
Abstract
The tumour suppressor p53 is one of the most important cancer genes. Previous findings have shown that p53 expression can influence DNA adduct formation of the environmental carcinogen benzo[a]pyrene (BaP) in human cells, indicating a role for p53 in the cytochrome P450 (CYP) 1A1-mediated biotransformation of BaP in vitro. We investigated the potential role of p53 in xenobiotic metabolism in vivo by treating Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice with BaP. BaP-DNA adduct levels, as measured by 32P-postlabelling analysis, were significantly higher in liver and kidney of Trp53(−/−) mice than of Trp53(+/+) mice. Complementarily, significantly higher amounts of BaP metabolites were also formed ex vivo in hepatic microsomes from BaP-pretreated Trp53(−/−) mice. Bypass of the need for metabolic activation by treating mice with BaP-7,8-dihydrodiol-9,10-epoxide resulted in similar adduct levels in liver and kidney in all mouse lines, confirming that the influence of p53 is on the biotransformation of the parent compound. Higher BaP-DNA adduct levels in the livers of Trp53(−/−) mice correlated with higher CYP1A protein levels and increased CYP1A enzyme activity in these animals. Our study demonstrates a role for p53 in the metabolism of BaP in vivo, confirming previous in vitro results on a novel role for p53 in CYP1A1-mediated BaP metabolism. However, our results also suggest that the mechanisms involved in the altered expression and activity of the CYP1A1 enzyme by p53 in vitro and in vivo are different.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The TP53 tumour suppressor gene, which encodes the protein p53, is often described as the guardian of the genome and is the most commonly mutated gene in human tumours (Olivier et al. 2010). As gatekeeper, p53 regulates cell growth by inhibiting proliferation or promoting apoptosis (Taneja et al. 2011). As caretaker, it controls cellular processes to maintain genomic integrity, including repair to remove DNA damage (Taneja et al. 2011). Disruption of the normal p53 response by TP53 mutation leads to increased risks of tumour development. TP53 is mutated in over 50 % of sporadic tumours, and various environmental carcinogens have been found to be associated with characteristic mutational signatures in TP53 (Olivier et al. 2010). In addition to somatic mutations in the TP53 gene, germline mutations have been found to cause predisposition to cancer, and TP53 polymorphisms have been shown to increase cancer susceptibility (Whibley et al. 2009). Besides its role in DNA damage response, p53 has also been found to regulate metabolic pathways, thereby linking p53 not only to cancer, but also to other diseases such as diabetes and obesity (Maddocks and Vousden 2011).
Previously, we used a panel of isogenic human colorectal carcinoma HCT116 cell lines that differed only with respect to their endogenous TP53 status in order to investigate the metabolism and DNA damage induced by the environmental carcinogens benzo[a]pyrene (BaP) and 3-nitrobenzanthrone (3-NBA) (Hockley et al. 2008; Wohak et al. 2014). We found that HCT116 TP53(−/−) and TP53(+/–) cells formed significantly lower BaP-DNA adduct levels than TP53(+/+) cells. In contrast, no difference in adduct formation was observed in HCT116 cells exposed to BaP-7,8-diol-9,10-epoxide (BPDE), the activated metabolite of BaP, indicating that p53 expression is linked to the cytochrome P450 (CYP)-mediated metabolic activation of BaP (compare Supporting Figure 1a). There were also significantly lower levels of BaP metabolites detected in the culture media of HCT116 TP53(−/−) and TP53(+/–) cells relative to TP53(+/+) cells, which was accompanied by a greater induction of CYP1A1 protein and CYP1A1 mRNA in TP53(+/+) cells than in the other cell lines (Wohak et al. 2014). We found that BaP-induced CYP1A1 expression was regulated through a p53 response element (p53RE) in the regulatory region of CYP1A1, thereby providing a novel pathway for the induction of CYP1A1 by polycyclic aromatic hydrocarbons (PAHs) such as BaP (Wohak et al. 2014). Interestingly, DNA adduct formation by 3-NBA was not different in HCT116 TP53(+/+) and TP53(−/−) cells (Hockley et al. 2008), suggesting that NAD(P)H:quinone oxidoreductase (NQO1), which is the principal enzyme activating 3-NBA (compare Supporting Figure 1b) (Arlt et al. 2005; Stiborova et al. 2010), is not regulated by p53.
Transgenic and knockout mouse models have been used to study tumour suppressor function through phenotypic analysis of the whole organism and by examining a variety of primary cell types (Taneja et al. 2011). The opportunity to study multiple tissues is particularly useful for Trp53 because p53 function is highly cell type specific (Donehower 2014; Kenzelmann Broz and Attardi 2010; Kucab et al. 2010; Lozano 2010). Much of the work carried out on the role of CYP enzymes in xenobiotic metabolism has been done in vitro (Nebert 2006; Nebert and Dalton 2006). However, extrapolation from in vitro data to in vivo pharmacokinetics requires additional factors to be considered such as route of administration, absorption, renal clearance and tissue-specific CYP expression (Nebert 2006; Nebert et al. 2013). For example, previous studies have revealed an apparent paradox, whereby hepatic CYP enzymes appear to be more important for detoxification of BaP in vivo, despite being involved in its metabolic activation in vitro (Arlt et al. 2008, 2012; Nebert et al. 2013).
To evaluate the impact of the cellular Trp53 status on the metabolic activation of BaP and 3-NBA, we have compared metabolism and DNA adduct formation of BaP and 3-NBA in Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice. DNA adduct formation in vivo and in vitro was investigated by 32P-postlabelling analysis. Tissue-specific expression and activity of xenobiotic-metabolising enzymes (XMEs) involved in BaP and 3-NBA metabolism were compared with DNA adduct formation in the same tissue. Nucleotide excision repair (NER) capacity was assessed phenotypically in selected tissues using a modified comet assay. Urinary BaP metabolites and the Cyp-mediated formation of BaP metabolites ex vivo in hepatic microsomes were measured by high-performance liquid chromatography (HPLC).
Materials and methods
Carcinogens
Benzo[a]pyrene (BaP; CAS number 50-32-8; purity ≥96 %) was obtained from Sigma-Aldrich. 3-Nitrobenzanthrone (3-NBA; CAS number 17117-34-9) was prepared as previously reported (Arlt et al. 2002). (±)-Anti-benzo[a]pyrene-trans-7,8-dihydrodiol-9,10-epoxide (BPDE) was synthesised at the Biochemical Institute for Environmental Carcinogens, Prof. Dr. Gernot Grimmer-Foundation, Germany.
