
Abstract
Phase separation, also known as biomolecule condensate, participates in physiological processes such as transcriptional regulation, signal transduction, gene expression, and DNA damage repair by creating a membrane-free compartment. Phase separation is primarily caused by the interaction of multivalent non-covalent bonds between proteins and/or nucleic acids. The strength of molecular multivalent interaction can be modified by component concentration, the potential of hydrogen, posttranslational modification, and other factors. Notably, phase separation occurs frequently in the cytoplasm of mitochondria, the nucleus, and synapses. Phase separation in vivo is dynamic or stable in the normal physiological state, while abnormal phase separation will lead to the formation of biomolecule condensates, speeding up the disease progression. To provide candidate suggestions for the clinical treatment of nervous system diseases, this review, based on existing studies, carefully and systematically represents the physiological roles of phase separation in the central nervous system and its pathological mechanism in neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The interior and exterior of the classical organelle are physically distinct because the cell membranes typically prevent biomolecules from passing through, and the composition of the organelle is controlled by the unique membrane transport system [1]. However, some cell compartments are not bound to the membrane. Montgomery and Wilson discovered membrane-free chambers in the 1930s [2]. As the name suggests, there are a number of membrane-free compartments within cells that either lack membranes entirely or are not surrounded by the membrane [3]. Additionally, many of them are produced by proteins binding to nucleic acids or other polypeptides that function as polymeric scaffolds [4]. That dynamic aggregation of biological macromolecules in cells is regarded as phase separation. According to multiple studies, phase separation could be a mechanism that directs the creation of membrane-less organelles or recruits proteins into membrane-less organelles [5]. For example, phase separation offers cells an isolated chamber that can not only accelerate the reaction but also prevent the interference of irrelevant substances in the cytoplasm or nucleus [6]. The normal occurrence of enzymatic reactions in cells depends critically on phase separation. As such, enzymes and their substrates are assembled into a specific location at a certain time to finish a series of biological processes [6]. Furthermore, subcellular contents are spatiotemporally organized in response to environmental cues for physiological functionality [7].
There is growing evidence demonstrating the dynamic formation process of various membrane-less organelles in living cells, which is significantly influenced by phase separation, particularly liquid-liquid phase separation (LLPS) [8]. LLPS is a reversible process by which a given molecule spontaneously separates into two phases, a dilute phase and a highly concentrated and condensed phase, when a molecule disperses uniformly in the liquid state after reaching a necessary threshold concentration [9, 10]. Biomolecules dwell in phase-separated droplets, like oil drops in water, in their concentrated and diluted states [11]. The molecular underpinning of LLPS is the emergence of multivalent and low-affinity interactions, including electrostatic, hydrophobic, and other affinities [12]. Notably, the impacting factors include critical concentration as well as intracellular external conditions like temperature, pH, and ionic strength [13,14,15,16]. Moreover, it has been proposed that all biopolymers can undergo LLPS when subjected to certain conditions [1, 17].
The physical LLPS mechanism aids in the formation of many membrane-free organelles. These membrane-less organelles are called biomolecular condensates. Condensates, such as stress granules (SGs), processing bodies (P-bodies), Cajal bodies, and nucleoli, have recently provided increasing proof of playing critical roles in biological activities [2]. Since they lack membranes, these feasible roles undergo activation and deactivation in response to minute environmental changes, such as temperature or pH variations, via the emergence and disassembly of the condensates [18, 19]. Additionally, their membrane-free quality is closely related to the quick movement of molecules across phases, allowing for unhindered transit through the barrier and a rapid chemical equilibrium between compartments with various properties. For example, a stable protein concentration gradient in the cytoplasm can be produced by spatially separated phosphorylation and dephosphorylation processes, which can then influence the protein diffusion rate [20]. While modulation to the protein concentration gradient may allow for dynamic assembly and disassembly of membrane-free organelles [21, 22], the interaction of molecules inside droplets and the surrounding fluid is visible through fluorescence recovery after photobleaching (FRAP). The rapid breakdown of liquid compartments may result from the entry of proteins that are concentrated in protein droplets or other regulators into droplet phases [23]. Likewise, enhanced thermodynamic and kinetic favorability of specific reactions could occur in some droplets as a result of a little rise in component concentration [24]. Notably, reactions in the cytoplasm may halt as cytoplasmic constituents become exhausted when they segregate into the condensed phase [25]. Biomolecular condensates are viewed as a quicker spatially structured and compartmentalized regulated form of intracellular substance as a result of their rapid responsiveness [26]. Many of these structures are common to a large proportion of cells, like the membrane-less structures located within the nucleus. The nuclear bodies nucleolus, Cajal bodies, and promyelocytic leukemia nuclear bodies (PML NBs) are just a few of the many nuclear bodies that have been defined thus far [13]. Cajal bodies have been thought to be involved in the ribonucleoprotein assembly process, which is necessary for housekeeping functions within the nucleus [27]. In addition to contributing to the subcellular localization and functional regulation of certain ribonucleoprotein telomerase components, Cajal bodies are also involved in cellular stress responses like RNA interference and DNA damage repair pathways [27]. PML NBs, a macromolecular polyprotein complex, have the property of phase separation of liquid droplets. PML NBs could interact with chromatin and alter the availability of chromatin-related components by regulating transcription factor activity [28]. Nevertheless, the LLPS-driven membrane-less compartment exists as well in the cytoplasm. For example, the P-bodies and SGs are composed of mRNAs and proteins and include translation initiation-stalled mRNA-protein complexes (mRNPs). Moreover, eukaryotic cells produce stress granules, which are condensates as a response to stress [29, 30]. Numerous non-translated mRNAs, translation initiation factors, and several proteins that influence mRNA functions are found in it. Researchers discovered that protein and RNA spontaneously condense on one side of the cell to produce P granules. Importantly, this droplet-like structure can exhibit fusion, wetting, flow, and other liquid characteristics. P granules rapidly dissolve or consolidate as the concentration of linked components changes [31]. As an illustration, consider the sequestration of mTORC1, which is stored in structures that resemble P granules and are known as stress granules. Consequently, stress granules disintegrate as a result of DYRK3 kinase activity, releasing mTORC1 for signaling [22].
The existence of phase separation in cells allows compartments with diverse chemical characteristics to maintain chemical balance via the fast movement of molecules, participating in RNA metabolism, ribosome biosynthesis, DNA damage response, and signal transduction [32]. Currently, there are three possibilities in which the abnormalities of phase separation can cause illness. Genetic mutations or environmental effects may impair the production of condensate by (a) directly modifying the molecular process of condensate assembly, (b) influencing the activity of a master regulator of condensation, or (c) changing the fundamental physicochemical circumstances within cells [33]. Many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD), are connected to LLPS disorders. Consequently, this paper covers the physiological functions of phase separation, their significance for the central nervous system, and the pathological underpinnings of illnesses in the central nervous system. Notably, phase separation could provide researchers with a novel outlook for comprehending and treating human diseases.
The physiological functions of phase separation
Phase separation can provide independent units for the occurrence of enzymatic reactions, protect the orderly progression of enzymatic reactions, and facilitate intracellular transcription regulation [34]. LLPS plays indispensable roles in biological processes, like signaling transduction, gene regulation and transport, DNA damage repairing, and inter-synaptic signaling [2].
Transcriptional regulation
In eukaryotes, transcriptional regulation is an important mechanism in regulating gene expressions. Enhancers are cis-regulatory elements of the genome that influence expression patterns of target genes in a tissue- and cell-type-specific manner [35]. “Super-enhancers” refer to an expansive domain constituted of collections of enhancer elements linked to critical cell-type-specific genes and cell identities, which are essential to the biological functions of these cells [36, 37]. In vitro studies have revealed that cyclin T1 (CycT1) and dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) could combine to create droplets [38]. In contrast, the histidine-rich domain (HRD) could recruit polymerase (Pol) II into the droplets by interacting with the C-terminal domain (CTD) of the largest subunit of RNA polymerase II of human RNA Pol II, promoting phosphorylation and transcriptional elongation of Pol II [38]. Octamer-binding transcription factor 4 (OCT4) is a crucial transcription factor in embryonic stem cells (ESCs), containing a DNA binding domain and a transcriptional activation binding domain. OCT4 interacts with the transcriptional mediator complex subunit 1 (MED1) through the transcriptional structural domain to form phase-separated condensates at super-enhancers of ESCs, stimulating the gene expressions [39]. Phase separation, related to genomic activity, interacts with autosomal regions with lower chromatin density. Sanulli demonstrated that Swi6, pombe HP1 protein, significantly boosts the accessibility and dynamics of hidden histone residues within a nucleosome, which contribute to dynamic exposure of buried nucleosomal regions and reducing these dynamics could hamper the ability of Swi6 to condense chromatin into liquid droplets [40]. Eukaryotic chromatin is heavily condensed and divided into chromosomes and flexibly accessible to being regulated. Similar to how chromatin acts in cells, eukaryote-wide histone H1 and inter-nucleosome linker lengths drive chromatin phase separation, regulate droplet properties, and interact together to generate condensates of uniform density [41]. Additionally, chromatin phase separation was capable of being de-antagonized by histone acetylation of protein p300, resulting in droplet lysis in vitro and fewer droplets in the nucleus [41]. These findings indicated that phase separation was vital in the regulation of the transcription process in organisms. Furthermore, phase separation condensates in mitochondria can modulate DNA transcription [42]. Phase-separated condensates of mitochondrial transcription factor A (TFAM) will aggregate to generate circular mitochondrial DNA (mtDNA), resulting in TFAM-mtDNA droplets that significantly concentrate transcriptional enzymes for effective transcription, including TFB2M and mitochondrial RNA polymerase (POLRMT), moreover, a co-phase separation that can suppress transcription is constituted of TFAM-mtDNA and mitochondrial transcription termination factor (MTERF1) [42].
