
Abstract
Purpose
This review article is intended to provide a perspective overview of potential strategies to overcome radiation resistance of tumors through the combined use of immune checkpoint and DNA repair inhibitors.
Methods
A literature search was conducted in PubMed using the terms (“DNA repair* and DNA damage response* and intracellular immune response* and immune checkpoint inhibition* and radio*”) until January 31, 2023. Articles were manually selected based on their relevance to the topics analyzed.
Results
Modern radiotherapy offers a wide range of options for tumor treatment. Radiation-resistant subpopulations of the tumor pose a particular challenge for complete cure. This is due to the enhanced activation of molecular defense mechanisms that prevent cell death because of DNA damage. Novel approaches to enhance tumor cure are provided by immune checkpoint inhibitors, but their effectiveness, especially in tumors without increased mutational burden, also remains limited. Combining inhibitors of both immune checkpoints and DNA damage response with radiation may be an attractive option to augment existing therapies and is the subject of the data summarized here.
Conclusion
The combination of tested inhibitors of DNA damage and immune responses in preclinical models opens additional attractive options for the radiosensitization of tumors and represents a promising application for future therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiotherapy (RT) is one of the main therapeutic options in most cancer entities, with approximately 50% of cancer patients receiving RT as part of their cancer care [1]. Intrinsic or adapted radioresistance is therefore a tremendous challenge, because such a large proportion of cancer patients is actively treated by RT. A deeper understanding of how radioresistance occurs and the development of new strategies to overcome radioresistance is therefore an urging field of research.
The discovery of immune checkpoint inhibitors (ICIs) has improved the therapeutic options for many cancer patients across multiple tumor entities. The two most targeted structures for immunotherapy are cytotoxic T‑lymphocyte antigen‑4 (CTLA-4) and programmed cell death 1/ligand 1 (PD-1/PD-L1). The first U.S. Food and Drug Administration (FDA) approval of a CTLA‑4 antibody (ipilimumab) in 2011 was followed by the approval of two PD‑1 antibodies (pembrolizumab and nivolumab) in 2014, all exclusively for treatment of advanced-stage melanoma [2,3,4,5]. Since then, the application of ICIs has been greatly expanded to more than 15 tumor entities. Although the response rate to immunotherapy was between 20 and 50% in initial clinical trials, only around 15–20% of patients are expected to respond to immunotherapy in the long term [6, 7]. The response to ICI treatment is largely dependent on the immunological state of the tumor. Currently, it is believed that especially tumors with a high tumor mutational burden (TMB), PD-L1 expression, or high infiltration of tumor-infiltrating lymphocytes (TILs)—and thus designated as immunologically “hot”—will benefit from immunotherapy [8]. Like most drugs, the emergence of resistance, initially or adaptive, is a widely investigated challenge in the treatment with ICIs. As a result, research is moving toward combination treatments. Recently, a combination treatment of patients with ICIs and DNA damage response (DDR) inhibitors together with RT has been proposed [9, 10].
In addition to initiating immune-related processes at the local tumor, the combination of RT with ICI can also enhance the so-called abscopal effect of RT. This phenomenon is observed when one tumor site is irradiated but distant tumor sites also show a response despite no ionizing radiation (IR) being directly applied. It is believed that immune signaling mediates this effect [11] and that it might be enhanced by combined ICI treatment, as this phenomenon is extremely rare [12, 13]. The interplay between these currently developing therapeutic options is therefore the focus of the present review.
The intrinsic cancer cell immune response
The immune response in the context of cancer is a complex, multidimensional network. The starting point for the immune response of the tumor microenvironment is DNA damage in the tumor cell. DNA damage is increasingly triggered by combination therapies including radiation, activates the DDR, and can be clinically exploited by enhancing the immune response of the tumor microenvironment. The resulting increased DNA damage leads to accumulation of cytosolic double-strand DNA (dsDNA), thereby activating the intracellular immune response via the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway. This is part of the innate immune system, which is the immediate response after pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) detection. In contrast to the adaptive immune system, it is a general mechanism that triggers unspecific inflammatory responses and does not require pathogen-specific antigens. As a result, inflammatory signaling gets activated, leading to recruitment of immune effector cells like dendritic cells (DCs), natural killer (NK) cells, and CD8+ T cells [14]. This immunogenic change in the tumor microenvironment can have positive impact, as immunologically hot tumors might be responsive to ICIs [15]. However, also immunosuppressive and tumorigenesis-favoring effects, including promotion of metastasis formation, have been observed [16, 17] and discussed detailed in Sect. “Immune signaling in response to ionizing radiation—drawbacks.” In this review the focus is on the intrinsic cancer cell immune signaling and how the interconnection of the involved pathways can be elucidated for novel therapeutic options.
The cGAS/STING pathway
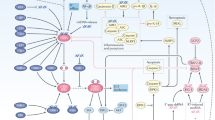
One of the intracellular immune signaling cascades that is activated by cytosolic DNA is the cGAS/STING pathway (Fig. 1). This mechanism was first described in response to and for defense against pathogenic infections by bacteria or viruses [18]. It is believed to be the main activated pathway in response to cytosolic DNA. Other different pathways have been shown to be involved in PAMP and DAMP detection, including the Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-1), and mitochondrial antiviral-signaling protein (MAVS). Since recognition involves detection of free dsDNA in the cytosol, the cGAS/STING pathway can also be triggered by the cell’s own DNA. Cytosolic DNA arises from multiple sources, including pathogens, micronuclei, and mitochondrial DNA [19]. Binding of dsDNA to cGAS induces conformational changes in the protein, activating its catalytically active site to synthesize cyclic GMP-AMP (cGAMP) from ATP and GTP [20, 21]. Recently, it was shown that the function of cGAS signaling depends on the length of the recognized DNA fragments [22]. The second messenger cGAMP activates STING upon binding, leading to STING translocation from the endoplasmic reticulum (ER) membrane to the Golgi. In its new location, STING recruits TANK-binding kinase 1 (TBK1) and IкB kinases (IKK) [23], and thereby activates interferon regulatory factor 3 (IRF3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) release by phosphorylation of IкBα [24, 25]. IRF3 and NF-кB translocate into the nucleus, where they activate the expression of type I interferons (IFNs), which in turn bind autocrine to IFN receptor 1 and 2 (IFNAR1/2), leading to activation of the Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling pathway [26]. The phosphorylated STAT1/2 heterodimer translocates into the nucleus and activates the expression of IFN-stimulated genes (ISGs), further driving the innate immune response and connecting it to the adaptive immune response [27]. IFN signaling further leads to recruitment and maturation of DCs, which then prime CD8+ T cells for tumor infiltration [28, 29].