Carcinogen treatment of Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice
Trp53(+/+), Trp53(+/–) and Trp53(−/−) male C57BL/6 mice were generated as reported (Jacks et al. 1994). Trp53(+/−) and Trp53(−/−) mice carry a mutation which removes approximately 40 % of the coding capacity of Trp53 and completely eliminates synthesis of p53 protein. More information about the strains can be found at (http://jaxmice.jax.org/strain/002101.html). All animal experiments were conducted in accordance with the law at the Leiden University Medical Center, Leiden, the Netherlands, after approval by the institutional ethics committee. Animals were kept under controlled specific pathogen-free conditions (23 °C, 40–50 % humidity) under a 12-h light–dark cycle. Food and water were available ad libitum. Genotyping of the animals was performed as described (Jacks et al. 1994) (see Supporting Figure 2). Groups of male Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice (3 months old; 25–30 g; n = 4/group) were treated with a single dose of 125 mg/kg body weight (bw) of BaP by intraperitoneal (i.p.) injection according to treatment protocols used previously to study BaP metabolism (Arlt et al. 2008, 2012). We chose i.p. injection as the administration route to achieve a high induction of hepatic CYP-mediated BaP metabolism. Similarly, groups (n = 4) of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice were injected i.p. with a single dose of 2 mg/kg bw of 3-NBA according to a previous study investigating 3-NBA metabolism (Arlt et al. 2005). Based on dose-finding experiments in Trp53(+/+) mice using single i.p. injections of 1.25, 6.25 or 12.5 mg/kg bw of BPDE, groups (n = 4) of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice were treated i.p. with 1.25 mg/kg bw of BPDE. Control mice (n = 4) received solvent (corn oil) only. Animals were killed 24 h after treatment, and their liver, lung, kidney, colon, small intestine, bladder, glandular stomach, forestomach and spleen were removed, snap-frozen in liquid nitrogen and stored at −80 °C until further analysis. Urine was collected for the preceding 24 h.
Detection of DNA adducts by 32P-postlabelling
DNA was isolated from tissues by a standard phenol–chloroform extraction method. DNA adduct analysis was performed by thin-layer chromatography 32P-postlabelling analysis (Phillips and Arlt 2007, 2014). For DNA from BaP- and BPDE-treated animals, the nuclease P1 digestion enrichment method was used (Arlt et al. 2008), while for DNA from 3-NBA-treated animals, the butanol extraction method was employed (Arlt et al. 2002). DNA samples (4 μg) were digested with micrococcal nuclease (288 mU; Sigma) and calf spleen phosphodiesterase (1.2 mU; MP Biomedical) and then enriched and labelled as reported.
Measurement of nucleotide excision repair (NER) capacity
The ability of NER-related enzymes present in isolated tissue extracts to detect and incise substrate DNA containing BPDE-DNA adducts was measured using a modified comet assay (Langie et al. 2006). Tissue protein extracts were prepared as described previously (Güngör et al. 2010), and protein concentrations were optimised for analysis of liver and kidney samples (0.2 mg/mL). The ex vivo repair incubation and electrophoresis were performed according to the published protocol (Langie et al. 2006). Dried slides stained with ethidium bromide (10 µg/mL) were viewed with a Zeiss Axioskop fluorescence microscope. Comets were scored using the Comet III system (Perceptive Instruments, UK). Fifty nucleoids were assessed per slide, and each sample was analysed in duplicate. All samples were measured blindly. Tail intensity (% tail DNA), defined as the percentage of DNA migrated from the head of the comet to the tail, was used to calculate repair capacity of the tissue extracts as reported previously (Langie et al. 2006).
Preparation of microsomal and cytosolic samples
Microsomal and cytosolic fractions were isolated from the livers and lungs of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice. Tissue frozen in liquid nitrogen was ground up in a Teflon container with a steel ball in a dismembrator to a frozen powder. This was transferred into a Potter-Elvehjem homogenizer and the Teflon receptacle rinsed with 1/15 M sodium phosphate buffer with 0.5 % potassium chloride pH 7.4. The powder was homogenised and transferred to a centrifuge tube and the potter rinsed with phosphate buffer. The homogenates were spun for 30 min at 18,000×g. Supernatant were transferred to an ultracentrifuge tube and spun at 100,000×g for 60 min at 4 °C. The resulting supernatants formed the cytosols, which was levered off the sediment gently, while the sediments (microsomes) were taken up in phosphate buffer. Protein concentration in the fractions was measured using bicinchoninic acid protein assay (Wiechelman et al. 1988) with bovine serum albumin and stored in small aliquots at −80 °C until analysis.
Microsomal BaP-DNA adduct formation
Incubation mixtures in a final volume of 750 µL consisted of 50 mM potassium phosphate buffer (pH 7.4), 1 mM NADPH, 0.5 mg of microsomal protein, 0.5 mg calf thymus DNA and 0.1 mM BaP [dissolved in 7.5 µL dimethylsulfoxide (DMSO)]. The reaction was initiated by adding NADPH. Microsomal incubations were carried out at 37 °C for 90 min. Microsomal-mediated BaP-DNA adduct formation was linear up to 120 min as reported previously (Arlt et al. 2008). Control incubations were carried out (i) without microsomes, (ii) without NADPH, (iii) without DNA and (iv) without BaP. After incubation, DNA was isolated by a standard phenol/chloroform extraction method.
Microsomal BaP metabolite formation
In a final volume of 500 µL, the incubation mixture contained 100 mM potassium phosphate buffer (pH 7.4), NADPH-generating system (1 mM NADP+, 10 mM d-glucose-6-phosphate, 1 μ/mL d-glucose-6-phosphate dehydrogenase), 0.5 mg microsomal protein and 50 µM BaP (dissolved in 5 µL DMSO). The reaction was initiated by adding 50 µL of the NADPH-generating system. Microsomal incubations were carried out at 37 °C for 20 min. Control incubations were carried out (1) without microsomes, (2) without NADPH-generating system and (3) without BaP. After incubation, 5 µL of 1 mM phenacetin in methanol was added as internal standard. The BaP mixtures were extracted with ethyl acetate (2 × 1 mL), the solvent was evaporated to dryness, and the residue was dissolved in 25 µL methanol for HPLC analysis.
HPLC analysis of BaP metabolites
HPLC analysis was performed on a Nucleosil® C18 reversed phase column (250 × 4 mm, 5 µm; Macherey Nagel, Germany), using a Dionex system consisting of a pump P580, a UV/VIS Detector UVD 170S/340S, an ASI-100 Automated Sample Injector, a termobox COLUMN OVEN LCO 101 and an In-Line Mobile Phase Degasser Degasys DG-1210 Dionex controlled with ChromeleonTM 6.11 build 490 software. HPLC conditions were as follows: 50 % acetonitrile in HPLC water (v/v), with a linear gradient to 85 % acetonitrile in 35 min, then an isocratic elution with 85 % acetonitrile for 5 min, a linear gradient from 85 % acetonitrile to 50 % acetonitrile in 5 min, followed by an isocratic elution of 50 % acetonitrile for 5 min. Detection was by UV at 254 nm. BaP metabolite peaks were collected and analysed by NMR and/or mass spectrometry as described (Stiborova et al. 2014). The structures of BaP metabolites analysed are given in Supplementary Figure 8. The metabolite peak areas were calculated relative to the peak area of the internal standard.
BaP metabolites in urine
Urine samples (0.4–1.7 mL) collected from Trp53(+/+) and Trp53(−/−) mice treated with BaP were mixed with four volumes of methanol and centrifuged for 4 min at 1000 rpm, and the supernatants were then evaporated to dryness. The residues were dissolved in 100 µL of methanol and analysed by HPLC as described above. Urine samples for Trp53(+/–) mice were lost during analysis.
