
Abstract
The Clinician’s Guide to Prevention and Treatment of Osteoporosis was developed by an expert committee of the National Osteoporosis Foundation (NOF) in collaboration with a multispecialty council of medical experts in the field of bone health convened by NOF. Readers are urged to consult current prescribing information on any drug, device, or procedure discussed in this publication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Executive summary
Osteoporosis is a silent disease until it is complicated by fractures—fractures that occur following minimal trauma or, in some cases, with no trauma. Fractures are common and place an enormous medical and personal burden on the aging individuals who suffer them and take a major economic toll on the nation. Osteoporosis can be prevented, diagnosed, and treated before fractures occur. Importantly, even after the first fracture has occurred, there are effective treatments to decrease the risk of further fractures. Prevention, detection, and treatment of osteoporosis should be a mandate of primary care providers.
Since the National Osteoporosis Foundation (NOF) first published the Guide in 1999, it has become increasingly clear that many patients are not being given appropriate information about prevention and many patients are not receiving appropriate testing to diagnose osteoporosis or establish osteoporosis risk. Most importantly, many patients who have osteoporosis-related fractures are not being diagnosed with osteoporosis and are not receiving any of the Food and Drug Administration (FDA)-approved, effective therapies.
This Guide offers concise recommendations regarding prevention, risk assessment, diagnosis, and treatment of osteoporosis in postmenopausal women and men age 50 and older. It includes indications for bone densitometry and fracture risk thresholds for intervention with pharmacologic agents. The absolute risk thresholds at which consideration of osteoporosis treatment is recommended were guided by a cost-effectiveness analysis.
Synopsis of major recommendations to the clinician
Recommendations apply to postmenopausal women and men age 50 and older.
Universal recommendations
-
Counsel on the risk of osteoporosis and related fractures.
-
Advise on a diet that includes adequate amounts of total calcium intake (1000 mg/day for men 50–70; 1200 mg/day for women 51 and older and men 71 and older), incorporating dietary supplements if diet is insufficient.
-
Advise on vitamin D intake (800–1000 IU/day), including supplements if necessary for individuals age 50 and older.
-
Recommend regular weight-bearing and muscle-strengthening exercise to improve agility, strength, posture, and balance; maintain or improve bone strength; and reduce the risk of falls and fractures.
-
Assess risk factors for falls and offer appropriate modifications (e.g., home safety assessment, balance training exercises, correction of vitamin D insufficiency, avoidance of central nervous system depressant medications, careful monitoring of antihypertensive medication, and visual correction when needed).
-
Advise on cessation of tobacco smoking and avoidance of excessive alcohol intake.
Diagnostic assessment
-
Measure height annually, preferably with a wall-mounted stadiometer.
-
Bone mineral density (BMD) testing should be performed:
-
In women age 65 and older and men age 70 and older
-
In postmenopausal women and men above age 50–69, based on risk factor profile
-
In postmenopausal women and men age 50 and older who have had an adult age fracture, to diagnose and determine degree of osteoporosis
-
At dual-energy X-ray absorptiometry (DXA) facilities using accepted quality assurance measures
-
-
Vertebral imaging should be performed:
-
In all women age 70 and older and all men age 80 and older if BMD T-score is ≤−1.0 at the spine, total hip, or femoral neck
-
In women age 65 to 69 and men age 70 to 79 if BMD T-score is ≤−1.5 at the spine, total hip, or femoral neck
-
In postmenopausal women and men age 50 and older with specific risk factors:
-
Low-trauma fracture during adulthood (age 50 and older)
-
Historical height loss (difference between the current height and peak height at age 20) of 1.5 in. or more (4 cm)
-
Prospective height loss (difference between the current height and a previously documented height measurement) of 0.8 in. or more (2 cm)
-
Recent or ongoing long-term glucocorticoid treatment
-
-
If bone density testing is not available, vertebral imaging may be considered based on age alone.
-
-
Check for secondary causes of osteoporosis.
-
Biochemical markers of bone turnover can aid in risk assessment and serve as an additional monitoring tool when treatment is initiated.
Monitoring patients
-
Perform BMD testing 1 to 2 years after initiating medical therapy for osteoporosis and every 2 years thereafter.
-
More frequent BMD testing may be warranted in certain clinical situations.
-
The interval between repeat BMD screenings may be longer for patients without major risk factors and who have an initial T-score in the normal or upper low bone mass range.
-
-
Biochemical markers can be repeated to determine if treatment is producing expected effect.
Pharmacologic treatment recommendations
-
Initiate pharmacologic treatment:
-
In those with hip or vertebral (clinical or asymptomatic) fractures
-
In those with T-scores ≤−2.5 at the femoral neck, total hip, or lumbar spine by DXA
-
In postmenopausal women and men age 50 and older with low bone mass (T-score between −1.0 and −2.5, osteopenia) at the femoral neck, total hip, or lumbar spine by DXA and a 10-year hip fracture probability ≥3 % or a 10-year major osteoporosis-related fracture probability ≥20 % based on the USA-adapted WHO absolute fracture risk model (Fracture Risk Algorithm (FRAX®); www.NOF.org and www.shef.ac.uk/FRAX)
-
-
Current FDA-approved pharmacologic options for osteoporosis are bisphosphonates (alendronate, ibandronate, risedronate, and zoledronic acid), calcitonin, estrogen agonist/antagonist (raloxifene), estrogens and/or hormone therapy, tissue-selective estrogen complex (conjugated estrogens/bazedoxifene), parathyroid hormone 1–34 (teriparatide), and receptor activator of nuclear factor kappa-B (RANK) ligand inhibitor (denosumab).
-
No pharmacologic therapy should be considered indefinite in duration. After the initial treatment period, which depends on the pharmacologic agent, a comprehensive risk assessment should be performed. There is no uniform recommendation that applies to all patients and duration decisions need to be individualized.
-
In adults age 50 and older, after a fracture, institute appropriate risk assessment and treatment measures for osteoporosis as indicated. Fracture liaison service (FLS) programs, where patients with recent fractures may be referred for care coordination and transition management, have demonstrated improvement in the quality of care delivered.
Osteoporosis: impact and overview
Scope of the problem
Osteoporosis is the most common bone disease in humans, representing a major public health problem as outlined in Bone Health and Osteoporosis: A Report of the Surgeon General (2004) [1]. It is characterized by low bone mass, deterioration of bone tissue and disruption of bone architecture, compromised bone strength, and an increase in the risk of fracture. According to the WHO diagnostic classification, osteoporosis is defined by BMD at the hip or lumbar spine that is less than or equal to 2.5 standard deviations below the mean BMD of a young-adult reference population. Osteoporosis is a risk factor for fracture just as hypertension is for stroke. The risk of fractures is highest in those with the lowest BMD; however, the majority of fractures occur in patients with low bone mass rather than osteoporosis, because of the large number of individuals with bone mass in this range.
Osteoporosis affects an enormous number of people, of both sexes and all races, and its prevalence will increase as the population ages. Based on data from the National Health and Nutrition Examination Survey III (NHANES III), NOF has estimated that more than 9.9 million Americans have osteoporosis and an additional 43.1 million have low bone density [2]. About one out of every two Caucasian women will experience an osteoporosis-related fracture at some point in her lifetime, as will approximately one in five men [1]. Although osteoporosis is less frequent in African Americans, those with osteoporosis have the same elevated fracture risk as Caucasians.
Medical impact
Fractures and their complications are the relevant clinical sequelae of osteoporosis. The most common fractures are those of the vertebrae (spine), proximal femur (hip), and distal forearm (wrist). However, most fractures in older adults are due at least in part to low bone mass, even when they result from considerable trauma. A recent fracture at any major skeletal site in an adult older than 50 years of age should be considered a significant event for the diagnosis of osteoporosis and provides a sense of urgency for further assessment and treatment. The most notable exceptions are those of the fingers, toes, face, and skull, which are primarily related to trauma rather than underlying bone strength. Fractures may be followed by full recovery or by chronic pain, disability, and death [3].
Hip fractures are associated with an 8 to 36 % excess mortality within 1 year, with a higher mortality in men than in women [4]; additionally, hip fractures are followed by a 2.5-fold increased risk of future fractures [5]. Approximately 20 % of hip fracture patients require long-term nursing home care, and only 40 % fully regain their pre-fracture level of independence [1]. Although the majority of vertebral fractures are initially clinically silent, these fractures are often associated with symptoms of pain, disability, deformity, and mortality [3]. Postural changes associated with kyphosis may limit activity, including bending and reaching.
Multiple thoracic fractures may result in restrictive lung disease, and lumbar fractures may alter abdominal anatomy, leading to constipation, abdominal pain, distention, reduced appetite, and premature satiety. Vertebral fractures, whether clinically apparent or silent, are major predictors of future fracture risk, up to 5-fold for subsequent vertebral fracture and 2- to 3-fold for fractures at other sites. Wrist fractures are less disabling but can interfere with some activities of daily living as much as hip or vertebral fractures.
Pelvic fractures and humerus fractures are also common and contribute to increased morbidity and mortality. Fractures can also cause psychosocial symptoms, most notably depression and loss of self-esteem, as patients grapple with pain, physical limitations, and lifestyle and cosmetic changes.
Economic toll
Annually, two million fractures are attributed to osteoporosis, causing more than 432,000 hospital admissions, almost 2.5 million medical office visits, and about 180,000 nursing home admissions in the USA [1]. Medicare currently pays for approximately 80 % of these fractures, with hip fractures accounting for 72 % of fracture costs. Due in part to an aging population, the cost of care is expected to rise to $25.3 billion by 2025 [6].
Despite the availability of cost-effective and well-tolerated treatments to reduce fracture risk, only 23 % of women age 67 or older who have an osteoporosis-related fracture receive either a BMD test or a prescription for a drug to treat osteoporosis in the 6 months after the fracture [7].
Basic pathophysiology
Bone mass in older adults equals the peak bone mass achieved by age 18–25 minus the amount of bone subsequently lost. Peak bone mass is determined largely by genetic factors, with contributions from nutrition, endocrine status, physical activity, and health during growth [8].
