
Abstract
Some observational studies indicate a link between blood lead and kidney function although results remain controversial. In this study, Mendelian randomisation (MR) analysis was applied to obtain unconfounded estimates of the casual association of genetically determined blood lead with estimated glomerular filtration rate (eGFR) and the risk of chronic kidney disease (CKD). Data from the largest genome-wide association studies (GWAS) on blood lead, eGFR and CKD, from predominantly ethnically European populations, were analysed in total, as well as separately in individuals with or without type 2 diabetes mellitus. Inverse variance weighted (IVW) method, weighted median (WM)-based method, MR-Egger, MR-Pleiotropy RESidual Sum and Outlier (PRESSO) as well as the leave-one-out method were applied. In a general population, lifetime blood lead levels had no significant effect on risk of CKD (IVW: p = 0.652) and eGFR (IVW: p = 0.668). After grouping by type 2 diabetes status (no diabetes vs. diabetes), genetically higher levels of blood lead had a significant negative impact among subjects with type 2 diabetes (IVW = Beta: −0.03416, p = 0.0132) but not in subjects without (IVW: p = 0.823), with low likelihood of heterogeneity for any estimates (IVW p > 0.158). MR-PRESSO did not highlight any outliers. Pleiotropy test, with very negligible intercept and insignificant p-value, indicated a low likelihood of pleiotropy for all estimations. The leave-one-out method demonstrated that links were not driven by a single SNP. Our results show, for the first time, that among subjects with type 2 diabetes, higher blood lead levels are potentially related to less favourable renal function. Further studies are needed to confirm our results.
Key messages
What is already known about this subject?
-
Chronic kidney disease is associated with unfavourable lifestyle behaviours and conditions such as type 2 diabetes.
-
Observational studies have reported an association between blood lead and reduced estimated glomerular filtration rate, but the relationship between lead exposure and renal function remains controversial.
What is the key question?
-
Using Mendelian randomisation with data from 5433 individuals from the UK and Australian populations, does genetically determined blood lead have a potentially causal effect on estimated glomerular filtration rate and the risk of chronic kidney disease?
What are the new findings?
-
Blood lead levels have a potentially causal effect on reduced renal function in individuals with type 2 diabetes.
-
In subjects without diabetes, no such causal relationship was identified.
How might this impact on clinical practice in the foreseeable future?
-
This highlights the risk of elevated blood lead, for example, due to environmental exposure, amongst those with type 2 diabetes, which may predispose them to impaired renal function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic kidney disease (CKD) is an age-associated decline in renal function, diagnosed by impaired glomerular filtration rate (GFR) or increased urinary albumin excretion (albuminuria) [1]. Up to 13% of the global population is estimated to suffer some degree of CKD with increasing age associated positively with reduced renal function such that over one-third of those 70 years or older are affected [2, 3]. Chronic kidney disease is a frequently observed comorbidity in multiple cardiometabolic conditions such as type 2 diabetes, hypertension (HT), obesity and cardiovascular disease (CVD) [4,5,6,7,8,9,10,11,12,13,14], considerably adding to the burden of these conditions. As the aforementioned conditions are also components of metabolic syndrome (MetS) [15], it is not surprising that CKD is also frequently associated with this diagnosis [16, 17] which is estimated to affect 20–25% of western populations [18, 19]. Of particular interest in patients with diabetes is the development of diabetic nephropathy, with diabetes being a primary cause of end-stage renal disease in 40–60% of cases, globally [20]. Furthermore, recent research has illustrated that environmental lead exposure may accelerate progressive diabetic nephropathy, and that reductions in body lead levels by chelation therapy can reduce this rate of progression [21].
Similar to MetS and its constituent conditions, the incidence of CKD is associated with unfavourable dietary patterns and lifestyle behaviours such as low levels of physical activity [22,23,24,25]. Interestingly, a number of observational studies have found an association between blood lead levels and reduced estimated glomerular filtration rate (eGFR) [26,27,28,29], although not to a clinically significant degree, and this finding is not consistently observed [30,31,32]. Lead exposure may also be associated with a slight hyperfiltration state, which has been found to attenuate the age-related decline in baseline creatinine clearance, a measure of GFR and even increased eGFR [32]. Thus, the relationship between lead exposure and renal function remains controversial, and further investigation is required. While randomised controlled trials (RCTs) are reliable determinants of causal inferences in nutrition science, not all exposure-outcome interactions can be tested. This is due to both a cost and time perspective and also because of ethical considerations brought about by exposing participants to presumed risk factors, in this case, lead.