Expression of Cyp1a1 and Nqo1 by Western blotting
Microsomal and cytosolic proteins were separated using NuPage 4–12 % Bis–Tris sodium-dodecyl sulphate (SDS)-polyacrylamide gels (Life Technologies) and Western-blotted as previously reported (Hockley et al. 2006). Chicken polyclonal antibody raised against recombinant rat CYP1A1 protein (Arlt et al. 2008) has been shown to recognise murine Cyp1a1. In microsomal samples, Cyp1a1 was probed with chicken anti-rat CYP1A1 at 1:5000, and peroxidase-conjugated goat anti-chicken (ab6877, Abcam, 1:10,000) was used as secondary antibody. Rat recombinant CYP1A1 and CYP1A2 (in Supersomes™, Gentest Corp.) were used as positive controls to identify protein bands in microsomal samples. In cytosolic samples, an affinity-purified rabbit antibody was used to detect Nqo1 (N5288, rabbit pAb, 1:10,000; Sigma) and peroxidase-conjugated goat anti-rabbit antibody (CST7076, Cell Signalling Technology, 1:10,000) was used as secondary antibody. Human recombinant NQO1 (Sigma) was used as positive control to identify the nqo1 band in cytosols. Gapdh was detected with mouse mAb #MAB374 (1:10,000; Millipore) and β-actin with mouse mAb ab6276 (1:10,000; Abcam) using peroxidase-conjugated goat anti-mouse as secondary antibody #170-5047 (1:5000; Bio-Rad). All proteins were visualised using the enhanced chemiluminescent SuperSignal West Pico detection reagent according to the manufacturer’s instruction (#34080; Thermo Scientific).
Measurement of Cyp1a1 enzyme activity
Microsomal samples were characterised for Cyp1a activity using 7-ethoxyresorufin O-deethylation (EROD) activity (Mizerovska et al. 2011). Enzyme activity was determined by following the conversion of 7-ethoxyresorufin into resorufin using fluorescent measurement on a Synergy HT Plate Reader (Bio-TEK) using an excitation wavelength of 530 nm and an emission wavelength of 580 nm.
Measurement of Nqo1 enzyme activity
Nqo1 enzyme activity in cytosolic samples was measured with menadione (2-methyl-1,4-naphthoquinone) as substrate as described previously (Mizerovska et al. 2011). Enzyme activity was determined by following the conversion of cytochrome c at 550 nm on a Synergy HT Plate Reader (Bio-TEK).
Expression of p53 by Western blotting
For the preparation of whole hepatic protein, extracts of liver tissues (30 mg) of Trp53(+/+) mice were homogenised in 300 µL of Tissue Protein Extraction Reagent (T-PER™, Life Technologies) buffer supplemented with 1 % protease inhibitor (Halt™, Life Technologies). Samples were sonicated and centrifuged for 20 min at 13,000g (4 °C), and the supernatant was saved. The protein concentration was measured as described above. For Western blotting, 25 µg of protein was separated by SDS–polyacrylamide electrophoresis as described above. The following antibodies were used: anti-p53 (rabbit pAb, NCL-p53-CM5p, 1:5000; Leica Biosystems) and anti-Gadph (mouse mAb #MAB374, 1:10,000; Millipore). Membranes were washed and incubated with peroxidase-conjugated goat anti-rabbit or goat anti-mouse as secondary antibodies (#170-5046 and #170-5047, 1:5000; Bio-Rad).
Results
DNA adduct formation in vivo
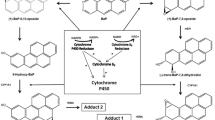
In the majority of tissues, the BaP-DNA adduct pattern consisted of a single adduct spot (spot 1), previously identified as 10-(deoxyguanosin-N 2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-BaP (dG-N 2-BPDE) (Supporting Figure 3a). For lung, colon and small intestine additional adduct spots were detected. In all three tissues, a minor adduct (spot 2) was detected that was previously suggested to be derived from reaction of 9-hydroxy-BaP-4,5-epoxide with guanine (Stiborova et al. 2014), while for colon and small intestine, an additional major adduct (spot 3) was found that has not yet been structurally identified. The same adduct profiles were observed in all three mouse lines. A scheme showing the formation of adducts 1 and 2 is given in Supporting Figure 6. No DNA adducts were detected in control animals (data not shown).
BaP-DNA adduct levels ranged from 25 to 100 adducts per 108 nucleotides (Fig. 1a). Adduct levels were significantly higher (~ twofold) in livers of Trp53(−/−) compared to Trp53(+/+) mice (106 ± 25 versus 48 ± 27 adducts per 108 nucleotides; p < 0.05), while adduct formation in kidney was significantly higher (~ twofold) in both Trp53(+/–) and Trp53(−/−) relative to Trp53(+/+) mice (73 ± 31 and 70 ± 20 versus 27 ± 6 adducts per 108 nucleotides, respectively; p < 0.05). A similar trend was observed in lung, fore stomach and spleen, although the difference in adduct levels did not reach statistical significance. In contrast, adduct levels in liver, lung and kidney did not significantly change after treatment with BPDE in all mouse lines (Fig. 1b). In these tissues, the adduct pattern consisted of adduct spot 1 only (see Supporting Figure 3b). As BPDE does not require CYP-mediated metabolic activation to bind to DNA, these findings suggest that the differences in DNA adduct formation observed in liver and kidney with the parent compound BaP are the consequence of the different capacities of the Trp53 mouse lines to metabolically activate BaP.

DNA adduct levels measured by 32P-postlabelling in various organs of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice after exposure to BaP (a) or BPDE (b). Values are the mean ± SD (n = 4). Statistical analysis was performed by one-way ANOVA followed by Tukey post hoc test [*p < 0.05; different from Trp53(+/+) mice]
The adduct pattern induced by 3-NBA consisted of up to four adduct spots (spots 1–4; Supporting Figure 4). These were characterised previously as 2-(2′-deoxyadenosin-N 6-yl)-3-aminobenzanthrone (dA-N 6-3-ABA; spot 1), 2-(2′-deoxyguanosin-N 2-yl)-3-aminobenzanthrone (dG-N 2-3-ABA; spot 3) and N-(2′-deoxyguanosin-8-yl)-3-aminobenzanthrone (dG-C8-N-3-ABA; spot 4), while spot 2 is an as-yet-uncharacterised deoxyadenosine adduct (Arlt et al. 2001, 2006). Adduct levels ranged from 2 to 40 adducts per 108 nucleotides, but there were no significant differences between mouse lines in DNA adduct formation in any of the tissues investigated (Fig. 2). These results suggest that in contrast to BaP metabolism, Trp53 status has no impact on 3-NBA metabolism in vivo, which is in accord with experiments on human cells in vitro (Hockley et al. 2008; Simoes et al. 2008).

DNA adduct levels measured by 32P-postlabelling in various organs of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice after exposure to 3-NBA. Values are the mean ± SD (n = 4). Statistical analysis was performed by one-way ANOVA followed by Tukey post hoc test; no significant differences were observed
DNA repair capacity in liver and kidney
As p53-dependent pathways affecting global NER have been identified (Ford 2005; Sengupta and Harris 2005), we assessed whether mouse Trp53 status influences NER activity. Tissue extracts from liver and kidney were examined for their ability to repair BPDE-induced DNA adducts using a modified comet assay (Langie et al. 2006). We found that in the liver, the repair capacity was ~50 % higher in Trp53(−/−) than in Trp53(+/+) mice, while no difference in the repair capacity between mouse lines was observed in kidney (Supporting Figure 12).
