stress and genotoxicity of the emerging mycotoxin alternariol and its monomethyl ether in esophageal cells. Arch Toxicol 91:1213–1226
Yanes O, Tautenhahn R, Patti GJ, Siuzdak G (2011) Expanding coverage of the metabolome for global metabolite profiling. Anal Chem 83:2152–2161
Acknowledgements
Open access funding provided by University of Vienna. We would like to thank Hannes Mikula, Philipp Skrinjar and Johannes Fröhlich (Vienna University of Technology, Austria), Joachim Podlech (Karlsruhe Institute of Technology, Germany), and Michael Rychlik (TUM, Germany) for providing valuable reference materials. Moreover, we highly acknowledge Vincent Lüttig, Julia Beisl, and the staff of the Mass Spectrometry Center at the Faculty of Chemistry at UNIVIE for skillful technical support and all colleagues supportive during the experiment. This work was financed by the University of Vienna and the Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical considerations
According to the animal welfare act 2012, BGBI. I Nr. 114/2012 (TVG 2012), the present study (GZ: BMWFW-66.009/0120-WF/II/3b/2014) was approved by the Austrian Federal Ministry of Education, Science and Research (Austria).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Puntscher, H., Aichinger, G., Grabher, S. et al. Bioavailability, metabolism, and excretion of a complex Alternaria culture extract versus altertoxin II: a comparative study in rats. Arch Toxicol 93, 3153–3167 (2019). https://doi.org/10.1007/s00204-019-02575-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02575-7