
Abstract
Aims/hypothesis
Tissue macrophage accumulation is thought to induce insulin resistance during obesity and stimulate the progression of diabetic nephropathy. Monocyte chemoattractant protein-1 (MCP-1) is a potent stimulator of macrophage recruitment. It is increased in adipose tissue during obesity and in diabetic kidneys, suggesting that inflammation of these tissues may be MCP-1-dependent. Based on these findings, the aim of this study was to examine whether a deficiency in MCP-1 would alter the development of type 2 diabetes and its renal complications.
Materials and methods
The role of MCP-1 in the progression of type 2 diabetes and its associated renal injury was assessed in obese db/db mice that were deficient in the gene encoding MCP-1 (Ccl2).
Results
The incidence and development of type 2 diabetes were similar in Ccl2 +/+ and Ccl2 −/− db/db mice between 8 and 32 weeks of age. Body mass, hyperglycaemia, hyperinsulinaemia, glucose and insulin tolerance, plasma triacylglycerol and serum NEFA were not different between these strains. Pathological changes in epididymal adipose tissue, including increases in macrophage accumulation and Tnfa mRNA and reductions in Adipoq mRNA, were unaffected by the absence of MCP-1. In contrast, kidney macrophage accumulation and the progression of diabetic renal injury (albuminuria, histopathology, renal fibrosis) were substantially reduced in Ccl2 −/− compared with Ccl2 +/+ db/db mice with equivalent diabetes.
Conclusions/interpretation
Our study demonstrates that MCP-1 promotes type 2 diabetic renal injury but does not influence the development of obesity, insulin resistance or type 2 diabetes in db/db mice. MCP-1 plays a critical role in inflammation of the kidney, but not adipose tissue, during the progression of type 2 diabetes.
Similar content being viewed by others


Introduction
Recent studies have implicated tissue macrophage accumulation in the development of obesity-related insulin resistance [1, 2] and type 2 diabetic nephropathy [3, 4]. Macrophage accrual in visceral adipose tissue is a feature of chronic obesity which is associated with the onset of insulin resistance [1, 2]. Indeed, animal studies have shown a correlation between macrophage numbers in epididymal fat and obesity, adipocyte size and reduced insulin sensitivity [1, 5]. Current perception is that macrophages may interact with adipocytes, resulting in the secretion of factors that act in a paracrine or endocrine manner to promote insulin resistance in adipose tissue, liver and muscle. During obesity, macrophages are the major source of TNF-α in adipose tissue, and TNF-α is known to reduce insulin signalling and insulin-responsive glucose transport in adipocytes [6, 7]. Macrophages in diabetic animals also produce nitric oxide [4], which can promote insulin resistance in muscle [8]. Furthermore, mice lacking IκB kinase-β in myeloid cells are protected against systemic insulin resistance induced by obesity, suggesting that activation of nuclear factor-κβ in adipose macrophages may contribute to the induction of insulin resistance [9]. In diabetic kidneys, the leucocytic infiltrate consists almost exclusively of macrophages and their accrual correlates with renal injury [4, 10]. In vitro studies have shown that elements of the diabetic milieu, such as hyperglycaemia and advanced glycation end-products, can stimulate macrophages to secrete substances which cause injury, cell proliferation or induce fibrotic responses in kidney cells [10, 11]. Furthermore, additional elements characteristic of type 2 diabetes, including hyperinsulinaemia, hyperlipidaemia and elevated levels of circulating adipokines, may also influence inflammatory responses in the kidney. Therefore, macrophages may be important promoters of both type 2 diabetes and its complications.
Monocyte chemoattractant protein-1 (MCP-1) is a chemokine that is known to affect the accumulation and function of macrophages [12–14]. MCP-1 levels are increased in plasma and adipose tissue in mouse models of obesity [15, 16], and correlate with the number of CD11b+ monocytes in these animals [16]. Primary cultures of human preadipocytes constitutively produce MCP-1 but these levels are reduced upon maturation [17]. Stimulation with TNF-α or insulin increases expression of the gene encoding MCP-1 (Ccl2) in adipocytes differentiated in vitro [15, 17]. Addition of MCP-1 to differentiated adipocytes in vitro decreases insulin-stimulated glucose uptake and the expression of several adipogenic genes, suggesting that MCP–1 may also affect adipocyte function [17]. In diabetic kidneys, Ccl2 expression increases with macrophage accumulation and disease progression [4] and appears to be dependent on both the diabetic milieu [18] and activation of the renin–angiotensin system [19]. In addition, urine MCP-1 levels correlate with albuminuria during human diabetic nephropathy [18, 20]. Therefore, MCP-1 may play a key role in the development of both type 2 diabetes and its associated nephropathy.
Our study examined whether MCP-1 plays a role in adipose and renal inflammation during the development of type 2 diabetes. Ccl2 +/+ and Ccl2 −/− db/db mice were used to determine the functional importance of MCP-1 in the development of obesity, inflammation, glucose and insulin tolerance and hyperglycaemia. A selected subgroup of these mice with equivalent diabetes in both strains was used to establish the role of MCP-1 in diabetic renal injury.
