
Abstract
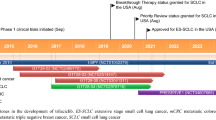
Cyclin-dependent kinases (CDKs) play a major role in regulating transitions within the cell cycle. Given the roles of CDK4/6 in promoting oncogenesis, selective inhibition of CDK4/6 has emerged as a novel approach for the treatment of breast cancer and various other tumors. While first and second generation CDK4/6 inhibitors were instrumental in targeting cell cycle pathways, they had numerous drawbacks such as limited selectivity and off-target effects. For that reason, a third generation of inhibitors was introduced and provided improved selectivity towards CDK4/6 leading to fewer side effects. To date, four compounds have been approved by the FDA as selective inhibitors of CDK4/6: palbociclib, ribociclib, abemaciclib, and trilaciclib. In this mini review, we summarize the biological, clinical, and chemical aspects of trilaciclib, a first-in-class CDK4/6 inhibitor notable for its dual role in cell cycle regulation and myelopreservation. Trilaciclib was granted FDA approval on February 2021, to improve the outcome of patients with metastatic-stage small cell lung cancer (SCLC) by protecting bone marrow suppression during chemotherapy.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Small-cell lung cancer (SCLC) is an extremely aggressive type of neuroendocrine lung tumor, characterized by genomic instability, rapid proliferation rates, and strong propensity for early metastasis [1]. The poor prognosis of SCLC is associated with increased proliferative rate and vascularity [2]. It is estimated that SCLC is responsible for causing 15% of global lung cancer incidence. The current approach to treat SCLC relies solely on chemotherapy, which can damage hematopoietic stem and progenitor cells (HSPCs) in bone marrow (BM) leading to chemotherapy-induced myelosuppression (CIM) [3]. CIM is characterized by reduced production of red blood cells, white blood cells, and platelets, which compromises long-term control of disease and the survival rate [4]. The effects of CIM can significantly impact patient health, increasing the risk of bleeding, fatigue, and infections, often requiring frequent transfusions and hospitalizations [5].
One mechanism to minimize myelosuppression is the reduction in treatment dose intensity; however, this reduces the effectiveness and eventually compromises disease control. The adverse effects associated with certain chemotherapeutic compounds can also be reduced with the aid of small molecules. For example, amifostine has been used to protect the kidneys from the undesired side-effects caused by cisplatin (a chemotherapeutic agent used in patients with advanced ovarian cancer) [6]. Likewise, dexrazoxane reduces risk of heart failure in women receiving doxorubicin for the treatment of breast cancer [7]. Unfortunately, there is concern that many such chemo-protectants, when given concomitantly, can diminish the effectiveness of chemotherapy. An alternative method to combat the myelotoxicity of chemotherapy includes the use of growth factors such as granulocyte-colony-stimulating factors and erythropoiesis-stimulating factors, G-CSF/EPO. Hematopoietic growth factors, a family of regulatory proteins, play a crucial role in the differentiation and survival of blood progenitor cells. Even though the commercial availability of these growth factors has attracted widespread application in cancer treatment, their use is restricted as they are expensive and not readily available to all patients in need, highlighting the need for alternative approaches that can proactively prevent CIM.
Cyclin-dependent kinases (CDKs) are essential proteins that regulate key stages of the cell cycle, including DNA replication and cell division. Among the CDK family, CDK4 and CDK6 are particularly crucial in driving cells from G1 to S phase. Due to their roles in cell cycle regulation, CDK4/6 play a pivotal role in the proliferation of HSPCs in the bone marrow. Temporarily arresting the cell cycle in these cells using a potent, selective CDK4/6 inhibitor can protect blood cell counts and improve survival in mice exposed to chemotherapy or high radiation doses [8, 9]. Co-administration of a CDK4/6 inhibitor with chemotherapy agents that kill proliferating cells could widen the therapeutic window by selectively inhibiting the proliferation of normal cell and not tumor cells [10]. While many tumors rely on CDK4/6 for their growth, some tumors can grow independent of CDK4/6. These include SCLC, triple-negative breast cancer, bladder cancer, human papillomavirus (HPV)-associated head and neck cancer, and prostate cancer. The standard treatment for many patients with these tumors involves myelosuppressive chemotherapy. Temporarily arresting the cell cycle in HSPCs using a CDK4/6 inhibitor during chemotherapy could potentially protect the bone marrow and immune system from the cytotoxic effect of chemotherapy without compromising its anti-tumor efficacy.
