
Abstract
The cyclin-dependent kinase (CDK) 4/6 inhibitors belong to a new class of drugs that interrupt proliferation of malignant cells by inhibiting progression through the cell cycle. Three such inhibitors, palbociclib, ribociclib, and abemaciclib were recently approved for breast cancer treatment in various settings and combination regimens. On the basis of their impressive efficacy, all three CDK4/6 inhibitors now play an important role in the treatment of patients with HR+, HER2− breast cancer; however, their optimal use still needs to be established. The three drugs have many similarities in both pharmacokinetics and pharmacodynamics. However, there are some differences on the basis of which the choice for a particular CDK4/6 inhibitor for an individual patient can be important. In this article, the clinical pharmacokinetic and pharmacodynamic profiles of the three CDK4/6 inhibitors are reviewed and important future directions of the clinical applicability of CDK4/6 inhibitors will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
CDK4/6 inhibitors play an eminent role in the treatment of advanced HR+, HER2− breast cancer and are of potential value in the (neo)adjuvant setting. |
Biomarkers need to be identified to optimize the therapy in patients treated with CDK4/6 inhibitors and to select patients with other types of (breast) cancer who could potentially benefit. |
Combinations with other drug modalities with a different pharmacodynamic profile need to be defined to further enhance the efficacy, overcome resistance, and widen the applicability of CDK4/6 inhibitors. |
1 Introduction
Of all patients with stage IV breast cancer, approximately 75% are hormone receptor-positive (HR+) and human epidermal growth factor receptor 2 negative (HER2−) [1,2,3]. As advanced or metastatic breast cancer represents an incurable disease, the main purpose of treatment is to delay disease progression, preferably with anticancer drugs that are patient friendly in their use and toxicity [4]. Patients with HR+, HER2− breast cancer are often treated with single-agent endocrine therapy. However, endocrine resistance always develops, resulting in the need for cytotoxic chemotherapy [5,6,7].
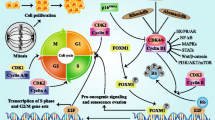
Recently, a novel drug class, the cyclin dependent kinase (CDK) 4/6 inhibitors, has been introduced as a treatment option for patients with HR+, HER2− advanced breast cancer, either as first-line therapy combined with an aromatase inhibitor or as second-line therapy in combination with fulvestrant [8,9,10,11,12,13]. The CDK-RB1-E2F pathway targeted by CDK4/6 inhibitors is essential for progression through the cell cycle and is disrupted in the majority of cancers [14,15,16,17,18]. In breast cancer, the activation of estrogen receptors as well as other proliferation-inducing signals stimulate the complexation of CDK4/6 with cyclin D1 [19]. Binding of CDK4/6 to cyclin D1 induces phosphorylation of the retinoblastoma (Rb) tumor suppressor protein, releasing its inhibitory effect and thereby providing the starting signal for cell division [16, 18, 20,21,22]. Normally, CDK4 and CDK6 are inhibited by the protein p16. However, in cancer this mechanism of cell cycle control is often disrupted [19, 23]. Furthermore, cyclin D1, the binding partner of CDK4/6, is often overexpressed in patients with HR+, HER2− breast cancer leading to continuous activation of the cyclin D1–CDK4/6 complex [24,25,26]. Inhibition of CDK4/6 induces complete dephosphorylation of Rb, resulting in sequestration of the transcription factor E2F and subsequent inhibition of cell cycle progression [27,28,29].
The effectiveness of CDK4/6 inhibitors can be increased by combining them with drugs that prevent the downstream estrogen-dependent stimulation of the cancer cell. Inhibition of the estrogen pathway—by endocrine therapy—results in downregulation of cyclin D1 and reduced complexation of CDK4 and CDK6 [30]. Therefore, the selective CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib are given in combination with endocrine therapy (aromatase inhibitors or fulvestrant) in the treatment of HR+, HER2− breast cancer [31,32,33].
So far, no head-to-head comparison has been carried out between the three different CDK4/6 inhibitors in HR+, HER2− advanced breast cancer. At present, CDK4/6 inhibitors are prescribed based on individual physicians’ experience with these CDK4/6 inhibitors, differences in toxicity profiles, costs, or the preference policy of the hospital. Insights into pharmacological profiles of these three CDK4/6 inhibitors may help to rationalize the selection of the most optimal inhibitor for the individual patient. In this review, the pharmacokinetics, pharmacodynamics, efficacy, and tolerability of palbociclib, ribociclib, and abemaciclib for the treatment of breast cancer are discussed. Furthermore, future directions of the clinical applicability of the CDK4/6 inhibitors are discussed such as the potential role of biomarkers in determining treatment strategy in specific patient groups, the combination with other drug modalities, and the use of these therapies in other types of (breast) cancer (Fig. 1).