Materials and methods
Animal model
Obese (db/db) and lean heterozygote control (db/+) mice were created from breeding pairs of C57BL/6 db/+ mice obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and were genotyped by PCR with restriction enzyme analysis for the mutated leptin receptor. Ccl2 −/− mice, created by targeted gene disruption [21], were backcrossed eight times onto the C57BL/6J strain. These Ccl2 −/− mice were then crossed with C57BL/6 db/+ mice to create db/+ Ccl2 −/− mice, which were validated by PCR-based genotyping. The latter were then interbred to create homozygous db/db Ccl2 −/− mice. Only male C57BL/6 db/db mice were used for experiments due to their higher incidence of diabetes [4]. Experimental db/db mice were maintained on a normal diet and were examined from the initial appearance of hyperglycaemia (8 weeks of age) until the development of overt diabetes and renal injury was evident in half of all wild-type males (32 weeks of age). Although these animals develop more slowly progressing diabetes compared with the hybrid C57BL/KsJ db/db strain [22], their genetic purity is maintained after intercrossing with C57BL/6 knock-out strains, which makes them a more appropriate strain for identifying the effects of molecular deficiencies on a defined genetic background [11].
The development of obesity and hyperglycaemia was examined in groups of Ccl2 +/+ and Ccl −/− db/db mice (n = 40–50) between 8 and 32 weeks of age. From each of these groups, 20 mice were randomly selected for assessment of glucose and insulin tolerance and fasting insulin levels at 8 and 32 weeks. Blood glucose levels were measured every 4 weeks by tail vein sampling using a Medisense glucometer (Abbott Laboratories, Bedford, MA, USA), and diabetes was defined as random morning blood glucose ≥16 mmol/l (300 mg/dl).
For assessment of adipose inflammation, groups of Ccl2 +/+ and Ccl2 −/− db/db mice (n = 10) were randomly selected and killed at 8 and 32 weeks to collect epididymal fat pads. For analysis of diabetic renal injury, subgroups of Ccl2 +/+ and Ccl2 −/− db/db mice (n = 10) with equivalent hyperglycaemia and obesity were selected and killed at 8 and 32 weeks. Groups of non-diabetic db/+ mice (n = 10) were killed at 8 and 32 weeks to obtain control tissue.
Approval for these animal studies was obtained from the Monash Medical Centre Animal Ethics Committee in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 7th edition (2004).
Genotyping
To genotype db/db mice, DNA was extracted from tails with proteinase K digestion [12]. PCR analysis of genomic DNA was performed to identify the intact MCP-1 gene (Ccl2) or the neomycin resistance cassette (Neo) used in the disruption of Ccl2. The Ccl2 PCR involved thermal cycling conditions of 94°C for 2 min, followed by 35 cycles of 94°C for 30 s, 55°C for 60 s and 72°C for 60 s. The Neo PCR used 35 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 120 s. The PCR primers used were: Ccl2 (5′ ACA GCT TCT TTG GGA CAC C 3′, 5′ GGA GCA TCC ACG TGT TGG C 3′); Neo (5′ATT GAA CAA GAT GGA TTG CAC 3′, 5′ CGT CCA GAT CAT CCT GAT C 3′). The resulting PCR products (Ccl2, 869 bp, Neo, 500 bp) were isolated by electrophoresis on a 2.2% agarose gel and detected with ethidium bromide (Sigma, St Louis, MO, USA).
Biochemical analysis
Urine was collected from mice housed in metabolic cages for 24 h at 8, 16, 24 and 32 weeks of age. At the end of experimentation, heparinised whole blood and serum were collected from anaesthetised mice after fasting (6 h) by cardiac puncture. HbA1c levels and plasma levels of creatinine (creatininase method), triacylglycerol and cholesterol were analysed by the Biochemistry Department at the Monash Medical Centre. ELISA kits were used to measure plasma insulin (Linco Research, St Charles, MO, USA), urine albumin (Bethyl Laboratories, Montgomery, TX, USA) and urine MCP-1 (R&D Systems, Minneapolis, MN, USA). Serum NEFA were assessed with a colorimetric assay kit (Wako Pure Chemical Industries, Osaka, Japan).
Glucose and insulin tolerance tests
To determine glucose tolerance, mice were injected intraperitoneally with d-glucose (1 g/kg) after a 12 h fast. For examining insulin tolerance, mice were injected intraperitoneally with human insulin (2 U/kg; Actrapid, Novo Nordisk, Bagsvaerd, Denmark) after a 6 h fast. For both tests, blood samples were collected at 0, 30, 60, 90 and 120 min and blood glucose was measured using the glucose oxidase method.