DNA adduct formation of BaP ex vivo
We investigated the ability of hepatic microsomes isolated from both control and BaP-treated animals to catalyse BaP-DNA adduct formation ex vivo (Fig. 3; Supporting Figure 5). While Trp53 status had no influence on DNA adduct formation by BaP with hepatic microsomes isolated from untreated animals (Supporting Figure 5), BaP-induced adduct levels were significantly higher with microsomal samples from Trp53(+/–) and Trp53(−/−) relative to Trp53(+/+) mice pretreated with BaP (Fig. 3). The adduct pattern induced by BaP ex vivo consisted of adduct spots 1 and 2 (see inserts Fig. 3 and Supporting Figure 5). Interestingly, adduct levels of adduct 2 were higher than adduct 1, the dG-N 2-BPDE adduct, and, in addition, adduct 2 was significantly higher in experiments with Trp53(+/–) and Trp53(−/−) microsomes than with Trp53(+/+) microsomes.

BaP-DNA adducts, measured by 32P-postlabelling, formed ex vivo by hepatic microsomes isolated from BaP-pretreated Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice. Values are the mean ± range (n = 4); duplicate incubations and each sample were determined by two independent post-labelled analyses. Statistical analysis was performed by one-way ANOVA followed by Tukey post hoc test [**p < 0.01; different from hepatic microsomes isolated from Trp53(+/+) mice]. Insert autoradiographic profiles of DNA adducts formed in hepatic microsomes isolated from Trp53(+/+) mice; the origins, at the bottom left-hand corners, were cut off before exposure
BaP metabolite profile ex vivo
As our data suggested that Trp53 status affects the NADPH-dependent metabolic activation of BaP in hepatic microsomes, metabolite profiles were determined by HPLC analysis. First, we investigated whether the hepatic microsomes isolated from BaP-treated mice contained residual BaP and/or its metabolites. No amounts of BaP and BaP metabolites were detectable in these microsomal fractions (Supporting Figure 7c–e). To determine the BaP metabolite profile, hepatic microsomes isolated from Trp53(+/+), Trp53(+/–) and Trp53(−/−) were incubated with BaP and subsequently analysed by HPLC analysis. A representative HPLC chromatogram of BaP metabolites formed in ex vivo incubations containing microsomes, NADPH and BaP is shown in Supporting Figure 7a.
Hydroxylated BaP metabolites, BaP-dihydrodiols and BaP-diones were identified (see Supporting Figure 8). Previous studies have shown that many of these BaP metabolites are formed by CYP1A1 in combination with microsomal epoxide hydrolase (Bauer et al. 1995; Kim et al. 1998; Luch and Baird 2005). No BaP metabolites were detected in control incubations without microsomes, without NADPH-generating system or without BaP (Supporting Figure 7b, c). With hepatic microsomes isolated from untreated mice, Trp53 status had no influence on the BaP metabolite profile and the amounts of BaP metabolites formed (Supporting Figure 9). However, pretreating mice with BaP led to a significant increase in BaP metabolite formation ex vivo (Fig. 4). Moreover, amounts of BaP-7,8-dihydrodiol, BaP-1,6-dione, BaP-3,6-dione, BaP-9-ol and BaP-3-ol were significantly higher in microsomes of pretreated Trp53(+/–) and Trp53(−/−) mice compared with pretreated Trp53(+/+) mice. These findings correlated with ex vivo BaP-DNA adduct formation using the same hepatic microsomes (compare Fig. 3).

Formation of BaP metabolites by hepatic microsomes isolated from BaP-treated Trp53(+/+) (a), Trp53(+/–) (b) and Trp53(−/−) mice (c). Relative peak areas of BaP metabolites were measured by HPLC analysis at 254 nm. Values are the mean ± SD (n = 3). Statistical analysis was performed by one-way ANOVA followed by Tukey post hoc test [*p < 0.05, **p < 0.01, ***p < 0.005; different from BaP-treated Trp53(+/+) mice]. Structures of the BaP metabolites detected by HPLC are shown in Supplementary Figure 8. Mx, an unknown BaP metabolite
Urine analysis of Trp53(+/+) and Trp53(−/−) mice treated with BaP
A representative HPLC chromatogram of urinary BaP metabolites is shown in Supporting Figure 13a. The amounts of BaP-4,5-dihydrodiol, BaP-7,8-dihydrodiol, BaP-3,6-dione and BaP-3-ol were significantly lower in urine of Trp53(−/−) compared to Trp53(+/+) mice (Supporting Figure 13b), although most of the differences were small (~15–20 % reduction). BaP metabolites can be conjugated to glucuronides and sulphates (Luch and Baird 2005), which are excreted in the urine and faeces, but only unconjugated BaP metabolites (see Supporting Figure 8) were determined in the present work.
Influence of Trp53 status on xenobiotic-metabolising enzymes
We next studied the expression and activity of enzymes metabolising BaP. Metabolic activation of BaP is catalysed mainly by CYP1A1, but also CYP1B1 (Supporting Figure 1a), which leads via BPDE to the formation of dG-N 2-BPDE, the major BaP-derived DNA adduct detected in vivo. For these investigations, we selected liver, as Trp53 status impacted on hepatic BaP-DNA adduct formation (see Fig. 1a), and also lung, because it is the main target organ of 3-NBA genotoxicity (Arlt 2005).
Treatment of mice with BaP led to a large induction of Cyp1a1 protein levels in liver (Fig. 5a). However, induction was much greater in hepatic microsomes of BaP-treated Trp53(+/–) and Trp53(−/−) mice than of Trp53(+/+) mice, which correlates with the levels of BaP-DNA adducts in the livers of these animals. In this context, it may be noteworthy that no induction of p53 protein levels was observed after BaP treatment in the livers of Trp53(+/+) mice (Supporting Figure 10). The increase in Cyp1a1 protein levels was associated with a significant increase in EROD activity (up to ~ twofold; p < 0.05), a measure of CYP1A enzyme activity, in BaP-treated Trp53(+/–) and Trp53(−/−) compared with Trp53(+/+) mice (Fig. 6a). In contrast, Trp53 status had no effect on EROD activity in hepatic microsomes isolated from control (untreated) mice (see Supporting Figure 11a), which was in line with no changes being observed in ex vivo BaP-DNA adduct formation (compare Supporting Figure 5) and BaP metabolite formation ex vivo using the same microsomes (compare Supporting Figure 9). Cyp1a1 protein levels were increased in pulmonary microsomes isolated from BaP-treated Trp53(−/−) compared to Trp53(+/+) (Fig. 5a), but no significant change in EROD activity was observed (Fig. 6b). Lung microsomes isolated from control (untreated) mice showed variability in EROD activity between preparations and/or experiments (compare Supporting Figure 11b, d) which may also be linked to low basal activity in these microsomes. No changes for Cyp1a1 protein levels, which were close to background levels (Fig. 5a), or enzyme activity (Fig. 6c, d) were observed in hepatic and pulmonary microsomes isolated from mice treated with 3-NBA between Trp53 genotypes.