Alternatively, Mendelian randomisation (MR) analysis uses functional polymorphisms (single nucleotide polymorphisms (SNPs)) associated with specific changes in exposures (e.g. lead) as genetic instruments and can provide unbiased and robust evidence on mechanisms of disease pathogenesis. Thus, MR studies can overcome this shortcoming of RCTs [33]. Unlike conventional observational studies and risk factor–based epidemiology, MR studies are considerably less prone to confounding, residual bias and reverse causation [34]. Therefore, we used MR analysis to obtain unconfounded estimates of the casual association of genetically determined blood levels of lead with renal function.
Methods
Study design
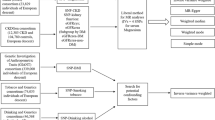
A two-sample MR study design was used, in which summary statistics from different genome wide association studies (GWAS) were analysed for the exposures (blood lead) and outcomes (renal function), to estimate the effects of exposure on outcome [35]. Essentially, we applied genetic predictors of blood lead to extensively genotyped case–control studies of renal function (eGFR and the risk of CKD) to obtain estimates of the association of exposure to our clinical outcomes.
Genetic predictors of exposures
We retrieved summary data for the association between SNPs and circulating lead from the GWAS carried out by the Queensland Institute of Medical Research (QIMR), Australia (n = 2603, mean age 47.2 years, 59% women), and from the Avon Longitudinal Study of Parents and Children (ALSPAC) (2830 unrelated mothers, mean age 28.4 years) [36]. Genotyping, quality control and imputation procedures are described elsewhere [36]. If a SNP was unavailable for the outcome GWAS summary statistics, we identified proxy SNPs with a minimum linkage disequilibrium (LD) r2 = 0.8. We used 13 independent SNPs with a p-value < 5 × 10−6. To minimize bias in effect estimates induced by correlation between SNPs, we restricted our genetic instrument to independent SNPs not in linkage disequilibrium (p = 0.0001). We refer to a set of SNPs that proxy blood lead as “genetic instruments.”
Genetic predictors of outcomes
Genetic associations with renal function were obtained from the largest available extensively genotyped study based on a meta-analysis (n = 133,413 individuals with replication in up to 42,166 individuals) (full details of all studies included are available in the original article) [37]. eGFR was estimated using the four-variable modification of diet in renal disease (MDRD) equation [37]. CKD was defined as eGFR < 60 ml/min/1.73 m2. Type 2 diabetes was defined as fasting glucose ≥ 126 mg/dl, antidiabetic drug treatment or by self-reported history. Kidney function and type 2 diabetes were assessed simultaneously.
For GWAS analysis, a centralized analysis plan was applied with each study regressing sex- and age-adjusted residuals of the logarithm of eGFR on SNP dosage levels. Furthermore, logistic regression of CKD was performed on SNP dosage levels adjusting for sex and age. For all traits, adjustment for appropriate study-specific features, such as study site and genetic principal components, was included in the regression and family-based studies appropriately accounted for relatedness. There was no overlap between the exposure sample size and outcome sample size.
Statistics
We combined the effect of instruments using the inverse variance weighted (IVW) method as implemented in the TwoSampleMR package running under R. Heterogeneity was assessed using Q value for IVW. To address the potential effect of pleiotropic variants on the final effect estimate, we performed sensitivity analysis including weighted median (WM) and MR-Egger. Sensitivity analysis was conducted using the leave-one-out method to identify instruments that might drive the MR results. The WM estimate provides correct estimates as long as SNPs accounting for ≥ 50% of the weight are valid instruments. Inverse variance is used to weight the variants, and bootstrapping is applied to estimate the CIs [35]. MR-Egger is able to make estimates even under the assumption that all SNPs are invalid instruments, as long as the assumption of instrument strength independent of direct effect (InSIDE) is satisfied [35]. However, the InSIDE assumption cannot be easily verified. Average directional pleiotropy across genetic variants was assessed from the p value of the intercept term from MR-Egger [35]. Causal estimates in MR-Egger are less precise than those obtained by using IVW MR [38]. Analysis using MR-Egger has a lower false-positive rate, but a higher false-negative rate, than IVW, i.e. it has a lower statistical power [39].