Real-time RT-PCR analysis
RNA was extracted from snap-frozen epididymal fat and whole kidney using a RiboPure RNA isolation kit (Ambion, Austin, TX, USA). cDNA was prepared from total RNA by reverse transcription using random hexamer primers and Superscript II (Invitrogen, Carlsbad, CA, USA).
Quantitative real-time RT-PCR was performed using primers and 6-carboxyfluorescein (FAM)-labelled minor groove binder (MGB) probes for mouse Ccl2, Tnfa and Adipoq. In each sample, ribosomal 18S was amplified using a commercial kit containing a VIC-labelled probe plus primers (Applied Biosystems, Foster City, CA, USA). PCR was performed over 50 cycles on a Rotor-Gene 3000 system (Corbett Research, Sydney, NSW, Australia) using a RealMastermix Probe kit (Eppendorf, Hamburg, Germany), UDG (New England Biolabs, Beverly, MA, USA) and VIC plus FAM-labelled primer/probe combinations. Each PCR involved thermal cycling conditions of 37°C for 10 min, 95°C for 5 min, followed by 50 cycles of 95°C for 15 s, 60°C for 20 s and 68°C for 20 s. The primer pairs and probes used were: Ccl2 (forward, 5′ GAC CCG TAA ATC TGA AGC TAA 3′; reverse, 5′ CAC ACT GGT CAC TCC TAC AGA A 3′; probe, 5′ ACA ACC ACC TCA AGC AC 3′); Tnfa (forward, 5′ GGC TGC CCC GAC TAC GT 3′; reverse, 5′ TTT CTC CTG GTA TGA GAT AGC AAA TC 3′; probe, 5′ TCA CCC ACA CCG TCA G 3′); and Adipoq (forward, 5′ CAT TCC GGG ACT CTA CTA CT 3′; reverse, 5′CTG ATA CTG GTC GTA GGT GAA G 3′; probe, 5′ GAA AGA TGT GAA GGT GA 3′). The relative abundance of each mRNA was calculated using the comparative Ct (ΔCt) method and normalised against 18S.
To verify deficiency of Ccl2 mRNA in Ccl2 −/− db/db mice, qualitative RT-PCR analysis was performed on cDNA samples prepared from the epididymal fat mRNA of Ccl2 +/+ and Ccl2 −/− db/db mice. Each PCR was run for 35 cycles using the same Ccl2 and 18S primers and thermocycling conditions as in real-time RT-PCR. The resulting PCR products (Ccl2, 79 bp; 18S, 100 bp) were isolated by electrophoresis on a 2.2% agarose gel and detected with ethidium bromide (Sigma).
Histopathology
Formalin-fixed kidney sections (2 μm) were stained with periodic acid–Schiff reagent to identify structure and haematoxylin to distinguish cell nuclei. Digital images of the tissue sections were obtained from microscopy (magnification ×400). Glomerular cellularity, glomerular volume and tubular atrophy were determined by image analysis as previously described [4]. All scoring was performed on blinded slides.
Antibodies
Antibodies used in this study were: rat anti-mouse CD68 (FA-11; Serotec, Oxford, UK); rat anti-mouse F4/80 (Serotec); rat anti-mouse CD169 (a macrophage activation marker [23]; Serotec); goat anti-mouse collagen IV (Santa Cruz Biotechnology, Santa Cruz, CA, USA); and fluorescein-conjugated anti-α-smooth muscle actin (1A4, Sigma). Normal rabbit and goat serum and isotype-matched irrelevant rat IgGs were used as negative controls.
Immunochemistry staining
Immunoperoxidase staining was used to detect expression of CD68, CD169 and collagen IV in 2% paraformaldehyde–lysine–periodate-fixed kidney cryostat sections and F4/80 and α-smooth muscle actin in formalin-fixed sections [24]. Activated macrophages were detected by two-colour immunostaining for CD68 and CD169 [24].
Quantitation of immunohistochemistry
Adipose macrophage accumulation, expressed as cells per mm2, was assessed by counting the number of F4/80+ macrophages in 20 fields (magnification ×250) in both epididymal fat pads of each mouse. Kidney macrophage accrual was assessed by immunostaining with CD68, because most glomerular macrophages are not detected with the F4/80 antibody [25]. Glomerular macrophage accumulation was determined by counting the number of immunostained CD68+ cells in 20 hilar glomerular tuft cross-sections per animal (magnification ×400). Immunostained interstitial macrophages, expressed as CD68+ cells per mm2, were counted in 25 consecutive interstitial fields (magnification ×250), representing 30–40% of kidney cortex in the cross-section. Expression of collagen IV and α-smooth muscle actin was assessed as the percentage area stained within the glomerular tuft or the cortical interstitium, using image analysis software (Image Pro Plus; Media Cybernetics, Silver Spring, MD, USA). All scoring was performed on blinded slides.