The process of bone remodeling that maintains a healthy skeleton may be considered a preventive maintenance program, continually removing older bone and replacing it with new bone. Bone loss occurs when this balance is altered, resulting in greater bone removal than replacement. The imbalance occurs with menopause and advancing age. With the onset of menopause, the rate of bone remodeling increases, magnifying the impact of the remodeling imbalance. The loss of bone tissue leads to disordered skeletal architecture and an increase in fracture risk.
Figure 1 shows the changes within cancellous bone as a consequence of bone loss. Individual trabecular plates of bone are lost, leaving an architecturally weakened structure with significantly reduced mass. Increasing evidence suggests that rapid bone remodeling (as measured by biochemical markers of bone resorption or formation) increases bone fragility and fracture risk.

Bone loss leads to an increased risk of fracture that is magnified by other aging-associated declines in functioning. Figure 2 shows the factors associated with an increased risk of osteoporosis-related fractures. These include general factors that relate to aging and sex steroid deficiency, as well as specific risk factors, such as use of glucocorticoids, which cause decreased bone formation and bone loss, reduced bone quality, and disruption of microarchitectural integrity. Fractures result when weakened bone is overloaded, often by falls or certain activities of daily living.

Pathogenesis of osteoporosis-related fractures, from Cooper and Melton, with modification [10]
Approach to the diagnosis and management of osteoporosis
NOF recommends a comprehensive approach to the diagnosis and management of osteoporosis. A detailed history and physical examination together with BMD assessment, vertebral imaging to diagnose vertebral fractures, and, when appropriate, the WHO 10-year estimated fracture probability are utilized to establish the individual patient’s fracture risk [11]. Therapeutic intervention thresholds are based on NOF’s economic analysis that takes into consideration the cost-effectiveness of treatments and competition for resources in the USA [12, 13]. The clinician’s clinical skills and past experience, incorporating the best patient-based research available, are used to determine the appropriate therapeutic intervention. The potential risks and benefits of all osteoporosis interventions should be reviewed with patients and the unique concerns and expectations of individual patients considered in any final therapeutic decision.
Risk assessment
All postmenopausal women and men age 50 and older should be evaluated for osteoporosis risk in order to determine the need for BMD testing and/or vertebral imaging. In general, the more risk factors that are present, the greater is the risk of fracture. Osteoporosis is preventable and treatable, but because there are no warning signs prior to a fracture, many people are not being diagnosed in time to receive effective therapy during the early phase of the disease. Many factors have been associated with an increased risk of osteoporosis-related fracture (Table 1).
Since the majority of osteoporosis-related fractures result from falls, it is also important to evaluate risk factors for falling (Table 2). The most important of these are personal history of falling, muscle weakness and gait, selected medications, balance, and visual deficits [15]. Dehydration is also a risk factor for falls.
Several of these risk factors have been included in the WHO 10-year fracture risk model (Table 3). As suggested by the WHO [11], this set of risk factors increases fracture risk independently of BMD and can be combined with BMD measurements to assess an individual patient’s risk of future fracture.
Diagnostic assessment
Consider the possibility of osteoporosis and fracture risk based on the presence of the risk factors and conditions outlined in Tables 1 and 3. Metabolic bone diseases other than osteoporosis, such as hyperparathyroidism or osteomalacia, may be associated with low BMD. Many of these diseases have very specific therapies, and it is appropriate to complete a history and physical examination before making a diagnosis of osteoporosis on the basis of a low BMD alone. In patients in whom a specific secondary, treatable cause of osteoporosis is being considered (Table 1), relevant blood and urine studies (see below) should be obtained prior to initiating therapy. Any adulthood fracture may be an indication of osteoporosis and should be evaluated accordingly. Consider hip and vertebral fractures as indications of osteoporosis unless excluded by the clinical evaluation and imaging. Fractures present a sense of urgency as they signify increased fracture risk over the subsequent 5 years [16]. Patients with recent fractures, multiple fractures, or very low BMD should be evaluated for secondary etiologies.
Osteoporosis affects a significant number of men, yet the condition often goes undetected and untreated. The evaluation of osteoporosis in men requires special consideration as some of the laboratory testing to assess underlying causes in men differs from those in women. Screening BMD and vertebral imaging recommendations for men are outlined in Table 8. The 2012 Endocrine Society’s Osteoporosis in Men: An Endocrine Society Clinical Practice Guideline provides a detailed approach to the evaluation and treatment of osteoporosis in men [17].
Diagnosis
The diagnosis of osteoporosis is established by measurement of BMD or by the occurrence of adulthood hip or vertebral fracture in the absence of major trauma (such as a motor vehicle accident or multiple story fall). Laboratory testing is indicated to exclude secondary causes of osteoporosis [1, 14, 17] (Table 4).
BMD measurement and classification
DXA measurement of the hip and spine is the technology used to establish or confirm a diagnosis of osteoporosis, predict future fracture risk, and monitor patients. Areal BMD is expressed in absolute terms of grams of mineral per square centimeter scanned (g/cm2) and as a relationship to two norms: compared to the BMD of an age-, sex-, and ethnicity-matched reference population (Z-score) or compared to a young-adult reference population of the same sex (T-score). The difference between the patient’s BMD and the mean BMD of the reference population, divided by the standard deviation (SD) of the reference population, is used to calculate T-scores and Z-scores. Peak bone mass is achieved in early adulthood, followed by a decline in BMD. The rate of bone loss accelerates in women at menopause and continues to progress at a slower pace in older postmenopausal women (see Fig. 3) and in older men. An individual’s BMD is presented as the standard deviation above or below the mean BMD of the reference population, as outlined in Table 5. The BMD diagnosis of normal, low bone mass (osteopenia), osteoporosis, and severe or established osteoporosis is based on the WHO diagnostic classification (Table 5) [18].

Z- and T-scores in women, from ISCD Bone Densitometry Clinician Course, Lecture 5 (2008), with permission of the International Society for Clinical Densitometry
BMD testing is a vital component in the diagnosis and management of osteoporosis. BMD has been shown to correlate with bone strength and is an excellent predictor of future fracture risk. Instead of a specific threshold, fracture risk increases exponentially as BMD decreases. Although available technologies measuring central (lumbar spine and hip) and peripheral skeletal sites (forearm, heel, fingers) provide site-specific and global (overall risk at any skeletal site) assessment of future fracture risk, DXA measurement at the hip is the best predictor of future hip fracture risk. DXA measurements of the lumbar spine and hip must be performed by appropriately trained technologists on properly maintained instruments. DXA scans are associated with exposure to trivial amounts of radiation.
In postmenopausal women and men age 50 and older, the WHO diagnostic T-score criteria (normal, low bone mass, and osteoporosis) are applied to BMD measurement by central DXA at the lumbar spine and femoral neck [18]. BMD measured by DXA at the one-third (33 %) radius site can be used for diagnosing osteoporosis when the hip and lumbar spine cannot be measured or are unusable or uninterpretable [19]. In premenopausal women, men less than 50 years of age, and children, the WHO BMD diagnostic classification should not be applied. In these groups, the diagnosis of osteoporosis should not be made on the basis of densitometric criteria alone. The International Society for Clinical Densitometry (ISCD) recommends that instead of T-scores, ethnic or race-adjusted Z-scores should be used, with Z-scores of −2.0 or lower defined as either “low bone mineral density for chronological age” or “below the expected range for age” and those above −2.0 being “within the expected range for age” [19].
Who should be tested?
The decision to perform bone density assessment should be based on an individual’s fracture risk profile and skeletal health assessment. Utilizing any procedure to measure bone density is not indicated unless the results will influence the patient’s treatment decision. The US Preventive Services Task Force recommends testing of all women age 65 and older and younger women whose fracture risk is equal to or greater than that of a 65-year-old white woman who has no additional risk factors [20].
Table 6 outlines the indications for BMD testing. BMD measurement is not recommended in children or adolescents and is not routinely indicated in healthy young men or premenopausal women unless there is a significant fracture history or there are specific risk factors for bone loss.
Vertebral imaging
A vertebral fracture is consistent with a diagnosis of osteoporosis, even in the absence of a bone density diagnosis, and is an indication for pharmacologic treatment with osteoporosis medication to reduce subsequent fracture risk [18, 21]. Most vertebral fractures are asymptomatic when they first occur and often are undiagnosed for many years. Proactive vertebral imaging is the only way to diagnose these fractures. The finding of a previously unrecognized vertebral fracture may change the diagnostic classification, alter future fracture risk calculations, and affect treatment decisions [22].
Independent of BMD, age, and other clinical risk factors, radiographically confirmed vertebral fractures (even if completely asymptomatic) are a sign of impaired bone quality and strength and a strong predictor of new vertebral and other fractures. The presence of a single vertebral fracture increases the risk of subsequent fractures 5-fold and the risk of hip and other fractures 2- to 3- fold [23]. Vertebral imaging can be performed using a lateral thoracic and lumbar spine X-ray or lateral vertebral fracture assessment (VFA), available on most modern DXA machines. VFA can be conveniently performed at the time of BMD assessment, while conventional X-ray may require referral to a standard X-ray facility.
Indications for vertebral imaging
Because vertebral fractures are so prevalent in older individuals and most produce no acute symptoms, vertebral imaging tests are recommended for the individuals defined in Table 7. Once a first vertebral imaging test is done, it only needs to be repeated if prospective height loss is documented or new back pain or postural change occurs [3, 24]. A follow-up vertebral imaging test is also recommended in patients who are being considered for a medication holiday, since stopping medication would not be recommended in patients who have recent vertebral fractures.
Biochemical markers of bone turnover
Bone remodeling (or turnover) occurs throughout life to repair fatigue damage and microfractures in bone and to maintain mineral homeostasis. Biochemical markers of bone remodeling [e.g., resorption markers—serum C-telopeptide (CTX) and urinary N-telopeptide (NTX)—and formation markers—serum bone-specific alkaline phosphatase (BSAP), osteocalcin (OC), and aminoterminal propeptide of type I procollagen (PINP)] are best collected in the morning while patients are fasting.