Mechanism of action of CDK4/6 inhibitors. The CDK4/6-cyclin D1 complex induces phosphorylation of the retinoblastoma (Rb) tumor-suppressor protein. Free transcription factor E2F stimulates cell transition from the G1 to the S phase and cell division. AKT protein kinase B, CDK4/6 cyclin dependent kinase 4 and 6, ER estrogen receptor, E2F transcription factor family, HER2 human epidermal growth factor receptor-2, PIK3 phosphoinositide 3-kinase, Rb retinoblastoma tumor suppressor protein, mTOR mammalian target of rapamycin
2 Pharmacokinetics
2.1 Pharmacokinetic Profiles
Palbociclib (Ibrance®, Pfizer, New York, USA) received US FDA approval in 2015, with a recommended starting dosage of 125 mg once daily in a ‘3 weeks on and 1 week off’ schedule in combination with a non-steroidal aromatase inhibitor or the selective estrogen receptor degrader (SERD) fulvestrant [32]. After rapid absorption of palbociclib, the maximum concentration (Cmax) is achieved within 6–12 h and the drug is being eliminated with an elimination half-life of 24–34 h. Steady-state will be achieved after 4–5 elimination half-lives, which is important to predict the onset of action or improvement of toxicity after treatment discontinuation. Ribociclib (Kisqali®, Novartis, Basel, Switzerland) received US FDA approval in 2017, with a recommended starting dosage of 600 mg once daily in a ‘3 weeks on/1 week off’ schedule in combination with an aromatase inhibitor or fulvestrant [33]. Ribociclib has a high absorption rate and reaches Cmax within 1–4 h upon intake. The elimination half-life of ribociclib is 30–55 h. Finally, abemaciclib (Verzenios®, Eli Lilly, Indianapolis, USA) received US FDA approval in 2017, with a recommended starting dosage of 150 mg twice daily in a continuous dosing schedule combined with an aromatase inhibitor or fulvestrant. In addition, abemaciclib is also approved for administration as monotherapy with a starting dosage of 200 mg twice daily [31]. Abemaciclib achieves its Cmax within 8 h upon intake and the drug elimination half-life is 17–38 h. Pharmacokinetic parameters of palbociclib, ribociclib, and abemaciclib are depicted in Table 1.
2.2 Similarities and Differences in Pharmacokinetics
2.2.1 Absorption
The pharmacokinetic profiles of the three inhibitors are strikingly similar. After rapid absorption and distribution, all three CDK4/6 inhibitors are metabolized mainly by CYP3A4. Abemaciclib is given in a continuous schedule of two doses per day, whereas palbociclib and ribociclib are given once daily in ‘3 weeks on/1 week off’ schedules. Only for abemaciclib, preclinical studies have displayed saturation of drug absorption, which supported the development and registration of a twice-daily dosing regimen to improve drug absorption [34, 35]. Furthermore, preclinical studies showed that continuous administration of abemaciclib reduced tumor growth more efficiently compared with an intermittent schedule [34,35,36]. The efficacy of palbociclib and ribociclib in continuous dosing schedules compared with the efficacy of the current intermittent schedule is unknown, but hematological toxicities urged the development of an intermittent schedule.
2.2.2 Distribution
From a biopharmaceutical point of view, a distinguishing feature of abemaciclib in comparison with the other CDK4/6 inhibitors is its theoretical ability to penetrate breast tissue and the blood–brain barrier more efficiently due to its higher lipophilicity (cLog P 5.5 versus 2.7 and 2.3, see Table 1). In support of this theoretical advantage, preclinical data from abemaciclib in human xenograft models showed decreased tumor growth in the brain, and when compared with palbociclib and ribociclib, abemaciclib had the highest unbound brain-to-plasma ratio, suggesting effective penetration [38]. In addition, a clinical study indicated that systemic treatment with abemaciclib resulted in similar concentrations of abemaciclib in both plasma and cerebrospinal fluid [39]. In addition, both palbociclib and ribociclib are substrates for breast cancer resistance protein (BCRP; ABCG2) and P-glycoprotein (P-gP; ABCB1), which is an additional explanation for their limited brain penetration capacity observed in preclinical studies [40, 41]. The efficacy of the three different CDK4/6 inhibitors in treating brain metastases is an important unanswered question in daily clinical practice. Prospective trials are ongoing to evaluate brain penetration and efficacy of palbociclib (NCT02774681), ribociclib (NCT02933736), and abemaciclib (NCT02308020) in the treatment of brain metastases.