cGAS/STING activation by radiation-induced DNA damage. a Cytosolic dsDNA migrates in micronuclei or directly in the cytosol and is recognized by cGAS which catalyzes cGAMP synthesis after dsDNA binding. cGAMP binds to STING, which translocates from the ER to the Golgi, recruits IKK and TBK1 and activates IRF3 and IкBα, resulting in release of NF-кB. IRF3 and NF-кB translocate into the nucleus and induce transcription of IFN type I and other proinflammatory chemokines. IFN binds to IFNAR1/2 receptors, activates JAK, and STAT1/2 is phosphorylated. The heterodimer translocates to the nucleus and induces expression of ISGs. b Activation of DDR kinases ATR, WEE1, ATM, and DNA-PK PARP1 after IR induces DNA repair. Accumulation of cytosolic dsDNA and formation of micronuclei leads to activation of overall cGAS/STING metabolism. ATR activation affects PD-L1 surface expression via CHK1 and STAT1/3-IRF1 signaling. ATM ataxia-telangiectasia mutated protein, ATR ataxia-telangiectasia and Rad3-related protein, cGAMP cyclic GMP-AMP, cGAS cyclic GMP-AMP synthase, CHK1 checkpoint kinase 1, DDR DNA damage response, DNA-PK DNA-dependent protein kinase, ER endoplasmic reticulum, IFN interferon, IFNAR1/2 interferon receptor 1/2, IKK IκB kinases, IRF1/3 interferon regulatory factor 1/3, ISG interferon-stimulated gene, JAK Janus kinase, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, PARP1 poly-ADP-ribose polymerase 1, PD-L1 programmed death ligand 1, STAT1/2/3 signal transducer and activator of transcription 1/2/3, STING stimulator of interferon genes, TBK1 TANK binding kinase 1; Wee1 Wee1 G2 checkpoint kinase. Adapted from “Blank Pathway (Linear),” by BioRender.com (2023)
Immune signaling in response to ionizing radiation—benefits
Upon DNA damage induction by ionizing radiation (IR), the integrity of the DNA is severely disrupted, resulting in genomic instability, including formation of micronuclei. Direct leakage of DNA out of the nucleus or micronuclei formation subsequently activates cGAS/STING signaling [30, 31]. Release of IFN‑1 as a consequence of cGAS/STING activation is the main driver of the antitumor immune signaling induced by IR [32] and enhances the attraction of DCs which process the tumor-associated antigens [33]. Upon maturation and migration to the lymph nodes, the DCs present those tumor antigens via the major histocompatibility complex (MHC) to the CD8+ T cells. This then allows for CD8+ T cells to infiltrate the tumor and recognize tumor-specific cells and potentially eradicate them [34]. Gupta et al. showed that RT specifically enhances tumor-specific CD8+ T cell activation by DCs, indicating how RT contributes to activation of the antitumor immune response [35]. Additionally, tumor-specific CD8+ T cell infiltration is enhanced by the release of chemokines due to activated ISG expression in response to JAK signaling. Two of the main inflammatory chemokines responsible for T cell attraction are C‑X‑C motif chemokine ligand 10 (CXCL10) and C‑C motif chemokine ligand 5 (CCL5) [36].
The cGAS/STING pathway has been suggested to also play an important role in DC activation internally. After recruitment of DCs to the immunogenic tumor site, the cGAS/STING pathway can be triggered inside the DCs, further enhancing antitumor IFN-1 signaling [37]. Different mechanisms have been proposed for how activation of the cGAS/STING pathway in non-tumor cells is mediated after IR: (i) phagosomal escape due to alkalinization of phagosomes in DCs [38]; (ii) transfer of the second messenger cGAMP from tumor cells to DCs [39], possibly via gap junctions [40]; (iii) direct exosomal transfer of tumor cell dsDNA to DCs [28]; (iv) uptake of oxidized tumor mitochondrial DNA [41].
Immune signaling in response to ionizing radiation—drawbacks
Radiotherapy induces not only beneficial antitumor effects of the immune system but can also support immune escape mechanisms [42]. Experiments in mice showed that induction of IFN-1 signaling was dependent on the fractionation and dose of RT. Single high-dose radiation did not show the same response and abscopal effect after combined RT and anti-CTLA‑4 treatment as fractionated RT with anti-CTLA‑4 antibodies [43]. This indicates that treatment planning for combination therapies has to be designed carefully. A dose dependence was also demonstrated by Vanpouille-Box and colleagues in 2017, where a single dose of 20 Gy upregulated three prime repair exonuclease 1 (TREX1) expression, while fractionation of 3 × 8 Gy upregulated ISG expression in patient-derived xenografts of a transcription factor p53 (TP53)- and GTPase K‑Ras (KRAS)-mutated lung adenocarcinoma [44]. TREX1 is a cytoplasmic DNA exonuclease which degrades the dsDNA arising in the cytoplasm due to IR; thus, high expression would suppress cGAS/STING activation, inhibiting the intracellular antitumor immune signaling cascade [44]. The same group later showed that TREX1 can also be transmitted via exosomal shuttling, inhibiting cGAS/STING activation in DCs [28]. However, this mechanism appears to be dependent on the cell system studied. Other studies have shown that tumors, which suppress cGAS/STING signaling due to mutations or epigenetic silencing, can also escape induction of the IFN-1 response [45, 46].