Biochemical markers of bone turnover may [25]:
-
Predict risk of fracture independently of bone density in untreated patients
-
Predict rapidity of bone loss in untreated patients
-
Predict extent of fracture risk reduction when repeated after 3–6 months of treatment with FDA-approved therapies
-
Predict magnitude of BMD increases with FDA-approved therapies
-
Help determine adequacy of patient compliance and persistence with osteoporosis therapy
-
Help determine duration of “drug holiday” and when and if medication should be restarted. (Data are quite limited to support this use, but studies are underway.)
Use of WHO FRAX® in the USA
FRAX® was developed to calculate the 10-year probability of a hip fracture and the 10-year probability of a major osteoporotic fracture (defined as clinical vertebral, hip, forearm, or proximal humerus fracture), taking into account femoral neck BMD and the clinical risk factors shown in Table 3 [11]. The FRAX® algorithm is available at www.nof.org as well as at www.shef.ac.uk/FRAX. It is also available on newer DXA machines or with software upgrades that provide the FRAX® scores on the bone density report.
The WHO algorithm used in this Guide was calibrated to US fracture and mortality rates; therefore, the fracture risk figures herein are specific for the US population. Economic modeling was performed to identify the 10-year hip fracture risk above which it is cost-effective, from the societal perspective, to treat with pharmacologic agents. The US-based economic modeling is described in one report [12], and the US-adapted WHO algorithm and its clinical application are illustrated in a companion report [13].
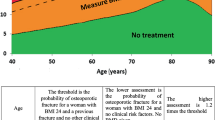
The latter analyses generally confirm the previous NOF conclusion that it is cost-effective to treat individuals with a prior hip or vertebral fracture and those with a DXA femoral neck T-score ≤−2.5. Previous analyses have established that a lumbar spine T-score ≤−2.5 also warrants treatment [26].
FRAX underestimates fracture risk in patients with recent fractures, multiple osteoporosis-related fractures, and those at increased risk for falling. FRAX® is most useful in patients with low femoral neck BMD. Utilizing FRAX® in patients with low BMD at the lumbar spine but a relatively normal BMD at the femoral neck underestimates fracture risk in these individuals. Specifically, the WHO algorithm has not been validated for the use of lumbar spine BMD. NOF recommends treatment of individuals with osteoporosis of the lumbar spine as well as the hip.
Application of US-adapted FRAX® in the USA
-
FRAX® is intended for postmenopausal women and men age 50 and older; it is not intended for use in younger adults or children.
-
The FRAX® tool has not been validated in patients currently or previously treated with pharmacotherapy for osteoporosis. In such patients, clinical judgment must be exercised in interpreting FRAX® scores. Patients who have been off osteoporosis medications for 1 to 2 years or more might be considered untreated [27].
-
FRAX® can be calculated with either femoral neck BMD or total hip BMD, but, when available, femoral neck BMD is preferred. The use of BMD from nonhip sites is not recommended.
-
The WHO determined that for many secondary causes of osteoporosis, fracture risk was mediated primarily through impact on BMD [28]. For this reason, when femoral neck BMD is inserted into FRAX®, the secondary causes of osteoporosis button are automatically inactivated.
The therapeutic thresholds proposed in this Guide are for clinical guidance only and are not rules. All treatment decisions require clinical judgment and consideration of individual patient factors, including patient preferences, comorbidities, risk factors not captured in the FRAX® model (e.g., frailty, falls), recent decline in bone density, and other sources of possible under- or overestimation of fracture risk by FRAX®.
The therapeutic thresholds do not preclude clinicians or patients from considering intervention strategies for those who do not have osteoporosis by BMD (WHO diagnostic criterion of T-score ≤−2.5), do not meet the cut points after FRAX®, or are not at high enough risk of fracture despite low BMD. Conversely, these recommendations should not mandate treatment, particularly in patients with low bone mass above the osteoporosis range. Decisions to treat must still be made on a case-by-case basis.
Additional bone densitometry technologies
The following bone mass measurement technologies included in Table 8 are capable of predicting both site-specific and overall fracture risk. When performed according to accepted standards, these densitometric techniques are accurate and highly reproducible [19]. However, T-scores from these technologies cannot be used according to the WHO diagnostic classification because they are not equivalent to T-scores derived from DXA.
Universal recommendations for all patients
Several interventions to preserve bone strength can be recommended to the general population. These include an adequate intake of calcium and vitamin D, lifelong participation in regular weight-bearing and muscle-strengthening exercise, cessation of tobacco use, identification and treatment of alcoholism, and treatment of risk factors for falling.
Adequate intake of calcium and vitamin D
Advise all individuals to obtain an adequate intake of dietary calcium. Providing adequate daily calcium and vitamin D is a safe and inexpensive way to help reduce fracture risk. Controlled clinical trials have demonstrated that the combination of supplemental calcium and vitamin D can reduce the risk of fracture [29]. A balanced diet rich in low-fat dairy products, fruits, and vegetables provides calcium as well as numerous nutrients needed for good health. If adequate dietary calcium cannot be obtained, dietary supplementation is indicated up to the recommended daily intake.
Lifelong adequate calcium intake is necessary for the acquisition of peak bone mass and subsequent maintenance of bone health. The skeleton contains 99 % of the body’s calcium stores; when the exogenous supply is inadequate, bone tissue is resorbed from the skeleton to maintain serum calcium at a constant level.
NOF supports Institute of Medicine (IOM) recommendations that men age 50–70 consume 1000 mg/day of calcium and that women age 51 and older and men age 71 and older consume 1200 mg/day of calcium [30]. There is no evidence that calcium intake in excess of these amounts confers additional bone strength. Intakes in excess of 1200 to 1500 mg/day may increase the risk of developing kidney stones, cardiovascular disease, and stroke. The scientific literature is highly controversial in this area [31–34].
Table 9 illustrates a simple method for estimating the calcium content of a patient’s diet. The average daily dietary calcium intake in adults age 50 and older is 600 to 700 mg/day. Increasing dietary calcium is the first-line approach, but calcium supplements should be used when an adequate dietary intake cannot be achieved.
Vitamin D plays a major role in calcium absorption, bone health, muscle performance, balance, and risk of falling. NOF recommends an intake of 800 to 1000 international units (IU) of vitamin D per day for adults age 50 and older. Institute of Medicine Dietary Reference Intakes for vitamin D are 600 IU/day until age 70 and 800 IU/day for adults age 71 years and older [30].
Chief dietary sources of vitamin D include vitamin D-fortified milk (400 IU/quart, although certain products such as soy milk are not always supplemented with vitamin D), some fortified juices and cereals (40 to 50 IU/serving or more), salt water fish, and liver. Some calcium supplements and most multivitamin tablets also contain vitamin D. Supplementation with vitamin D2 (ergocalciferol) or vitamin D3 (cholecalciferol) may be used. Vitamin D2 is derived from plant sources and may be used by individuals on a strict vegetarian diet.
Many older patients are at high risk for vitamin D deficiency, including patients with malabsorption (e.g., celiac disease) or other intestinal diseases (e.g., inflammatory bowel disease, gastric bypass surgery), chronic renal insufficiency, patients on medications that increase the breakdown of vitamin D (e.g., some antiseizure drugs), housebound patients, chronically ill patients and others with limited sun exposure, individuals with very dark skin, and obese individuals. There is also a high prevalence of vitamin D deficiency in patients with osteoporosis, especially those with hip fractures, even in patients taking osteoporosis medications [35, 36].
Since vitamin D intakes required to correct vitamin D deficiency are so variable among individuals, serum 25(OH)D levels should be measured in patients at risk of deficiency. Vitamin D supplements should be recommended in amounts sufficient to bring the serum 25(OH)D level to approximately 30 ng/ml (75 nmol/L) and a maintenance dose recommended to maintain this level, particularly for individuals with osteoporosis. Many patients with osteoporosis will need more than the general recommendation of 800–1000 IU/day. The safe upper limit for vitamin D intake for the general adult population was increased to 4000 IU/day in 2010 [30].
Treatment of vitamin D deficiency
Adults who are vitamin D deficient may be treated with 50,000 IU of vitamin D2 or vitamin D3 once a week or the equivalent daily dose (7000 IU vitamin D2 or vitamin D3) for 8–12 weeks to achieve a 25(OH)D blood level of approximately 30 ng/ml. This regimen should be followed by maintenance therapy of 1500–2000 IU/day or whatever dose is needed to maintain the target blood level [37, 38].
Regular weight-bearing and muscle-strengthening exercise
Recommend regular weight-bearing and muscle-strengthening exercise to reduce the risk of falls and fractures [39–42]. Among the many health benefits, weight-bearing and muscle-strengthening exercise can improve agility, strength, posture, and balance, which may reduce the risk of falls. In addition, exercise may modestly increase bone density. NOF strongly endorses lifelong physical activity at all ages, both for osteoporosis prevention and overall health, as the benefits of exercise are lost when people stop exercising.
Weight-bearing exercise (in which bones and muscles work against gravity as the feet and legs bear the body’s weight) includes walking, jogging, Tai Chi, stair climbing, dancing, and tennis. Muscle-strengthening exercise includes weight training and other resistive exercises, such as yoga, Pilates, and boot camp programs. Before an individual with osteoporosis initiates a new vigorous exercise program, such as running or heavy weight-lifting, a clinician’s evaluation is appropriate.
Fall prevention
Major risk factors for falling are shown in Table 2. In addition to maintaining adequate vitamin D levels and physical activity, as described above, several strategies have been demonstrated to reduce falls. These include, but are not limited to, multifactorial interventions such as individual risk assessment, Tai Chi and other exercise programs, home safety assessment, and modification especially when done by an occupational therapist, and gradual withdrawal of psychotropic medication if possible. Appropriate correction of visual impairment may improve mobility and reduce risk of falls.