Signal transduction
Phase separation occurs frequently in the downstream signal transduction pathways and the molecular mechanism of cell-surface receptors. Transmembrane receptors on the surface of cells typically cluster from the nanometer to the micrometer scale to initiate signal transduction [43, 44]. Phase separation is a critical cellular mechanism of activity on biological membranes [45]. Transitions between ordered and disordered phases are feasible in lipid bilayer model membranes, and phase separations can occur in membranes comprised of multiple lipid species [46]. Multiple complex components within the cell membrane form a certain order in a phase-separated form to regulate the physiological functions of the cell so that the corresponding cellular functions can be performed at the appropriate time and space. Lipid phase separation in membranes impacts the transport of lipids and proteins as well as the local composition, dynamics, and allosteric modulation of membrane proteins [47]. Moreover, protein–protein collisions cause steric pressure [48], destabilizing the lipid phase separation and causing proteins and lipids to be evenly distributed on the membrane surface, maintaining the dynamic balance of the components of membrane.
LLPS has been proven to facilitate the clustering of transmembrane proteins with their cytoplasmic binding partners at membranes [49]. Linker for the activation of T cells (LAT) includes several tyrosines (Y) that are phosphorylated following T cell receptor (TCR) triggering. Three of them (Y171, Y191, and Y226) are known to attract the cytosolic adaptor protein, growth factor receptor-bound protein 2 (Grb2), via their Src homology 2 (SH2) domains. Moreover, Grb2 has two SH3 domains that interact with proline-rich areas in the nucleotide exchange factor’s C-terminal domain, Son of Sevenless (SOS) [49]. They can interact with the phosphorylated LAT and engage in phase separation [50], which promotes downstream signaling pathways. Notably, phase separation occurs in the nephrin/non-catalytic region of tyrosine kinase (Nck)/neural Wiskott-Aldrich syndrome protein (N-WASP) [51] system during membrane signaling in renal podocytes, and the degree of phase separation is regulated by the relative concentrations between molecules. Phase separation also significantly up-regulates the activity of N-WASP by increasing its membrane dwell time and enhances its ability to stimulate actin-related protein 2/3-mediated actin assembly. SOS is a guanine nucleotide exchange factor (GEF) in the LAT-Grb2-SOS signaling pathway, an essential Ras activator that is autoinhibited in the cytosol [52]. Huang et al. identified several phases involved in the process of full-length SOS stimulating the Ras GEF activity. After Grb2 recruits SOS to the membrane, the dwell time is approximately 4 ~ 6 s on the membrane. Phase separation can be observed to merge with rising SOS concentrations and then repel SOS to make it leave the cell membrane rapidly [53]. Following Grb2-mediated membrane recruitment, structural changes within SOS uncover the allosteric Ras-binding site and free autoinhibition of SOS guanine nucleotide exchange activity. In this process, the SOS condensate produced by phase separation could stay on the membrane for a considerable time [50]. This ensures that the interference signal in the upstream signal pathway will not significantly activate the downstream signal pathway under the conditions of low activity level. G protein-coupled receptor kinase interactor (GIT) and PIX are Arf-specific GTPase-activating proteins (GAPs) and Rho-specific guanine nucleotide exchange factors (GEFs), respectively. The GAP-ANK-SHD sequence of the GIT and PIX was able to form high-concentration condensate GIT2/β-Pix, which improved the ability to activate the GTP enzyme [54]. Furthermore, the multivalent interaction developed in the cytoplasm allowed for this phase separation behavior. GIT/PIX condensates can also target various cellular compartments via phase separation [54].
Many studies have illustrated how the interaction between the GIT1/β-Pix complex with paxillin promotes phase separation [55]. Through GIT1-mediated binding to paxillin, GIT1/β-Pix condensates generated by endogenous enzymes can be directed to focal adhesions, playing a critical role in regulating cell migration. When Shank family scaffold proteins bind to β-Pix in neurons [56, 57], β-Pix is concentrated at postsynaptic densities (PSDs) of excitatory synapses to regulate cell growth and synapse development. By serving as a scaffold complex [58], PIX and GIT regulate the activity of the Hippo (Hpo) kinase and encourage Hpo dimerization and autophosphorylation of the Hpo activation loop. Additionally, Hippo is the critical regulator of the Hippo pathway [59]. Mammalian Ste20-like kinases 1/2 (MST1/2, homologues of Hpo) and mitogen-activated protein kinase kinase kinase kinase (MAP4K) family members contribute to phosphorylate large tumor suppressor 1/2 (LATS1/2, homologues of Wts)/transcriptional co-activator with PDZ-binding motif (TAZ)[60]. Yes-associated protein (YAP)/TAZ is phosphorylated and inhibited by LATS1/2 to regulate cell growth and proliferation. The Laforin-Mst1/2 complex is assembled due to LLPS occurring during the aggregation of excessive glycogen, inducing excessive cell growth and proliferation as well as inhibiting apoptosis [61]. Phase separation is also necessary for insulin signaling to initiate the metabolism of glucose and glycogen. Insulin receptor substrates 1/2 (IRS) are known to recruit phosphatidylinositol 3-kinase (PI3K)-3-phosphoinositide-dependent protein kinase-1 (PDK1)-protein kinase B (PKB) to the plasma membrane after developing phase separation in the cytoplasm and nucleus [62]. Consequently, PKB can detach from the plasma membrane since being phosphorylated by PDK1 and the mechanistic target of rapamycin C2 (mTORC2) to access the cytosol and nucleus and phosphorylate its downstream targets. Accumulating results indicate that the phase separation of biomacromolecules can activate membrane receptors and their downstream signals [63]. Researchers have a great demand for effective and safe phase separation tools. The “biomimetic protein phase separation technology” designed and implemented by Li et al. [64] may flexibly manage and visualize the aggregation of cell membrane receptors and the subsequent signaling of those results. This is anticipated to lead to the development of a new paradigm for initiating receptor multimerization.
DNA protection and damage repair
Defects in the DNA damage response (DDR) can induce genomic instability, potentially leading to tumors and neurodegeneration [40]. There are three major phases in the repair of DNA damage: (1) Firstly, DNA damage sites are sensed and recruited by intracellular DNA damage sensor proteins such as poly (ADP-ribose) polymerase 1 (PARP1) and MRE11-RAD50-NBS1 (MRN) complex. (2) Subsequently, these proteins further recruit downstream signal sensor proteins such as ataxia-telangiectasia mutated and rad3 related (ATR) and ataxia-telangiectasia mutated (ATM), meanwhile amplifying the signal. (3) Ultimately, activated effector proteins, such as p53, arrest the cell cycle and repair damaged DNA through DNA repair pathways including non-homologous end joining (NHEJ) and homologous recombination (HR) [65]. p53-binding protein 1 (53BP1) contains an intrinsically disordered region (IDR) at its C-terminus, which is the molecular structural basis of phase separation. Kilic et al.’s team confirmed that 53BP1 facilitates the assembly of downstream effectors and draws DNA damage repair proteins by optoDroplet experiments [66]. 53BP1 acts as master scaffold to generate functional complexes with double-strand break (DSB)–responsive factors at damaged chromatin. Through interactions with the MRN complex, DSBs trigger the stimulation of ATM and ATM-induced 53BP1 phosphorylation to phase separation [66, 67]. Non-POU domain-containing octamer-binding protein (NONO) can undergo spontaneous phase separation in vitro, which is recruited to DNA damage sites and participates in the NHEJ DNA damage repair process [68]. Furthermore, condensate formation through phase separation of NONO protein can recruit epidermal growth factor receptor (EGFR) and DNA-dependent protein kinase (DNA-PK) into condensate and enhance the interaction between EGFR and DNA-PK, promoting phosphorylation of T2609-DNA-PK and thus enhancing NHEJ-mediated DNA damage repair [69].