Chronic activation of IFN-1 signaling in a tumor can result in immunosuppressive behavior [47]. In the study of Benci et al., the authors showed that continuous IFN signaling was associated with STAT1-related epigenomic and transcriptomic modifications, which lead to PD-L1-independent adaptive resistance [47]. Another study from Bakhoum et al. in 2018 showed that chromosomal instability in tumor cells can lead to activation of the cGAS/STING pathway. This activation was associated with metastasis formation driven by the noncanonical NF-кB signaling pathway [16]. The same pathway was shown to be activated in DCs and negatively affect IR-induced IFN-1 signaling. It was further shown that inhibition of the noncanonical NF-кB pathway led to tumor regression by treatment with IR [48].
However, these identified adverse effects of RT for the immune response provided deeper insights into how it may be possible to overcome nonresponsive phenotypes or acquired resistances.
Genomic instability as a predictive biomarker for immune therapies
Immunotherapy is currently transforming existing tumor treatment. ICIs have achieved tremendous therapeutic success in numerous tumor types, including cancers traditionally considered nonimmunogenic. However, a significant proportion of patients do not respond to these therapies. Therefore, early selection of susceptible patients is critical, and the development of predictive biomarkers is one of the major challenges in ICI development. Starting from the immunogenic side, the expression of PD-L1 and infiltrating T lymphocytes are the most frequently used biomarkers of potential response to tumor therapy [49]. Recent publications suggest that the genomic landscape of the tumor, its mutational burden, and tumor-specific neoantigens are potential determinants of the response to ICI and may therefore influence immunotherapy outcomes. In addition, tumor-associated defects in DNA repair mechanisms have been associated with improved survival and durable clinical benefit from ICI. Thus, the TMB and associated tumor-specific neoantigens appear to be important predictive approaches to anticipate potential clinical benefits of ICI, as they closely reflect the repair capacity of tumor cells and their intrinsic genomic instability. Initially, studies showed that TMB is correlated with the clinical benefit of anti-PD‑1 and anti-CTLA‑4 therapy in several tumor types, including malignant myeloma, non-small cell lung cancer (NSCLC), and various tumors with DNA repair deficits [50]. Overall, a direct correlation between DNA repair deficiency, TMB, predicted neoantigen load, and clinical activity of ICI is suggested.
Tumor mutational burden
A correlation between TMB and response to ICI has been enabled by recent advances in next-generation sequencing (NGS) technology, particularly in whole-exome sequencing and RNA sequencing. High mutation load, defined as > 100 nonsynonymous single-nucleotide variants (nsSNV) per exome, was associated with clinical benefit in melanoma patients treated with anti-CTLA‑4 therapy [51, 52]. Rizvi and colleagues correlated high TMB (defined as > 178 nsSNVs per exome) and durable clinical benefit in two independent cohorts of NSCLC patients receiving pembrolizumab [53]. Of note, the study reported a significantly increased overall response rate (ORR) in tumors with a smoking-related molecular signature, which are potential determinants of response to ICI. In addition, mutations in DNA repair genes, including DNA polymerase delta 1, DNA polymerase epsilon catalytic subunit, mismatch repair gene MutS homolog 2 (MSH2), breast cancer gene 2 (BRCA2), RAD51 gene homolog C (RAD51C), and RAD17 checkpoint clamp loader compound (RAD17) were observed in responders with the highest mutational burden. This supports the notion that DNA repair defects may increase tumor immunogenicity by promoting somatic mutations. Consistently, later findings showed higher response rates to anti-PD‑1 therapy in mismatch repair (MMR)-deficient tumors and in BRCA2-mutated melanoma [50].
Major challenges that remain to be addressed to improve the robustness of TMB include the definition of optimal tumor purity and sequencing depth, as well as the threshold for defining “high” and “low” mutation burden. Indeed, there is a significant overlap in the mutational range between responders and nonresponders [51, 52]. Some patients still benefit from ICI despite very low mutation rates, and conversely, high TMB does not always correlate with response. This is best illustrated in relapsed or refractory Hodgkin’s lymphoma, which is very sensitive to PD‑1 blockade despite having virtually no mutation [54]. Mutational signatures, which are functional slices of past and current disease biology related to DNA damage and DNA repair, may provide an additional genomic determinant of response to ICI. Their use, combined with assessment of TMB and detection of mutations in DNA repair genes, may therefore allow better grouping of patients and identify ICI-sensitive tumors. Importantly, the mutational landscape analyses described above provide only an instantaneous and descriptive picture of a tumor genome. Mutational signatures may even in some cases exclusively reflect past DNA repair deficiencies and may not be relevant markers of the current DNA repair status of the tumor. It is therefore critical to evaluate the potential of these mutations to functionally enhance the antitumor immune response through the generation of immunogenic neoantigens.