Heterogeneity between individual genetic variant estimates was assessed by the use of the Q′ heterogeneity statistic [40]. The Q′ statistic uses modified 2nd-order weights that are a derivation of a Taylor series expansion, taking into account the uncertainty in both numerator and denominator of the instrumental variable ratio [40].
To assess the instrumental variable analysis “exclusion-restriction” assumption, we used Ensembl release (http://useast.ensembl.org/index.html) that contains a base of SNP phenotypes and PhenoScanner (Ensembl gives SNP phenotypes, PhenoScanner also gives phenotypes of correlated SNPs.).
Sensitivity analysis
As sensitivity analysis, we used MR-Egger and MR pleiotropy residual sum and outlier (MR-PRESSO) test [40]. MR-Egger and MR-PRESSO may provide correct estimates as long as the instrument strength independent of direct effect assumption is satisfied [40]. MR-Egger can be imprecise, particularly if the associations for SNPs on exposure are similar, or the number of genetic instruments is low [40]. A non-null MR-Egger intercept suggests that the IVW estimate is invalid. MR-Egger does not explicitly identify outliers. MR-PRESSO detects, and if necessary, corrects for potentially pleiotropic outliers [40]. The MR-PRESSO framework detects effect estimates that are outliers and removes them from the analysis by regressing the variant-outcome associations on variant-exposure associations. A global heterogeneity test is then implemented to compare the observed distance between residual sums of squares of all variants to the regression line with the distance expected under the null hypothesis of no pleiotropy [41]. Furthermore, MR-Robust Adjusted Profile Score (RAPS) was applied. This method can correct for pleiotropy using robust-adjusted profile scores. We consider as results causal estimates that agreed in direction and magnitude across MR methods, passed nominal significance in IVW MR, and did not show evidence of bias from horizontal pleiotropy using heterogeneity tests. All analyses were done using the R software (version 3.4.2 R Core Team, 2017).
Ethics
This investigation uses published or publicly available summary data. No original data were collected for this manuscript. Ethical approval for each of the studies included in the present analysis can be found in the original publications (including informed consent from each participant). The study conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
Results
Demographic characteristics of the study participants are shown in Online Resource 1. The genetic instruments and observed phenotypes are shown in Online Resource 2, and the instrument associations for blood lead levels are shown in Online Resource 3. The instruments have F-statistics higher than threshold, making significant bias from use of weak instruments unlikely [42]. The results, expressed as beta-coefficient for blood lead per 1 standard deviation (SD) increase in outcomes, are presented in Table 1.
Genetically higher blood lead levels had no significant effect on risk of CKD (IVW = Beta: 0.02677, p = 0.652: Table 1; Fig. 1) or level of eGFR (IVW = Beta: −0.001514, p = 0.668, Table 1) in this sample. After grouping subjects based on type 2 diabetes status (no type 2 diabetes vs. type 2 diabetes), genetically determined levels of blood lead had no significant impact on subjects without type 2 diabetes (IVW = Beta: 0.0008706, p = 0.823: Table 1; Fig. 2). However, in subjects with type 2 diabetes, a significant effect on eGFR was observed (IVW = Beta: − 0.03416, p = 0.0132: Table 1; Fig. 3).