Statistical analysis
Statistical differences between two groups were analysed by the unpaired Student’s t test and differences between multiple groups were assessed by one-way ANOVA using Tukey’s multiple comparison test. Correlation analyses were performed using Pearson’s coefficient. Data were recorded as the means±SEM and values of p < 0.05 were considered significant. All analyses were performed using GraphPad Prism 4.0 (GraphPad Software, San Diego, CA, USA).
Results
MCP-1/Ccl2 expression is increased in obese db/db mice
Qualitative PCR analysis of tail-derived genomic DNA (Fig. 1a) and cDNA transcribed from epididymal fat mRNA (Fig. 1b) demonstrated that Ccl2 was absent in Ccl2 −/− db/db mice. Further analysis by real-time RT-PCR showed that Ccl2 mRNA was elevated six- to sevenfold in the epididymal fat of obese wild-type db/db mice at the onset of diabetes (8 weeks of age) compared with lean db/+ control mice, and this elevated Ccl2 expression was still evident in db/db mice at 32 weeks of age (Fig. 1c). Levels of Ccl2 mRNA in the epididymal fat of db/db mice at 8 and 32 weeks did not correlate with either hyperglycaemia (p = 0.44) or HbA1c level (p = 0.99). In contrast to fat levels, kidney Ccl2 mRNA levels were not different between db/+ and wild-type db/db mice at 8 weeks of age. However, at 32 weeks, db/db mice showed a sixfold increase in kidney Ccl2 mRNA compared with db/+ mice (Fig. 1d). Analysis of db/db mice at 8 and 32 weeks showed that kidney Ccl2 mRNA levels correlated with the levels of blood glucose (p = 0.007) and HbA1c (p = 0.007).

Ccl2 expression in db/db mice. a PCR analysis of genomic DNA obtained from tail samples demonstrates that Ccl2 is present in wild-type db/db mice (Ccl2, lanes 2 and 3) and is absent in Ccl2 −/− db/db mice (Ccl2, lanes 4 and 5). Additional PCR analysis of these DNA samples showed that Ccl2 −/− db/db mice express the neomycin resistance cassette used in the disruption of the Ccl2 gene (Neo, lanes 4 and 5). Lane 1 exhibits molecular mass markers (2,000, 1,200, 800, 400 bp). b Qualitative RT-PCR analysis of epididymal fat mRNA shows that a 79 bp fragment of Ccl2 cDNA is absent in Ccl2 −/− db/db mice but is present in wild-type db/db mice at 8 weeks of age. An additional PCR performed on the same reverse transcription products showed that a 100 bp fragment of 18S cDNA was expressed equally by the two strains. Further quantitation of Ccl2 mRNA levels was performed by real-time RT-PCR in (c) the epididymal white adipose tissue (WAT) and (d) the kidneys of db/+ mice (empty bars) and wild-type db/db mice (solid bars) at 8 and 32 weeks of age. Data are means ± SEM; n = 10. a p < 0.01 vs age-matched db/+ control mice
MCP-1 deficiency does not alter obesity or the incidence of diabetes in db/db mice Ccl2 +/+ and Ccl2 −/− db/db mice were equally obese between 16 and 32 weeks of age, compared with lean db/+ control mice (Fig. 2a). The epididymal fat mass of the two db/db strains was similar at 8 weeks and increased compared with db/+ control mice (Table 1). The development of overt diabetes (non-fasting blood glucose >16 mmol/l) first appeared in some obese db/db mice at 8 weeks (Fig. 2b) and was detected in half of all males at 32 weeks. Ccl2 +/+ and Ccl2 −/− db/db mice developed diabetes at the same rate and in similar proportions and showed similar impairment of glucose and insulin tolerance (Figs. 2b and 3; Table 1). There was also no difference in blood glucose levels, HbA1c, plasma insulin, plasma triacylglycerol, plasma cholesterol and serum NEFA in Ccl2 +/+ and Ccl −/− db/db mice at 8 and 32 weeks (Table 1).

Effect of MCP-1 on obesity and incidence of diabetes in db/db mice. Male Ccl2 +/+ and Ccl2 −/− db/db mice between 8 and 32 weeks of age were assessed every 4 weeks for a body weight (db/+ [circles, dotted line], Ccl2 +/+ db/db [triangles, solid line], Ccl2 −/− db/db [inverted triangles, solid line]) and b the percentage incidence of diabetes (Ccl2 +/+ db/db [solid line], Ccl2 −/− db/db [dotted line]). Data are means ± SEM; n = 40–50. a p < 0.001 vs age-matched db/+ control mice; b p < 0.001 vs age-matched Ccl2 +/+ db/db mice

Effect of MCP-1 on glucose and insulin tolerance in db/db mice. Male Ccl2 +/+ and Ccl2 −/− db/db mice were assessed at 8 and 32 weeks of age for a, b glucose tolerance and c, d insulin tolerance (db/+ [circles, dotted line], Ccl2 +/+ db/db [triangles, solid line], Ccl2 −/− db/db [inverted triangles, solid line]). Data are means ± SEM; n = 20
MCP-1 deficiency does not affect adipose inflammation in diabetic db/db mice
Adipose macrophages were rarely observed in db/+ mice (Fig. 4a). In contrast, macrophages were frequently detected in epididymal fat sections of db/db mice at 8 and 32 weeks (Fig. 4b-d), although their accrual did not correlate with Ccl2 mRNA levels in the epididymal fat (p = 0.63). Our examination found similar accumulation of adipose macrophages in Ccl2 +/+ and Ccl2 −/− db/db mice at both these time points (Fig. 4b–d). Compared with lean db/+ control mice, obese db/db mice had a fourfold increase in Tnfa mRNA in epididymal fat at 8 weeks, which became a fivefold increase at 32 weeks (Fig. 5a). Adipoq mRNA levels in epididymal fat were not different in db/+ and db/db mice at 8 weeks; however, there was a 66% reduction in Adipoq mRNA in db/db compared with db/+ mice at 32 weeks (Fig. 5b). These observed changes in Tnfa and Adipoq mRNA levels in db/db mice were not affected by the absence of MCP-1.