Phase separation mediates synaptic-related physiological activities
Formation of presynaptic active region complex
It was observed that in neurons, electron-dense particles appear in presynaptic zones visualized by electron microscopy. Optical super-resolution microscopy allowed imaging of Rab3-interacting molecule (RIM), RIM binding protein (RIM-BP), and ETS-like transcription factor (ELKS) to form dense clusters in the active region. Notably, the deletion of RIMs and RIM-BP together significantly reduces the ability of active zone–specific presynaptic dense projections to assemble [70]. Wu et al. further demonstrate that the main components of presynaptic active region complex are a set of scaffold proteins related to synaptic vesicles (SVs) and fusion, including, RIM, RIM-BP, ELKS, and calmodulin-dependent serine protein kinase (CASK). Importantly, they could form condensates through phase separation [71,72,73,74,75]. Likewise, the recombination method in vitro confirmed that the purified RIM and RIM-BP mixtures could be phase separated at physiological concentrations. Here, researchers showed how phase separation allowed RIM and RIM-BP to build dynamic and condensed assemblies through multivalent bindings. The specific mechanism was that RIM1a-PRMs (proline-rich motif; “S” for SH3 domain binding) interacts with all three Src homology 3 (SH3) domains of RIM-BP, through C-terminal-tail-mediated directly binding to both RIM and RIM-BP [76]; voltage-gated Ca2+ channels (VGCCs) could be enriched to the RIM and RIM-BP condensates [71]. Finally, VGCCs-RIM-BP-RIM facilitated phase separation. The scaffold protein condensate between ELKS and Liprins was similar to RIM/BP-RIM, which also anchored to the presynaptic membrane by binding to proteins [71].
Formation of postsynaptic dense density
Protein-rich subcompartment, the postsynaptic density (PSD), an aggregation responsible for receiving, decoding, and storing signals sent by presynaptic axonal termini, is located beneath the postsynaptic plasma membranes of each synaptic junction [77]. Synaptic strength was positively correlated with the number of proteins in the PSD of the postsynaptic membrane. The main components of excitatory PSDs (ePSDs) are scaffold proteins, including PSD-95, SAP90/PSD-95-associated protein (SAPAP), Shank3, and Homer [78,79,80,81]. ePSDs can enhance the stability of synaptic structure and provide structural support for functional enzyme SynGAP. Gephyrin-binding proteins in inhibitory PSDs (iPSDs) are neither highly accumulated in synapses like gephyrin nor are multidomain scaffold proteins [82]. Most iPSDs are thought to reside on the dendritic shaft, while ePSDs are mainly confined to the protrusions along the dendritic spines [83]. Additionally, the scaffold protein in PSD is highly concentrated and moves dynamically. However, its diffusion rate is much slower, indicating that the phase separations of the scaffold protein and PSD are usually correlated. Zeng and team’s in vitro experiments found that when scaffold proteins PSD-95, SAPAP1, Shank3, and Homer3 were mixed at a molar ratio of 1:1:1:1, the mixture would phase separate and these four proteins well co-localized and condensed into spherical droplets, resulting in a decreasing speed [84]. In vivo experiments of mice also demonstrated that the multivalent interaction between transmembrane AMPA receptor regulatory protein (TARP) and PSD-95 in TARP gamma-8 mutant decreases synaptic transmission function in hippocampal neurons, proving that ePSD formation mediated by phase separation can functionally promote AMPAR aggregation and synaptic transmission [76]. Epilepsy, intellectual disability (ID), and autism spectrum disorder (ASD) are all correlated with SynGAP mutations [85]. Furthermore, SynGAP acts non-enzymatically in PSD and synaptic activity is highly sensitive to the dose of SynGAP. When synapses are stimulated, SynGAP disperses from PSD condensate in living neurons. Thus, activation of small G proteins such as Ras is a negative modulator of synaptic strength. Therefore, in vitro experiments showed that the homotrimer of SynGAP and PSD-95 could undergo phase separation, but the monomer SynGAP could not generate LLPS, but not the monomer SynGAP [86] (Fig. 1).

Phase separation at the synapse. A Formation of presynaptic active region complex and postsynaptic dense density. B, C Domain organization and interaction network of major scaffold proteins mediating phase separation of the ePSD (B) and the active zone (C). Two-way arrows denote interactions between proteins within each network and self-associations of domains. Other types of binding are indicated by one-way arrows. AZ active zone, PSD postsynaptic density, SV synaptic vesicle
Phase separation participates in neuronal development
Symmetric cell division
The mitotic spindle is an organelle rich in microtubules (MTs) and is essential for the accurate segregation of sister chromatids into daughter cells. The mitotic spindle performs functions depending on three key components: spindle poles, spindle microtubules, and kinetochore microtubules [87]. The poles of the mitotic spindle arise from centrosome amplification, while meiotic spindle is the absence of centrosome [88]. The centrosome serves as the primary microtubule-organizing center (MTOC) in most animal cells [89]. Centrosomal proteins regulate centrosome maturation and separation during phase separation. Polo-like kinase 4 (Plk4) assemblies concentrate tubulin and act as an MTOC to promote centrosome maturation and stability. During centrosome formation and maturation, Plk4 is firstly localized to centriole by exhibiting a ring-like pattern of localization around the Cep152 scaffold in early G1. Plk4 can also phase separate and construct a spherical condensate by using its autophosphorylated non-catalytic cryptic polo-box (CPB). Likewise, Plk4 condensate can behave as an assembly element for centriole biogenesis since CPB phosphorylation encourages Plk4 to escape from the Cep152 tether while binding to downstream STIL [90]. Notably, when the centrosome proteins Plk1, SPD-2 (mammalian homologue: Cep192), TPXL-1 (mammalian homologue: TPX2), and Zyg-9 (mammalian homologue: CKAP5) were incorporated into the centrosome surfactant-associated protein D-5 (SPD-5) condensate, the phase separation of the condensate was enhanced. As mammalian oocytes lack traditional centrosomes, the activity of transforming acidic coiled-coil protein-3 (TACC3) and its related receptors and enzymes is reduced [91]. Thus, these effects will destroy the liquid-like meiotic spindle domain (LISD), resulting in significant diminishing of K-fibers and interpolar microtubule density, as well as spindle volume, and the delay in separation [92].
Asymmetric cell division
Neuroblast (NB) is the main member in adult neural stem cells, which proliferates into neural precursor cells, neurons, and various glial cells [93]. Once NBs transform into an asymmetric cell division cycle, they will yield an apical–basal structure. Importantly, NBs divide into a larger cell that retains NB characteristics and a smaller ganglion mother cell (GMC) through interaction between protein aggregates and stellate microtubules, which is crucial in nervous system development [94]. In the asymmetric cell division (ACD) of Drosophila neural stem cells, related complexes are formed to establish of cell apical–basal polarity. When proteins and RNA within each pole split apart obliquely, this enables daughter cells of different fates. Moreover, these complexes begin to assemble at the interphase of mitosis and agglutinate at the metaphase and dissolve at the anaphase. This dynamic process indicates that the development of these complexes is closely related to phase separation [95]. Previous in vivo and in vitro research on Numb and Pon showed that phase separation is the driver of Numb polarization condensation. Multivalent Numb/Pon interaction network in vitro shows that many spherical droplets can rapidly fuse into larger droplets, and the formation of condensate is characterized by phase separation [96]. During ACD process, Par protein complexes Par3, Par6, and aPKC are also assembled by phase separation and then regulate the establishment of top–bottom cell polarity axis and neuron differentiation [96].
Pathological roles of phase separation in the central nervous system
Liquid-like droplets gradually transform into irreversible solid in pathological conditions during aging. Additionally, aged condensate droplets do not fuse but are more prone to clumping and sometimes forming a network of amyloid fibrils and amyloid-like aggregates [97]. Many neurodegenerative disorders are caused by aggregation and deposition of abnormal condensates [98]. As such, neurodegenerative diseases are characterized by progressive loss of cognitive and motor function, such like AD, PD, ALS, HD, frontotemporal lobar degeneration (FTLD), and traumatic brain injury [99]. Thus, identifying the cause and mechanism of protein-aggregation is crucial for precision therapy. Importantly, the concept of phase separation represents a new research idea for studying the disorder of protein self-assembly in neurodegenerative diseases.
AD
AD is the most common cause of dementia. The pathological features of AD are the deposition of diffuse neuritic plaque marked by extracellular amyloid beta (Aβ) and the aggregation of hyperphosphorylated tau protein (p-tau) in cells to form neurofibrillary tangles (NFTs) composed of paired helical filaments [100]. Tau protein is a highly soluble microtubule-associated protein. Phase separation of tau protein can concentrate tubulin and nucleate microtubule bundles and promote assembly of microtubules in healthy neurons [101]. However, the aggregation and deposition of p-Tau protein in AD have neurotoxic effects [102]. Under physiological conditions, LLPS will not appear when tau protein is not phosphorylated. It was found that both hyperphosphorylated tau protein and non-phosphorylated tau protein in the brain of AD represented LLPS. Regardless of its source, the droplets of Tau protein in patients will also become gel-like in minutes and start to spontaneously form tau aggregates over days [100], which are competent in accelerating the progress of AD [103]. Notably, tau rich in Ser and Thr is easily phosphorylated, and phosphorylated tau (p-tau441) is the major driving force for tau LLPS [100, 104, 105]. Moreover, this reaction is driven by a complex combination of electrostatic and hydrophobic interactions within tau protein per se [106]. The acetylation of tau proteins like K3, K18, K259, K290, K321, K353, and S356 inhibited the phase separation of tau proteins, because acetylation eliminates positive charge on Lys residues and weakens intermolecular electrostatic interactions [107]. However, the acetylation of tau at K174 leads to monomeric tau accumulation in vivo and enhances phase separation [108]. Added studies have also identified additional regulators of tau protein phase separation, including heparin, positively charged polyamine, RNA binding proteins, elevated metal ions (like zinc ions), and increased phosphorylation levels [109, 110]. These factors facilitate the formation and aging of tau droplets, and their dynamic decline progresses and evolves into the hardening and deposition of phase separation aggregates as time and temperature increase. Moreover, these neurofibrillary tangles (NFTs) containing filamentous tau protein deposits propagate and further diffuse between neurons. Furthermore, a mutation in the MAPT gene, which codes for tau protein, will result in tau protein misfolding. Thus, an abnormal ubiquitin-proteasome system, improper autophagy degradation system, and an alteration to the ubiquitin protein K290 will all cause the deposition of phase-separated condensate, accelerating the progression of AD [111] (Fig. 2).