Western blot analysis of Cyp1a1 (a) and Nqo1 protein expression (b) in hepatic and pulmonary cytosols isolated from Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice exposed to BaP or 3-NBA. Representative images of the Western blotting are shown, and at least duplicate analysis was performed from independent experiments. β-Actin protein expression was used as a loading control of the microsomal fractions and Gadph for the cytosolic fractions, and a representative blot is shown

EROD activity (a–d) in hepatic (a, c) and pulmonary microsomes (b, d) isolated from Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice exposed to BaP or 3-NBA. Nqo1 enzyme activity (e–h) was determined in hepatic (e and g) and pulmonary cytosols (f and h) isolated from Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice exposed to BaP or 3-NBA. Values are the mean ± SD (n = 4). Statistical analysis was performed by one-way ANOVA followed by Tukey post hoc test [*p < 0.05; different from Trp53(+/+) mice]
The principal enzyme that activates 3-NBA by nitroreduction is NQO1 (Supporting Figure 1b) (Arlt et al. 2005; Stiborova et al. 2010). The resulting N-hydroxy-3-aminobenzanthrone (N–OH-3-ABA) can spontaneously form reactive nitrenium ions capable of forming DNA adducts. Alternatively, N–OH-3-ABA can be further activated by N-acetyltransferases or sulfotransferases, leading to the formation of the same reactive nitrenium ions (Arlt 2005). It has been shown that in vivo cytosolic nitroreduction of 3-NBA is more important than nitroreduction by microsomal NAD(P)H:cytochrome P450 oxidoreductase (Arlt et al. 2003, 2005). As we did not observe any differences in DNA adduct formation with 3-NBA, we measured the expression and activity of Nqo1 in hepatic and pulmonary cytosols. In addition, as BaP-derivatives can also be metabolised by NQO1 (Luch and Baird 2005), we also determined expression and activity of Nqo1 in mice exposed to BaP.
All cytosolic samples contained Nqo1 (Fig. 5b). In both liver and lung, BaP treatment led to an induction of Nqo1 (Fig. 5b) that was independent of the Trp53 genotype of the animals. These findings were in line with increased Nqo1 enzyme activity in these animals after BaP treatment (compare Supporting Figure 11e, f); however, Trp53 status had no effect on Nqo1 enzyme activity in either organ, with or without BaP pretreatment of the animals. In hepatic cytosols, Nqo1 protein levels were unchanged in 3-NBA-treated mice (Fig. 5b). Nqo1 protein levels were increased in the lungs of 3-NBA-treated Trp53(+/+) mice, which is in concordance with previous studies showing that 3-NBA exposure can induce NQO1 (Stiborova et al. 2006, 2008), but not in 3-NBA-treated Trp53(+/–) and Trp53(−/−) mice. Exposure to 3-NBA and Trp53 genotype had no impact on Nqo1 enzyme activity in hepatic and pulmonary cytosols (Fig. 6g, h; Supporting Figure 11g, h); however, strong variability in Nqo1 activity was seen in lung cytosols isolated from 3-NBA-treated Trp53(+/+) mice (Supporting Figure 11h). Collectively, these results suggest that p53 expression has no impact on Nqo1-mediated bioactivation of 3-NBA, which correlates with the lack of influence of Trp53 genotype on 3-NBA-DNA adduct formation in vivo (Fig. 2).
Discussion
We used Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice to investigate the effect of p53 on BaP metabolism and DNA adduct formation induced by BaP in vivo. The BaP dose used in this study, 125 mg/kg bw, has been shown to be carcinogenic and able to induce mutagenicity in multiple organs after repeated administration (Hakura et al. 1998). We found that BaP-induced DNA adduct formation in liver and kidney was significantly higher in Trp53(−/−) mice than in Trp53(+/+) mice after acute BaP treatment. Similar trends in DNA adduct formation were seen in other tissues (e.g. lung), although the difference in adduct formation did not reach statistical significance. This is in contrast to another study examining the effect of pentachlorophenol on DNA adduct formation in Trp53(+/+) and Trp53(−/−) mice exposed to BaP (Ress et al. 2002). In that study, no influence of Trp53 status on BaP-DNA adduct formation was evident in the tissues investigated, namely liver and lung. The discrepancy between the two studies could be attributable to the different dosing regimens (i.e. a higher BaP dose was used in our study), but otherwise remains unexplained at present. CYP1A1 is considered to be one of the key enzymes responsible for the metabolic activation of BaP in organisms (Luch and Baird 2005). Our data showed that higher BaP-DNA adduct levels in the livers of Trp53(−/−) mice relative to Trp53(+/+) mice correlated with higher hepatic Cyp1a protein levels and increased Cyp1a enzyme activity in these animals.
The amounts of BaP metabolites and BaP-DNA adduct levels formed in incubations using hepatic microsomes isolated from BaP-pretreated Trp53(−/−) and Trp53(+/–) mice were higher than when using those from BaP-pretreated Trp53(+/+) mice. This indicates again that Cyp activity was induced in Trp53(−/−) mice which correlated with higher BaP-DNA adduct formation in this mouse line. However, studying BaP-DNA adduct formation ex vivo, we found that adduct 2, but not formation of dG-N 2-BPDE, was higher in experiments with Trp53(+/–) and Trp53(−/−) microsomes than with Trp53(+/+) microsomes, whereas dG-N 2-BPDE levels were twice as high in livers of Trp53(−/−) mice relative to Trp53(+/+) mice in vivo. The presence of both adducts in the microsomal incubation ex vivo can be explained by different metabolic pathways, leading to the formation of the precursors of each adduct (see Supporting Figure 6). The levels of adduct 2 correlated well with Cyp1a enzyme activity in ex vivo incubations with hepatic microsomes because they are solely dictated by the Cyp1a-mediated formation of 9-hydroxy-BaP-4,5-epoxide (Stiborova et al. 2014). In contrast, formation of dG-N 2-BPDE strongly depends on the activity of microsomal epoxide hydrolase (mEH) which catalyses the hydration of BaP-7,8-epoxide to BaP-7,8-dihydrodiol which is the precursor of BPDE (see Supporting Figure 6). The formation of BaP-7,8-dihydrodiol in vivo should be more effective than ex vivo because, in addition to mEH present in hepatic microsomes used in the ex vivo incubations, other EH isoenzymes can catalyse the hydration reaction in vivo. Nevertheless, our ex vivo incubations demonstrate that Cyp-mediated BaP-DNA adduct formation is dependent on Trp53 status.
Studies investigating xenobiotic metabolism and/or carcinogen-DNA adduct formation in transgenic mice with altered Trp53 status are sparse and have mainly used Trp53(+/–) mice (Ariyoshi et al. 2001; Carmichael et al. 2001; Mori et al. 2001; Sanders et al. 2001). In one study, DNA adduct levels induced by diethylstilbestrol (DES) were ~twofold higher in Trp53(+/–) mice than in Trp53(+/+) mice (Carmichael et al. 2001). Small differences in the protein expression of several Cyp enzymes including Cyp1a1/2, Cyp1b1, Cyp2b9 and Cyp3a11 were observed, and it was concluded that these differences in the expression of XMEs between Trp53(+/–) and Trp53(+/+) mice could have contributed to the higher DES-induced DNA adduct levels seen in Trp53(+/–) mice. In this context, it is noteworthy that we also found higher DNA adduct levels in Trp53(+/–) mice relative to Trp53(+/+) mice after BaP exposure. Another study found that hepatic Cyp1a protein induction was higher in female Trp53(+/–) mice than in Trp53(+/+) mice after treatment with another PAH, 3-methylcholanthrene (Ariyoshi et al. 2001). This finding is in line with our results, showing that Cyp1a protein levels and Cyp1a enzyme activity are increased in the livers of Trp53(+/–) animals after BaP exposure. Other studies found no difference in the Cyp-mediated metabolism of N-butyl-N-(4-hydroxy-butyl)nitrosamine, benzene, ethoxyquin or methacrylonitrile between Trp53(+/+) and Trp53(+/–) mice (Mori et al. 2001; Sanders et al. 2001).