Macrophage accumulation in adipose tissue in db/db mice. Epididymal fat from male Ccl2 +/+ and Ccl2 −/− db/db mice and control db/+ mice was assessed for immunostaining of F4/80+ macrophages at 8 and 32 weeks of age. Macrophages (arrows) were rarely detected in a the adipose tissue of db/+ mice at 8 weeks of age. In contrast, many macrophages were detected in the adipose tissue of Ccl2 +/+(b) and Ccl2 −/− db/db (c) mice at 8 weeks. Quantification of immunostaining (d) showed no difference in macrophage accumulation in the epididymal fat of Ccl2 +/+ db/db mice (solid bars) and Ccl2 −/− db/db mice (chequered bars) at 8 and 32 weeks of age. However, adipose macrophage numbers were elevated in both db/db strains compared with age-matched db/+ mice (open bars). Magnification: a–c, ×400. Data are means ± SEM; n = 10. a p < 0.001 vs age-matched db/+ control mice. WAT white adipose tissue

Effect of MCP-1 on adipose tissue expression of inflammation-related cytokines in db/db mice. Real-time quantitative PCR was used to analyse levels of the mRNA transcripts of a Tnfa and b Adipoq in the epididymal fat of db/+ mice (open bars), Ccl2 +/+ db/db mice (solid bars) and Ccl2 −/− db/db mice (chequered bars) at 8 and 32 weeks of age. Data are means ± SEM; n = 10. a p < 0.001 vs age-matched db/+ control mice
MCP-1 deficiency reduces renal inflammation in diabetic db/db mice
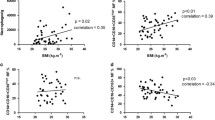
Renal inflammation and injury were assessed in selected groups of Ccl2 +/+ and Ccl2 −/− db/db mice with equivalent diabetes (Table 2). Kidney macrophage accumulation increased significantly in wild-type db/db mice following the development of diabetes and was pronounced at 32 weeks when kidney levels of MCP-1 were elevated (Figs. 6 and 7). Urine MCP-1 levels obtained at 32 weeks for wild-type db/+ (5 ± 1 pg/24 h) and db/db mice (39 ± 16 pg/24 h; p < 0.05 vs db/+) correlated with the number of glomerular macrophages (p = 0.005) and interstitial macrophages (p = 0.04) detected by CD68 immunostaining. Similarly, kidney Ccl2 mRNA levels detected in db/db mice at 8 and 32 weeks also correlated with the accrual of glomerular macrophages (p = 0.003) and interstitial macrophages (p = 0.04). Comparison of Ccl2 +/+ and Ccl2 −/− diabetic db/db mice showed significant differences in renal inflammation (Fig. 7). At 32 weeks, the numbers of total CD68+ macrophages and activated CD169+CD68+ macrophages were markedly reduced in the glomeruli and interstitium of Ccl2 −/− compared with Ccl2 +/+ diabetic kidneys (Fig. 7).