Phase separation of tau protein in neurons. In healthy neurons, physiological tau phosphorylation detaches tau from microtubules and can lead to the formation of p-tau droplets in neurons. In diseased neurons, stable droplets and aggregates deplete p-tau from the pool available for microtubule binding. This results in uncontrolled microtubule fragmentation, disruption of proteo-homeostasis, cell stress, and eventually neuronal death
HD
HD is a progressive and fatal neurodegenerative disease; the clinical features include progressive motor dysfunction, cognitive decline, and psychiatric disturbance[112]. HD is caused by a CAG repeat expansion in the huntingtin (HTT) gene. It is caused by a CAG repeat expansion in exon 1 of the HTT gene that encodes an abnormally long polyglutamine (polyQ) tract. This mutated gene is translated to mutant huntingtin (mHTT), the causative agent of the disease [113]. When mHTT is cleaved, the polyglutamine-containing N-terminal fragment is released, and in individuals with HD, this fragment forms inclusions [114]. According to Peskett et al., protein fragments can lead to invertible liquid-like assemblies and transform into solid-like fibrillar assemblies, when the polyglutamine tract arrives at disease-associated lengths [115]. Notably, an N-terminal amphipathic region, a polyQ region, and a proline-rich region constitute huntingtin. mHTT is unstable, and the main form includes monomers, soluble oligomers, and insoluble polymers both in vivo and in vitro [114]. Likewise, polyQ length and duration equally impact how rapidly mHTT oligomers and polymers are shaped from monomers [116]. Moreover, the severity and timing of the onset of HD relate to polyQ region expansion and this change determines whether LLPS occurs in mHTT [117]. Posey et al. reported that purified N-terminal fragments of Huntington protein (Htt-NTF) can be divided into three phases, according to their saturation concentration, size, and shape. M phase is composed of soluble monomers and oligomers. S phase is mainly made up larger soluble aggregates of about 25 nm in diameter and the key components of F phase are insoluble protofibrillar aggregates [118]. Liquid-like condensates of mHTT may have progressed into fibril when polyQ length extends beyond the threshold for HD in vitro [119]. Notably, HTT could interact with several protein arginine methyltransferases (PRMT) through its N-terminal domain [120]. PRMT4 and PRMT6 are the main enzymes specifically methylate HTT and their alteration can cause HTT more difficult to solubilize in cells. Consequently, the extended HTT1-586 fragment can form liquid-like assemblies, which converts to solid assemblies when the R200/205 methylation site is altered, affecting the disease course [119, 120].
ALS/FTD
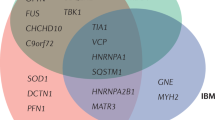
ALS is a progressive and fatal neurodegenerative disease characterized by upper motor neurons (UMN), including cranial motor nuclei in the pons and medulla and frontal cortex, and lower motor neurons (LMN), such as motor neurons in the anterior horn of the spinal cord damage, resulting in progressive muscle weakness and atrophy, with fasciculations, hyperreflexia, and spasticity [121, 122]. The pathology is characterized by degeneration and death of motor neurons like pyramidal cells and Baez cells with intracellular inclusion bodies and proliferation of glial cells [122, 123]. Previous studies also showed that mutations in C9orf72, SOD1, TARDBP/TARDBP/TAR DNA binding protein (TDP-43) and fused in sarcoma are the most frequent genetic forms of ALS [124,125,126,127,128]. Other pathogenic mutations include HNRNPA1, HNRNPA2B1, VCP, OPTN, PFN1, ANG, SETX, and MATR3 [127, 129,130,131,132,133,134,135]. Moreover, some genetic variants do not cause ALS per se, but enhance ALS susceptibility, such as ATXN2 gene amplification, SMN1 gene duplication, TIA1 mutation, and UBQLN2 mutation [136,137,138].
Clinically, FTLD is characterized by progressive deficits in behavior, personality, and/or language. The main dramatic behavioral symptoms for this disease include disinhibition, loss of empathy and comprehension, and socially unacceptable behavior [139]. Consequently, relatively localized degeneration of the frontal and temporal lobes is represented in the patient’s brain. Common genetic variants in FTLD patients include MAPT mutation encoding tau protein and C9ORF72 hexanucleotide repeat expansion [140, 141]. Rare variants associated with FTLD include VCP, CHMP2B, TBK1, HNRNPA1, and ATXN2 [142, 143].
ALS and FTLD overlap clinical and neuropathological characteristics, and both are relevant to C9orf72 amplification and RBP gene variants (TDP-43, FUS, ATXN2, EWSR1, TAF15, HNRNPA1, HNRNPA2B1, TIA1, and MATR3) [135, 144, 145]. The underlying mechanisms of these genetic variants are altering the driving force and subcellular localization of protein aggregation which in turn incite abnormal posttranslational modifications (PTMs) and disrupt the protein quality control system, inducing protein misfolding and aggregation.
Diffuse expression and partial pathological clustering of TAR DNA-binding protein 43(TDP-43) and its 35-kd and 25-kd cleavage fragments and FUS proteins are common in the brain of ALS/FTLD patients [146]. The commonly found proteins hnRNPA0, hnRNPA1, hnRNPA2B1, TIA1, ATXN2, PABPC1, and eIF2α in SGs are also present in this pathological RNA binding protein aggregate [12, 147]. Thus, this indicates that SGs are precursors of pathological aggregates. Notably, SGs are complexes composed of ribonucleoprotein particles. Under stressful pressure during physiological states, nuclear RBPs (TIA1, FUS, TDP43, hnRNPA1, hnRNPA2B1, EWSR1, and ATXN2), responsible for regulating mRNA splicing, RNA helicase activity, and RNA polymerase elongation, are transferred to the cytoplasm [148, 149]. After undergoing phase separation to form SG droplets under multivalent interactions between tyrosine residues of their prion-like domains and arginine residues of their RNA-binding domains, it can recruit mRNA and maintain a reversible steady state. SGs can also sort mRNAs and execute mRNA restart, storage, or degradation programs, respectively. Moreover, eIF4G in SGs interacts with TRAF2, thereby avoiding apoptosis and acting as a neuronal protector [150]. When the stress factor is gone, SGs are solubilized with the action of decomposing enzyme such as VCP [151]. Consequently, the internal dynamics of SGs are altered under chronic long-term stress conditions. For example, hyperphosphorylation of mutations in the prion-like domain of TDP-43 prevents nuclear RBPs from returning to the cytoplasm and re-solubilizing SGs. Simultaneously, this process also gives rise to recruit more mRNAs and nuclear RBPs to interfere with the synthesis and transport of RNA by SGs [152, 153]. ALS-linked mutations in the low-complexity structural domains of FUS, hnRNPA2B1, EWS, TAF15, MATR3, and TIA1 lead to increased local concentrations of these proteins, enhancing amyloid interactions and forming pathological aggregations of non-functional oligomers and protofibrils, causing difficulty in degrading both autophagy and ubiquitination pathways, which in turn damages neurons [137, 149, 154]. Furthermore, lipoamide promotes phase separation to generate liquid-like cohesions, thus slowing down the fibrosis progression of FUS proteins and providing a new idea for the treatment of ALS/FTLD from the drive of protein aggregation [155].