There is a lack of evidence that the use of hip protectors by community-dwelling adults provides statistically significant reduction in the risk of hip or pelvis fractures. Also, there is no evidence that the use of hip protectors reduces the rate of falls. In long-term care or residential care settings, some studies have shown a marginally significant reduction in hip fracture risk. There are no serious adverse effects of hip protectors; however, adherence to long-term use is poor [43]. There is additional uncertainty as to which hip protector to use, as most of the marketed products have not been tested in randomized clinical trials.
Cessation of tobacco use and avoidance of excessive alcohol intake
Advise patients to stop tobacco smoking. The use of tobacco products is detrimental to the skeleton as well as to overall health [44–47]. NOF strongly encourages a smoking cessation program as an osteoporosis intervention.
Recognize and treat patients with excessive alcohol intake. Moderate alcohol intake has no known negative effect on bone and may even be associated with slightly higher bone density and lower risk of fracture in postmenopausal women. However, alcohol intake of more than two drinks per day for women or three drinks a day for men may be detrimental to bone health, increases the risk of falling, and requires further evaluation for possible alcoholism [48].
Pharmacologic therapy
All patients being considered for treatment of osteoporosis should also be counseled on risk factor reduction including the importance of calcium, vitamin D, and exercise as part of any treatment program for osteoporosis. Prior to initiating treatment, patients should be evaluated for secondary causes of osteoporosis and have BMD measurements by central DXA, when available, and vertebral imaging studies when appropriate. Biochemical marker levels should be obtained if monitoring of treatment effects is planned. An approach to the clinical assessment of individuals with osteoporosis is outlined in Table 10.
The percentage of risk reductions for vertebral and nonvertebral fractures cited below are those cited in the FDA-approved prescribing information. In the absence of head-to-head trials, direct comparisons of risk reduction among drugs should be avoided.
Who should be considered for treatment?
Postmenopausal women and men age 50 and older presenting with the following should be considered for treatment:
-
A hip or vertebral fracture (clinically apparent or found on vertebral imaging). There are abundant data that patients with spine and hip fractures will have reduced fracture risk if treated with pharmacologic therapy. This is true for fracture patients with BMD in both the low bone mass and osteoporosis range [49–58]. In patients with a hip or spine fracture, the T-score is not as important as the fracture itself in predicting future risk of fracture and antifracture efficacy from treatment.
-
T-score ≤−2.5 at the femoral neck, total hip, or lumbar spine. There is abundant evidence that the elevated risk of fracture in patients with osteoporosis by BMD is reduced with pharmacotherapy [52, 57, 59–70].
-
Low bone mass (T-score between −1.0 and −2.5 at the femoral neck or lumbar spine) and a 10-year probability of a hip fracture ≥3 % or a 10-year probability of a major osteoporosis-related fracture ≥20 % based on the US-adapted WHO algorithm [13, 15, 71, 72].
Although FRAX calculated fracture risk prediction has been confirmed in multiple studies, there are relatively few data confirming fracture risk reductions with pharmacotherapy in this group of patients.
US FDA-approved drugs for osteoporosis
Current FDA-approved pharmacologic options for the prevention and/or treatment of postmenopausal osteoporosis include, in alphabetical order: bisphosphonates (alendronate, alendronate plus D, ibandronate, risedronate and zoledronic acid), calcitonin, estrogens (estrogen and/or hormone therapy), estrogen agonist/antagonist (raloxifene), tissue-selective estrogen complex (conjugated estrogens/bazedoxifene), parathyroid hormone (PTH [1–34], teriparatide), and the receptor activator of nuclear factor kappa-B (RANK) ligand (RANKL) inhibitor denosumab. Please see prescribing information for specific details of their use.
The antifracture benefits of FDA-approved drugs have mostly been studied in women with postmenopausal osteoporosis. There are limited fracture data in glucocorticoid-induced osteoporosis and in men. FDA-approved osteoporosis treatments have been shown to decrease fracture risk in patients who have had fragility fractures and/or osteoporosis by DXA. Pharmacotherapy may also reduce vertebral fractures in patients with low bone mass (osteopenia) without fractures, but the evidence supporting overall antifracture benefit is not as strong. Thus, the clinician should assess the potential benefits and risks of therapy in each patient and the effectiveness of a given osteoporosis treatment on reduction of vertebral and nonvertebral fractures.
Note that the intervention thresholds do not take into account the nonskeletal benefits or risks associated with specific drug use. NOF does not advocate the use of drugs not approved by the FDA for prevention and treatment of osteoporosis. Examples of these drugs are listed in Table 11 for information only.
Bisphosphonates
Drug efficacy
Alendronate, brand name: Fosamax®, Fosamax Plus D, Binosto™, and generic alendronate
Alendronate sodium is approved by the FDA for the prevention (5 mg daily and 35 mg weekly tablets) and treatment (10 mg daily tablet, 70 mg weekly tablet, 70 mg weekly tablet with 2,800 or 5,600 IU of vitamin D3, and 70 mg effervescent tablet) of postmenopausal osteoporosis. Alendronate is also approved for treatment to increase bone mass in men with osteoporosis and for the treatment of osteoporosis in men and women taking glucocorticoids [73].
Alendronate reduces the incidence of spine and hip fractures by about 50 % over 3 years in patients with a prior vertebral fracture or in patients who have osteoporosis at the hip site [49, 59]. It reduces the incidence of vertebral fractures by 48 % over 3 years in patients without a prior vertebral fracture [74].
Ibandronate, brand name: Boniva® and generic ibandronate
Ibandronate sodium is approved by the FDA for the treatment (150 mg monthly tablet and 3 mg every 3 months by intravenous injection) of postmenopausal osteoporosis. Ibandronate is available as a generic preparation in the USA. The oral preparations are also approved for the prevention of postmenopausal osteoporosis.
Ibandronate reduces the incidence of vertebral fractures by about 50 % over 3 years, but reduction in risk of nonvertebral fracture with ibandronate has not been documented [50].
Risedronate, brand name: Actonel®, Atelvia™, and generic risedronate
Risedronate sodium is approved by the FDA for the prevention and treatment (5 mg daily tablet; 35 mg weekly tablet; 35 mg weekly delayed release tablet; 35 mg weekly tablet packaged with six tablets of 500 mg calcium carbonate; 75 mg tablets on two consecutive days every month; and 150 mg monthly tablet) of postmenopausal osteoporosis. Risedronate is also approved for treatment to increase bone mass in men with osteoporosis and for the prevention and treatment of osteoporosis in men and women who are either initiating or taking glucocorticoids [75].
Risedronate reduces the incidence of vertebral fractures by 41 to 49 % and nonvertebral fractures by 36 % over 3 years, with significant risk reduction occurring within 1 year of treatment in patients with a prior vertebral fracture [51, 52].
Zoledronic acid, brand name: Reclast®
Zoledronic acid is approved by the FDA for the prevention and treatment (5 mg by intravenous infusion over at least 15 min once yearly for treatment and once every 2 years for prevention) of osteoporosis in postmenopausal women. It is also approved to improve bone mass in men with osteoporosis and for the prevention and treatment of osteoporosis in men and women expected to be on glucocorticoid therapy for at least 12 months. Zoledronic acid is also indicated for the prevention of new clinical fractures in patients (both women and men) who have recently had a low-trauma (osteoporosis-related) hip fracture [58].
Zoledronic acid reduces the incidence of vertebral fractures by 70 % (with significant reduction at 1 year), hip fractures by 41 %, and nonvertebral fractures by 25 % over 3 years in patients with osteoporosis defined by prevalent vertebral fractures and osteoporosis by BMD of the hip [66].
Drug administration
Alendronate (generic and Fosamax) and risedronate (Actonel) tablets must be taken on an empty stomach, first thing in the morning, with 8 oz of plain water (no other liquid). Binosto must be dissolved in 4 oz of room temperature water taken on an empty stomach, first thing in the morning. Delayed release risedronate (Atelvia) tablets must be taken immediately after breakfast with at least 4 oz of plain water (no other liquid). After taking these medications, patients must wait at least 30 min before eating, drinking, or taking any other medication. Patients should remain upright (sitting or standing) during this interval.
Ibandronate must be taken on an empty stomach, first thing in the morning, with 8 oz of plain water (no other liquid). After taking this medication, patients must remain upright and wait at least 60 min before eating, drinking, or taking any other medication. Ibandronate, 3 mg/3 ml prefilled syringe, is given by intravenous injection over 15 to 30 s, once every 3 months. Serum creatinine should be checked before each injection.
Zoledronic acid, 5 mg in 100 ml, is given once yearly or once every 2 years by intravenous infusion over at least 15 min. Patients should be well hydrated and may be pre-treated with acetaminophen to reduce the risk of an acute phase reaction (arthralgia, headache, myalgia, fever). These symptoms occurred in 32 % of patients after the first dose, 7 % after the second dose, and 3 % after the third dose.
Drug safety
Side effects are similar for all oral bisphosphonate medications and include gastrointestinal problems such as difficulty swallowing and inflammation of the esophagus and stomach.
All bisphosphonates can affect renal function and are contraindicated in patients with estimated GFR below 30–35 ml/min. Zoledronic acid is contraindicated in patients with creatinine clearance less than 35 mL/min or in patients with evidence of acute renal impairment. Healthcare professionals should screen patients prior to administering zoledronic acid in order to identify at-risk patients and should assess renal function by monitoring creatinine clearance prior to each dose of zoledronic acid [76]. Eye inflammation can also occur. Any such complication should be reported to the healthcare provider as soon as possible.
There have been rare reports of osteonecrosis of the jaw (ONJ) with long-term use of bisphosphonates for osteoporosis, though ONJ is much more common following high-dose intravenous bisphosphonate treatment for patients with cancer. The risk of ONJ appears to increase with duration of treatment beyond 5 years [77].
Although rare, low-trauma atypical femur fractures may be associated with the long-term use of bisphosphonates (e.g., >5 years of use). Pain in the thigh or groin area, which can be bilateral, often precedes these unusual fractures. Patients should be evaluated closely for these unusual fractures, including proactive questioning regarding thigh and groin pain. For patients with thigh and groin pain, a stress fracture in the subtrochanteric region or femoral shaft of the femur may be present. Bilateral X-ray of the femurs should be ordered when an atypical femur fracture is suspected, followed by an MRI or a radionuclide bone scan when clinical suspicion is high enough [78]. Surgical fixation is required in some cases, whereas medical conservative treatment is appropriate in other cases. Bisphosphonates should be stopped if atypical femur fractures have occurred.