Forest plot of overall and individual SNP effects on CKD. SNP single nucleotide polymorphism; CKD chronic kidney disease

Scatter plot of the association of the effect of SNP-determined blood lead on eGFR in individuals without T2DM. Each black point represents an SNP, plotted by the estimate of SNP on blood lead level (x-axis, nmol/L) and the estimate of SNP on eGFR (y-axis, mL/min). The slopes of each line represent the potential causal associations for each method. SNP single nucleotide polymorphism; T2DM type 2 diabetes mellitus; eGFR estimated glomerular filtration rate

Scatter plot of the association of the effect of SNP-determined blood lead on eGFR in individuals with T2DM. Each black point represents an SNP, plotted by the estimate of SNP on blood lead level (x-axis, nmol/L) and the estimate of SNP on eGFR (y-axis, mL/min). The slopes of each line represent the potential causal associations for each method. SNP single nucleotide polymorphism; T2DM type 2 diabetes mellitus; eGFR estimated glomerular filtration rate
Heterogeneity results and pleiotropy bias are also shown in Table 1. Estimation based on both MR Egger and IVW was higher than 0.05, which indicated no chance of heterogeneity (all IVW p > 0.158, all MR Egger p > 0.175). Further, the results of the MR-PRESSO did not indicate any outliers for all the estimates. The horizontal pleiotropy test, with very negligible Egger regression intercept, also indicated a low likelihood of pleiotropy for all of our estimations (all p > 0.139). The results of the MR-RAPS were identical with the IVW estimates, highlighting again a low likelihood of pleiotropy. The results of the leave-one-out method demonstrated that the links were not driven by single SNPs.
Discussion
In this study, we have analysed a set of genetic variants that were demonstrated to be associated with blood lead levels in order to determine their relationship with renal function. Mendelian randomisation analyses showed that higher blood lead might be linked with less favourable renal function but only amongst individuals with type 2 diabetes.
Lead is commonly used for industrial purposes, and chronic exposure to lead, either through industrial or environmental means, has been responsible for numerous cases of lead toxicity or plumbism [43,44,45]. Concerns over the toxicity of lead have led to the phasing out of some of its use in industry and consumer goods [46,47,48]. In particular, lead in petrol and paint is believed to have been one of the principle contributors to increased blood lead levels in humans and was phased out of use in the USA from the late 1970s [49].
While the relation between lead exposure and CVD is well established [50, 51], the role of blood lead levels in the development of CKD and reduced renal function remains controversial. Indeed, cross-sectional studies of lead-exposed workers often report changes in markers of kidney function, such as increases in creatinine clearance, without clinically significant reductions in eGFR or diagnosis of renal failure [28, 29, 52, 53]. In a sample of 803 Korean lead workers, blood lead levels were significantly associated with increased uric acid (UA) levels (which is known to be nephrotoxic) in the oldest tertile of workers with serum creatinine greater than the median [28]. Similarly, in a sample of 229 Chinese lead battery factory workers, there was an increasing trend in the dose–response relationship between blood lead levels and indicators of renal function of blood-urea nitrogen (BUN) and UA [29]. However, only those with longer periods of occupational lead exposure had a higher possibility of reduced renal function. Cardenas et al. [52] compared data from 50 Belgian, lead-exposed workers with age-matched controls and reported no indication of significantly increased proteinuria in those exposed to lead. However, blood lead was associated with altered urinary excretion of 6-keto-PGF and thromboxane, eicosanoids which may contribute to the pathologies involved in renal failure and hypertension [54]. Pollock and Ibels [53] presented a case study of 6 men exposed to lead from paint in Australia and suffering from lead intoxication. While some measures related to renal function, such as serum uric acid, urinary protein and creatinine clearance, were abnormal in some cases, these were not consistently observed in the majority of the cases presented. Thus, it can be seen that while lead exposure may have effects on renal-related parameters, a conclusive relationship between lead and CKD in otherwise healthy populations cannot be drawn. Furthermore, such cross-sectional data is not sufficient to determine a causal relationship between lead exposure and CKD, and thus, sufficiently controlled, longitudinal studies as well as mechanistic evidence for a causal effect would be needed. However, the use of MR analysis can overcome the limitations of observational studies as MR is a powerful tool for the detection of causation [34]. As such, the results of this study provide evidence that small, life-long changes in genetically determined blood lead do not impact the development of CKD in individuals without type 2 diabetes.
As such, our study did find an association between genetically determined blood lead and decreased eGFR in those presenting with type 2 diabetes. Renal tubule damage is a common feature of type 2 diabetes and is considered to be a pathway to glomerular dysfunction associated with proteinuria and the development of CKD in those with type 2 diabetes [55]. It could be speculated that the nephrotoxic effects of substances such as UA, which are elevated in lead-exposed individuals, might contribute to the development of diminished kidney function in those already experiencing renal tubule damage due to type 2 diabetes [28, 29]. This might explain why high blood lead is only seen to contribute to CKD in those with type 2 diabetes, i.e. those with pre-existing damage to renal tubules.