Link between DNA repair and the immune response
Understanding the mechanism underlying DNA damage-derived signal transduction is critical for overcoming refractory cancer, especially when cancer immunotherapy is used in combination with DNA damage-dependent radio-/chemotherapy. Several lines of evidence suggest a link between DNA damage signaling and modulation of the immune response. In this process, DNA damage-triggered signals within the cell of origin are transmitted to the cell surface and neighboring cells, modulating immune and inflammatory responses. The interplay between PD-L1 expression, microsatellite instability (MSI), and accumulation of mutations in the cancer genome leads to production of neoantigens and presentation of the human leukocyte antigen (HLA)–neoantigen complex in cancer cells. HLA neoantigen presentation promotes immune activity in the tumor environment and characterizes a so-called hot tumor [55]. Several studies have shown that PD-L1 expression is upregulated in cancer cells in response to increased DNA damage [56,57,58,59,60]. This may be triggered by loss of individual DNA repair proteins responsible for double strand break (DSB) repair, such as BRCA2 or Ku70/80, after irradiation [58]. Likewise, defects in other DNA repair pathways can lead to upregulation of PD-L1 [56]. The common mechanism leading to upregulation of PD-L1 occurs through activation of the DNA damage response. This directly affects activation of the STAT1/3-IRF1 signaling cascade, downstream of which PD-L1 mRNA transcription is activated [58]. Supporting this observation, it was shown that DNA repair-deficient breast tumors were associated with CD4+ and CD8+ lymphocyte infiltration and that cells from these DNA repair-deficient breast tumors expressed the chemokines CXCL10 and CCL5 more strongly [59].
In terms of RT delivery, this means that a wide range of diverse DNA damage, including base damage, single-strand breaks (SSBs), DSBs, and DNA crosslinks (ICLs), is induced [61]. DNA damage induced by IR activates the DDR, which subsequently regulates the appropriate DNA repair pathway choice. The DDR represents a complex network consisting of cell cycle control, DNA repair, and inactivation of multiple interconnected signaling pathways and mechanisms, all aiming to maintain cell viability. Once DNA damage is detected, cell cycle checkpoints are activated to arrest the cell cycle for DNA repair prior to cell division. This allows the cell to survive genomic instability and replication stress through successful DNA repair, or to initiate permanent arrest or cell death. The DDR consists of a series of pathways with different protein sets specialized for specific types of damage and classified as sensors, transducers, and effectors. Over the past few years, it has become more evident that failure or defectiveness of individual components of the DDR machinery leads to fragmentation of DNA, which enters the cytoplasm and thereby contributes to immune signaling in the tumor [31, 57, 59, 62, 63].
Recognition of DNA damage
Double-strand breaks are the most lethal event in a cell and are therefore rapidly recognized by the MRE11-RAD50-NBS1 (MRN) complex, which interacts with chromatin and promotes activation of ataxia-telangiectasia mutated (ATM) kinase by subsequent autophosphorylation at Ser1981 [64]. The ATM kinase, discovered in 1995 by Y. Shiloh [65], is an essential sensor of DNA damage by activating hundreds of different substrates, including TP53 and checkpoint kinase 2 (CHK2) [66]. In addition, ATM enables phosphorylation of histone H2AX to form H2AX foci [67], which is essential for the repair of DSBs.
Single-strand breaks are recognized by another repair pathway, the RAD9-HUS1-RAD1 complex, which in cooperation with RAD17, replication factor C (RFC) 2, RFC3, RFC4, and RFC5 activates the ataxia-telangiectasia and Rad3-related (ATR) kinase [68]. ATR-interacting protein (ATRIP)-dependent recruitment of ATR to replication protein A (RPA)-bound single-stranded DNA is initiated upon ATR activation [69, 70], leading to phosphorylation of checkpoint kinase 1 (CHK1) [71].
CHK1 and CHK2 then further transmit the signaling by phosphorylation of various downstream effectors. CHK2 suppresses phosphatase cell division cycle 25 A (CDC25A), which abrogates suppressive phosphorylation of cyclin E/cyclin-dependent kinase (CDK) 2 and cyclin A/CDK2 complexes, preventing entry into the S‑phase of the cell cycle [66]. CHK1 regulates the G2/M checkpoint by activating G2 checkpoint kinase (WEE1), which then phosphorylates CDK1, reducing its activity and preventing entry into mitosis [72]. In addition, CHK1 modulates the S‑phase checkpoint by facilitating degradation of CDC25A phosphatase, whose activity is critical for removing the suppressive phosphate groups of CDK4 and CDK2 kinases and ensuring cell cycle progression [73].
Another important kinase responding to DNA damage is the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), one of three related kinases in the phosphatidylinositol 3‑kinase-related kinase (PIKK) family. These kinases are activated after DNA damage and phosphorylate many downstream targets to activate DNA damage checkpoints and stimulate DNA repair [74,75,76]. PIKKs function in overlapping DNA damage signaling networks. Regarding DSB repair, despite extensive crosstalk between PIKKs, the generally accepted view is emerging that DNA-PKcs play a direct and central role in DNA repair through nonhomologous end-joining (NHEJ), while ATM and ATR play a critical role in promoting repair through homologous recombination (HR) of open DSBs and DSBs at collapsed replication forks, respectively (summarized in [77]).
DNA repair pathways to eliminate IR-induced DNA damage
Double-strand breaks are mainly repaired by two processes: NHEJ and HR. NHEJ joins ends without requiring a repair template; therefore, it is error prone and typically results in small deletions or insertions at repair sites. NHEJ functions throughout the entire cell cycle and is the dominant DSB repair pathway [78]. HR requires a homologous repair template and is thought to be generally error free [79,80,81]. HR is largely restricted to the S/G2 phases of the cell cycle and is mediated by the recombinase RAD51, which is loaded onto the DNA by interaction with the BRCA1, BRCA2, and partner and localizer of BRCA2 (PALB2) complex [82, 83]. HR is important for precise repair of open DSBs, those directly induced by IR, and is critical for repair of replication-associated DSBs, including those that arise when replication forks encounter radiation-induced single-strand lesions [84].