Trilaciclib (G1T28) received FDA approval in February 2021 to improve the outcome of patients with metastatic (extensive stage) SCLC by protecting bone marrow suppression during chemotherapy (Table 1). Metastatic SCLC typically refers to the spread or involvement of the cancer beyond the lung to distant organs or tissues. Trilaciclib is a first-in-class competitive CDK4/6 inhibitor that was developed by G1 Therapeutics to provide myeloprotection. Trilaciclib induces reversible G1-arrest in CDK4/6-dependent cells (such as HSPCs and lymphocytes). Due to this arrest, HSPCs are prevented from transitioning into cell cycle stages where they are susceptible to chemotherapy induced DNA damage, while CDK4/6-independent cells (such as SCLC cells) remain susceptible to the effects of the cytotoxic treatment [11]. This approach may lead to a reduced decline and quicker recovery of circulating blood cells, prevention of bone marrow depletion, and preservation of immune cell number and function, potentially resulting in a stronger immune response against the tumor. After the conclusion of chemotherapy, HSPCs are again able to resume their crucial function of blood cell production (Fig. 1).

Mechanism of action of trilaciclib
Role of cyclin-dependent kinases 4/6 in cancer
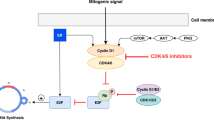
CDKs are critical regulatory enzymes governing progression through the cell cycle via complex molecular interactions [12]. The cell cycle is divided into two main stages: mitosis (M) and interphase (further broken down into G1, S, and G2 subphases) [13]. All phases of the cell cycle are regulated by oscillations in the levels of cyclin proteins, which bind and activate their cognate CDKs in cyclin-CDK complexes [14]. In mammalian cells, 21 different CDKs are regulated in part by interactions with 29 different cyclins [15, 16]. CDKs can be divided into two distinct categories: cell cycle regulators (CDK1/2/4/6) and transcriptional regulators (CDK7/8/9/11) [15]. CDK4/6 are activated by binding to Cyclin D (D1, D2, or D3), to promote progression through the G1/S checkpoint. The G1/S checkpoint is a critical decision point controlling cell proliferation as once cells enter S phase and begin DNA replication, they become committed to dividing. CDK4/6 drive progression through the G1/S checkpoint by phosphorylating the tumor suppressor retinoblastoma protein (RB) [17]. When the G1/S checkpoint is active, RB binds to the transcription factor E2F and prevents its translocation to the nucleus, thereby inhibiting its function [18]. Phosphorylation of RB by CDK4/6 causes dissociation of RB from E2F, allowing for E2F to enter the nucleus and activate a transcriptional program that drives entry into S-phase (Fig. 2). As part of this program, E2F promotes transcription of Cyclin E, which binds and activates CDK2 [19]. This initiates a positive feedback loop in which CDK2 continues to phosphorylate RB allowing further E2F-mediated transcription [18]. From here, CDK2-Cyclin E continues to promote progression through S phase until Cyclin E is degraded and CDK2 switches to bind Cyclin A [20]. In approximately 10% of human cancer, including SCLC, triple-negative breast cancer, urothelial cancer, and prostate cancer, the RB pathway is often dysregulated [21,22,23]. This can occur through the accumulation of cyclin D-CDK 4/6 complexes or the loss of functional RB protein [24]. When RB is inactive, CDK 4/6 is no longer required for cell cycle progression, leading to uncontrolled cell replication, a hallmark of cancer [22].

Regulation of the cell cycle by CDKs
CDK4/6 is hyperactivated to promote uncontrolled proliferation in many cancers, including breast, ovarian, and cervical cancers, through a variety of mechanisms, including amplifications, mutations and translocations in the genes encoding CDK4/6 or Cyclin D, mutational activation of upstream regulators that induce Cyclin D expression (e.g. PI3K/Akt and Ras/Raf/MEK/ERK signaling), and loss of negative regulators (e.g. p16INK4A) [12, 25, 26]. These genetic alterations uncouple CDK4/6 activation from normal regulatory inputs and can drive entry into S-phase even in the absence of extracellular mitogenic signals. In addition to driving progression into S-phase, E2F transcribes a wide range of genes that modulate metabolism, apoptosis, and DNA repair, further enhancing cancer cell proliferation and survival [27]. CDK4/6 also activate the master metabolic regulator mTOR Complex 1 (mTORC1) by directly phosphorylating and inhibiting the TSC complex, which is an essential upstream negative regulator of mTORC1 [28,29,30]. CDK4/6 are therefore important drivers of cancer cell growth and cell proliferation. This has led to the use of CDK4/6 inhibitors for the treatment of some cancers with hyperactive CDK4/6. Metastatic SCLC cells, however, are generally resistant to CDK4/6 inhibition, which allows trilaciclib to be used to prevent myelosuppression without reducing the effectiveness of chemotherapies that target rapidly proliferating cells.