Calcitonin
Drug efficacy
Brand name: Miacalcin® or Fortical® and generic calcitonin
Salmon calcitonin is FDA-approved for the treatment of osteoporosis in women who are at least 5 years postmenopausal when alternative treatments are not suitable.
Miacalcin nasal spray has not been shown to increase bone mineral density in early postmenopausal women.
Calcitonin reduces vertebral fracture occurrence by about 30 % in those with prior vertebral fractures but has not been shown to reduce the risk of nonvertebral fractures [54, 79]. Due to the possible association between malignancy and calcitonin-salmon use, the need for continued therapy should be re-evaluated on a periodic basis.
Drug administration
Two hundred international units delivered as a single daily intranasal spray. Subcutaneous administration by injection also is available.
Drug safety
Intranasal calcitonin can cause rhinitis, epistaxis, and allergic reactions, particularly in those with a history of allergy to salmon. The FDA has reviewed long-term post-marketing data concerning calcitonin and the very small increase in the risk of certain cancers. A meta-analysis of 21 randomized, controlled clinical trials with calcitonin-salmon (nasal spray and investigational oral forms) suggests an increased risk of malignancies in calcitonin-salmon-treated patients compared to placebo-treated patients. The overall incidence of malignancies reported in the 21 trials was higher among calcitonin-salmon-treated patients (4.1 %) compared with placebo-treated patients (2.9 %). The data were not sufficient for further analyses by specific type of malignancy. Although a definitive causal relationship between the calcitonin-salmon use and malignancies cannot be established from this meta-analysis, the benefits for the individual patient should be carefully evaluated against all possible risks [80, 81].
Estrogen/hormone therapy (ET/HT)
Drug efficacy
ET brand names:e.g., Climara®, Estrace®, Estraderm®, Estratab®, Ogen®, Premarin®, Vivelle®; HT brand names: e.g., Activella®, Femhrt®, Premphase®, Prempro®
Estrogen/hormone therapy is approved by the FDA for the prevention of osteoporosis, relief of vasomotor symptoms, and vulvovaginal atrophy associated with menopause. Women who have not had a hysterectomy require HT, which also contains progestin to protect the uterine lining.
The Woman’s Health Initiative (WHI) found that 5 years of HT (Prempro®) reduced the risk of clinical vertebral fractures and hip fractures by 34 % and other osteoporotic fractures by 23 % [69].
Drug administration
ET/HT is available in a wide variety of oral as well as transdermal preparations including estrogen only, progestin only, and combination estrogen–progestin. ET/HT dosages include cyclic, sequential, and continuous regimens. If and when treatment is stopped, bone loss can be rapid and alternative agents should be considered to maintain BMD.
Drug safety
The WHI reported increased risks of myocardial infarction, stroke, invasive breast cancer, pulmonary emboli, and deep vein thrombosis during 5 years of treatment with conjugated equine estrogen and medroxyprogesterone acetate (Prempro®) [69]. Subsequent analyses of these data showed no increase in cardiovascular disease in women starting treatment within 10 years of menopause [82]. In the estrogen only arm of WHI, no increase in breast cancer incidence was noted over 7.1 years of treatment. Other doses and combinations of estrogen and progestins were not studied and, in the absence of comparable data, their risks should be assumed to be comparable. Because of the risks, ET/HT should be used in the lowest effective doses for the shortest duration to treat moderately severe menopausal symptoms and should be considered primarily for women within the first few years of menopause. When ET/HT use is considered solely for prevention of osteoporosis, the FDA recommends that approved nonestrogen treatments should first be carefully considered. When ET/HT treatments are stopped, bone loss can be rapid and alternative agents should be considered to maintain BMD.
Estrogen agonist/antagonist (formerly known as SERMs): Raloxifene
Drug efficacy
Raloxifene, brand name: Evista® and generic raloxifene
Raloxifene is approved by the FDA for both prevention and treatment of osteoporosis in postmenopausal women.
Raloxifene reduces the risk of vertebral fractures by about 30 % in patients with a prior vertebral fracture and by about 55 % in patients without a prior vertebral fracture over 3 years [55]. Reduction in risk of nonvertebral fracture with raloxifene has not been documented. Raloxifene is also indicated for the reduction in risk of invasive breast cancer in postmenopausal women with osteoporosis [83–86]. Raloxifene does not reduce the risk of coronary heart disease.
Drug administration
Available in a 60-mg tablet form to be taken with or without food.
Drug safety
Raloxifene increases the risk of deep vein thrombosis to a degree similar to that observed with estrogen. It can also increase hot flashes and cause leg cramps.
Tissue-selective estrogen complex: conjugated estrogens/bazedoxifene (conjugated estrogens paired with estrogen agonist/antagonist)
Drug efficacy
Conjugated estrogens/bazedoxifene, brand name: Duavee®
Conjugated estrogens/bazedoxifene is approved by the FDA for women who suffer from moderate-to-severe hot flashes (vasomotor symptoms) associated with menopause and to prevent osteoporosis after menopause.
The medication combines conjugated estrogen with an estrogen agonist/antagonist (bazedoxifene). The bazedoxifene component reduces the risk of endometrial hyperplasia (excessive growth of the lining of the uterus) that can occur with the estrogen component of the drug. Therefore, progestins do not need to be taken with conjugated estrogens/bazedoxifene.
Use of this combination drug significantly increased mean lumbar spine BMD (treatment difference, 1.51 %), at 12 months compared to placebo in women who had been postmenopausal between 1 and 5 years. Treatment with conjugated estrogens/bazedoxifene also increased total hip BMD. The treatment difference in total hip BMD at 12 months was 1.21 % [87–90].
Drug administration
Available as a tablet containing conjugated estrogens and bazedoxifene 0.45 mg/ 20 mg, to be taken once daily without regard to meals.
Drug safety
Conjugated estrogens/bazedoxifene is intended only for postmenopausal women who still have a uterus. Like other products containing estrogen, it should be used for the shortest duration consistent with treatment goals and risks for the individual woman. When using this drug only for the prevention of osteoporosis, such use should be limited to women who are at significant risk of osteoporosis and only after carefully considering alternatives that do not contain estrogen.
Side effects of conjugated estrogens/bazedoxifene include muscle spasms, nausea, diarrhea, dyspepsia, upper abdominal pain, oropharyngeal pain, dizziness, and neck pain. Because this product contains estrogen, it is approved with the same Boxed Warning and other Warnings and Precautions that have been approved with estrogen products.
Parathyroid hormone: teriparatide
Drug efficacy
PTH(1-34), teriparatide, brand name: Forteo®
Teriparatide is approved by the FDA for the treatment of osteoporosis in postmenopausal women and men at high risk for fracture. It is also approved for treatment in men and women at high risk of fracture with osteoporosis associated with sustained systemic glucocorticoid therapy [91]. Teriparatide reduces the risk of vertebral fractures by about 65 % and nonvertebral fragility fractures by about 53 % in patients with osteoporosis, after an average of 18 months of therapy [57].
Drug administration
Teriparatide is an anabolic (bone-building) agent administered by 20 μg daily subcutaneous injection. If and when treatment is stopped, bone loss can be rapid and alternative agents should be considered to maintain BMD. Treatment duration is recommended not to exceed 18 to 24 months.
Drug safety
Side effects of teriparatide include leg cramps, nausea, and dizziness. Because it caused an increase in the incidence of osteosarcoma in rats (high doses, long duration treatment in the rodent), patients with an increased risk of osteosarcoma (e.g., patients with Paget’s disease of bone and those having prior radiation therapy of the skeleton), bone metastases, hypercalcemia, or a history of skeletal malignancy should not receive teriparatide therapy. It is common practice to follow teriparatide treatment with an antiresorptive agent, usually a bisphosphonate, to maintain or further increase BMD.
RANKL/RANKL inhibitor: denosumab
Drug efficacy
Denosumab, brand name Prolia®
Denosumab is approved by the FDA for the treatment of osteoporosis in postmenopausal women at high risk of fracture. Denosumab reduces the incidence of vertebral fractures by about 68 %, hip fractures by about 40 %, and nonvertebral fractures by about 20 % over 3 years [56]. Denosumab is also indicated to increase bone mass in men at high risk of fracture, treat bone loss in women with breast cancer on aromatase inhibitor therapies, and to treat bone loss in men receiving gonadotropin-reducing hormone treatment for prostate cancer who are at high risk for fracture.
Drug administration
Administered by a health professional, 60 mg every 6 months as a subcutaneous injection.
Drug safety
Denosumab may cause hypocalcemia. Hypocalcemia must be corrected before starting denosumab. Denosumab increased the risk of serious skin infections (cellulitis) and skin rash. Denosumab has been rarely associated with the development of ONJ, both when used to treat osteoporosis and to treat patients with cancer (at much higher doses), although it is much more common in the latter setting. Denosumab has also been associated rarely with the development of atypical femur fractures. If and when denosumab treatment is stopped, bone loss can be rapid and alternative agents should be considered to maintain BMD.
Sequential and combination therapy
When osteoporosis is diagnosed in young individuals, choices of osteoporosis medication may change over time to take advantage of the best benefit to risk ratio at each stage of life (sequential monotherapy). For more severe osteoporosis, sequential treatment with anabolic therapy followed by an antiresorptive agent is generally preferred to concomitant combination therapy. However, combination therapy with teriparatide and an antiresorptive can be considered in a few clinical settings in patients with very severe osteoporosis such as spine and hip fractures. There are few indications for combining two antiresorptive treatments, but such options could be considered in the short term in women who are experiencing active bone loss while on low dose HT for menopausal symptoms or raloxifene for breast cancer prevention.