We believe this to be the first study to report that there is a relationship between genetically determined blood lead levels and reduced eGFR in individuals with diabetes. Indeed, diabetes is a frequent comorbidity in CKD and is believed to contribute to the development of impaired renal function [7, 20]. It has been observed that individuals with earlier onset type 2 diabetes, and consequently longer duration of diabetes, have a 2.6-fold higher risk of CKD, compared to those with later-onset diabetes [56]. Lead is known to contribute to oxidative stress in those exposed to high levels [57, 58], and more specifically, lead has been reported to lead to oxidative stress and apoptosis in in vitro human mesangial cells which may be a possible mechanism for lead-induced nephrotoxicity [59]. Similarly, lead exposure is known to affect the immune system resulting in altered cytokine metabolism and a proinflammatory response [60]. We propose that as the diabetic state is associated with metabolic derangement such as elevated oxidative stress [61] as well as elevated levels of proinflammatory cytokines [62, 63] and renal tubule damage [55], lead exposure may accelerate and augment these detrimental processes (which may not be present in those without diabetes) and lead to renal dysfunction more readily in subjects with type 2 diabetes. Further research is needed to investigate the mechanisms of the blood lead–related renal dysfunction amongst those with diabetes.
A major strength of our study is the large sample population with access to individual participant data of high validity for eGFR and CKD status, and with the relevant SNPs available for blood lead concentration. Additionally, the use of MR methods allows us to examine the potential causal effects of blood lead, largely without the disadvantages of confounding or reverse causation. We checked for known pleiotropy using Ensembl and found few known phenotypes of the genetic predictors of blood lead apart from multiple associations for rs550057 (ABO) (Online Resource 2). A potential limitation of this study is the use of a predominantly white, ethnically European population which limits the generalizability of the results. As such, ethnically diverse GWAS and MR studies are necessary to generalize MR results to people of different ancestries. Furthermore, while this MR analysis provides evidence on the effect of smaller life-long, genetically determined blood lead levels, it may not necessarily apply to short-term larger changes in blood lead, due to environmental factors. Another potential concern with MR analysis is the risk of stratification bias, which would only be an issue if type 2 diabetes resulted from both elevated blood lead levels and the presence of CKD. Finally, due to the limited number of shared SNPs identified by both the QIMR and ALSPAC studies (n = 3) (Online Resources 4 and 5), it is not possible to perform a sensitivity analysis to determine differences between the results of both datasets. As such, future research should endeavour to perform such sensitivity analyses, as sufficient data on relevant SNPs becomes available.
In conclusion, this investigation found evidence to support a potential causal association between genetically determined blood lead levels on renal function in individuals with type 2 diabetes. However, in subjects without diabetes, no such causal relationship was identified. While further investigation is required to investigate the link between lead exposure and indices of renal function in those with diabetes, this novel data also contributes to the current understanding that the relationship between lead exposure and CKD in non-diabetic individuals may simply be associative.
Availability of data and material
The datasets analysed in this study are publicly available summary statistics.