An overview of CDK inhibitors
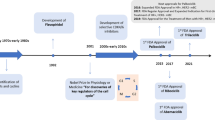
CDK inhibitors are classified into pan-CDK (first and second generation) and selective CDK (third generation) inhibitors (Table 2). Flavopiridol, the first CDK inhibitor to advance to human clinical trials, belongs to a group of first-generation CDK inhibitors, which also includes roscovitine [31]. It is a structural mimic of a natural alkaloid found in Dysoxylum binectariferum, a plant native to India. Flavoperidol inhibits multiple CDKs including CDK1, CDK2, CDK4/6, and CDK7, resulting in G1 arrest [32]. It is given intravenously and results in side-effects such as diarrhea and neutropenia. Roscovitine is a purine derivative, which inhibits CDK1, CDK2, CDK5, CDK7, and CDK9 (IC50 ~ 0.2–0.7 µM) but lacks potency against CDK 4/6 and CDK8 (IC50 > 100 µM) [33]. A phase-I study, conducted in patients with solid tumors, showed that roscovitine was not well-tolerated on a daily dosing regimen, and the patients exhibited severe side effects such as hypokalemia and elevation of liver enzymes [34]. Overall, first-generation CDK inhibitors demonstrated limited clinical benefits due to their off-target side effects, which are mainly attributed due to their action on several kinases [35, 36].
As first-generation inhibitors showed largely disappointing results in terms of clinical efficacy, safety, and tolerability, second-generation inhibitors were developed to improve selectivity. This class of inhibitors includes dinaciclib and riviciclib. As compared to flavopiridol (IC50 = 12 and 14 nM against CDK2 and CDK5, respectively), dinaciclib showed enhanced selectivity for CDK2 and CDK5 (IC50 = 1 nM for both CDK2 and CDK5). Dinaciclib is also a potent inhibitor of CDK1 and CDK9 with IC50 values 3 nM and 4 nM respectively. It is less effective at inhibiting CDK4/6, with IC50 values between 60 and 100 nM [37]. Riviciclib is a novel CDK1, CDK4, and CDK9 inhibitor with IC50 values of 79, 63, and 20 nM, respectively. It showed 40-fold selectivity toward CDK4, compared with CDK2 [38]. Even though second-generation inhibitors are relatively selective, they demonstrated inadequate balance between efficacy and toxicity in clinical studies. The toxicity of these inhibitors was mainly due to the multi-target activity against CDK1 and CDK9. This ultimately led to highly selective third generation CDK inhibitors with a wide therapeutic window and fewer off-target toxicities [39, 40].
Third generation selective CDK4/6 inhibitors include the FDA-approved drugs palbociclib, ribociclib, abemaciclib, and trilaciclib. Palbociclib is a pyridopyrimidine derivative that inhibits the activities of the cyclin D-CDK4/6 complex at concentrations of 11 nM (CDK4) and 15 nM (CDK6) [41]. Palbociclib was the first selective CDK4/6 inhibitor approved by the FDA. It was granted FDA approval in February 2016 to treat postmenopausal women with hormone receptor positive (HR-positive) and human epidermal growth factor receptor 2-negative (HER2-negative) advanced or metastatic breast cancer [42]. Abemaciclib, a pyrimidine-benzimidazole derivative, arrests cells in G1 and inhibits CDK4/6 activity at nanomolar doses (IC50 = 2 nM and 10 nM against cyclin D1-CDK4 and cyclin D1/2/3-CDK6, respectively) [43]. In addition to CDK4/6 inhibition, abemaciclib also inhibits CDK9 (IC50 = 57 nM) [44]. Abemaciclib was approved by the FDA in October of 2017 as the first CDK4/6 inhibitor for adjuvant treatment of early breast cancer. Ribociclib shows an enhanced CDK4/6 selectivity (IC50 = 10 nM and 39 nM for cyclin D1-CDK4 and cyclinD1/2/3-CDK6, respectively) [26]. Ribociclib received FDA approval in March of 2017, in combination with an aromatase inhibitor (for example, letrozole) to treat breast cancer in postmenopausal women [45].