Duration of treatment
No pharmacologic therapy should be considered indefinite in duration. All nonbisphosphonate medications produce temporary effects that wane upon discontinuation. If these treatments are stopped, benefits rapidly disappear. In contrast, bisphosphonates may allow residual effects even after treatment discontinuation. Therefore, it may be possible to discontinue bisphosphonates and retain residual benefits against fracture at least for several years.
Evidence of efficacy beyond 5 years is limited, whereas rare safety concerns such as ONJ and atypical femur fractures become more common beyond 5 years [67, 92]. Since there is no extensive evidence base to guide treatment duration decisions, duration decisions need to be individualized [93]. After the initial 3- to 5-year treatment period, a comprehensive risk assessment should be performed. This should include interval clinical history, particularly with respect to intercurrent fracture history and new chronic diseases or medications, as well as height measurement, BMD testing, and vertebral imaging if there has been any documented height loss during the treatment period. It is reasonable to discontinue bisphosphonates after 3 to 5 years in people who appear to be at modest risk of fracture after the initial treatment period. In contrast, for those who appear to be at high risk for fracture, continued treatment with a bisphosphonate or an alternative therapy should be considered [94].
Monitoring patients
It is important to ask patients whether they are taking their medications and to encourage continued and appropriate compliance with their osteoporosis therapies to reduce fracture risk. It is also important to review their risk factors and encourage appropriate calcium and vitamin D intakes, exercise, fall prevention, and other lifestyle measures. Furthermore, the need for continued medication to treat osteoporosis should be reviewed annually. Duration of treatment must be individualized. Some patients may be able to discontinue treatment temporarily after several years of therapy, particularly after bisphosphonate administration [95, 96]. Other patients will need to continue treatment. If treatment is discontinued, serial monitoring should include clinical assessment for fractures, falling, any interval chronic disease occurrence and consideration of serial BMD testing, use of biochemical markers, and vertebral imaging in some patients.
Accurate yearly height measurement is a critical determination of osteoporosis treatment efficacy. Patients who lose 2 cm (or 0.8 in.) or more in height either acutely or cumulatively should have a repeat vertebral imaging test to determine if new or additional vertebral fractures have occurred since the prior vertebral imaging test.
Serial central DXA testing is an important component of osteoporosis management. Measurements for monitoring patients should be performed in accordance with medical necessity, expected response, and in consideration of local regulatory requirements. NOF recommends that repeat BMD assessments generally agree with Medicare guidelines of every 2 years but recognizes that testing more frequently may be warranted in certain clinical situations.
The following techniques may be used to monitor the effectiveness of treatment:
Central DXA
Central DXA assessment of the hip or lumbar spine is the “gold standard” for serial assessment of BMD. Biological changes in bone density are small compared to the inherent error in the test itself, and interpretation of serial bone density studies depends on appreciation of the smallest change in BMD that is beyond the range of error of the test. This least significant change (LSC) varies with the specific instrument used, patient population being assessed, measurement site, technologist’s skill with patient positioning and test analysis, and the confidence intervals used [97]. Changes in the BMD of less than 3–6 % at the hip and 2–4 % at the spine from test to test may be due to the precision error of the testing itself. Information on how to assess precision and calculate the LSC is available at www.ISCD.org.
QCT
Volumetric BMD of the lumbar spine can be used to monitor age-, disease, and treatment-related BMD changes in men and women. Precision of acquisition should be established by phantom data and analysis precision by re-analysis of patient data.
pDXA, pQCT, and QUS
Peripheral skeletal sites do not respond with the same magnitude as the spine and hip to medications and thus are not appropriate for monitoring response to therapy at this time.
Biochemical markers of bone turnover
Suppression of biochemical markers of bone turnover after 3–6 months of treatment and biochemical marker increases after 1–3 months of anabolic therapy have been predictive of greater BMD responses and in some cases fracture risk reduction in large clinical trials. Biochemical marker changes in individuals must exceed the LSC in order to be clinically meaningful. The LSC is specific to the biomarker being utilized, which is calculated by multiplying the “precision error” of the specific biochemical marker (laboratory provided) by 2.77 (95 % confidence level). Biological variability can be reduced by obtaining samples in the early morning after an overnight fast. Serial measurements should be made at the same time of day at the same laboratory.
Vertebral imaging
Once the first vertebral imaging test has been performed to determine prevalent vertebral fractures (indications above), repeat testing should be performed to identify incident vertebral fractures if there is a change in the patient’s status suggestive of new vertebral fracture, including documented prospective height loss, undiagnosed back pain, postural change, or a possible finding of new vertebral deformity on chest X-ray. If patients are being considered for a temporary cessation of drug therapy, vertebral imaging should be repeated to determine that no vertebral fractures have occurred in the interval off treatment. A new vertebral fracture on therapy indicates a need for more intensive or continued treatment rather than treatment cessation [95].
Implementation of FLS secondary fracture prevention programs
FLS programs have been implemented successfully in a number of closed and open settings over the last 15 years, both in the USA (including the American Orthopedic Association Own the Bone program) as well as abroad. These programs have accomplished a reduction in secondary fracture rates as well as health care cost savings [98, 99]. In the USA, Kaiser Permanente’s Healthy Bones program has reduced the expected hip fracture rate by 38 % since 1998 [100]; Geisinger Health System achieved $7.8 million in cost savings over 5 years [101].
A Fracture Liaison Service is a coordinated care system headed by an FLS coordinator (a nurse practitioner, physician’s assistant, nurse, or other health professional) who ensures that individuals who suffer a fracture receive appropriate diagnosis, treatment, and support [102]. The FLS uses established protocols to find and assess fracture patients. The program creates a population database of fracture patients and establishes a process and timeline for patient assessment and follow-up care. An FLS coordinator is frequently based in a hospital and requires support from a qualified physician or physician team.
Physical medicine and rehabilitation
Physical medicine and rehabilitation can reduce disability, improve physical function, and lower the risk of subsequent falls in patients with osteoporosis. Rehabilitation and exercise are recognized means to improve function, such as activities of daily living. Psychosocial factors also strongly affect functional ability of the patient with osteoporosis who has already suffered fractures.
Recommendations from the Health Professional’s Guide to Rehabilitation of the Patient with Osteoporosis [14]:
-
Evaluate and consider the patient’s physical and functional safety as well as psychological and social status, medical status, nutritional status, and medication use before prescribing a rehabilitation program.
-
Evaluate the patient and her/his current medication use and consider possible interactions and risk for altered mental status. Intervene as appropriate.
-
Provide training for the performance of safe movement and safe activities of daily living, including posture, transfers, lifting, and ambulation in populations with or at high risk for osteoporosis. Intervene as appropriate, e.g., with prescription for assistive device for improved balance with mobility.
-
Implement steps to correct underlying deficits whenever possible, i.e., improve posture and balance and strengthen quadriceps muscles to allow a person to rise unassisted from a chair; promote use of assistive devices to help with ambulation, balance, lifting, and reaching.
-
Evaluate home environment for risk factors for falls and intervene as appropriate.
-
Based on the initial condition of the patient, provide a complete exercise recommendation that includes weight-bearing aerobic activities for the skeleton, postural training, progressive resistance training for muscle and bone strengthening, stretching for tight soft tissues and joints, and balance training.
-
Advise patients to avoid forward bending and exercising with trunk in flexion, especially in combination with twisting.
-
As long as principles of safe movement are followed, walking and daily activities, such as housework and gardening, are practical ways to contribute to maintenance of fitness and bone mass. Additionally, progressive resistance training and increased loading exercises, within the parameter of the person’s current health status, are beneficial for muscle and bone strength. Proper exercise may improve physical performance/function, bone mass, muscle strength, and balance, as well as reduce the risk of falling.
-
Avoid long-term immobilization and recommend partial bed rest (with periodic sitting and ambulating) only when required and for the shortest periods possible.
-
In patients with acute vertebral fractures or chronic pain after multiple vertebral fractures, the use of trunk orthoses (e.g., back brace, corset, posture training support devices) may provide pain relief by reducing the loads on the fracture sites and aligning the vertebra. However, long-term bracing may lead to muscle weakness and further de-conditioning.
-
Effective pain management is a cornerstone in rehabilitation from vertebral fractures. Pain relief may be obtained by the use of a variety of physical, pharmacological, and behavioral techniques with the caveat that the benefit of pain relief should not be outweighed by the risk of side effects such as disorientation or sedation which may result in falls.
-
Individuals with recent, painful vertebral fractures that fail conservative management may be candidates for interventions, such as kyphoplasty or vertebroplasty, when performed by experienced practitioners.
Conclusions and remaining questions
The Guide has focused on the prevention, diagnosis, and treatment of osteoporosis in postmenopausal women and men age 50 and older using the most common existing diagnostic and treatment methods available. Many additional issues urgently need epidemiologic, clinical, and economic research. For example:
-
How can we better assess bone strength using noninvasive technologies and thus further refine or identify patients at high risk for fracture?
-
Can we expand the WHO FRAX™ algorithm to incorporate information on lumbar spine BMD and to consider multiple fractures and recency of fractures in quantitative risk assessment.
-
Can we develop a fracture risk calculator for patients who have already initiated pharmacologic therapy.
-
How can children, adolescents, and young adults maximize peak bone mass?
-
What are the precise components (type, intensity, duration, frequency) of an effective exercise program for osteoporosis prevention and treatment?
-
What should be done to identify and modify risk factors for falling, and what would be the magnitude of effect on fracture risk in a population?
-
How effective are different FDA-approved treatments in preventing fractures in patients with moderately low bone mass? Do benefits exceed risks?
-
What approaches are most effective in treating osteoporosis in disabled populations?
-
How can we make the diagnosis of vertebral fractures more accurate and consistent, particularly mild fractures?
-
How long should antiresorptive therapies be continued, and are there long-term side effects as yet unknown?
-
Are combination therapies useful and, if so, which drug combinations are best and when should they be used?
-
Can we identify agents or medications that will return bone mass and bone structure to normal even in those starting with severe osteoporosis?
-
Should we treat patients to a certain goal and then reconsider type and/or dose of therapy? If so, what should that goal be?