Macrophage accumulation and histopathology in diabetic db/db mice. Immunostaining detected only a small number of CD68+ macrophages in a a db/+ kidney at 32 weeks of age. In comparison, CD68+ macrophages were frequently observed in the glomeruli and interstitium of b a diabetic Ccl2 +/+ db/db kidney at 32 weeks of age. c The number of CD68+ macrophages was reduced in a diabetic Ccl2 −/− db/db kidney at the same age. Histological staining with periodic acid–Schiff reagent and haematoxylin shows normal kidney structure (d) in a non-diabetic db/+ mouse at 32 weeks. e In comparison, there is significant damage to glomeruli (hypertrophy, hypercellularity, mesangial periodic acid–Schiff deposits) and tubules (dilatation, atrophy; asterisks) in a diabetic db/db mouse at 32 weeks, which is attenuated in f an equally diabetic Ccl2 −/− db/db mouse at the same age. Magnification in a–f, ×400

Effect of MCP-1 on kidney macrophage accrual and activation in diabetic db/db mice. Immunostaining found similar numbers of CD68+ macrophages in a the glomeruli and b the interstitium of db/+ kidneys (circles, dotted line) and db/db kidneys at 8 weeks of age. In comparison, macrophage accumulation was markedly increased in the diabetic kidneys of Ccl2 +/+ db/db mice (triangles, solid line) at 32 weeks of age. Macrophage accumulation was reduced in Ccl2 −/− db/db mice (inverted triangles, solid line). An increased proportion of kidney macrophages in diabetic Ccl2 +/+ db/db mice (solid bars) was found to express CD169 in c the glomeruli and d the interstitium compared with non-diabetic db/+ control mice (open bars); however, this was significantly reduced in diabetic Ccl2 −/− db/db mice (chequered bars). Data are means ± SEM; n = 10. a p < 0.05, b p < 0.01, c p < 0.001 vs db/+; d p < 0.05, e p < 0.01 vs Ccl2 +/+ db/db mice. gcs glomerular cross-section
MCP-1 deficiency decreases kidney damage in diabetic db/db mice
Urine albumin excretion (UAE) was normal in non-diabetic db/+ and diabetic db/db mice at 8 weeks of age (Fig. 8). Between 16 and 32 weeks, UAE increased progressively in wild-type diabetic db/db mice and was 16-fold greater than in non-diabetic control mice at 32 weeks (Fig. 8). In comparison, UAE increased at a much slower rate in diabetic Ccl2 −/− db/db mice and was 60% lower than levels seen in wild-type diabetic db/db mice at 24 and 32 weeks (Fig. 8). At 32 weeks, plasma creatinine was increased twofold in wild-type db/db mice (45 ± 3.2 μmol/l) compared with db/+ mice (23.3 ± 1.0 μmol/l), indicating a decline in renal function. However, the increase in plasma creatinine observed in wild-type db/db mice was reduced by 65% in Ccl −/− db/db mice (30.9 ± 2.4 μmol/l) at the same age (p = 0.0019 vs wild-type db/db).

Effect of MCP-1 on renal injury in diabetic db/db mice. The rate of urine albumin excretion (UAE) was similar in lean db/+ and obese db/db mice at 8 weeks of age. In comparison, the UAE was increased 15- to 16-fold in diabetic Ccl2 +/+ db/db mice (triangles, solid line) at 24 and 32 weeks of age relative to db/+ control mice (circles, dotted line), and this was substantially reduced in diabetic Ccl2 −/− db/db mice (inverted triangles, solid line). Data are means ± SEM; n = 10. a p < 0.01, b p < 0.001 vs db/+; c p < 0.05, d p < 0.01 vs Ccl2 +/+ db/db mice
Analysis of kidney sections at 32 weeks identified glomerular hypertrophy, glomerular hypercellularity and increased glomerular immunostaining of collagen IV in wild-type diabetic db/db mice compared with db/+ control mice (Table 3). Tubular atrophy and tubulointerstitial immunostaining for α-smooth muscle actin and collagen IV were also increased in these wild-type diabetic db/db mice. In contrast, each of these histological markers of renal injury was significantly reduced in Ccl2 −/− compared with Ccl2 +/+ diabetic db/db mice (Table 3).
Discussion
Using a gene knock-out approach, our study demonstrates that the development of type 2 diabetes in db/db mice is independent of MCP-1. Deficiency of MCP-1 had no effect on the incidence of diabetes or the levels of obesity, hyperglycaemia, hyperinsulinaemia and hyperlipidaemia. In addition, the impairment of glucose and insulin tolerance observed during the early and advanced stages of disease were unaffected by the absence of MCP-1, suggesting that MCP-1 does not contribute to peripheral insulin resistance. In contrast, MCP-1 deficiency provided marked protection against the development of renal injury in diabetic db/db mice.
This study indicates that chronic inflammation in white adipose tissue is independent of MCP-1 in db/db mice. Levels of Ccl2 mRNA were increased five- to sixfold in the epididymal fat of wild-type db/db mice at 8 and 32 weeks compared with lean db/+ mice, and correlated with body weight and epididymal fat mass. This suggests that obesity induces MCP-1 production in visceral adipose tissue. However, at 8 and 32 weeks of age, epididymal fat from Ccl2 +/+ and Ccl2 −/− db/db mice had a similar increase in macrophage numbers and Tnfa mRNA and a similar decrease in Adipoq mRNA, suggesting equivalent inflammation. These findings are also consistent with the development of glucose and insulin tolerance being similar in the two strains. In comparison, a human study recently showed that pioglitazone treatment improved insulin sensitivity in subjects with impaired glucose tolerance in association with reductions in MCP-1 and macrophages in subcutaneous adipose tissue, suggesting that these events were related [26]. However, on the basis of our present findings, it now appears likely that pioglitazone may have affected insulin sensitivity, MCP-1 levels and macrophage accrual through different mechanisms.