The myelopreservation effects of trilaciclib sharply contrast with the myelosuppressive effects of palbociclib, abemaciclib, and ribociclib, which are currently approved for treating hormone receptor-positive breast cancer. Oral CDK4/6 inhibitors are typically administered continuously to inhibit CDK4/6-dependent tumor proliferation, leading to prolonged blockade of HSPC proliferation in the bone marrow and resulting in myelosuppression. In contrast, Trilaciclib is administered intravenously and intermittently, specifically before chemotherapy, to induce reversible G1 arrest of CDK4/6-dependent healthy stem cells (HSPCs), while leaving CDK4/6-independent SCLC cells unaffected. After the conclusion of chemotherapy, HSPCs are able to resume their critical function of blood cell production. This targeted approach helps prevent damage to HSPCs, allowing for precise control over the duration of HSPC cycle arrest and avoiding the prolonged myelosuppression effects often associated with oral CDK4/6 inhibitors [46].
The discovery of trilaciclib
Trilaciclib is a conformationally restricted tricyclic analog of ribociclib that was created using rational structure-based drug design (Fig. 3A) [47]. With an aim to produce potent and selective CDK4/6 inhibitors, G1 Therapeutics developed a structurally diverse set of compounds with a N-(pyridin-2-yl) pyrimidin-2-amine core ring system. The N-(pyridin-2-yl) pyrimidin-2-amine core is found in all previous examples of selective CDK4/6 inhibitors. To determine selectivity, a series of compounds were tested against cyclin A/CDK2 and cyclin E/CDK2, and biochemical profiling was done against cyclin D1/CDK4 and cyclin D3/CDK6 to access potency. Compounds with over 100-fold selectivity for cyclin D1/CDK4 versus cyclin E/CDK2 were screened for their potential to induce G1 arrest in normal H268 fibroblast cells with a functioning RB pathway. In addition to CDK4 kinase activity, candidate compounds were also tested against CDK6 kinase activity using palbociclib as a reference compound. All compounds were found to be more potent with respect to palbociclib, with Trilaciclib demonstrating an appropriate balance of efficacy, safety, and selectivity.

A The discovery of trilaciclib; (B) SAR analysis of trilaciclib; activities are against CDK4/cycD1 complex; selectivities are for CDK4 over CDK2
Next, the effects of substitution on the core tricyclic lactam ring of trilaciclib were studied (Fig. 3B) [48]. The potency was greatly diminished when the cyclohexane ring was replaced with a cyclopentane ring or any other alkyl substituents. In studying the central heterocyclic ring structure, an N-methyl piperazine ring system yielded maximum inhibition. The N-methyl piperazine is required for an appropriate balance of selectivity and potency. Lastly, the presence of the amide group is necessary for inhibition as loss of the carbonyl group had a negative impact on the selectivity and efficacy.
Preclinical assessment of trilaciclib
Trilaciclib selectively targeted both CDK4/cyclin D1 and CDK6/cyclinD3 at concentrations of 1 and 4 nmol/L, respectively in microfluidic kinase detection assays. Inhibition of CDK4 was more than three orders of magnitude greater than CDK2/cyclin A, CDK2/cyclin E, CDK5/p25, CDK5/p35, and CDK7/cyclin H/Mat1 inhibition. Additionally, trilaciclib was approximately 50-fold more selective for CDK4 than CDK9/cyclin T. Trilaciclib inhibited the proliferation of the CDK 4/6-dependent cell line HS68, with an EC50 of 30 nmol/L, whereas no G1 arrest was observed in the CDK4/6- independent A2058 cell line. Phospho-RB Western blot analysis demonstrated that, in RB-dependent cell lines, trilaciclib blocked RB phosphorylation for 16 h after exposure, whereas the CDK4/6- independent, A2058 cell line exhibited no RB or phospho-RB expression, as expected. To demonstrate that trilaciclib induces transient and reversible G1 arrest, HS68 cells were treated with trilaciclib at 300 nmol/L for 24 h, and at various time-intervals, cell-cycle analysis was performed. 16 h of trilaciclib treatment induced G1 arrest, and subsequent washout resulted in cells reentering the cell cycle indicating that trilaciclib effects are reversible. To assess whether the trilaciclib-induced G1 arrest protects CDK4/6 dependent cells from chemotherapy-induced damage (i.e., apoptosis or DNA damage), HS68 cells were pretreated with trilaciclib or control for 16 h, at which time an array of chemotherapies, such as carboplatin, doxorubicin, etoposide, and camptothecin, were added. A dose-dependent decrease in γH2AX foci, a marker of DNA damage, was observed with increasing trilaciclib concentrations suggesting a decrease in chemotherapy-induced DNA damage. A similar dose-dependent decrease in caspase-3/7 activation was also observed suggesting an attenuation of apoptosis. Finally, trilaciclib induced suppression of proliferation was also observed in a dose-dependent manner in murine and canine HSPCs [47].