In addition to HR and NHEJ, a “backup” DSB repair pathway has been discovered in tumors. It has similar mechanisms to the two main DSB repair pathways but is genetically distinct and referred to as alternative end-joining (a-EJ) or microhomology-mediated end-joining [85]. The a‑EJ engages similar initiation processes and factors to HR or NHEJ regarding the connection of open DNA ends, as the MRN complex is involved in a‑EJ initiation. The polymerase theta has been shown to be essential for this pathway [86], as well as poly(ADP-ribose) polymerase 1 (PARP1) [3]. These backup repair pathways cause gene deletions, translocations, and rearrangements in cancer cells. Currently, there is increasing interest in a‑EJ signaling pathways as potential therapeutic targets due to cancer cell specificity [86, 87].
Irradiation-induced base damage and SSBs are rapidly and efficiently repaired by base excision repair (BER). X‑ray cross-complementing protein 1 is the facilitator of BER, as it interacts with DNA ligase III, polymerase β, and PARP1 [88].
Nucleotide excision repair (NER) is conducted by the cell to remove bulky DNA lesions [89]. Removal of these lesions is mediated by several different complexes, and deficiencies of involved proteins lead to severe diseases like xeroderma pigmentosa or Cockayne syndrome [90].
The MMR pathway is required to detect and repair base–base mismatches of the DNA during replication or in response to induced DNA damage. The MSH heterodimers of MSH2/MSH6, detecting base–base mismatches and small insertions or deletions, and MSH2/MSH3, detecting also larger insertions or deletions, together with PMS1, PMS2, MLH1, and MLH3 are the recognition sensors of MMR [91]. Interactions with additional proteins like proliferating-cell nuclear antigen, exonuclease 1, RPA, and RFC then cause excision of the mismatches and repair. Mutations of proteins involved in MMR have been shown to lead to MSI [91].
DNA crosslinks between and within DNA strands represent a dangerous form of damage that blocks vital cellular processes such as transcription and replication. The Fanconi anemia (FA) pathway is responsible for repairing these aberrations in the DNA structure [92, 93]. Fanconi anemia is a heterogeneous genetic disease involving 22 different genes that can be divided into three main groups: the FA core complex, the I‑D2 complex, and the downstream FA proteins [94]. The FA pathway allows for unblocking of the replication fork by inducing formation of a DSB and coordinating the action of three critical repair mechanisms: translesion synthesis bypasses the lesion, and after removal of toxic adducts by NER, the gap is closed by HR [93].
Translational aspects of DNA repair and immune signaling connection
Several studies indicate a correlation between DNA damage and the immune response. Particularly in tumor cells with deficiencies in the DNA repair pathways HR and MMR, increased immunogenicity has been shown to correlate with response to ICIs [95, 96]. MMR deficiency, which is known to lead to MSI, has already been identified as a prognostic marker for the use of ICIs [95]. However, MSI is mainly observed in three cancer entities: endometrial, gastric, and colorectal cancer. Identification of additional DNA repair defects to predict response to ICIs is pending.
These observations further imply that targeting the DDR in combination with ICI may be a promising target for intensified therapy. Triple combinations of DDR inhibitors, ICI, and RT are currently being investigated, as RT-induced DNA damage could be even more effective in causing cancer cell death when DDR is inhibited and immune response signaling is maximized.
Combined therapy of ICI with PARP inhibition
Mutations in the HR protein BRCA1 were associated with higher infiltration of T lymphocytes in a cGAS/STING pathway-dependent manner in breast cancer cells [59]. This was due to increased release of the chemokines CXCL10 and CCL5 upon cGAS/STING activation. The same study showed that PD-L1 expression was enhanced by DNA damage in S phase, also dependent on cGAS/STING [59]. Loss of BRCA2 was observed to lead to increased activation of innate immune signaling. This led to chronic upregulation of ISG expression and could be enhanced by PARP1 inhibition [9]. However, it was also observed that a deficiency in BRCA1 or 2 did not lead to the same modulation of the immune response, indicating the complexity of the connection between the DDR and the immune response [97]. Inhibition of PARP1 has been shown to be effective in treatment of tumors carrying mutations in the BRCA1 or BRCA2 genes due to synthetic lethality. In these tumors, the accumulation of SSBs upon treatment with PARP1 inhibitors leads to blockage of replication forks and the formation of DSBs [98,99,100]. More recently, it has become evident that PARP1 inhibition can directly activate the cGAS/STING pathway independent of the BRCA status of the tumor [101]. PARP1 inhibition caused a time-dependent upregulation of the chemokines CXCL10 and CCL5, which was reversed in cells with knockdowns in the key proteins of the cGAS/STING pathway. These observations in cell lines were confirmed by in vivo studies, where CXCL10 and CCL5 upregulation led to increased CD8+ T cell infiltration. A combination treatment of PARP1 inhibition and ICI potentiated the therapeutic effects in colon and ovarian cancer mice models [101]. Another study investigating the effects of PARP1 inhibition on intracellular immune signaling revealed that activation of cGAS/STING is dependent on PARP1 trapping, as treatment of PARP1-deficient cells with the PARP1 inhibitor talazoparib did not show any activation of the cGAS/STING pathway [102].
In the study from Sato et al. it was shown that PD-L1 upregulation was caused in an ATM/ATR/CHK1-dependent manner [58]. Cells deficient in BRCA2 or Ku80 showed increased PD-L1 expression after IR or PARP1 inhibition, but this effect was suppressed again upon CHK1 inhibition. They further revealed that the upregulation required signaling via STAT1/3 and IRF1 [58].
Combined therapy of ICI with DDR inhibition and radiation
Increasingly, the combination of DDR inhibitors and ICIs with radiation is being investigated in preclinical tumor models (Fig. 1). Current progress in the clinical implementation of immunostimulatory DNA-damaging treatment regimens in combination with radio-/chemotherapy and the necessary future directions to optimize the immunosensitizing potential of DNA damage response inhibitors were summarized by [103].