-
How should therapeutic agents be sequentially prescribed in order to maximize benefits and minimize risks over the lifespan of the patient?
NOF is committed to continuing the effort to answer these and other questions related to this debilitating disease, with the goal of eliminating osteoporosis as a threat to the health of present and future generations. For additional resources on osteoporosis and bone health, visit www.nof.org.
References
Office of the Surgeon General (US) (2004) Bone health and osteoporosis: a report of the Surgeon General. Office of the Surgeon General (US), Rockville (MD). Available from: http://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed March 2014
Wright NC, Looker A, Saag K, Curtis JR, Dalzell ES, Randall S, Dawson-Hughes B (2014) The recent prevalence of osteoporosis and low bone mass based on bone mineral density at the femoral neck or lumbar spine in the United States. J Bone Miner Res. doi: 10.1002/jbmr.2269
Lewiecki EM, Laster AJ (2006) Clinical review: clinical applications of vertebral fracture assessment by dual-energy x-ray absorptiometry. J Clin Endocrinol Metab 91(11):4215–4222
Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C (2009) Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 20(10):1633–1650
Colón-Emeric C, Kuchibhatla M, Pieper C et al (2003) The contribution of hip fracture to risk of subsequent fractures: data from two longitudinal studies. Osteoporos Int 11:879–883
Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King AB, Tosteson A (2007) Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 22(3):465–475
National Committee for Quality Assurance. HEDIS & performance measurement. Proposed changes to existing measure for HEDIS®1 2015: osteoporosis management in women who had a fracture (OMW). Washington, DC. Available at www.ncqa.org/HEDISQualityMeasurement.aspx. Accessed 2 March 2014
Khosla S, Riggs BL (2005) Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin N Am 34:1015–1030
Dempster DW, Shane E, Horbert W, Lindsay R (1986) A simple method for correlative light and scanning electron microscopy of human iliac crest bone biopsies: qualitative observations in normal and osteoporotic subjects. J Bone Miner Res 1(1):15–21
Cooper C, Melton LJ 3rd (1992) Epidemiology of osteoporosis. Trends Endocrinol Metab 3(6):224–229
Kanis JA, on behalf of the World Health Organization Scientific Group (2007) Assessment of osteoporosis at the primary health care level. Technical Report. World Health Organization Collaborating Center for Metabolic Bone Diseases. University of Sheffield, UK
Tosteson ANA, Melton LJ 3rd, Dawson-Hughes B, National Osteoporosis Foundation Guide Committee et al (2008) Cost-effective osteoporosis treatment thresholds: the United States perspective. Osteoporos Int 19(4):437–447
Dawson-Hughes B, Tosteson ANA, Melton LJ 3rd, National Osteoporosis Foundation Guide Committee et al (2008) Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the U.S.A. Osteoporos Int 19(4):449–458
National Osteoporosis Foundation (2003) Health professional’s guide to rehabilitation of the patient with osteoporosis. National Osteoporosis Foundation, Washington, DC
American Geriatrics Society, British Geriatrics Society and American Academy of Orthopaedic Surgeons Panel on Falls Prevention (2001) Guideline for the prevention of falls in older persons. J Am Geriatr Soc 49:664–672
van den Bergh JP, van Geel TA, Geusens PP (2012) Osteoporosis, frailty and fracture: implications for case finding and therapy. Nat Rev Rheumatol 8(3):163–172
Watts NB, Adler RA, Bilezikian JP, Endocrine Society et al (2012) Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 97(6):1802–1822. (Also available on the Endocrine Society’s website http://www.endo-society.org/guidelines/Current-Clinical-Practice-Guidelines.cfm)
Kanis JA, Melton LJ 3rd, Christiansen C, Johnston CC, Khaltaev N (1994) The diagnosis of osteoporosis. J Bone Miner Res 9(8):1137–1141
International Society for Clinical Densitometry. 2013 Official Positions—Adult. http://www.iscd.org/official-positions/2013-iscd-official-positions-adult/. Accessed Feb 2014.
Nelson HD, Haney EM, Chou R, Dana T, Fu R, Bougatsos C (2010) Screening for osteoporosis: systematic review to update the 2002 US Preventive Services Task Force Recommendation. Evidence Syntheses No. 77. AHRQ Publication No. 10-05145-EF-1. Agency for Healthcare Research and Quality, (US), Rockville
Melton LJ, Chrischilles EA, Cooper C, Lane AW, Riggs BL (2005) How many women have osteoporosis? J Bone Miner Res 20(5):886–892
Lenchik L, Rogers LF, Delmas PD, Genant HK (2004) Diagnosis of osteoporotic vertebral fractures: importance of recognition and description by radiologists. Am J Roentgenol 183(4):949–958
Ross PD, Davis JW, Epstein RS, Wasnich RD (1991) Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 114(11):919–923
Schousboe JT, Ensrud KE, Nyman JA, Kane RL, Melton LJ 3rd (2005) Potential cost-effective use of spine radiographs to detect vertebral deformity and select osteopenic post-menopausal women for amino-bisphosphonate therapy. Osteoporos Int 16(12):1883–1893
Burch J, Rice S, Yang H, Neilson A, Stirk L, Francis R, Holloway P, Selby P, Craig D (2014) Systematic review of the use of bone turnover markers for monitoring the response to osteoporosis treatment: the secondary prevention of fractures, and primary prevention of fractures in high-risk groups. Health Technol Assess (Winchester, England) 18(11):1–206
National Osteoporosis Foundation (2005) Physician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation, Washington, DC
National Osteoporosis Foundation (NOF) and International Society for Clinical Densitometry (ISCD). Recommendations to DXA Manufacturers for FRAX® Implementation. Available at http://www.nof.org/files/nof/public/content/resource/862/files/392.pdf. Accessed 28 Jan 2013
Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E (2008) FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 19(4):385–397
Larsen ER, Mosekilde L, Foldspang A (2004) Vitamin D and calcium supplementation prevents osteoporotic fractures in elderly community dwelling residents: a pragmatic population-based 3-year intervention study. J Bone Miner Res 19(3):370–378
Institute of Medicine (US) Committee to review dietary reference intakes for vitamin D and calcium (2011). In: Ross AC, Taylor CL, Yaktine AL et al (eds) Dietary reference intakes for calcium and vitamin D. National Academies Press (US), Washington (DC). Available from: http://www.ncbi.nlm.nih.gov/books/NBK56070/. Accessed March 2014
Prentice RL, Pettinger MB, Jackson RD et al (2013) Health risks and benefits from calcium and vitamin D supplementation: Women’s Health Initiative clinical trial and cohort study. Osteoporos Int 24(2):567–580
Reid IR, Bolland MJ (2012) Calcium supplements: bad for the heart? Heart 98(12):895–896
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR (2011) Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 19:342
Moyer VA, U.S. Preventive Services Task Force (2013) Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 158(9):691–696
LeBoff MS, Hawkes WG, Glowacki J, Yu-Yahiro J, Hurwitz S, Magaziner J (2008) Vitamin D-deficiency and post-fracture changes in lower extremity function and falls in women with hip fractures. Osteoporos Int 19(9):1283–1290
Holick MF, Siris ES, Binkley N et al (2005) Prevalence of vitamin D inadequacy among postmenopausal north American women receiving osteoporosis therapy. J Clin Endocrinol Metab 90(6):3215–3224
Looker AC, Pfeiffer CM, Lacher DA, Schleicher RL, Picciano MF, Yetley EA (2008) Serum 25-hydroxyvitamin D status of the US population: 1988–1994 compared to 2000–2004. Am J Clin Nutr 88(6):1519–1527
Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF (2000) Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 72(3):690–693
Granacher U, Gollhofer A, Hortobágyi T, Kressig RW, Muehlbauer T (2013) The importance of trunk muscle strength for balance, functional performance and fall prevention in seniors: a systematic review. Sports Med 43(7):627–641
Sherrington C, Whitney JC, Lord SR, Herbert RD, Cumming RG, Close JC (2008) Effective exercise for the prevention of falls: a systematic review and meta-analysis. J Am Geriatr Soc 56(12):2234–2243
Choi M, Hector M (2012) Effectiveness of intervention programs in preventing falls: a systematic review of recent 10 years and meta-analysis. J Am Med Dir Assoc 13(2):188.13–188.e21
Gillespie LD, Robertson MC, Gillespie WJ, Sherrington C, Gates S, Clemson LM, Lamb SE (2012) Interventions for preventing falls in older people living in the community. Cochrane Database Syst Rev 12(9), CD007146. doi:10.1002/14651858.CD007146.pub3
Gillespie WJ, Gillespie LD, Parker MJ (2010) Hip protectors for preventing hip fractures in older people. Cochrane Database Syst Rev 10, CD001255
Ayo-Yusuf OA, Olutola BG (2014) Epidemiological association between osteoporosis and combined smoking and use of snuff among South African women. Niger J Clin Pract 17:174–177
Waugh EJ, Lam MA, Hawker GA, McGowan J, Papaioannou A, Cheung AM et al (2009) Risk factors for low bone mass in healthy 40–60 year old women: a systematic review of the literature. Osteoporos Int 20:1–21
Benson BW, Shulman JD (2005) Inclusion of tobacco exposure as a predictive factor for decreased bone mineral content. Nicotine Tob Res 7:719–724
Krall EA, Dawson-Hughes B (1999) Smoking increases bone loss and decreases intestinal calcium absorption. J Bone Miner Res 14:215–220
Maurel DB, Boisseau N, Benhamou CL, et al (2012) Alcohol and bone: review of dose effects and mechanisms. Osteoporos Int 23(1):1–16
Black DM, Cummings SR, Karpf DB et al (1996) Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 348(9041):1535–1541
Chesnut CH 3rd, Skag A, Christiansen C et al (2004) Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res 19(18):1241–1249
Harris ST, Watts NB, Genant HK et al (1999) Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. JAMA 282(14):1344–1352
Reginster J, Minne HW, Sorensen OH et al (2000) Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int 11(1):83–91
Kanis JA, Barton IP, Johnell O (2005) Risedronate decreases fracture risk in patients selected solely on the basis of prior vertebral fracture. Osteoporos Int 16(5):475–482