Haploinsufficiency in TP53 has been shown to promote tumour development (Berger and Pandolfi 2011) as reduction in p53 gene dosage can impact on a cell’s ability to respond to DNA damage (Berger et al. 2011). As such, survival of Trp53(+/–) mice shows an intermediate survival to that of Trp53(−/−) and Trp53(+/+) mice, and tumours that develop in Trp53(+/–) mice do not always display loss of the remaining wild-type allele (Berger et al. 2011). Interestingly, in kidney, Trp53(+/–) mice behaved like Trp53(−/−), showing higher BaP-DNA adduct levels relative to Trp53(+/+) mice, while adduct formation in the livers of Trp53(+/–) and Trp53(+/+) mice was similar but lower compared to Trp53(−/−) mice. However, both hepatic Cyp1a protein levels and Cyp1a enzyme activity were higher in BaP-treated Trp53(+/–) and Trp53(−/−) mice than in Trp53(+/+) mice, suggesting that in Trp53(+/–) animals, other factors besides Cyp1a expression may influence BaP-DNA adduct formation in the liver.
NER is considered to be the main DNA repair pathway for bulky DNA adducts (Kucab et al. 2015), and p53-dependent pathways affecting global genomic NER have been identified (Ford 2005; Sengupta and Harris 2005). A modified comet assay measuring the tissue-specific NER capacity in untreated (control) animals showed no impact of Trp53 status in kidney but an significant increase in NER capacity in Trp53(−/−) mice relative to Trp53(+/+) mice in liver. Although it is possible that BaP treatment may impact on NER capacity, a recent in vivo study has shown no transcriptomic responses related to NER genes in mice exposed to BaP (van Kesteren et al. 2013). Therefore, our results suggest that tissue-specific NER capacity did not contribute to the higher BaP-DNA adduct levels observed in Trp53(−/−) mice than in Trp53(+/+) mice, in both liver and kidney. This conclusion was strengthened by the fact that DNA adduct formation in livers and kidneys of Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice exposed to the corresponding reactive metabolite of BaP, BPDE, resulted in similar adduct levels in all mouse lines. As treatment with BPDE bypasses the need for metabolic activation, these results again indicated that the level of p53 expression impacts on the metabolic activation of the parent compound, BaP, but not on DNA repair.
Previous studies conducted in a panel of isogenic human cells differing only with respect to their endogenous TP53 status showed that complete loss of p53 function (i.e. TP53(−/−) cells) resulted in considerably lower BaP-DNA adduct levels compared to TP53(+/+) cells after BaP exposure (Hockley et al. 2008; Wohak et al. 2014). CYP1A1 protein expression was induced to a much greater extent in TP53(+/+) cells than in TP53(+/–) and TP53(−/−) cells. There were also significantly lower levels of BaP metabolites in the culture medium of TP53(+/–) and TP53(−/−) cells correlating with lower BaP-DNA adduct formation in these cell lines. It was also shown that exposure to BPDE resulted in similar adduct levels in all cell lines and that NER capacity did not contribute to the observed differences in BaP-DNA adduct formation as the repair capacity was the same in all cell lines (Wohak et al. 2014). Collectively, these results demonstrate the role of p53 in the CYP1A1-mediated metabolism of BaP in human cells.
However, the impact of p53 function on BaP metabolism is different in vitro and in vivo. Whereas loss of p53 function results in a decrease in BaP-DNA adduct formation in vitro, loss of p53 function in vivo leads to an increase in BaP-DNA adduct formation. Other studies (Arlt et al. 2008; Nebert et al. 2013) have revealed a paradox, whereby CYP enzymes (particularly CYP1A1) appear to be more important for detoxification of BaP in vivo, despite being involved in its metabolic activation in vitro, demonstrating that XMEs can have different effects on carcinogen metabolism in vitro and in vivo. Nevertheless, the mechanism involved in the altered expression and activity of Cyp1a enzymes by p53 function in mice is presently unclear. It may not always be possible to extrapolate from in vitro data to in vivo pharmacokinetics as additional factors need to be considered such as route of administration, absorption, renal clearance and tissue-specific CYP expression (Arlt et al. 2008). Additionally, it will be necessary to examine whether there are species-dependent differences between cultured human and mouse cells in the p53-dependent BaP metabolism in vitro.
Many studies have shown that BaP can induce CYP1A1 through binding to the aryl hydrocarbon receptor (Ahr), a transcription factor that also regulates the expression of a number of phase I and phase II XME genes (Shimizu et al. 2000; Wang et al. 2011). However, when BaP was administered i.p. to Ahr(−/−) mice, total BaP-DNA adduct levels were similar to those in Ahr(+/+) mice, although the formation of individual adduct spots determined by 32P-postlabelling varied (Kondraganti et al. 2003). In contrast, in other studies, BaP-induced adduct levels in the livers of Ahr(−/−) mice were significantly higher than those in Ahr(+/+) mice after oral administration (Sagredo et al. 2006, 2009). Thus, these studies provide evidence of a mechanism of BaP biotransformation that is Ahr independent. Whether this mechanism involves p53 function in vivo is currently untested and would require the creation of a Ahr(−/−)/Trp53(−/−) double-knockout mouse line.
We recently found that BaP-induced CYP1A1 expression in human cells can be regulated through p53 binding to a p53 response element (p53RE) in the regulatory region of CYP1A1, thereby enhancing its transcription and thus explaining the role of p53 function in BaP metabolism in vitro as described above (Wohak et al. 2014). Others have shown that p53 induces the activity of CYP3A4 in human cells via its binding to p53REs and the subsequent transcriptional enhancement of CYP3A4 (Goldstein et al. 2013). However, the induction of Cyps like Cyp1a1 via p53 binding to a p53RE in the Cyp1a1 promoter region fails to explain the impact of p53 on BaP metabolism in vivo (present study), as Cyp1a protein levels and Cyp1a enzyme activity were actually higher in Trp53(−/−) mice which lack p53 function suggesting that the mechanism is different in vitro and in vivo, at least in the rodent model.
It remains to be seen whether this observation is specific to BaP or a more general phenomenon. However, bioactivation and DNA adduct formation of 3-NBA in vivo were not influenced by Trp53 status (present study) which is in concordance with previous findings that 3-NBA induces similar levels of DNA adducts in human TP53(+/+) and TP53(−/−) cell lines (Hockley et al. 2008; Simoes et al. 2008). This observation indicates that the cellular impact of p53 on carcinogen metabolism depends on the agent studied and/or that only certain XMEs depend on p53 function. The most efficient enzyme to activate 3-NBA to DNA adducts is NQO1 (Arlt et al. 2005; Stiborova et al. 2010), and thus, NQO1 seems to be an enzyme not affected by p53 expression. This is in line with the fact that overall Nqo1 protein levels and Nqo1 enzyme activity were not significantly different in the Trp53(+/+), Trp53(+/–) and Trp53(−/−) mouse lines, after both BaP and 3-NBA treatment. Future studies will be required to address this question, thereby testing other environmental and dietary carcinogens both in vitro and in vivo.