Altered subcellular localization of RNA-binding proteins can also contribute to disease. Mutations in the nuclear localization signal of FUS lead to protein mislocalization in the cytoplasm [156]. As such, the concentration of FUS in the cytoplasm exceeds the critical value for phase separation, and the concentration of RNA in the cytoplasm is so low that it cannot suffer phase separation properly. Thus, FUS gradually tends to fibrillate [157]. In addition, the cytoplasmic PQC system, unlike the nucleus, lacks nuclear input receptors in the cytoplasm, which cannot specifically bind to the nuclear localization signal in FUS and inhibit its phase separation and aggregation. This results in the accumulation of FUS inclusion bodies in neurons. Repeated amplification of the C9ORF72 gene GGGGCC leads to over-translation of the dipeptide repeat protein poly(proline-arginine) (poly-PR) and the dipeptide repeat protein poly(glycine-arginine) (poly-GR). Poly-PR and poly-GR are extremely toxic and interfere with important components of the nucleolar fluid [158]. The phase separation of nucleophosmin 1 (NPM1) and its abnormal accumulation in the nucleolus accelerate the disease progression of ALS/FTD [159,160,161]. Poly-PR and Poly-GR, which are rich in arginine, are capable of stronger multivalent interactions with NPM1 and rRNA and also will isolate rRNA and competitively bind NPM1, causing NPM1 to delocalize from the nucleolus [162]. Consequently, disturbances in nucleolus dynamics and organization can inhibit ribosome biogenesis and ultimately lead to cell death. Reducing transcription factor p53 in neurons can reduce poly-PR and poly-GR production and improve axonal degeneration, also providing new ideas for treating ALS/FTD caused by C9ORF72 amplification from inhibiting highly specific transcriptional programs [163]. In addition to pathogenic mutations, abnormal PTMs on RNA-binding proteins also contribute to ALS/FTD-associated pathological phase changes. Aberrant phosphorylation of TDP-43 inclusion bodies is common in sporadic ALS/FTD. Moreover, phosphorylation at TDP-43 serine residues 409 and 410 promotes TDP-43 phase separation, but FUS phosphorylation inhibits its phase separation [164]. The binding of TDP-43 and FUS to poly (ADP-ribose) (PAR) on RNA-binding proteins promotes the phase separation of FUS, TDP-43, and hnRNP A1 [165]. More importantly, PAR binding promotes the recruitment and targeting of TDP-43 and hnRNPA1 to SGs, causing excessive accumulation of RBPs in SGs and accelerating SG sclerosis [166]. Furthermore, UBQLN2 is extensively involved in PQC as a ubiquitin-bridging egg [167]. Mutant UBQLN2 prevents the misfolded protein from binding to Hsp70, resulting in impaired targeting of the proteasome, thereby preventing degradation of the misfolded protein via the ubiquitination pathway. Thus, the misfolded protein concentration exceeds the phase separation threshold to form cytotoxic amyloid fibrils [111] (Fig. 3).

The LLPS in descending corticospinal neurons. A In neurodegenerative diseases, accumulation of oxidative stress may induce PARP-1 hyperactivation. In addition, aggregates formed by different amyloid proteins may increase the activity of PARP-1 and the levels of cellular PAR, which together lead to neuronal cell death. B Phases in the stress granule cycle. C ALS-associated dipeptide repeats (R-rich DPRs) alter NPM1 phase separation, leading to NPM1 sequestration and driving droplet dissolution in vitro and NPM1 delocalization from nucleoli
PD
Parkinson’s disease (PD) is a long-term, progressive disease of the central nervous system that is characterized by a movement dysfunction brought on by a particular type of dopamine (DA) neuron degeneration in the substantia nigra pars compacta [168]. The substantia nigra and striatum lost dopaminergic neurons, which decreased from 550,000 to 100,000, and the current findings may also be an indication of decreased GABA production. These are the key neuropathological characteristics of PD, and all of them reduce the formation of non-motor and motor symptoms and have a negative impact on patient quality of life [169]. The Lewy body (LB), a cytoplasmic aggregate of α-synuclein (αSyn), was considered the gold standard for definitive diagnosis [97]. The phase separation and droplet formation of αSyn in physiological conditions have been demonstrated, and this may be one of the key factors underlying Sal’s neuroprotective properties [170, 171]. A53T, E46K, H50Q, and A53V are four missense point mutations in the SNCA gene, producing αSyn, which have been correlated to family types of PD [172, 173]. It can also be detected that αSyn condensate undergoes a liquid-to-solid phase transition. As such, they form cytotoxic αSyn oligomers and protofibrils, leading to neuronal dysfunction and cell death [171]. αSyn has three major domains: the N-terminal, which can bind to lipids such as membranes and PEGs; the C-terminal, which is rich in negative charges; and the hydrophobic non-amyloidal component region (NAC), which is the core region for the formation of αSyn oligomers and protofibrils [174]. Under physiological conditions, the N-terminal and NAC low-complexity domains interact multivalently [175], driving αSyn to experience spontaneous phase separate, accumulating in the presynaptic membrane, and playing an important role in vesicle transport [176]. Under pathological conditions (high critical concentration and/or presence of factors that promote spontaneous α-Syn phase separation), αSyn condensate frequently undergoes liquid-solid phase transition [174]. Acidic environments, higher concentrations of metal cations [18, 177, 178] (e.g., copper, manganese, calcium, and trivalent iron) and liposomes [179], and abnormal PTMs, mainly Ser129 phosphorylation (locate at the C-terminus) [180], promote the progressive formation of αSyn dimers, oligomers, and protofibrils. Moreover, the interaction of αSyn with tau has been shown to synergistically promote fibrosis [180]. The C-terminus of αSyn has been detected in some PD patients to bind to the proline-rich P2 region of tau, while mutations in the SNCA gene (mainly at the N-terminal) may promote phase separation by enhancing the sensitivity of αSyn to the harmful environment [181]. Surprisingly, αSyn oligomers are the key substance causing neuronal damage, and misfolded αSyn oligomers that are phagocytosed by normal neurons can act as a template to induce misfolding [171]. Additionally, αSyn can reduce ferrous ions to ferric ions and redistribute iron ions, leading to iron metabolism disorders, causing cellular oxidative stress and neuronal death [182]. Unfortunately, there is no curative approach to PD. However, researchers have now constructed and optimized the structure of a small ubiquitin-like modifier (SUMO)1 variant to obtain SUMO1, the segment including the hydrophobic binding pocket residues 15–55 [183], to interact multivalently with two SUMO-interacting motifs (SIM) located in the control of αSyn to exert stronger phase separation, inhibit αSyn aggregation, and improve the pathological manifestation of PD (Fig. 4).

The process of α-syn aggregation. A When mutations occur in A53T, E46K, H50Q, and A53V, the balance between α-syn generation and clearance is disrupted and the monomers aggregate to form oligomers. The oligomers also tend to form protofibrils and eventually fibrils. B Factors modulating misfolding and aggregation of a-Syn
Conclusion
This review focuses on several neurodegenerative diseases in which we comprehensively expound the underlying mechanism of phase separation involved in normal physiological activities and the occurrence of human disease [184]. Overall, aberrant phase separation can lead to the formation of aggregates, likely accelerating disease progression. Yet, there are still issues that have not been resolved. For example, how we could prevent phase separation from happening with disease progression remains unanswered. Phase separation mainly takes place within cells. However, the identification of phase separation becomes a major challenge due to the complexity of intracellular components. In addition, the indigenous milieu for these cells in vivo was not entirely replicated by the in vitro environment. Thus, data obtained in vitro should be validated with in vivo experiments, which will contribute to our understanding of the role of phase separation in neurodegenerative diseases. Notably, phase separation detection technology essentially restricts us from understanding mechanisms in vitro and in vivo. There are still several horizons for the development of phase separation in the field of physiological and pathological processes, as well as the development of targeted drugs. Therefore, we believe that with the continuous exploration and progress in many biomedical disciplines, the mystery of phase separation will eventually be fully unraveled and applied.
Data availability
Not applicable.