Combined therapy of ICI with ATR inhibition and radiation
Because of its exclusive importance in regulating the replication stress level, most of the available studies involve inhibition of ATR. The ATR kinase inhibitor AZD6738 was observed to attenuate radiation-induced CD8+ T cell depletion and enhance CD8+ T cell activity in mouse models of KRAS-mutated cancer in combination with conformal RT. Mechanistically, ATR inhibition by AZD6738 appears to block radiation-induced PD-L1 upregulation on tumor cells, thereby reducing the number of tumor-infiltrating regulatory T cells (Tregs). Of note, AZD6738 in combination with conformal RT can induce immunological memory in treatment-responsive mice [104]. Feng et al. observed that inhibitors of the DNA damage response kinase ATR can significantly enhance innate immune responses triggered by IR. They showed that both the cGAS/STING-dependent DNA-sensing pathway and the MAVS-dependent RNA-sensing pathway are responsible for type I IFN signaling induced by IR. The authors suggested that DNA fragments released due to DNA damage may either activate the cGAS/STING pathway or be transcribed, thereby initiating MAVS-dependent RNA sensing and signaling. Both observations suggest that different pathways are involved in type I IFN signaling in response to DNA damage and may thus represent a promising new combination therapy against cancer [105]. Also, an enhanced tumor-inhibitory effect of ATR inhibition in combination with fractionated RT was shown in an immunocompetent mouse model for human papillomavirus (HPV)-positive malignancies. Here, significant radiosensitization by the ATR inhibitor AZD6738 was observed, accompanied by a marked increase in DNA damage and immune cell infiltration. In parallel, increased numbers of CD3 and NK cells were observed. ATR inhibition plus IR resulted in a gene expression signature consistent with the observed type I/II IFN response. Increased MHC I levels were monitored on tumor cells, with transcriptional level data indicating increased antigen processing and presentation in the tumor. In vivo, significant modulation of cytokine gene expression (particularly CCL2, CCL5, and CXCL10) was observed. In vitro data also indicate that CCL2, CCL5, and CXCL10 were increasingly expressed by tumor cells after ATR inhibition plus RT [106]. In an HPV-negative mouse model of oral squamous cell carcinoma, Patin et al. observed that inhibition of ATR enhanced IR-induced inflammation of the tumor microenvironment, with NK cells playing a central role in maximizing treatment efficacy. It was found that ICI can further enhance the antitumor activity of NK cells [107].
Sheng et al. also observed immune-stimulatory effects of the ATR inhibitor AZD6738 in combination with IR and ICI in hepatocellular carcinoma. It was found that AZD6738 increased IR-stimulated CD8+ T cell infiltration and reversed the immunosuppressive effect of IR on the number of Tregs in mouse xenografts. Moreover, the addition of AZD6738 enhanced infiltration; increased cell proliferation and the ability to produce IFN‑γ from CD8+ T cells derived from TILs; and caused a decreasing trend in the number of TILs and Tregs, and depleted T cells in mouse xenografts. This significantly improved the immunological microenvironment of the tumor [108].
Combined therapy of ICI with ATM inhibition and radiation
For inhibition of DDR signaling via ATM, it was shown that activation of the innate immune response through enhanced induction of DNA damage could increase the efficacy of ICI [109]. The inhibition of ATM alone was already able to increase tumoral type I IFN expression independently of the cGAS/STING pathway. This was done in a TBK1 and proto-oncogenic tyrosine protein kinase Src-dependent manner. The combination of ATM inhibition and IR increased TBK1 activity even more markedly and, accordingly, increased IFN production and antigen presentation. In addition, silencing of ATM increased PD-L1 expression and enhanced the sensitivity of pancreatic tumors to PD-L1-blocking antibodies. This was associated with an increase in tumor CD8+ T cells and established immune memory [109]. The authors also found that low ATM expression inversely correlated with PD-L1 expression in patients’ pancreatic tumors. Overall, these results indicate that the efficacy of ICI in pancreatic cancer is enhanced by ATM inhibition and further enhanced by IR, depending on the increased immunogenicity of the tumor [109].
Combined therapy of ICI with DNAPKcs inhibition and radiation
Inhibition of the DDR sensor kinase DNA-PKcs, which is responsible for NHEJ, was also demonstrated to have an immunomodulatory effect [110]. The combination of IR and DNA-PKcs inhibition was shown to enhance cytosolic dsDNA and tumor-associated type I IFN signaling independently of cGAS and STING. In parallel, PD-L1 expression was stimulated upon DNA-PKcs inhibition and IR. Simultaneous use of anti-PD-L1 in combination with IR and DNA-PKcs inhibitors potentiated antitumor immunity in pancreatic cancer models [110].
Combined therapy of ICI with WEE1 inhibition and radiation
As another interesting DDR protein for stimulating the immune response, WEE1 was investigated in combination with IR regarding its effects for cell killing by T lymphocytes and a sensitizing effect for ICI. In several models it was observed that the WEE1 inhibitor AZD1775 led to DNA damage accumulation and that the combination treatment improved tumor control in a syngeneic mouse model of oral cavity cancer (MOC1) in vivo. Combination treatment enhanced granzyme B‑dependent T lymphocyte killing by reversing additive G2/M cell cycle blockade. The combination of IR and AZD1775 improved CD8+ T cell-dependent control of MOC1 tumor growth and the rate of complete eradication of established tumors in the context of the PD-axis ICI. Functional assays demonstrated enhanced tumor antigen-specific immune responses in sorted T lymphocytes. The combination of IR and AZD1775 not only increased tumor-specific cytotoxicity, but also improved susceptibility to killing by T lymphocytes and response to PD-axis ICI [111].