In summary, we found that murine Cyp-mediated bioactivation of BaP in vivo is influenced by p53 function, thereby providing new fundamental insights into PAH-induced carcinogenesis. These results indicate that gene–environment interactions need to be taken into account with regard to xenobiotic metabolism. Our results are in line with studies demonstrating an emerging role for p53 in carcinogen metabolism in human cells in vitro. Whereas in human cells, BaP-induced CYP1A1 expression is regulated through p53 binding to a p53RE in the CYP1A1 regulatory region, thereby enhancing its transcription, the mechanism involved in the altered expression and activity of the Cyp1a1 enzyme by p53 in vivo in mice has yet to be identified. Future investigations will need to assess whether the metabolic activation of other environmental carcinogens depends on p53 in vivo and where the differences lie between murine and human p53.
References
Ariyoshi N, Imaoka S, Nakayama K, Takahashi Y, Fujita K, Funae Y, Kamataki T (2001) Comparison of the levels of enzymes involved in drug metabolism between transgenic or gene-knockout and the parental mice. Toxicol Pathol 29(Suppl):161–172
Arlt VM (2005) 3-Nitrobenzanthrone, a potential human cancer hazard in diesel exhaust and urban air pollution: a review of the evidence. Mutagenesis 20:399–410
Arlt VM, Bieler CA, Mier W, Wiessler M, Schmeiser HH (2001) DNA adduct formation by the ubiquitous environmental contaminant 3-nitrobenzanthrone in rats determined by (32)P-postlabeling. Int J Cancer 93:450–454
Arlt VM, Glatt H, Muckel E, Pabel U, Sorg BL, Schmeiser HH, Phillips DH (2002) Metabolic activation of the environmental contaminant 3-nitrobenzanthrone by human acetyltransferases and sulfotransferase. Carcinogenesis 23:1937–1945
Arlt VM, Stiborova M, Hewer A, Schmeiser HH, Phillips DH (2003) Human enzymes involved in the metabolic activation of the environmental contaminant 3-nitrobenzanthrone: evidence for reductive activation by human NADPH:cytochrome p450 reductase. Cancer Res 63:2752–2761
Arlt VM, Stiborova M, Henderson CJ, Osborne MR, Bieler CA, Frei E, Martinek V, Sopko B, Wolf CR, Schmeiser HH, Phillips DH (2005) Environmental pollutant and potent mutagen 3-nitrobenzanthrone forms DNA adducts after reduction by NAD(P)H:quinone oxidoreductase and conjugation by acetyltransferases and sulfotransferases in human hepatic cytosols. Cancer Res 65:2644–2652
Arlt VM, Schmeiser HH, Osborne MR, Kawanishi M, Kanno T, Yagi T, Phillips DH, Takamura-Enya T (2006) Identification of three major DNA adducts formed by the carcinogenic air pollutant 3-nitrobenzanthrone in rat lung at the C8 and N2 position of guanine and at the N6 position of adenine. Int J Cancer 118:2139–2146
Arlt VM, Stiborova M, Henderson CJ, Thiemann M, Frei E, Aimova D, Singh R, Gamboa da Costa G, Schmitz OJ, Farmer PB, Wolf CR, Phillips DH (2008) Metabolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo: experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis 29:656–665
Arlt VM, Poirier MC, Sykes SE, John K, Moserova M, Stiborova M, Wolf CR, Henderson CJ, Phillips DH (2012) Exposure to benzo[a]pyrene of Hepatic Cytochrome P450 Reductase Null (HRN) and P450 Reductase Conditional Null (RCN) mice: detection of benzo[a]pyrene diol epoxide-DNA adducts by immunohistochemistry and 32P-postlabelling. Toxicol Lett 213:160–166
Bauer E, Guo Z, Ueng YF, Bell LC, Zeldin D, Guengerich FP (1995) Oxidation of benzo[a]pyrene by recombinant human cytochrome P450 enzymes. Chem Res Toxicol 8:136–142
Berger AH, Pandolfi PP (2011) Haplo-insufficiency: a driving force in cancer. J Pathol 223(2):137–146
Berger AH, Knudson AG, Pandolfi PP (2011) A continuum model for tumour suppression. Nature 476:163–169
Carmichael PL, Mills JJ, Campbell M, Basu M, Caldwell J (2001) Mechanisms of hormonal carcinogenesis in the p53 +/- hemizygous knockout mouse: studies with diethylstilbestrol. Toxicol Pathol 29(Suppl):155–160
Donehower LA (2014) Insights into wild-type and mutant p53 functions provided by genetically engineered mice. Human Mutat 35:715–727
Ford JM (2005) Regulation of DNA damage recognition and nucleotide excision repair: another role for p53. Mutat Res 577:195–202
Goldstein I, Rivlin N, Shoshana OY, Ezra O, Madar S, Goldfinger N, Rotter V (2013) Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis 34:190–198
Güngör N, Haegens A, Knaapen AM, Godschalk RW, Chiu RK, Wouters EF, van Schooten FJ (2010) Lung inflammation is associated with reduced pulmonary nucleotide excision repair in vivo. Mutagenesis 25:77–82
Hakura A, Tsutsui Y, Sonoda J, Kai J, Imade T, Shimada M, Sugihara Y, Mikami T (1998) Comparison between in vivo mutagenicity and carcinogenicity in multiple organs by benzo[a]pyrene in the lacZ transgenic mouse (Muta Mouse). Mutat Res 398:123–130
Hockley SL, Arlt VM, Brewer D, Giddings I, Phillips DH (2006) Time- and concentration-dependent changes in gene expression induced by benzo(a)pyrene in two human cell lines, MCF-7 and HepG2. BMC Genom 7:260
Hockley SL, Arlt VM, Jahnke G, Hartwig A, Giddings I, Phillips DH (2008) Identification through microarray gene expression analysis of cellular responses to benzo(a)pyrene and its diol-epoxide that are dependent or independent of p53. Carcinogenesis 29:202–210
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4:1–7
Kenzelmann Broz D, Attardi LD (2010) In vivo analysis of p53 tumor suppressor function using genetically engineered mouse models. Carcinogenesis 31:1311–1318
Kim JH, Stansbury KH, Walker NJ, Trush MA, Strickland PT, Sutter TR (1998) Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis 19:1847–1853
Kondraganti SR, Fernandez-Salguero P, Gonzalez FJ, Ramos KS, Jiang W, Moorthy B (2003) Polycyclic aromatic hydrocarbon-inducible DNA adducts: evidence by 32P-postlabeling and use of knockout mice for Ah receptor-independent mechanisms of metabolic activation in vivo. Int J Cancer 103:5–11
Kucab JE, Phillips DH, Arlt VM (2010) Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J 277:2567–2583
Kucab JE, van Steeg H, Luiten M, Schmeiser HH, White PA, Phillips DH, Arlt VM (2015) TP53 mutations induced by BPDE in Xpa-WT and Xpa-Null human TP53 knock-in (Hupki) mouse embryo fibroblasts. Mutat Res, Fundam Mol Mech Mutagen 773:48–62
Langie SA, Knaapen AM, Brauers KJ, van Berlo D, van Schooten FJ, Godschalk RW (2006) Development and validation of a modified comet assay to phenotypically assess nucleotide excision repair. Mutagenesis 21:153–158