References
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18:285–298
Xia J (2022) Liquid-liquid phase separation: a new perspective to understanding aging and pathogenesis. Biosci Trends 16:359–362
Liu Y, Feng W, Wang Y, Wu B (2024) Crosstalk between protein post-translational modifications and phase separation. Cell Commun Signal 22:110
Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, Rosen MK (2016) Compositional control of phase-separated cellular bodies. Cell 166:651–663
You K, Huang Q, Yu C, Shen B, Sevilla C, Shi M, Hermjakob H, Chen Y, Li T (2020) PhaSepD B: a database of liquid-liquid phase separation related proteins. Nucleic Acids Res 48:D354–d359
Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD et al (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57:936–947
Hyman AA, Weber CA, Jülicher F (2014) Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30:39–58
Treen N, Shimobayashi SF, Eeftens J, Brangwynne CP, Levine M (2021) Properties of repression condensates in living Ciona embryos. Nat Commun 12:1561
Zhang JZ, Mehta S, Zhang J (2021) Liquid-liquid phase separation: a principal organizer of the cell’s biochemical activity architecture. Trends Pharmacol Sci 42:845–856
Uversky VN (2017) Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 44:18–30
Corpet A, Kleijwegt C, Roubille S, Juillard F, Jacquet K, Texier P, Lomonte P (2020) PML nuclear bodies and chromatin dynamics: catch me if you can! Nucleic Acids Res 48:11890–11912
Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L et al (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol 28:420–435
Zbinden A, Pérez-Berlanga M, De Rossi P, Polymenidou M (2020) Phase separation and neurodegenerative diseases: a disturbance in the force. Dev Cell 55:45–68
Cinar H, Fetahaj Z, Cinar S, Vernon RM, Chan HS, Winter RHA (2019) Temperature, hydrostatic pressure, and osmolyte effects on liquid-liquid phase separation in protein condensates: physical chemistry and biological implications. Chemistry (Weinheim an der Bergstrasse, Germany) 25:13049–13069
Adame-Arana O, Weber CA, Zaburdaev V, Prost J, Jülicher F (2020) Liquid phase separation controlled by pH. Biophys J 119:1590–1605
Ruff KM, Roberts S, Chilkoti A, Pappu RV (2018) Advances in Understanding stimulus-responsive phase behavior of intrinsically disordered protein polymers. J Mol Biol 430:4619–4635
Alberti S, Gladfelter A, Mittag T (2019) Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176:419–434
Lin Y, Protter DS, Rosen MK, Parker R (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell 60:208–219
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163:123–133
Wu H, Chen X, Shen Z, Li H, Liang S, Lu Y, Zhang M (2024) Phosphorylation-dependent membraneless organelle fusion and fission illustrated by postsynaptic density assemblies. Mol Cell 84:309–326.e307
Griffin EE, Odde DJ, Seydoux G (2011) Regulation of the MEX-5 gradient by a spatially segregated kinase/phosphatase cycle. Cell 146:955–968
Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L (2013) Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 152:791–805
Yang Y, Willis TL, Button RW, Strang CJ, Fu Y, Wen X, Grayson PRC, Evans T, Sipthorpe RJ, Roberts SL et al (2019) Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat Commun 10:3759
Stender EGP, Ray S, Norrild RK, Larsen JA, Petersen D, Farzadfard A, Galvagnion C, Jensen H, Buell AK (2021) Capillary flow experiments for thermodynamic and kinetic characterization of protein liquid-liquid phase separation. Nat Commun 12:7289
Jia TZ, Chandru K, Hongo Y, Afrin R, Usui T, Myojo K, Cleaves HJ 2nd (2019) Membraneless polyester microdroplets as primordial compartments at the origins of life. Proc Natl Acad Sci USA 116:15830–15835
Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, Drummond DA (2017) Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168:1028–1040.e1019
Trinkle-Mulcahy L, Sleeman JE (2017) The Cajal body and the nucleolus: “in a relationship” or “it’s complicated”? RNA Biol 14:739–751
Peng PH, Hsu KW, Wu KJ (2021) Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am J Cancer Res 11:3766–3776
Sheth U, Parker R (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science (New York, NY) 300:805–808
Kedersha NL, Gupta M, Li W, Miller I, Anderson P (1999) RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 147:1431–1442
Berry J, Weber SC, Vaidya N, Haataja M, Brangwynne CP (2015) RNA transcription modulates phase transition-driven nuclear body assembly. Proc Natl Acad Sci USA 112:E5237–5245
Bracha D, Walls MT, Brangwynne CP (2019) Probing and engineering liquid-phase organelles. Nat Biotechnol 37:1435–1445
Alberti S, Dormann D (2019) Liquid-liquid phase separation in disease. Annu Rev Genet 53:171–194
Bracha D, Walls MT, Wei MT, Zhu L, Kurian M, Avalos JL, Toettcher JE, Brangwynne CP (2018) Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 175:1467–1480.e1413
Cramer P (2019) Organization and regulation of gene transcription. Nature 573:45–54
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153:307–319
Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA (2013) Super-enhancers in the control of cell identity and disease. Cell 155:934–947
Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q (2018) Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558:318–323
Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175:1842–1855.e1816
Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575:390–394
Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, Rosen MK (2019) Organization of chromatin by intrinsic and regulated phase separation. Cell 179:470–484.e421
Long Q, Zhou Y, Wu H, Du S, Hu M, Qi J, Li W, Guo J, Wu Y, Yang L et al (2021) Phase separation drives the self-assembly of mitochondrial nucleoids for transcriptional modulation. Nat Struct Mol Biol 28:900–908
Iwahara J (2024) Transient helices with functional roles. Biophys J. https://doi.org/10.1016/j.bpj.2024.01.038
Yan X, Zhang M, Wang D (2024) Interplay between posttranslational modifications and liquid-liquid phase separation in tumors. Cancer Lett 584:216614
Woodward X, Kelly CV (2020) Single-lipid dynamics in phase-separated supported lipid bilayers. Chem Phys Lipid 233:104991
Elson EL, Fried E, Dolbow JE, Genin GM (2010) Phase separation in biological membranes: integration of theory and experiment. Annu Rev Biophys 39:207–226
Miller EJ, Ratajczak AM, Anthony AA, Mottau M, Rivera Gonzalez XI, Honerkamp-Smith AR (2020) Divide and conquer: how phase separation contributes to lateral transport and organization of membrane proteins and lipids. Chem Phys Lipid 233:104985
Imam ZI, Kenyon LE, Carrillo A, Espinoza I, Nagib F, Stachowiak JC (2016) Steric pressure among membrane-bound polymers opposes lipid phase separation. Langmuir : the ACS journal of surfaces and colloids 32:3774–3784
Case LB, Ditlev JA, Rosen MK (2019) Regulation of transmembrane signaling by phase separation. Annu Rev Biophys 48:465–494
Huang WY, Yan Q, Lin WC, Chung JK, Hansen SD, Christensen SM, Tu HL, Kuriyan J, Groves JT (2016) Phosphotyrosine-mediated LAT assembly on membranes drives kinetic bifurcation in recruitment dynamics of the Ras activator SOS. Proc Natl Acad Sci USA 113:8218–8223
Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF et al (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483:336–340
Iversen L, Tu HL, Lin WC, Christensen SM, Abel SM, Iwig J, Wu HJ, Gureasko J, Rhodes C, Petit RS et al (2014) Molecular kinetics. Ras activation by SOS: allosteric regulation by altered fluctuation dynamics. Science (New York, NY) 345:50–54
Huang WYC, Alvarez S, Kondo Y, Lee YK, Chung JK, Lam HYM, Biswas KH, Kuriyan J, Groves JT (2019) A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science (New York, NY) 363:1098–1103
Zhou W, Li X, Premont RT (2016) Expanding functions of GIT Arf GTPase-activating proteins, PIX Rho guanine nucleotide exchange factors and GIT-PIX complexes. J Cell Sci 129:1963–1974
Legg K (2011) Cell migration: keeping young and mobile with β-PIX. Nat Rev Mol Cell Biol 12:278
Zhu J, Zhou Q, Xia Y, Lin L, Li J, Peng M, Zhang R, Zhang M (2020) GIT/PIX condensates are modular and ideal for distinct compartmentalized cell signaling. Mol Cell 79:782–796.e786
Shin EY, Lee CS, Kim HB, Park JH, Oh K, Lee GW, Cho EY, Kim HK, Kim EG (2021) Kinesin-1-dependent transport of the βPIX/GIT complex in neuronal cells. BMB Rep 54:380–385
Dent LG, Poon CL, Zhang X, Degoutin JL, Tipping M, Veraksa A, Harvey KF (2015) The GTPase regulatory proteins Pix and Git control tissue growth via the Hippo pathway. Current biology : CB 25:124–130
Fu M, Hu Y, Lan T, Guan KL, Luo T, Luo M (2022) The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct Target Ther 7:376
Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo JS, Lu W, Lu S et al (2015) MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun 6:8357
Liu Q, Li J, Zhang W, Xiao C, Zhang S, Nian C, Li J, Su D, Chen L, Zhao Q et al (2021) Glycogen accumulation and phase separation drives liver tumor initiation. Cell 184:5559–5576.e5519
Zhou K, Chen Q, Chen J, Liang D, Feng W, Liu M, Wang Q, Wang R, Ouyang Q, Quan C et al (2022) Spatiotemporal regulation of insulin signaling by liquid-liquid phase separation. Cell Discov 8:64
Wang B, Zhang L, Dai T, Qin Z, Lu H, Zhang L, Zhou F (2021) Liquid-liquid phase separation in human health and diseases. Signal Transduct Target Ther 6:290
Li R, Li T, Lu G, Cao Z, Chen B, Wang Y, Du J, Li P (2022) Programming cell-surface signaling by phase-separation-controlled compartmentalization. Nat Chem Biol 18:1351–1360
Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (2008) PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283:1197–1208
Kilic S, Lezaja A, Gatti M, Bianco E, Michelena J, Imhof R, Altmeyer M (2019) Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J 38:e101379
Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I, Iannelli F et al (2017) Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat Cell Biol 19:1400–1411