References
Eckardt KU, Coresh J, Devuyst O et al (2013) Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382(9887):158–169. https://doi.org/10.1016/s0140-6736(13)60439-0
Hill NR, Fatoba ST, Oke JL et al (2016) Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS ONE 11(7):e0158765. https://doi.org/10.1371/journal.pone.0158765
Levey AS, Eckardt KU, Tsukamoto Y et al (2005) Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 67(6):2089–2100. https://doi.org/10.1111/j.1523-1755.2005.00365.x
Gansevoort RT, Correa-Rotter R, Hemmelgarn BR et al (2013) Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 382(9889):339–352. https://doi.org/10.1016/s0140-6736(13)60595-4
Garofalo C, Borrelli S, Minutolo R, Chiodini P, De Nicola L, Conte G (2017) A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int 91(5):1224–1235. https://doi.org/10.1016/j.kint.2016.12.013
Herzog CA, Asinger RW, Berger AK et al (2011) Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 80(6): 572–586. https://doi.org/10.1038/ki.2011.223
Iwai T, Miyazaki M, Yamada G et al (2018) Diabetes mellitus as a cause or comorbidity of chronic kidney disease and its outcomes: the Gonryo study. Clin Exp Nephrol 22(2):328–336. https://doi.org/10.1007/s10157-017-1451-4
Kim YJ, Hwang SD, Oh TJ et al (2017) Association between obesity and chronic kidney disease, defined by both glomerular filtration rate and albuminuria. Korean adults Metab Syndr Relat Disord 15(8):416–422. https://doi.org/10.1089/met.2017.0053
Kramer HJ, Nguyen QD, Curhan G, Hsu CY (2003) Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. JAMA 289(24):3273–3277. https://doi.org/10.1001/jama.289.24.3273
Matsushita K, van der Velde M, Astor BC et al (2010) Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 375(9731):2073–2081. https://doi.org/10.1016/s0140-6736(10)60674-5
Pugliese G, Penno G, Natali A et al (2020) Diabetic kidney disease: new clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on "The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function". J Nephrol 33(1): 9–35. https://doi.org/10.1007/s40620-019-00650-x
Rao MV, Qiu Y, Wang C, Bakris G (2008) Hypertension and CKD: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES), 1999–2004. Am J Kidney Dis 51(4 Suppl 2):S30-37. https://doi.org/10.1053/j.ajkd.2007.12.012
Seccia TM, Caroccia B, Calo LA (2017) Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J Hypertens 35(2): 205–212. https://doi.org/10.1097/hjh.0000000000001170
Wu CK, Yang CY, Tsai CT et al (2010) Association of low glomerular filtration rate and albuminuria with peripheral arterial disease: the National Health and Nutrition Examination Survey, 1999–2004. Atherosclerosis 209(1):230–234. https://doi.org/10.1016/j.atherosclerosis.2009.08.038
Cornier MA, Dabelea D, Hernandez TL et al (2008) The metabolic syndrome. Endocr Rev 29(7):777–822. https://doi.org/10.1210/er.2008-0024
Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD (2011) Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol 6(10):2364–2373. https://doi.org/10.2215/cjn.02180311
Rashidbeygi E, Safabakhsh M, Delshad Aghdam S, Mohammed SH, Alizadeh S (2019) Metabolic syndrome and its components are related to a higher risk for albuminuria and proteinuria: Evidence from a meta-analysis on 10,603,067 subjects from 57 studies. Diabetes Metab Syndr 13(1):830–843. https://doi.org/10.1016/j.dsx.2018.12.006
Beltrán-Sánchez H, Harhay MO, Harhay MM, McElligott S (2013) Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J Am Coll Cardiol 62(8): 697–703. https://doi.org/10.1016/j.jacc.2013.05.064
O’Neill S, O’Driscoll L (2015) Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obesity reviews : an official journal of the International Association for the Study of Obesity 16(1):1–12. https://doi.org/10.1111/obr.12229
Lim A (2014) Diabetic nephropathy - complications and treatment. Int J Nephrol Renovasc Dis 7:361–381. https://doi.org/10.2147/ijnrd.S40172
Lin JL, Lin-Tan DT, Yu CC, Li YJ, Huang YY, Li KL (2006) Environmental exposure to lead and progressive diabetic nephropathy in patients with type II diabetes. Kidney Int 69(11):2049–2056. https://doi.org/10.1038/sj.ki.5001505
Rodríguez-Monforte M, Sánchez E, Barrio F, Costa B, Flores-Mateo G (2017) Metabolic syndrome and dietary patterns: a systematic review and meta-analysis of observational studies. Eur J Nutr 56(3):925–947. https://doi.org/10.1007/s00394-016-1305-y