Lozano G (2010) Mouse models of p53 functions. Cold Spring Harb Perspect Biol 2:a001115
Luch A, Baird WM (2005) Metabolic activation and detoxification of polycyclic aromatic hydrocarbons. Imperial College Press, London
Maddocks OD, Vousden KH (2011) Metabolic regulation by p53. J Mol Med 89:237–245
Mizerovska J, Dracinska H, Frei E, Schmeiser HH, Arlt VM, Stiborova M (2011) Induction of biotransformation enzymes by the carcinogenic air-pollutant 3-nitrobenzanthrone in liver, kidney and lung, after intra-tracheal instillation in rats. Mutat Res 720:34–41
Mori Y, Koide A, Fuwa K, Wanibuchi H, Fukushima S (2001) Lack of change in the levels of liver and kidney cytochrome P-450 isozymes in p53 +/- knockout mice treated with N-butyl-N-(4-hydroxybutyl)nitrosamine. Mutagenesis 16:377–383
Nebert DW (2006) Comparison of gene expression in cell culture to that in the intact animal: relevance to drugs and environmental toxicants. Focus on “development of a transactivator in hepatoma cells that allows expression of phase I, phase II, and chemical defense genes”. Am J Physiol Cell Physiol 290:C37–C41
Nebert DW, Dalton TP (2006) The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev 6:947–960
Nebert DW, Shi Z, Galvez-Peralta M, Uno S, Dragin N (2013) Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences–Cyp1 knockout mouse lines as a paradigm. Mol Pharmacol 84:304–313
Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2:a001008
Phillips DH, Arlt VM (2007) The 32P-postlabeling assay for DNA adducts. Nat Protocol 2:2772–2781
Phillips DH, Arlt VM (2014) (32)P-postlabeling analysis of DNA adducts. Meth Mol Biol 1105:127–138
Ress NB, Donnelly KC, George SE (2002) The effect of pentachlorophenol on DNA adduct formation in p53 wild-type and knockout mice exposed to benzo[a]pyrene. Cancer Lett 178:11–17
Sagredo C, Ovrebo S, Haugen A, Fujii-Kuriyama Y, Baera R, Botnen IV, Mollerup S (2006) Quantitative analysis of benzo[a]pyrene biotransformation and adduct formation in Ahr knockout mice. Toxicol Lett 167:173–182
Sagredo C, Mollerup S, Cole KJ, Phillips DH, Uppstad H, Ovrebo S (2009) Biotransformation of benzo[a]pyrene in Ahr knockout mice is dependent on time and route of exposure. Chem Res Toxicol 22:584–591
Sanders JM, Burka LT, Chanas B, Matthews HB (2001) Comparative xenobiotic metabolism between Tg.AC and p53 +/- genetically altered mice and their respective wild types. Toxicol Sci 61:54–61
Sengupta S, Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 6:44–55
Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T (2000) Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci USA 97:779–782
Simoes ML, Hockley SL, Schwerdtle T, da Costa GG, Schmeiser HH, Phillips DH, Arlt VM (2008) Gene expression profiles modulated by the human carcinogen aristolochic acid I in human cancer cells and their dependence on TP53. Toxicol Appl Pharmacol 232:86–98
Stiborova M, Dracinska H, Hajkova J, Kaderabkova P, Frei E, Schmeiser HH, Soucek P, Phillips DH, Arlt VM (2006) The environmental pollutant and carcinogen 3-nitrobenzanthrone and its human metabolite 3-aminobenzanthrone are potent inducers of rat hepatic cytochromes P450 1A1 and -1A2 and NAD(P)H:quinone oxidoreductase. Drug Metab Dispos 34:1398–1405
Stiborova M, Dracinska H, Mizerovska J, Frei E, Schmeiser HH, Hudecek J, Hodek P, Phillips DH, Arlt VM (2008) The environmental pollutant and carcinogen 3-nitrobenzanthrone induces cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase in rat lung and kidney, thereby enhancing its own genotoxicity. Toxicology 247:11–22
Stiborova M, Martinek V, Svobodova M, Sístkova J, Dvorak Z, Ulrichova J, Simanek V, Frei E, Schmeiser HH, Phillips DH, Arlt VM (2010) Mechanisms of the different DNA adduct forming potentials of the urban air pollutants 2-nitrobenzanthrone and carcinogenic 3-nitrobenzanthrone. Chem Res Toxicol 23:1192–1201
Stiborova M, Moserova M, Cerna V, Indra R, Dracinsky M, Sulc M, Henderson CJ, Wolf CR, Schmeiser HH, Phillips DH, Frei E, Arlt VM (2014) Cytochrome b5 and epoxide hydrolase contribute to benzo[a]pyrene-DNA adduct formation catalyzed by cytochrome P450 1A1 under low NADPH:P450 oxidoreductase conditions. Toxicology 318:1–12
Taneja P, Zhu S, Maglic D, Fry EA, Kendig RD, Inoue K (2011) Transgenic and knockout mice models to reveal the functions of tumor suppressor genes. Clin Med Insights Oncol 5:235–257
Van Kesteren PCE, Zwart PE, Schaap MM, Pronk TE, van Herwijnen MHM, Kleinjans JCS, Bokkers BGH, Godschalk RWL, Zeilmaker MJ, van Steeg H, Luijten M (2013) Benzo[a]pyrene-induced transcriptomic responses in primary hepatocytes and in vivo liver: toxicokinetics is essential for in vivo-in vitro comparisons. Arch Toxicol 87:505–515
Wang T, Gavin HM, Arlt VM, Lawrence BP, Fenton SE, Medina D, Vorderstrasse BA (2011) Aryl hydrocarbon receptor activation during pregnancy, and in adult nulliparous mice, delays the subsequent development of DMBA-induced mammary tumors. Int J Cancer 128:1509–1523
Whibley C, Pharoah PD, Hollstein M (2009) p53 polymorphisms: cancer implications. Nature Rev 9:95–107
Wiechelman KJ, Braun RD, Fitzpatrick JD (1988) Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal Biochem 175:231–237
Wohak LE, Krais AM, Kucab JE, Stertmann J, Ovrebo S, Seidel A, Phillips DH, Arlt VM (2014) Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53 dependent mechanism. Arch Toxicol, Nov 15 (Epub ahead of print)
Acknowledgments
Work at King’s College London is supported by Cancer Research UK (Grant C313/A14329) and the Wellcome Trust (Grants 101126/Z/13/Z and 101126/B/13/Z). Annette Krais was supported by a fellowship of the German Research Foundation (DFG). Work at Charles University was supported by a UNCE 204025/2012 Center Grant and the Grant Agency of the Czech Republic (GACR; Grant 14-18344S in panel P301). David Phillips and Volker Arlt are members of the Wellcome Trust funded COMSIG (Causes of Mutational SIGnatures) consortium.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krais, A.M., Speksnijder, E.N., Melis, J.P.M. et al. The impact of p53 on DNA damage and metabolic activation of the environmental carcinogen benzo[a]pyrene: effects in Trp53(+/+), Trp53(+/–) and Trp53(−/−) mice. Arch Toxicol 90, 839–851 (2016). https://doi.org/10.1007/s00204-015-1531-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-015-1531-8