Wei Y, Luo H, Yee PP, Zhang L, Liu Z, Zheng H, Zhang L, Anderson B, Tang M, Huang S et al (2021) Paraspeckle protein NONO promotes TAZ phase separation in the nucleus to drive the oncogenic transcriptional program. Adv Sci (Weinheim, Baden-Wurttemberg, Germany) 8:e2102653
Fan XJ, Wang YL, Zhao WW, Bai SM, Ma Y, Yin XK, Feng LL, Feng WX, Wang YN, Liu Q et al (2021) NONO phase separation enhances DNA damage repair by accelerating nuclear EGFR-induced DNA-PK activation. Am J Cancer Res 11:2838–2852
Acuna C, Liu X, Südhof TC (2016) How to make an active zone: unexpected universal functional redundancy between RIMs and RIM-BPs. Neuron 91:792–807
Acuna C, Liu X, Gonzalez A, Südhof TC (2015) RIM-BPs mediate tight coupling of action potentials to Ca(2+)-triggered neurotransmitter release. Neuron 87:1234–1247
Held RG, Kaeser PS (2018) ELKS active zone proteins as multitasking scaffolds for secretion. Open Biol 8(2):170258. https://doi.org/10.1098/rsob.170258
Tan C, de Nola G, Qiao C, Imig C, Born RT, Brose N, Kaeser PS (2022) Munc13 supports fusogenicity of non-docked vesicles at synapses with disrupted active zones. Elife 11:e79077. https://doi.org/10.7554/eLife.79077
Astigarraga S, Hofmeyer K, Farajian R, Treisman JE (2010) Three Drosophila Liprins interact to control synapse formation. J Neurosci Off J Soc Neurosci 30:15358–15368
Gao R, Piguel NH, Melendez-Zaidi AE, Martin-de-Saavedra MD, Yoon S, Forrest MP, Myczek K, Zhang G, Russell TA, Csernansky JG et al (2018) CNTNAP2 stabilizes interneuron dendritic arbors through CASK. Mol Psychiatry 23:1832–1850
Wu X, Cai Q, Shen Z, Chen X, Zeng M, Du S, Zhang M (2019) RIM and RIM-BP form presynaptic active-zone-like condensates via phase separation. Mol Cell 73:971–984.e975
Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, Zhang M (2016) Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell 166:1163–1175.e1112
Zhu YC, Li D, Wang L, Lu B, Zheng J, Zhao SL, Zeng R, Xiong ZQ (2013) Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc Natl Acad Sci USA 110:9118–9123
Lee SE, Kim JA, Chang S (2018) nArgBP2-SAPAP-SHANK, the core postsynaptic triad associated with psychiatric disorders. Exp Mol Med 50:1–9
Jeong J, Li Y, Roche KW (2021) CaMKII phosphorylation regulates synaptic enrichment of Shank3. eNeuro 8(3):ENEURO.0481-20.2021. https://doi.org/10.1523/ENEURO.0481-20.2021
Clifton NE, Trent S, Thomas KL, Hall J (2019) Regulation and function of activity-dependenthomer in synaptic plasticity. Mol Neuropsychiatry 5(3):147–161. https://doi.org/10.1159/000500267
Bai G, Wang Y, Zhang M (2021) Gephyrin-mediated formation of inhibitory postsynaptic density sheet via phase separation. Cell Res 31:312–325
Villa KL, Berry KP, Subramanian J, Cha JW, Oh WC, Kwon HB, Kubota Y, So PT, Nedivi E (2016) Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89:756–769
Zeng M, Chen X, Guan D, Xu J, Wu H, Tong P, Zhang M (2018) Reconstituted postsynaptic density as a molecular platform for understanding synapse formation and plasticity. Cell 174:1172–1187.e1116
Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA et al (2012) Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151:709–723
Araki Y, Zeng M, Zhang M, Huganir RL (2015) Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85:173–189
Jiang H, He X, Wang S, Jia J, Wan Y, Wang Y, Zeng R, Yates J 3rd, Zhu X, Zheng Y (2014) A microtubule-associated zinc finger protein, BuGZ, regulates mitotic chromosome alignment by ensuring Bub3 stability and kinetochore targeting. Dev Cell 28:268–281
Prosser SL, Pelletier L (2017) Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol 18:187–201
Hori A, Barnouin K, Snijders AP, Toda T (2016) A non-canonical function of Plk4 in centriolar satellite integrity and ciliogenesis through PCM1 phosphorylation. EMBO Rep 17:326–337
Park JE, Zhang L, Bang JK, Andresson T, DiMaio F, Lee KS (2019) Phase separation of Polo-like kinase 4 by autoactivation and clustering drives centriole biogenesis. Nat Commun 10:4959
So C, Seres KB, Steyer AM, Mönnich E, Clift D, Pejkovska A, Möbius W, Schuh M (2019) A liquid-like spindle domain promotes acentrosomal spindle assembly in mammalian oocytes. Science 364(6447):eaat9557. https://doi.org/10.1126/science.aat9557
Montenegro Gouveia S, Zitouni S, Kong D, Duarte P, Ferreira Gomes B, Sousa AL, Tranfield EM, Hyman A, Loncarek J, Bettencourt-Dias M (2018) PLK4 is a microtubule-associated protein that self-assembles promoting de novo MTOC formation. J Cell Sci 132(4):jcs219501. https://doi.org/10.1242/jcs.219501
van Strien ME, van den Berge SA, Hol EM (2011) Migrating neuroblasts in the adult human brain: a stream reduced to a trickle. Cell Res 21:1523–1525
Choksi SP, Southall TD, Bossing T, Edoff K, de Wit E, Fischer BE, van Steensel B, Micklem G, Brand AH (2006) Prospero acts as a binary switch between self-renewal and differentiation in Drosophila neural stem cells. Dev Cell 11:775–789
Wen W, Zhang M (2018) Protein Complex Assemblies in Epithelial Cell Polarity and Asymmetric Cell Division. J Mol Biol 430:3504–3520
Shan Z, Tu Y, Yang Y, Liu Z, Zeng M, Xu H, Long J, Zhang M, Cai Y, Wen W (2018) Basal condensation of Numb and Pon complex via phase transition during Drosophila neuroblast asymmetric division. Nat Commun 9:737
Mukherjee S, Sakunthala A, Gadhe L, Poudyal M, Sawner AS, Kadu P, Maji SK (2023) Liquid-liquid phase separation of α-synuclein: a new mechanistic insight for α-synuclein aggregation associated with Parkinson’s disease Pathogenesis. J Mol Biol 435:167713
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science (New York, NY) 296:1991–1995
Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396
Savastano A, Flores D, Kadavath H, Biernat J, Mandelkow E, Zweckstetter M (2021) Disease-associated tau phosphorylation hinders tubulin assembly within tau condensates. Angew Chem Int Ed Engl 60:726–730
Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M (2017) Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun 8:275
Liu M, Dexheimer T, Sui D, Hovde S, Deng X, Kwok R, Bochar DA, Kuo MH (2020) Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci Rep 10:16551
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci Off J Soc Neurosci 33:1024–1037
Stathas S, Alvarez VE, Xia W, Nicks R, Meng G, Daley S, Pothast M, Shah A, Kelley H, Esnault C et al (2022) Tau phosphorylation sites serine202 and serine396 are differently altered in chronic traumatic encephalopathy and Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 18:1511–1522
Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S,Laskowski PR, MacKenzie D, Kamath T, Commins C, Vanderburg C, Roe AD, Fan Z, Molliex AM, Hernandez-Vega A, Muller D, Hyman AA, Mandelkow E, Taylor JP, Hyman BT (2018) Tauprotein liquid-liquid phase separation can initiate tau aggregation. EMBO J 37(7):e98049. https://doi.org/10.15252/embj.201798049
Koopman MB, Ferrari L, Rüdiger SGD (2022) How do protein aggregates escape quality control in neurodegeneration? Trends Neurosci 45:257–271
Boyko S, Surewicz WK (2022) Tau liquid-liquid phase separation in neurodegenerative diseases. Trends Cell Biol 32:611–623
Alquezar C, Arya S, Kao AW (2020) Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 11:595532
Kanaan NM, Hamel C, Grabinski T, Combs B (2020) Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun 11:2809
Ash PEA, Lei S, Shattuck J, Boudeau S, Carlomagno Y, Medalla M, Mashimo BL, SocorroG, Al-Mohanna LFA, Jiang L, Öztürk MM, Knobel M, Ivanov P, Petrucelli L, Wegmann S,Kanaan NM, Wolozin B (2021) TIA1 potentiates tau phase separation and promotes generation of toxicoligomeric tau. Proc Natl Acad Sci U S A 118(9):e2014188118. https://doi.org/10.1073/pnas.2014188118
Dao TP, Kolaitis RM, Kim HJ, O’Donovan K, Martyniak B, Colicino E, Hehnly H, Taylor JP, Castañeda CA (2018) Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell 69:965–978.e966
Podvin S, Reardon HT, Yin K, Mosier C, Hook V (2019) Multiple clinical features of Huntington’s disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration. J Neurol 266:551–564
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98
Aktar F, Burudpakdee C, Polanco M, Pei S, Swayne TC, Lipke PN, Emtage L (2019) The huntingtin inclusion is a dynamic phase-separated compartment. Life Sci Alliance 2(5):e201900489. https://doi.org/10.26508/lsa.201900489
Peskett TR, Rau F, O’Driscoll J, Patani R, Lowe AR, Saibil HR (2018) A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol Cell 70:588–601.e586
Yang J, Yang X (2020) Phase transition of huntingtin: factors and pathological relevance. Front Genet 11:754
Cable J, Brangwynne C, Seydoux G, Cowburn D, Pappu RV, Castañeda CA, Berchowitz LE, Chen Z, Jonikas M, Dernburg A et al (2019) Phase separation in biology and disease-a symposium report. Ann N Y Acad Sci 1452:3–11
Posey AE, Ruff KM, Harmon TS, Crick SL, Li A, Diamond MI, Pappu RV (2018) Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J Biol Chem 293:3734–3746
Pessina F, Gioia U, Brandi O, Farina S, Ceccon M, Francia S, di Fagagna FDA (2021) DNA damage triggers a new phase in neurodegeneration. TiG 37(337):354
Ratovitski T, Jiang M, O’Meally RN, Rauniyar P, Chighladze E, Faragó A, Kamath SV, Jin J, Shevelkin AV, Cole RN et al (2022) Interaction of huntingtin with PRMTs and its subsequent arginine methylation affects HTT solubility, phase transition behavior and neuronal toxicity. Hum Mol Genet 31:1651–1672
Štětkářová I, Ehler E (2021) Diagnostics of amyotrophic lateral sclerosis: Up to date. Diagnostics (Basel) 11(2):231. https://doi.org/10.3390/diagnostics11020231
Meyer T (1946) (2021) [Amyotrophic lateral sclerosis (ALS) - diagnosis, course of disease and treatment options]. Dtsch Med Wochenschr 146:1613–1618
Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9:617–628
Farg MA, Konopka A, Soo KY, Ito D, Atkin JD (2017) The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet 26:2882–2896