Trilaciclib in the clinical management of metastatic SCLC
In February 2021, FDA approved Trilaciclib as a supportive care along with platinum/etoposide or topotecan/etoposide regimen to decrease the incidence of chemotherapy-induced myelosuppression. Trilaciclib proactively preserves bone marrow progenitor cells from its chemotherapy-induced apoptosis by causing transient G1 arrest. The efficacy of trilaciclib as a myeloprotective agent was evaluated in a randomized, double-blind, placebo-controlled phase II study in patients with newly diagnosed metastatic SCLC [49]. 52 patients were given trilaciclib prior to carboplatin, etoposide, and atezolizumab (E/P/A), while 53 patients received placebo. As compared to patients who received placebo, prior administration of trilaciclib resulted in significant reduction in mean duration of severe neutropenia (0 versus 4 days; P < 0.0001) and in the occurrence of severe neutropenia (1.9% versus 49.1%; P < 0.0001). Additionally, administration of trilaciclib improved red blood cells (RBC) and platelet counts as well as improved health-related quality of life standards. Compared to the placebo, fewer grade 3/4 adverse effects were present, mainly due to less high-grade hematological toxicity. Simultaneously, phase Ib/II studies conducted to assess the safety and tolerability of trilaciclib with the etoposide/carboplatin combination demonstrated an improvement in the patient’s tolerability of chemotherapy as shown by myelopreservation across multiple hematopoietic lineages. As compared to placebo, patients who received trilaciclib showed an improved overall response rate (66.7% versus 56.8%; P = 0.3831), overall survival rate (10.9 versus 10.6 months; Hazard Ratio (HR) 0.87; P = 0.6107), and median Progression-Free Survival (6.2 versus 5.0 months; HR 0.71; P = 0.1695) resulting in fewer supportive care interventions and dose reductions [50].
Synthesis of trilaciclib
The initial synthesis of trilaciclib was accomplished in seven steps starting from 5-bromo-2,4-dichloropyrimidine (1, Scheme 1) [48]. Nucleophilic aromatic substitution with tert-butyl ((1-aminocyclohexyl) methyl) carbamate [2] was followed by the Sonogashira coupling with 3,3-diethoxyprop-1-yne to yield 3. The pyrrolopyrimidine ring system was formed via a two-step procedure involving treatment with TBAF followed by acid-catalyzed hydrolysis of the acetal intermediate. Oxidation of the aldehyde followed by amide bond formation yielded spirocycle 5, which underwent Buchwald-Hartwig amination to produce trilaciclib.

First generation synthesis of trilaciclib
To improve yield and scalability, a second-generation synthesis of trilaciclib was developed (Scheme 2). The 7-deazapurine scaffold was formed from nucleophilic enolate addition to aldehyde 8 to yield 9. Compound 9 could also be obtained through a multistep addition to ester 11. The final Buchwald coupling was replaced with an SNAr of aniline 15 to mesylate 9 yielding trilaciclib in 63.7% yield. Overall, this second-generation synthesis was a significant improvement over the first-generation synthesis as the route removed the requirement of chromatographic purifications, allowing large scale (>400 g) synthesis.

Second generation synthesis of trilaciclib
Docking mode of trilaciclib
The binding mode of trilaciclib with CDK4 has yet to be investigated; however, its interactions with CDK6 have been studied. The docking of trilaciclib with CDK6 shows formation of 2 hydrogen bonds: one between the carbonyl of Val101 and the exocyclic NH of the pyridine system and a second between the amide NH of Val101 and the nitrogen of the pyridine system (Fig. 4D) [51]. Palbociclib, ribociclib and abemaciclib showed almost similar interactions with Val101 at the hinge region of the binding site. Palbociclib and ribociclib share this common feature of H-bonding between their amide and the Asp-Phe-Gly (DFG) domain of the CDK4/6 pocket (Fig. 4A, B). As compared to abemaciclib, palbociclib and ribociclib are characterized by larger substituents (dimethylamide and acetyl functionality), which interact with the hydrophobic back pocket of CDK4/6’s ATP binding site. Consequently, due to this steric interaction, palbociclib and ribociclib have higher selectivity when compared to abemaciclib, which is more readily buried in ATP pocket (Fig. 4C). The crystallographic complexes of palbociclib, ribociclib, and abemaciclib with CDK6 revealed that the positively charged piperazine ring of each inhibitor faces the solvent-exposed ridge containing Asp104 and Thr107. In CDK1, CDK2, CDK3 and CDK5, a lysine residue replaces the CDK6-Thr107 interaction causing a possible electrostatic repulsion with the piperazine. This could be the source of the higher CDK6 selectivity for these three inhibitors [52]. Due to the presence of the same piperazine side chain on trilaciclib, a similar orientation may exist; however, no crystallography exists for trilaciclib.