Our data in db/db mice demonstrate that MCP-1 is redundant as a macrophage chemokine during adipose inflammation and that any direct functional effects of MCP-1 on adipocytes or adipose macrophage activation do not contribute to the development of type 2 diabetes. In comparison, two recent studies in a high-fat diet model of obesity have suggested that transgenic overexpression of Ccl2 in adipose tissue promotes adipose macrophage accumulation and insulin resistance [27, 28]. However, the aP2 promoter used in these experiments to drive transgenic Ccl2 expression is not specific for adipocytes and is also active in macrophages [29]. Since MCP-1 is known to activate macrophages [13, 14], it is entirely feasible that the increased recruitment of macrophages into adipose tissue in these studies is facilitated by secretion of MCP-1 and other chemokines/cytokines by constitutively activated macrophages. One of these studies also found that a single intramuscular treatment with a plasmid encoding a human mutant form of Ccl2 (7ND), which dimerises with wild-type MCP-1 and inhibits its activity, was sufficient to reduce insulin resistance in obese db/db mice [27]. Surprisingly, the effect of 7ND treatment on blood glucose levels and macrophage accumulation in adipose tissue was not examined in these mice. In addition, the selectivity of 7ND for the inhibition of MCP-1 compared with other related chemokines has yet to be established.
Studies of high-fat diet-induced obesity in mice deficient in chemokine CC motif receptor 2 (CCR2) are somewhat controversial, one study showing that these mice are unprotected from adipose inflammation and insulin resistance and another indicating protection [5, 30]. It is indeed possible that CCR2 deficiency may not duplicate the results of MCP-1 deficiency, because CCR2 is not an exclusive receptor for MCP-1, and other ligands capable of binding CCR2, such as MCP-3, are also increased in adipose tissue during obesity [30]. In addition, adipose levels of MCP-1 and macrophage expression of CCR2 may not be equivalent in different mouse models of obesity, which may lead to variations in the severity and mechanisms of inflammation in adipose tissue.
The finding that adipose inflammation in obese db/db mice is independent of MCP-1 suggests that other mechanisms are responsible for adipose macrophage accumulation. Additional macrophage chemokines, including macrophage inhibitory protein-1α, macrophage migration inhibitory factor and macrophage-colony stimulating factor (M-CSF), are produced by adipocytes and may be important for adipose macrophage recruitment and activation [17, 31, 32]. In addition, M-CSF is known to promote tissue macrophage accrual by inducing local proliferation [33], and this may be a contributing mechanism for macrophage accumulation in adipose tissue.
Examination of Ccl2 +/+ and Ccl2 −/− db/db mice with equivalent diabetes showed that kidney macrophage accumulation (CD68+ cells) and activation (CD68+CD169+ cells) were MCP-1-dependent. In addition, kidney and urine MCP-1 correlated with kidney macrophage accrual in the Ccl2 +/+ db/db mice, suggesting that inflammation in diabetic kidneys can be monitored clinically through urine MCP-1 measurement. These findings are supported by studies showing that hyperglycaemia and advanced glycation end-products stimulate MCP-1 production by kidney cells and that MCP-1 promotes renal inflammation during streptozotocin-induced type 1 diabetes [24].
Although both epididymal fat and the kidney produce elevated levels of Ccl2 in db/db mice, Ccl2 was only associated with inflammation in the kidney. Ccl2 was increased early in the epididymal fat of db/db mice, coinciding with the onset of obesity, but preceding the development of diabetes. In the epididymal fat of db/db mice, Ccl2 levels did not correlate with macrophage accumulation, suggesting that MCP-1 is not an important contributor to this event. In contrast, kidney Ccl2 levels in db/db mice did correlate with kidney macrophage accumulation and the development of diabetes and were found to be functionally important for the renal inflammation. These findings, along with other in vitro studies [24], suggest that MCP-1 production is induced by different mechanisms in epididymal fat (obesity) and the diabetic kidney (hyperglycaemia, advanced glycation end-products), and these different environments may influence whether or not MCP-1 promotes inflammation.
This study supports a role for MCP-1-dependent macrophage accumulation in promoting type 2 diabetic renal injury. A reduction in kidney macrophage accumulation and activation in diabetic Ccl2 −/− db/db mice was associated with a similar decline in glomerular pathology (hypertrophy, hypercellularity, and fibrosis) and interstitial pathology (tubular atrophy, myofibroblast accumulation and fibrosis). The significance of these effects was also detected clinically in terms of reductions in urine albumin excretion (62%) and plasma creatinine (65%) in diabetic Ccl2 −/− db/db mice. However, it should be noted that creatinine measurements in these obese mice are not a particularly reliable measure of renal function, since significant muscle wastage occurs during disease progression [11].