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, NY) 323:1208–1211
Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, Liu E, Donkervoort S, Zheng JG, Shi Y et al (2010) Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75:807–814
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP (2017) Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 88:540–549
Leblond CS, Kaneb HM, Dion PA, Rouleau GA (2014) Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol 262 Pt B:91–101
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A et al (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473
González-Pérez P, Cirulli ET, Drory VE, Dabby R, Nisipeanu P, Carasso RL, Sadeh M, Fox A, Festoff BW, Sapp PC et al (2012) Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 79:2201–2208
Yang L, Cheng Y, Jia X, Liu X, Li X, Zhang K, Shen D, Liu M, Guan Y, Liu Q et al (2021) Four novel optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in Mainland China. Neurobiol Aging 97:149.e141–149.e148
Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM et al (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488:499–503
Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413
Grunseich C, Patankar A, Amaya J, Watts JA, Li D, Ramirez P, Schindler AB, Fischbeck KH, Cheung VG (2020) Clinical and Molecular aspects of senataxin mutations in amyotrophic lateral sclerosis 4. Ann Neurol 87:547–555
Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666
Lattante S, Conte A, Zollino M, Luigetti M, Del Grande A, Marangi G, Romano A, Marcaccio A, Meleo E, Bisogni G et al (2012) Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 79:66–72
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A et al (2017) TIA1 Mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95:808–816.e809
Gellera C, Tiloca C, Del Bo R, Corrado L, Pensato V, Agostini J, Cereda C, Ratti A, Castellotti B, Corti S et al (2013) Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J Neurol Neurosurg Psychiatry 84:183–187
Van Mossevelde S, Engelborghs S, van der Zee J, Van Broeckhoven C (2018) Genotype-phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol 14:363–378
Le Ber I, Guedj E, Gabelle A, Verpillat P, Volteau M, Thomas-Anterion C, Decousus M, Hannequin D, Véra P, Lacomblez L et al (2006) Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain : a journal of neurology 129:3051–3065
Greaves CV, Rohrer JD (2019) An update on genetic frontotemporal dementia. J Neurol 266:2075–2086
Bang J, Spina S, Miller BL (2015) Frontotemporal dementia. Lancet (London, England) 386:1672–1682
Häkkinen S, Chu SA, Lee SE (2020) Neuroimaging in genetic frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis 145:105063
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science (New York, NY) 348:1151–1154
Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D, Sapp PC, Silani V, Bosco DA, Shaw CE et al (2011) Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 156b:285–290
Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S (2013) Overexpression of ALS-associated p. M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol 48:22–35
Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P (2000) Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151:1257–1268
Fang MY, Markmiller S, Vu AQ, Javaherian A, Dowdle WE, Jolivet P, Bushway PJ, Castello NA, Baral A, Chan MY et al (2019) Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103:802–819.e811
Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH et al (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88:678–690
Guzikowski AR, Chen YS, Zid BM (2019) Stress-induced mRNP granules: Form and function of processing bodies and stress granules. Wiley Interdiscip Rev RNA 10:e1524
Protter DSW, Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26:668–679
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY) 323:1205–1208
Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O’Meally R, Dignon GL, Conicella AE, Zheng W et al (2017) Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J 36:2951–2967
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B et al (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077
Jain A, Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546:243–247
Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci 10:35
Mitrea DM, Cika JA, Stanley CB, Nourse A, Onuchic PL, Banerjee PR, Phillips AH, Park CG, Deniz AA, Kriwacki RW (2018) Self-interaction of NPM1 modulates multiple mechanisms of liquid-liquid phase separation. Nat Commun 9:842
Kanekura K, Harada Y, Fujimoto M, Yagi T, Hayamizu Y, Nagaoka K, Kuroda M (2018) Characterization of membrane penetration and cytotoxicity of C9orf72-encoding arginine-rich dipeptides. Sci Rep 8:12740
Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F (2016) Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet 25:1803–1813
Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, Blum JA, Castruita PA, Linares GR, Ruan K et al (2021) p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 184:689–708.e620
Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y, Yoshida M, Hasegawa M (2016) Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep 6:23281
McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG, Shorter J, Bonini NM (2018) Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell 71:703–717.e709
Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS et al (2018) Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson's disease. Science 362(6414):eaat8407. https://doi.org/10.1126/science.aat8407
Sharkey LM, Sandoval-Pistorius SS, Moore SJ, Gerson JE, Komlo R, Fischer S, Negron-Rios KY, Crowley EV, Padron F, Patel R et al (2020) Modeling UBQLN2-mediated neurodegenerative disease in mice: shared and divergent properties of wild type and mutant UBQLN2 in phase separation, subcellular localization, altered proteostasis pathways, and selective cytotoxicity. Neurobiol Dis 143:105016
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V (2022) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22:657–673
Bezard E, Yue Z, Kirik D, Spillantini MG (2013) Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Movement disorders : official journal of the Movement Disorder Society 28:61–70
Wakabayashi K, Tanji K, Mori F, Takahashi H (2007) The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology : official journal of the Japanese Society of Neuropathology 27:494–506
Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, Josephs KA, Fasano A, Morgante F, Savica R et al (2019) Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 92:329–337
Long H, Zheng W, Liu Y, Sun Y, Zhao K, Liu Z, Xia W, Lv S, Liu Z, Li D, He KW, Liu C (2021) Wild-type α-synuclein inherits the structure and exacerbated neuropathology of E46K mutantfibril strain by cross-seeding. Proc Natl Acad Sci U S A 118(20):e2012435118. https://doi.org/10.1073/pnas.2012435118
Griffey CJ, Yamamoto A (2022) Living in α-syn: tackling aggregates in Parkinson’s disease. Neuron 110:351–352
Sawner AS, Ray S, Yadav P, Mukherjee S, Panigrahi R, Poudyal M, Patel K, Ghosh D, Kummerant E, Kumar A et al (2021) Modulating α-synuclein liquid-liquid phase separation. Biochemistry 60:3676–3696
Martin EW, Mittag T (2018) Relationship of sequence and phase separation in protein low-complexity regions. Biochemistry 57:2478–2487
Snead D, Eliezer D (2019) Intrinsically disordered proteins in synaptic vesicle trafficking and release. J Biol Chem 294:3325–3342
Rcom-H’cheo-Gauthier AN, Osborne SL, Meedeniya AC, Pountney DL (2016) Calcium: alpha-synuclein interactions in alpha-synucleinopathies. Front Neurosci 10:570
Xu B, Huang S, Liu Y, Wan C, Gu Y, Wang D, Yu H (2022) Manganese promotes α-synuclein amyloid aggregation through the induction of protein phase transition. J Biol Chem 298:101469
Man WK, Tahirbegi B, Vrettas MD, Preet S, Ying L, Vendruscolo M, De Simone A, Fusco G (2021) The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition. Nat Commun 12:927
Oueslati A (2016) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinson’s Dis 6:39–51
Mohite GM, Navalkar A, Kumar R, Mehra S, Das S, Gadhe LG, Ghosh D, Alias B, Chandrawanshi V, Ramakrishnan A et al (2018) The familial α-synuclein A53E mutation enhances cell death in response to environmental toxins due to a larger population of oligomers. Biochemistry 57:5014–5028
Ji ZS, Gao GB, Ma YM, Luo JX, Zhang GW, Yang H, Li N, He QY, Lin HS (2022) Highly bioactive iridium metal-complex alleviates spinal cord injury via ROS scavenging and inflammation reduction. Biomaterials 284:121481
Liang Z, Chan HYE, Lee MM, Chan MK (2021) A SUMO1-derived peptide targeting SUMO-interacting motif inhibits α-synuclein aggregation. Cell Chem Biol 28:180–190.e186
Zhang H, Ji X, Li P, Liu C, Lou J, Wang Z, Wen W, Xiao Y, Zhang M, Zhu X (2020) Liquid-liquid phase separation in biology: mechanisms, physiological functions and human diseases. Sci China Life Sci 63:953–985
Acknowledgements
This work was supported by Department of Anesthesiology, First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China. The authors thank AiMi Academic Services (www.aimieditor.com) for English language editing and review services.
Funding
This work was supported by the National Natural Science Foundation of China (82272225, 81860249) and the Wujieping Medical Foundation (320.6750.2021–03-1).
Author information
Authors and Affiliations
Contributions
Sheng Wang and Jin Hu had the idea for the article. Jiaxin Wang, Ruijia Tian, and Haoliang Zhang performed the literature search and drafted it. Qian Zhang and Hongrui Zhu critically revised the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Zhu, H., Tian, R. et al. Physiological and pathological effects of phase separation in the central nervous system. J Mol Med 102, 599–615 (2024). https://doi.org/10.1007/s00109-024-02435-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-024-02435-7