Binding modes of palbociclib (A), ribociclib (B), and abemaciclib (C). D Docking mode of trilaciclib as described in the literature [51]. Green and yellow represents hydrogen bonding sites at the hinge region; orange represents the region exposed to the solvent-exposed ridge; and blue depicts hydrogen bonding interactions at the DFG domain
Dosage and safety profile
Dosage regimen
Trilaciclib is manufactured as a 300 mg sterile, preservative-free, yellow, lyophilized cake in a single-dose vial for reconstitution and further dilution. The recommended dose of trilaciclib is 240 mg/m2 per dose, and it is administered as a 30-min intravenous infusion, 4 h prior to the start of chemotherapy on each day chemotherapy is administered [53, 54]. The subsequent dose of trilaciclib must be taken no later than 28 h after administering its previous dose. If the trilaciclib dose is missed, the patient should discontinue chemotherapy the day trilaciclib dose was missed, and then resume both trilaciclib and chemotherapy on the next scheduled day for chemotherapy. If trilaciclib is discontinued, the patient should wait 96 h from the last dose of trilaciclib before resuming the chemotherapy-only session [55].
Safety considerations
Multiple clinical trials pertaining trilaciclib reported that common adverse reactions have occurred in ≥10% of patients. These adverse reactions included fatigue, hypocalcemia, hypokalemia, hypophosphatemia, increased aspartate aminotransferase levels, headache, and pneumonia. Serious adverse reactions, including respiratory failure, hemorrhage, and thrombosis, occurred in >3% of patients. 9% of patients were required to permanently discontinue the infusion due to the occurrence of adverse reactions including pneumonia (2%), asthenia (2%), injection-site reactions, thrombocytopenia, cerebrovascular accident, ischemic stroke, infusion-related reaction, respiratory failure, and myositis (<1% each) [56]. Since trilaciclib administration can cause injection-site reactions, patients receiving IV infusion should be monitored for signs and symptoms of injection-site reactions, including infusion-site pain and erythema during infusion. Trilaciclib is toxic to the fetal embryo and thus, females of reproductive potential should use an effective method of contraception during treatment and for at least 3 weeks after the final dose [55]. Of note, trilaciclib is an inhibitor of organic cation transporter 2 (OCT2), multidrug and toxin extrusion protein 2 (MATE-1), and MATE-2K. Consequently, co-administration of trilaciclib may increase the concentration or net accumulation of OCT2, MATE1, and MATE-2K substrates in the kidney.
Benefits of trilaciclib over existing therapies
The regular dosing of CDK4/6 inhibitors causes myelosuppression due to the continual blockade of HSPC proliferation which eventually reduces the production of new blood cells [31, 57]. Trilaciclib did not show lingering myelosuppressive effect while inhibiting CDK4/6. In contrast to lineage-specific strategies (such as G-CSF/EPO), which are used to treat acute CIM, trilaciclib protects all blood cell lineages. Even though growth factor therapy induces transient recovery of blood cell count, there is an increased risk of bone marrow failure and leukemia [58, 59]. On the other hand, trilaciclib-mediated HSC preservation provides a method to protect cells without developing risk of BM failure and leukemia [10]. Trilaciclib also reduced the incidence of chemotherapy-induced late BM toxicity by regulating the cell cycle of rapidly differentiating HSCs while getting exposed to cytotoxic agents. Due to this, clinical concerns, which arose due to CIM-dose reduction and delay of treatment were prevented.
Conclusion
Chemotherapy-induced myelosuppression is currently a limiting factor in approaches to treat metastatic SCLC. As a result, co-administration of cytotoxic chemotherapy agents with CDK4/6 inhibitors can improve the therapeutic window by protecting normal cells from the damage caused by chemotherapy. The novel tricyclic lactam scaffold of trilaciclib was developed by G1 Therapeutics using molecular constriction of a known CDK 4/6 inhibitor. Trilaciclib induced reversible and transient G1-arrest of CDK4/6 dependent cells, and thus it was approved by FDA for its myeloprotective and anti-tumor efficacy. The current clinical use of trilaciclib highlights its efficacy and application over the known existing strategies to treat SCLC.