In summary, this study has demonstrated that MCP-1 promotes macrophage accumulation in kidneys but not in epididymal fat during the development of type 2 diabetes in db/db mice. Although MCP-1 had no impact on the physiological characteristics of type 2 diabetes, it played a significant role in the progression of diabetic renal injury. Therefore, neutralising MCP-1 activity should be viewed as an important therapeutic strategy in the treatment of type 2 diabetic nephropathy.
Abbreviations
- CCR2:
-
chemokine CC motif receptor 2
- M-CSF:
-
macrophage-colony stimulating factor
- MCP-1:
-
monocyte chemoattractant protein-1
- MCP-3:
-
monocyte chemoattractant protein-3
- UAE:
-
urine albumin excretion
References
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Furuta T, Saito T, Ootaka T et al (1993) The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis 21:480–485
Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH (2004) Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int 65:116–128
Weisberg SP, Hunter D, Huber R et al (2006) CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116:115–124
Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A (1993) Tumor necrosis factor-α suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 268:26055–26058
Stephens JM, Lee JL, Pilch PF (1997) Tumor necrosis factor-α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and glut4 expression without loss of insulin receptor-mediated signal transduction. J Biol Chem 272:971–976
Perreault M, Marette A (2001) Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7:1138–1143
Arkan MC, Hevener AL, Greten FR et al (2005) IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11:191–198
Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH (2004) Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant 19:2987–2996
Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH (2005) ICAM-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol 16:1711–1722
Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR (1999) MCP-1 dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Faslpr mice. J Exp Med 190:1813–1824
Biswas SK, Sodhi A, Paul S (2001) Regulation of nitric oxide production by murine peritoneal macrophages treated in vitro with chemokine monocyte chemoattractant protein 1. Nitric Oxide 5:566–579
Biswas SK, Sodhi A (2002) In vitro activation of murine peritoneal macrophages by monocyte chemoattractant protein-1: upregulation of CD11b, production of proinflammatory cytokines, and the signal transduction pathway. J Interferon Cytokine Res 22:527–538
Sartipy P, Loskutoff DJ (2003) Monocyte chemoattractant protein-1 in obesity and insulin resistance. Proc Natl Acad Sci USA 100:7265–7270
Takahashi K, Mizuarai S, Araki H et al (2003) Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. J Biol Chem 278:46654–46660
Gerhardt CC, Romero IA, Cancello R, Camoin L, Strosberg AD (2001) Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol Cell Endocrinol 175:81–92
Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K (2000) Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int 58:684–690
Amann B, Tinzmann R, Angelkort B (2003) ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care 26:2421–2425
Morii T, Fujita H, Narita T et al (2003) Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J Diabetes Complications 17:11–15
Lu B, Rutledge BJ, Gu L et al (1998) Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 187:601–608
Kumar S, McCue P, Dunn SR (2003) Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol 284:F1138–F1144
van den Berg TK, van Die I, de Lavalette CR et al (1996) Regulation of sialoadhesin expression on rat macrophages. Induction by glucocorticoids and enhancement by IFN-beta, IFN-gamma, IL-4 and lipopolysaccharide. J Immunol 157:3130–3138
Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollins BJ, Tesch GH (2006) Monocyte chemoattractant protein-1 promotes diabetic renal injury in streptozotocin-treated mice. Kidney Int 69:73–80
Masaki T, Chow F, Nikolic-Paterson DJ, Atkins RC, Tesch GH (2003) Heterogeneity of antigen expression explains controversy over glomerular macrophage accumulation in mouse glomerulonephritis. Nephrol Dial Transplant 18:178–181
Di Gregorio GB, Yao-Borengasser A, Rasouli N et al (2005) Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance and reduction by pioglitazone. Diabetes 54:2305–2313
Kanda H, Tateya S, Tamori Y et al (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494–1505
Kamei N, Tobe K, Suzuki R et al (2006) Overexpression of MCP-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem DOI 10.1074/jbc.M601284200
Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS (2005) The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J Biol Chem 280:12888–12895
Chen A, Mumick S, Zhang C et al (2005) Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes Res 13:1311–1320
Skurk T, Herder C, Kraft I, Muller-Scholze S, Haumer H, Kolb H (2005) Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology 146:1006–1011
Levine JA, Jensen MD, Eberhardt NL, O’Brien T (1998) Adipocyte macrophage-colony stimulating factor is a mediator of adipose tissue growth. J Clin Invest 101:1557–1564
Le Meur Y, Tesch GH, Hill PA et al (2002) Macrophage accumulation at a site of renal inflammation is dependent on the M-CSF/c-fms pathway. J Leuk Biol 72:530–537
Acknowledgements
This work was funded by project grants from the Diabetes Australia Research Trust and the National Health and Medical Research Council of Australia and fellowship support by Kidney Health Australia and the Australian and New Zealand Society of Nephrology.
Duality of interest
None of the authors has any duality of interest in connection with this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chow, F.Y., Nikolic-Paterson, D.J., Ma, F.Y. et al. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 50, 471–480 (2007). https://doi.org/10.1007/s00125-006-0497-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0497-8