Chesnut CH 3rd, Silverman S, Andriano K et al (2000) A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. PROOF Study Group. Am J Med 109(4):267–276
Ettinger B, Black DM, Mitlak BH et al (1999) Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 282(7):637–645. (Erratum in: JAMA 1999;282(22):2124)
Cummings SR, San Martin J, McClung MR et al (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361(8):756–765, Erratum in: N Engl J Med 2009;361(19):1914
Neer RM, Arnaud CD, Zanchetta JR et al (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344(19):1434–1441
Lyles KW, Colón-Emeric CS, Magaziner JS et al (2007) Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 357(18):1799–1809
Black DM, Thompson DE, Bauer DC et al (2000) Fracture risk reduction with alendronate in women with osteoporosis: the Fracture Intervention Trial. FIT Research Group. J Clin Endocrinol Metab 85(11):4118–4124
Black DM, Schwartz AV, Ensrud KE et al (2006) Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 296(24):2927–2938
Bone HG, Hosking D, Devogelaer JP, Alendronate Phase III Osteoporosis Treatment Study Group et al (2004) Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Engl J Med 350(12):1189–1199
Miller PD, McClung MR, Macovei L et al (2005) Monthly oral ibandronate therapy in postmenopausal osteoporosis: 1-year results from the MOBILE study. J Bone Miner Res 20(8):1315–1322
Reginster JY, Adami S, Lakatos P et al (2006) Efficacy and tolerability of once-monthly oral ibandronate in postmenopausal osteoporosis: 2 year results from the MOBILE study. Ann Rheum Dis 65(5):654–661
Eisman JA, Civitelli R, Adami S et al (2008) Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 35(3):488–497
McClung MR, Geusens P, Miller PD, Hip Intervention Program Study Group et al (2001) Effect of risedronate on the risk of hip fracture in elderly women. Hip Intervention Program Study Group. N Engl J Med 344(5):333–340
Black DM, Delmas PD, Eastell R, HORIZON Pivotal Fracture Trial et al (2007) Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 356(18):1809–1822
Sorensen OH, Crawford GM, Mulder H et al (2003) Long-term efficacy of risedronate: a 5-year placebo-controlled clinical experience. Bone 32(2):120–126
Cummings, SR, San Martin J, McClung MR et al (2009) FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361(8):756–765. (Erratum in: N Engl J Med. 2009;36(10):191)
Rossouw JE, Anderson GL, Prentice RL, Writing Group for the Women’s Health Initiative Investigators et al (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288(3):321–333
Anderson GL, Limacher M, Assaf AR, Women’s Health Initiative Steering Committee et al (2004) Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 291(14):1701–1712
Kanis JA, Johansson H, Oden A, Dawson-Hughes B, Melton LJ 3rd, McCloskey EV (2010) The effects of a FRAX revision for the USA. Osteoporos Int 21(1):35–40
Ettinger B, Black DM, Dawson-Hughes B, Pressman AR, Melton LJ 3rd (2010) Updated fracture incidence rates for the US version of FRAX. Osteoporos Int 21(1):25–33
Saag KG, Emkey R, Schnitzer TJ et al (1998) Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. NEJM 339(5):292–299
Cummings SR, Black DM, Thompson DE et al (1998) Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA 280(24):2077–2082
Eastell R, Devogelaer JP, Peel NF et al (2000) Prevention of bone loss with risedronate in glucocorticoid-treated rheumatoid arthritis patients. Osteoporos Int 11(4):331–337
U.S. Food and Drug Administration. Reclast (zoledronic acid): drug safety communication—new contraindication and updated warning on kidney impairment. Posted 09/01/2011. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm270464.htm. Accessed 28 Jan 2013
Khosla S, Burr D, Cauley J, American Society for Bone and Mineral Research et al (2007) Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 22(10):1470–1491
Shane E, Burr D, Abrahmsen B, American Society for Bone and Mineral Research et al (2014) Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 29(1):1–23
Binkley N, Bolognese M, Sidorowicz-Bialynicka A, Vally T, Trout R, Miller C, Buben CE, Gilligan JP, Krause DS, for the Oral Calcitonin in Postmenopausal Osteoporosis (ORACAL) Investigators (2012) A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: the oral calcitonin in postmenopausal osteoporosis (ORACAL) trial. J Bone Miner Res 27(8):1821–1829
Overman RA, Borse M, Gourlay ML (2013) Salmon calcitonin use and associated cancer risk. Ann Pharmacother 47(12):1675–1684
Food and Drug Administration. Questions and answers: changes to the indicated population for miacalcin (calcitonin-salmon). March 11, 2013. Available online: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm388641.htm
Manson JE, Allison MA, Rossouw JE et al (2007) Estrogen therapy and coronary-artery calcification. N Engl J Med 356:2591–2602
Cauley JA, Norton L, Lippman ME et al (2001) Continued breast cancer risk reduction in postmenopausal women treated with raloxifene: 4-year results from the MORE trial. Multiple outcomes of raloxifene evaluation. Breast Cancer Res Treat 65(2):125–134
Martino S, Cauley JA, Barrett-Connor E, CORE Investigators et al (2004) Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 96(23):1751–1761
Vogel VG, Costantino JP, Wickerham DL, National Surgical Adjuvent Breast and Bowel Project (NSABP) et al (2006) Effects of tamoxifen vs. raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 295(23):2727–2741
Barrett-Connor E, Mosca L, Collins P, Raloxifene Use for The Heart (RUTH) Trial Investigators et al (2006) Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 355(2):125–137
Gennari L, Merlotti D, De Paola V, Martini G, Nuti R (2008) Bazedoxifene for the prevention of postmenopausal osteoporosis. Ther Clin Risk Manag 4(6):1229–1242
Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G (2009) Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril 92(3):1045–1052
Mirkin S, Komm BS, Pan K, Chines AA (2013) Effects of bazedoxifene/conjugated estrogens on endometrial safety and bone in postmenopausal women. Climacteric 16(3):338–346
Pinkerton JV, Pickar JH, Racketa J, Mirkin S (2012) Bazedoxifene/conjugated estrogens for menopausal symptom treatment and osteoporosis prevention. Climacteric 15(5):411–418
Saag K, Shane E, Boonen S et al (2007) Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 357(20):2028–2039
Black DM, Reid IR, Boonen S et al (2012) The effect of 3 versus 6 years of zoledronic acid treatment of osteoporosis: a randomized extension to the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 27:243–254, Erratum in: J Bone Miner Res. 2012;27(12):2612
Boonen S, Ferrari S, Miller PD (2012) Postmenopausal osteoporosis treatment with antiresorptives: effects of discontinuation or long-term continuation on bone turnover and fracture risk—a perspective. J Bone Miner Res 27(5):963–974
Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ (2012) Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med 366(22):2051–2053
Cosman F, Cauley J, Eastell R, Boonen S, Palermo L, Reid I, Cummings SR, Black DM (2014) Reassessment of fracture risk in women after 3 years of treatment with zoledronic acid: when is it reasonable to discontinue treatment? JCEM (in press)
Bauer DC, Schwartz A, Palermo L, Cauley J, Hochberg M, Santora A, Cummings SR, Black DM (2014) Fracture prediction after discontinuation of 4 to 5 years of alendronate therapy: the FLEX study. JAMA Intern Med 174(7):1126–1134. doi:10.1001/jamainternmed.2014.1232
Bonnick SL (2013) Dual-energy x-ray absorptiometry: interpreting reports and serial measurements. Clin Obstet Gynecol 56(4):677–685
Dell R, Greene D (2010) Is osteoporosis disease management cost effective? Curr Osteoporos Rep 8(1):49–55
McLellan AR, Wolowacz SE, Zimovetz EA et al (2011) Fracture liaison services for the evaluation and management of patients with osteoporotic fracture: a cost-effectiveness evaluation based on data collected over 8 years of service provision. Osteoporos Int 22(7):2083–2098
Dell R, Greene D, Schelkun SR, Williams K (2008) Osteoporosis disease management: the role of the orthopaedic surgeon. J Bone Joint Surg Am 90(Suppl 4):188–194
Newman ED (2003) A schema for effective osteoporosis management: outcomes of the Geisinger Health System Osteoporosis Program. Dis Manag Health Outcome 11(10):611–616
Eisman JA, Bogoch ER, Dell R, Harrington JT, McKinney RE, McLellan A, Mitchell PJ, Silverman S, Singleton R, Siris E, for the ASBMR Task Force on Secondary Fracture Prevention (2012) Making the first fracture the last fracture: ASBMR task force report on secondary fracture prevention. J Bone Miner Res 27:2039–2046
Acknowledgments
The NOF acknowledges the members of the 2008 Clinican’s Guide Develoment Committee, Bess Dawson-Hughes, Sundeep Khosla, L. Joseph Melton III, Anna N.A. Tosteson, Murray Favus, and Sanford Baim, and the consultants to the 2014 Update Committee, Karl Insogna, Douglas Kiel, Harold Rosen, and John Schousboe.
Disclosure policy
It is the policy of NOF to ensure balance, independence, objectivity, and scientific rigor in all sponsored publications and programs. NOF requires disclosure of any financial interest or any other relationship that the committee members have with the manufacturer(s) of any commercial product(s). All contributors to this publication have disclosed any real or apparent interest that may have direct bearing on the subject matter of this program. All potential conflicts have been resolved to the satisfaction of the NOF. Medication information included in this guidance follows the US FDA-approved label.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
The Clinician’s Guide to Prevention and Treatment of Osteoporosis 2014 is endorsed by the following organizations: American Academy of Pain Medicine (AAPM), American Association of Clinical Endocrinologists (AACE), American Orthopaedic Association (AOA), American Osteopathic Association (AOA), American Society for Bone and Mineral Research (ASBMR), and International Society for Clinical Densitometry (ISCD).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Cosman, F., de Beur, S.J., LeBoff, M.S. et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int 25, 2359–2381 (2014). https://doi.org/10.1007/s00198-014-2794-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-014-2794-2