Abbreviations
- (BM):
-
Bone Marrow
- (CDK):
-
Cyclin-Dependent Kinase
- (CIM):
-
Chemotherapy Induced Myelosuppression
- (DIPEA):
-
N, N-Diisopropylethylamine
- (EPO):
-
Erythropoiesis-stimulating agents
- (G-CSF):
-
Granulocyte-colony stimulating factor
- (HER2-negative):
-
Human Epidermal Growth Factor Receptor 2-negative
- (HR-positive):
-
Hormone Receptor Positive
- (HSC):
-
Hematopoietic stem cells
- (HSPC):
-
Hematopoietic stem and progenitor cells
- (mPFS):
-
median Progression Free Survival
- (mOS):
-
median Overall Survival
- (MATE):
-
Multidrug and Toxin Extrusion protein
- (MOA):
-
Mechanism of Action
- (OCT2):
-
Organic Cation Transporter 2
- (PK):
-
Pharmacokinetics
- (RB):
-
Retinoblastoma protein
- (SCLC):
-
Small Cell Lung Cancer
- (TBAF):
-
Tetra-n-butylammonium fluoride
References
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17:725–37.
Tariq S, Kim SY, Cheng H, Monteiro DONJ. Update 2021: management of small cell lung cancer. Lung. 2021;199:579–87.
Dhillon S. Trilaciclib: first approval. Drugs. 2021;81:867–74.
Lyman GH. Chemotherapy dose intensity and quality cancer care. Oncol (Williston Park). 2006;20:16–25.
Ferrarotto R, Anderson I, Medgyasszay B, García-Campelo MR, Edenfield W, Feinstein TM, et al. Trilaciclib prior to chemotherapy reduces the usage of supportive care interventions for chemotherapy-induced myelosuppression in patients with small cell lung cancer: pooled analysis of three randomized phase 2 trials. Cancer Med. 2021;10:5748–56.
Kouvaris JR, Kouloulias VE, Vlahos LJ. Amifostine: the first selective-target and broad-spectrum radioprotector. Oncologist. 2007;12:738–47.
Macedo AVS, Hajjar LA, Lyon AR, Nascimento BR, Putzu A, Rossi L, et al. Efficacy of dexrazoxane in preventing anthracycline cardiotoxicity in breast cancer. JACC: CardioOncology. 2019;1:68–79.
Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–36.
Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–87.
He S, Sharpless NE, He S, Sharpless NE, Roberts PJ, Sorrentino JA, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017;9:eaal3986.
Powell K, Prasad V. Concerning FDA approval of trilaciclib (Cosela) in extensive-stage small-cell lung cancer. Transl Oncol. 2021;14:101206-.
Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell. 2017;32:761–76.e6.
Norbury C, Nurse P. Animal cell cycles and their control. Ann Rev Biochem. 1992;61:441–68.
Oudah K, Abdou N, Serya R, Abouzid K. An overview on the prospective CDKs inhibitors as anti-cancer drugs: review article. J Am Sci. 2017;13:6–23.
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122.
Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol. 2009;11:1275–6.
Ammazzalorso A, Agamennone M, De Filippis B, Fantacuzzi M. Development of CDK4/6 inhibitors: a five years update. Molecules. 2021;26:1488.
Rubin SM, Sage J, Skotheim JM. Integrating old and new paradigms of G1/S control. Mol Cell. 2020;80:183–92.
Sherr C. The pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–95.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46.
Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci USA. 2014;111:3110–5.
Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7.
Macleod KF. The RB tumor suppressor: a gatekeeper to hormone independence in prostate cancer? J Clin Invest. 2010;120:4179–82.
Donjerkovic D, Scott DW. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000;10:1–16.
Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. 2018;28:911–25.
Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–67.
Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer. 2022;22:356–72.
Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255–69.
Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A, Teleman AA. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31:107504.
Zacharek SJ, Xiong Y, Shumway SD. Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer Res. 2005;65:11354–60.
Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist. 2015;20:483–90.
Matranga CB, Shapiro GI. Selective sensitization of transformed cells to flavopiridol-induced apoptosis following recruitment to S-Phase1. Cancer Res. 2002;62:1707–17.
Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, et al. Roscovitine in cancer and other diseases. Ann Transl Med. 2015;3:135.
Gupta P, Narayanan S, Yang D-H Chapter 9 - CDK inhibitors as sensitizing agents for cancer chemotherapy. In: Chen Z-S, Yang D-H, editors. Protein kinase inhibitors as sensitizing agents for chemotherapy. 4: Academic Press; 2019. p. 125-49.
Blum KA, Ruppert AS, Woyach JA, Jones JA, Andritsos L, Flynn JM, et al. Risk factors for tumor lysis syndrome in patients with chronic lymphocytic leukemia treated with the cyclin-dependent kinase inhibitor, flavopiridol. Leukemia. 2011;25:1444–51.
Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs. 2013;22:723–38.
Ettl T, Schulz D, Bauer RJ. The renaissance of cyclin dependent kinase inhibitors. Cancers (Basel). 2022;14:293.