Garralda-Del-Villar M, Carlos-Chillerón S, Diaz-Gutierrez J et al (2018) Healthy lifestyle and incidence of metabolic syndrome in the SUN cohort. Nutrients 11(1). https://doi.org/10.3390/nu11010065
Hu EA, Steffen LM, Grams ME et al (2019) Dietary patterns and risk of incident chronic kidney disease: the Atherosclerosis Risk in Communities study. Am J Clin Nutr 110(3):713–721. https://doi.org/10.1093/ajcn/nqz146
Lee J, Walker ME, Gabriel KP, Vasan RS, Xanthakis V (2020) Associations of accelerometer-measured physical activity and sedentary time with chronic kidney disease: The Framingham Heart Study. PLoS ONE 15(6):e0234825. https://doi.org/10.1371/journal.pone.0234825
Spector JT, Navas-Acien A, Fadrowski J, Guallar E, Jaar B, Weaver VM (2011) Associations of blood lead with estimated glomerular filtration rate using MDRD, CKD-EPI and serum cystatin C-based equations. Nephrol Dial Transplant 26(9):2786–2792. https://doi.org/10.1093/ndt/gfq773
Harari F, Sallsten G, Christensson A et al (2018) Blood lead levels and decreased kidney function in a population-based cohort. Am J Kidney Dis 72(3):381–389. https://doi.org/10.1053/j.ajkd.2018.02.358
Weaver VM, Jaar BG, Schwartz BS et al (2005) Associations among lead dose biomarkers, uric acid, and renal function in Korean lead workers. Environ Health Perspect 113(1):36–42. https://doi.org/10.1289/ehp.7317
Wang VS, Lee MT, Chiou JY et al (2002) Relationship between blood lead levels and renal function in lead battery workers. Int Arch Occup Environ Health 75(8):569–575. https://doi.org/10.1007/s00420-002-0362-0
Buchet JP, Roels H, Bernard A, Lauwerys R (1980) Assessment of renal function of workers exposed to inorganic lead, calcium or mercury vapor. J Occup Med 22(11):741–750
Omae K, Sakurai H, Higashi T, Muto T, Ichikawa M, Sasaki N (1990) No adverse effects of lead on renal function in lead-exposed workers. Ind Health 28(2):77–83. https://doi.org/10.2486/indhealth.28.77
Roels H, Lauwerys R, Konings J et al (1994) Renal function and hyperfiltration capacity in lead smelter workers with high bone lead. Occup Environ Med 51(8):505–512. https://doi.org/10.1136/oem.51.8.505
Plotnikov D, Guggenheim JA (2019) Mendelian randomisation and the goal of inferring causation from observational studies in the vision sciences. Ophthalmic Physiol Opt 39(1):11–25. https://doi.org/10.1111/opo.12596
Smith GD, Ebrahim S (2003) “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32(1):1–22. https://doi.org/10.1093/ije/dyg070
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40(4):304–314. https://doi.org/10.1002/gepi.21965
Warrington NM, Zhu G, Dy V et al (2015) Genome-wide association study of blood lead shows multiple associations near ALAD. Hum Mol Genet 24(13):3871–3879. https://doi.org/10.1093/hmg/ddv112
Pattaro C, Teumer A, Gorski M et al (2016) Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat Commun 7:10023. https://doi.org/10.1038/ncomms10023
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44(2):512–525. https://doi.org/10.1093/ije/dyv080
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG (2017) Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 28(1):30–42. https://doi.org/10.1097/ede.0000000000000559
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J (2017) A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med 36(11):1783–1802. https://doi.org/10.1002/sim.7221
Verbanck M, Chen CY, Neale B, Do R (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 50(5):693–698. https://doi.org/10.1038/s41588-018-0099-7
Palmer TM, Lawlor DA, Harbord RM et al (2012) Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 21(3):223–242. https://doi.org/10.1177/0962280210394459
Bernard BP, Becker CE (1988) Environmental lead exposure and the kidney. J Toxicol Clin Toxicol 26(1–2):1–34. https://doi.org/10.3109/15563658808995395
Rosen JF (1995) Adverse health effects of lead at low exposure levels: trends in the management of childhood lead poisoning. Toxicology 97(1–3):11–17. https://doi.org/10.1016/0300-483x(94)02963-u
Goyer RA (1993) Lead toxicity: current concerns. Environ Health Perspect 100:177–187. https://doi.org/10.1289/ehp.93100177
Filella M, Bonet J (2017) Environmental impact of alkyl lead(IV) derivatives: Perspective after Their Phase-out Met Ions Life Sci 17. https://doi.org/10.1515/9783110434330-014