Combined therapy of ICI with STING antagonists and radiation
Very recent data in a preclinical model showed that not only cell surface ligands such as PD-L1 or PD-L2 are suitable for combined therapy of ICI and DDR inhibitors for enhanced radiation sensitization. It was observed that intracellular immune response proteins like STING antagonists also resulted in significant radiation sensitization with improved survival in a syngeneic genetically engineered mouse model and human pediatric high-grade glioma cells [112]. Significantly improved survival was observed when the PARP inhibitor pamiparib was combined with the STING antagonist H151 after IR. The CHK1 inhibitor was also shown to prolong survival in a mouse model when combined with H151 and IR [112].
Perspective
Defects in the DDR can trigger intracellular immune signaling endogenously. This effect can be enhanced by exogenously induced DNA damage via DDR inhibition, chemotherapeutics, or RT. The combination of DDR inhibitors and RT may be able to force tumors, which are not endogenously inflamed, to activate proinflammatory signaling. The intracellular immune signals are then transmitted to the tumor microenvironment by release of IFNs and chemokines, leading to recruitment of immune effector cells. This suggests that the combination of the recently tested inhibitors of DDR and immune response in preclinical models opens new options for radiation sensitization and thus represents an attractive option for promising use in cancer therapy.
References
Delaney G et al (2005) The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 104(6):1129–1137
Hodi FS et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Robert I, Dantzer F, Reina-San-Martin B (2009) Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J Exp Med 206(5):1047–1056
Hamid O et al (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369(2):134–144
Topalian SL et al (2012) Safety, activity, and immune correlates of anti-PD‑1 antibody in cancer. N Engl J Med 366(26):2443–2454
Haslam A, Prasad V (2019) Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open 2(5):e192535
Haslam A, Gill J, Prasad V (2020) Estimation of the percentage of US patients with cancer who are eligible for immune checkpoint inhibitor drugs. JAMA Netw Open 3(3):e200423
Li H, van der Merwe PA, Sivakumar S (2022) Biomarkers of response to PD‑1 pathway blockade. Br J Cancer 126(12):1663–1675
Reislander T, Groelly FJ, Tarsounas M (2020) DNA damage and cancer immunotherapy: a STING in the tale. Mol Cell 80(1):21–28
Hintelmann K, Petersen C, Borgmann K (2022) Radiotherapeutic strategies to overcome resistance of breast cancer brain metastases by considering Immunogenic aspects of cancer stem cells. Cancers (Basel) 15(1):211. https://doi.org/10.3390/cancers15010211
Demaria S et al (2004) Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 58(3):862–870
Postow MA, Harding J, Wolchok JD (2012) Targeting immune checkpoints: releasing the restraints on anti-tumor immunity for patients with melanoma. Cancer J 18(2):153–159
Hsieh RC et al (2022) ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci Immunol 7(72):eabl9330
Vivier E, Malissen B (2005) Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol 6(1):17–21
McLaughlin M et al (2020) Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 20(4):203–217
Bakhoum SF et al (2018) Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553(7689):467–472
Palucka AK, Coussens LM (2016) The basis of oncoimmunology. Cell 164(6):1233–1247
Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461(7265):788–792
Miller KN et al (2021) Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184(22):5506–5526
Zhang X et al (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep 6(3):421–430
Gao P et al (2013) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153(5):1094–1107
Luecke S et al (2017) cGAS is activated by DNA in a length-dependent manner. EMBO Rep 18(10):1707–1715
Fitzgerald KA et al (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4(5):491–496
Wu J et al (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339(6121):826–830
Abe T, Barber GN (2014) Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 88(10):5328–5341
de Weerd NA et al (2013) Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat Immunol 14(9):901–907
Schoggins JW et al (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472(7344):481–485
Diamond MS et al (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208(10):1989–2003
Fuertes MB et al (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J Exp Med 208(10):2005–2016
Mackenzie KJ et al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548(7668):461–465
Harding SM et al (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548(7668):466–470
Burnette BC et al (2011) The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res 71(7):2488–2496
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252
Deng L et al (2014) STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41(5):843–852
Gupta A et al (2012) Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol 189(2):558–566
Nagarsheth N, Wicha MS, Zou W (2017) Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17(9):559–572
Woo SR et al (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41(5):830–842
Klarquist J et al (2014) STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol 193(12):6124–6134
Marcus A et al (2018) Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 49(4):754–763.e4
Schadt L et al (2019) Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep 29(5):1236–1248.e7
Fang C et al (2021) Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell Mol Immunol 18(9):2211–2223
Weichselbaum RR et al (2017) Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol 14(6):365–379
Dewan MZ et al (2009) Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA‑4 antibody. Clin Cancer Res 15(17):5379–5388
Vanpouille-Box C et al (2017) DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8:15618
Xia T, Konno H, Barber GN (2016) Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res 76(22):6747–6759
Konno H et al (2018) Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37(15):2037–2051
Benci JL et al (2016) Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167(6):1540–1554.e12
Hou J et al (2016) HER2 reduces breast cancer radiosensitivity by activating focal adhesion kinase in vitro and in vivo. Oncotarget 7(29):45186–45198
Li L et al (2023) Immunotherapy for triple-negative breast cancer: combination strategies to improve outcome. Cancers (Basel) 15(1):321. https://doi.org/10.3390/cancers15010321
Chabanon RM et al (2016) Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res 22(17):4309–4321
Snyder A et al (2014) Genetic basis for clinical response to CTLA‑4 blockade in melanoma. N Engl J Med 371(23):2189–2199
Van Allen EM et al (2015) Genomic correlates of response to CTLA‑4 blockade in metastatic melanoma. Science 350(6257):207–211