Joshi KS, Rathos MJ, Joshi RD, Sivakumar M, Mascarenhas M, Kamble S, et al. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol Cancer Therapeutics. 2007;6:918–25.
Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–53.
Czudor Z, Balogh M, Bánhegyi P, Boros S, Breza N, Dobos J, et al. Novel compounds with potent CDK9 inhibitory activity for the treatment of myeloma. Bioorg Med Chem Lett. 2018;28:769–73.
Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18:568–76.
Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21:4760–6.
Shapiro GI, Rosen LS, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, et al. A first-in-human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J Clin Oncol. 2013;31:2500-.
Jorda R, Hendrychová D, Voller J, Řezníčková E, Gucký T, Kryštof V. How selective are pharmacological inhibitors of cell-cycle-regulating cyclin-dependent kinases? J Medicinal Chem. 2018;61:9105–20.
Shah A, Bloomquist E, Tang S, Fu W, Bi Y, Liu Q, et al. FDa approval: ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin Cancer Res. 2018;24:2999–3004.
Hart LL, Ferrarotto R, Andric ZG, Beck JT, Subramanian J, Radosavljevic DZ, et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: results from a randomized, double-blind, placebo-controlled phase II study. Adv Ther. 2021;38:350–65.
Bisi JE, Sorrentino JA, Roberts PJ, Tavares FX, Strum JC. Preclinical characterization of G1T28: a novel cdk4/6 inhibitor for reduction of chemotherapy-induced myelosuppression. Mol Cancer Therapeutics. 2016;15:783–93.
Strum JC, Bisi JE, Roberts PJ, Sorrentino J, Storrie-White H, inventors; G1 Therapeutics, Inc., USA. assignee. Anti-Neoplastic combinations and dosing regimens using CDK4/6 inhibitor compounds to treat RB-Positive Tumors patent US 2017/0182043 A1. 2016.
Daniel D, Kuchava V, Bondarenko I, Ivashchuk O, Reddy S, Jaal J, et al. Trilaciclib prior to chemotherapy and atezolizumab in patients with newly diagnosed extensive-stage small cell lung cancer: A multicentre, randomised, double-blind, placebo-controlled Phase II trial. Int J Cancer. 2020;148:2557–70.
Weiss JM, Csoszi T, Maglakelidze M, Hoyer RJ, Beck JT, Domine Gomez M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30:1613–21.
Yuan K, Wang X, Dong H, Min W, Hao H, Yang P. Selective inhibition of CDK4/6: a safe and effective strategy for developing anticancer drugs. Acta Pharm Sin B. 2021;11:30–54.
Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Therapeutics. 2016;15:2273–81.
Li C, Hart L, Owonikoko TK, Aljumaily R, Rocha Lima CM, Conkling PR, et al. Trilaciclib dose selection: an integrated pharmacokinetic and pharmacodynamic analysis of preclinical data and Phase Ib/IIa studies in patients with extensive-stage small cell lung cancer. Cancer Chemother Pharm. 2021;87:689–700.
Li C, Rich B, Bullock JM, Barrière O, Marier J-F, Beelen A. Population pharmacokinetics and exposure–response of trilaciclib in extensive-stage small cell lung cancer and triple-negative breast cancer. Br J Clin Pharmacol. 2023;89:1067–79.
COSELATM (trilaciclib): US prescribing information: G1 Therapeutics; 2021 [Available from: https://www.g1therapeutics.com/cosela/pi/.
Weiss J, Goldschmidt J, Andric Z, Dragnev KH, Gwaltney C, Skaltsa K, et al. Effects of trilaciclib on chemotherapy-induced myelosuppression and patient-reported outcomes in patients with extensive-stage small cell lung cancer: pooled results from three phase II randomizeda, double-blind, placebo-controlled studies. Clin Lung Cancer. 2021;22:449–60.
Vidula N, Rugo HS. Cyclin-dependent kinase 4/6 inhibitors for the treatment of breast cancer: a review of preclinical and clinical data. Clin Breast Cancer. 2016;16:8–17.
Hershman D, Neugut AI, Jacobson JS, Wang J, Tsai WY, McBride R, et al. Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007;99:196–205.
Hornung RL, Longo DL. Hematopoietic stem cell depletion by restorative growth factor regimens during repeated high-dose cyclophosphamide therapy. Blood. 1992;80:77–83.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, T.I., Joshi, J.N., Valvezan, A.J. et al. A review of trilaciclib, a first-in-class cyclin-dependent kinase 4/6 inhibitor, for the management of metastatic small-cell lung cancer. Med Chem Res (2024). https://doi.org/10.1007/s00044-024-03288-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00044-024-03288-y