Intrakamhaeng V, Clavier KA, Townsend TG (2019) Initiatives to reduce lead from electronic devices: evidence of success from the toxicity characteristic leaching procedure. J Air Waste Manag Assoc 69(9):1116–1121. https://doi.org/10.1080/10962247.2019.1640807
Thomas VG, Kanstrup N, Fox AD (2019) The transition to non-lead sporting ammunition and fishing weights: review of progress and barriers to implementation. Ambio 48(9):925–934. https://doi.org/10.1007/s13280-018-1132-x
Juberg DR, Kleiman CF, Kwon SC (1997) Position paper of the American Council on Science and Health: lead and human health. Ecotoxicol Environ Saf 38(3):162–180. https://doi.org/10.1006/eesa.1997.1591
Navas-Acien A, Guallar E, Silbergeld EK, Rothenberg SJ (2007) Lead exposure and cardiovascular disease–a systematic review. Environ Health Perspect 115(3):472–482. https://doi.org/10.1289/ehp.9785
Vaziri ND (2008) Mechanisms of lead-induced hypertension and cardiovascular disease. Am J Physiol Heart Circ Physiol 295(2):H454-465. https://doi.org/10.1152/ajpheart.00158.2008
Cárdenas A, Roels H, Bernard AM et al (1993) Markers of early renal changes induced by industrial pollutants. II. Application to workers exposed to lead. Br J Ind Med 50(1): 28–36. https://doi.org/10.1136/oem.50.1.28
Pollock CA, Ibels LS (1986) Lead intoxication in paint removal workers on the Sydney Harbour Bridge. Med J Aust 145(11–12):635–639
Stork JE, Rahman MA, Dunn MJ (1986) Eicosanoids in experimental and human renal disease. Am J Med 80(1a):34–45. https://doi.org/10.1016/0002-9343(86)90930-7
Thomas MC, Burns WC, Cooper ME (2005) Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis 12(2):177–186. https://doi.org/10.1053/j.ackd.2005.01.008
Liu JJ, Liu S, Gurung RL et al (2020) Risk of progressive chronic kidney disease in individuals with early-onset type 2 diabetes: a prospective cohort study. Nephrol Dial Transplant 35(1):115–121. https://doi.org/10.1093/ndt/gfy211
Lopes AC, Peixe TS, Mesas AE, Paoliello MM (2016) Lead exposure and oxidative stress: a systematic review. Rev Environ Contam Toxicol 236:193–238. https://doi.org/10.1007/978-3-319-20013-2_3
Dobrakowski M, Pawlas N, Kasperczyk A et al (2017) Oxidative DNA damage and oxidative stress in lead-exposed workers. Hum Exp Toxicol 36(7):744–754. https://doi.org/10.1177/0960327116665674
Jia Q, Ha X, Yang Z, Hui L, Yang X (2012) Oxidative stress: a possible mechanism for lead-induced apoptosis and nephrotoxicity. Toxicol Mech Methods 22(9):705–710. https://doi.org/10.3109/15376516.2012.718811
Metryka E, Chibowska K, Gutowska I et al (2018) Lead (Pb) exposure enhances expression of factors associated with inflammation. Int J Mol Sci 19(6). https://doi.org/10.3390/ijms19061813
Srivastava SK, Ramana KV, Bhatnagar A (2005) Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev 26(3):380–392. https://doi.org/10.1210/er.2004-0028
Lim AK, Tesch GH (2012) Inflammation in diabetic nephropathy. Mediators Inflamm 2012:146154. https://doi.org/10.1155/2012/146154
Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH (2007) Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 50(2):471–480. https://doi.org/10.1007/s00125-006-0497-8
Acknowledgements
We thank Professor Lakshminarayan Ranganath (University of Liverpool) for his invaluable comments on the manuscript.
Funding
This work was partially supported by the Institute for Health Research at Liverpool John Moores University via a studentship for RK.
Author information
Authors and Affiliations
Contributions
MM designed the study, acquired the data and performed the analyses. MM, RK and IGD interpreted the findings and drafted the manuscript, and all the authors contributed to critical reading and revision of the draft report. All the authors approved the final version to be published.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
RK has received speaker’s honoraria from the British Association for Parenteral and Enteral Nutrition and fees for media content creation from Myprotein. MM and IGD declare that they have no conflict of interest.
Disclaimer
The study funder was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mazidi, M., Kirwan, R. & Davies, I.G. Genetically determined blood lead is associated with reduced renal function amongst individuals with type 2 diabetes mellitus: insight from Mendelian Randomisation. J Mol Med 100, 125–134 (2022). https://doi.org/10.1007/s00109-021-02152-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02152-5