Rizvi NA et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD‑1 blockade in non-small cell lung cancer. Science 348(6230):124–128
Ansell SM et al (2015) PD‑1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372(4):311–319
Uchihara Y et al (2021) Modulation of immune responses by DNA damage signaling. DNA Repair (Amst) 104:103135
Permata TBM et al (2019) Base excision repair regulates PD-L1 expression in cancer cells. Oncogene 38(23):4452–4466
Sato H, Jeggo PA, Shibata A (2019) Regulation of programmed death-ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci 110(11):3415–3423
Sato H et al (2017) DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8(1):1751
Parkes EE et al (2016) Activation of STING-dependent innate immune signaling by S‑phase-specific DNA damage in breast cancer. J Natl Cancer Inst 109(1):djw199. https://doi.org/10.1093/jnci/djw199
van Vugt M, Parkes EE (2022) When breaks get hot: inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 8(3):174–189
Ward JF (2000) Complexity of damage produced by ionizing radiation. Cold Spring Harb Symp Quant Biol 65:377–382
Kwon J, Bakhoum SF (2020) The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov 10(1):26–39
Bhattacharya S et al (2017) RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res 45(8):4590–4605
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421(6922):499–506
Savitsky K et al (1995) A single ataxia telangiectasia gene with a product similar to PI‑3 kinase. Science 268(5218):1749–1753
Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282(5395):1893–1897
Bonner WM et al (2008) GammaH2AX and cancer. Nat Rev Cancer 8(12):957–967
Cimprich KA et al (1996) cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci U S A 93(7):2850–2855
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300(5625):1542–1548
Zou L, Liu D, Elledge SJ (2003) Replication protein A‑mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A 100(24):13827–13832
Zhao H, Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21(13):4129–4139
Liu J, Wang H, Balasubramanian MK (2000) A checkpoint that monitors cytokinesis in Schizosaccharomyces pombe. J Cell Sci 113(7):1223–1230
Xiao Z et al (2003) Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem 278(24):21767–21773
Carter T et al (1990) A DNA-activated protein kinase from HeLa cell nuclei. Mol Cell Biol 10(12):6460–6471
Lees-Miller JP, Yan A, Helfman DM (1990) Structure and complete nucleotide sequence of the gene encoding rat fibroblast tropomyosin 4. J Mol Biol 213(3):399–405
Lees-Miller SP, Chen YR, Anderson CW (1990) Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol 10(12):6472–6481
Blackford AN, Jackson SP (2017) ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66(6):801–817
Moore JK, Haber JE (1996) Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature 383(6601):644–646
Strathern JN, Shafer BK, McGill CB (1995) DNA synthesis errors associated with double-strand-break repair. Nat Genet 140(3):965–972
Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5(11):a12740
Nickoloff JA et al (2021) Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist 4(2):244–263
Baumann M, Krause M, Hill R (2008) Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer 8(7):545–554
Lord CJ, Ashworth A (2007) RAD51, BRCA2 and DNA repair: a partial resolution. Nat Struct Mol Biol 14(6):461–462
Arnaudeau C, Lundin C, Helleday T (2001) DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307(5):1235–1245
Chang HHY et al (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18(8):495–506
Mateos-Gomez PA et al (2015) Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518(7538):254–257
Ceccaldi R et al (2015) Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518(7538):258–262
Polo LM et al (2019) Efficient single-strand break repair requires binding to both poly(ADP-ribose) and DNA by the central BRCT domain of XRCC1. Cell Rep 26(3):573–581.e5
Marteijn JA et al (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15(7):465–481
Rapin I et al (2000) Cockayne syndrome and xeroderma pigmentosum. Neurology 55(10):1442–1449
Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18(1):85–98
Nojima K et al (2005) Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 65(24):11704–11711
Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493(7432):356–363
Peake JD, Noguchi E (2022) Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum Genet 141(12):1811–1836
Le DT et al (2015) PD‑1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372(26):2509–2520
Hsiehchen D et al (2020) DNA repair gene mutations as predictors of immune checkpoint inhibitor response beyond tumor mutation burden. Cell Rep Med 1(3):100034
Samstein RM et al (2021) Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat Cancer 1(12):1188–1203
Farmer H et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921
Tutt AN et al (2005) Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol 70:139–148
Bryant HE et al (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):913–917
Shen J et al (2019) PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 79(2):311–319
Kim C, Wang XD, Yu Y (2020) PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 9:e60637
Carlsen L, El-Deiry WS (2022) Anti-cancer immune responses to DNA damage response inhibitors: molecular mechanisms and progress toward clinical translation. Front Oncol 12:998388
Vendetti FP et al (2018) ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest 128(9):3926–3940
Feng X et al (2020) ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J 39(14):e104036
Dillon MT et al (2019) ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res 25(11):3392–3403
Patin EC et al (2022) Harnessing radiotherapy-induced NK-cell activity by combining DNA damage-response inhibition and immune checkpoint blockade. J Immunother Cancer 10(3):e4306. https://doi.org/10.1136/jitc-2021-004306
Sheng H et al (2020) ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer 8(1):e340. https://doi.org/10.1136/jitc-2019-000340
Zhang Q et al (2019) Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res 79(15):3940–3951
Xue Z et al (2022) PD-L1 deficiency sensitizes tumor cells to DNA-PK inhibition and enhances cGAS-STING activation. Am J Cancer Res 12(5):2363–2375
Patel P et al (2019) Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. OncoImmunology 8(11):e1638207
Haase S et al (2022) H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J Clin Invest 132(22):e154229. https://doi.org/10.1172/JCI154229
Funding
This project was funded by BMBF grant no.: 02NUK055B.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S. Classen, C. Petersen, and K. Borgmann declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Classen, S., Petersen, C. & Borgmann, K. Crosstalk between immune checkpoint and DNA damage response inhibitors for radiosensitization of tumors. Strahlenther Onkol 199, 1152–1163 (2023). https://doi.org/10.1007/s00066-023-02103-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00066-023-02103-8