2.2.3 Metabolism and Excretion
For all three CDK4/6 inhibitors, metabolization occurs hepatically, and was shown by both in vitro and in vivo studies to be primarily mediated by CYP3A4. Concomitant administration of CDK4/6 inhibitors and strong CYP3A4 inhibitors (i.e., itraconazole, ketoconazole, and ritonavir) can lead to an increase in exposure of CDK4/6 inhibitors in the blood and an increased chance of toxicity. The administration of palbociclib in combination with the strong CYP3A4 inhibitor itraconazole resulted in an increase in area under the concentration–time curve from zero to infinity (AUC0–inf) and Cmax of 87% and 34%, respectively; ribociclib in combination with ritonavir increased ribociclib exposure up to 3.2-fold; and based on an animal model, ketoconazole is predicted to increase the exposure of abemaciclib by up to 16-fold. The FDA therefore advises to avoid concomitant use of CDK4/6 inhibitors and strong CYP3A4 inhibitors [31,32,33].
Yu et al. published a physiologically based pharmacokinetic model—based on silico, in vitro, and in vivo pharmacokinetic data—in which they simulated the effects of the moderate CYP3A4 inhibitors verapamil and diltiazem on palbociclib pharmacokinetics. They estimated an increase in Cmax and AUC of 22% and 38% for verapamil and 23% and 42% for diltiazem, respectively. The authors concluded that the risk of drug–drug interactions for palbociclib co-administered with moderate CYP3A4 inhibitors is relatively modest and that no upfront dose adjustment is needed [42]. However, a 40% increase in exposure could still be clinically relevant, especially since a higher palbociclib exposure is associated with increased toxicity [42]. A clear relationship between exposure and toxicity is not described in literature for either ribociclib or abemaciclib. More research is needed to elucidate the plausible relationship between exposure and adverse events. Currently, a randomized pharmacokinetic cross-over trial is ongoing to evaluate the effect of the moderate CYP3A4 inhibitor erythromycin on the pharmacokinetics of palbociclib (Netherlands Trial Register NL7549) and results are expected in 2021.
2.2.4 Metabolism and Pharmacogenetics
Besides CYP3A4-mediated metabolism, palbociclib is also hepatically metabolized by the sulfotransferase enzyme SULT2A1 [43]. SULTs, such as SULT2A1, are highly expressed in the small intestine, liver, and adrenal cortex and metabolize orally administered drugs through sulfate conjugation. Pharmacogenetic variation in SULT2A1 activity could affect the drug’s biotransformation and thus its pharmacokinetics [44]. Variability in CYP3A4 and SULT2A1 drug metabolization can often be partly explained by genetic polymorphisms. The extent to which this applies for all three CDK4/6 inhibitors is unclear and requires further research. Sequencing of CYP3A4 and SULT2A1 genes in patients treated with CDK4/6 inhibitors may potentially identify subpopulations requiring dose adjustments. The incidence of any neutropenia and grade 3 neutropenia specifically is higher in the Asian population compared with non-Asian patients treated with palbociclib [10, 45], concordant with the observed higher mean steady-state concentration of palbociclib [45]. The reason for differences in pharmacokinetics between Asian and non-Asian populations is unknown, but could be related to genetic predispositions influencing metabolism or higher numbers of CDK receptors and/or sensitivity of the receptors [46].
3 Pharmacodynamics
CDK4 is a prominent oncogenic driver in breast cancer, while CDK6 plays a crucial role in differentiation of hematopoietic stem cells [47, 48]. Palbociclib, ribociclib, and abemaciclib display subtle differences in kinase selectivity. Abemaciclib is the most potent CDK4/6 inhibitor and is approximately five times more potent against CDK4 than CDK6, which leads to the expectation that abemaciclib exerts less hematological toxicity (Table 2) [34, 37, 49]. Preclinical drug-exposure experiments showed inhibition of CDK4 and CDK6 with half maximal inhibitory concentrations (IC50) of 9–11 nM and 15 nM (ratio IC50 CDK4:CDK6 is 1:1.5) for palbociclib, 10 nM and 39 nM (1:4) for ribociclib, and 2 nM and 9.9 nM (1:5), for abemaciclib, respectively [50, 51]. In contrast to palbociclib and ribociclib, abemaciclib was shown to be a potent inhibitor of CDK9 as well [50]. CDK9 inhibition could potentially modify the cascade of glycogen synthase kinase 3 (GSK3)-mediated effects of abemaciclib leading to the specific intestinal toxicity observed [52, 53].
4 Clinical Efficacy
4.1 Advanced Breast Cancer
The effects of the CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib in combination with non-steroidal aromatase inhibitors were studied in postmenopausal ER+, HER2− advanced breast cancer in the first-line setting in three large randomized phase III trials, PALOMA-2 (palbociclib), MONALEESA-2 (ribociclib), and MONARCH-3 (abemaciclib) [8,9,10]. All three CDK4/6 inhibitors significantly prolonged the progression free survival (PFS) with almost identical hazard ratios (HR) for PFS (Table 2). Overall survival (OS) data for these studies are not available yet. However, an OS benefit was found in the MONALEESA-7 trial, which investigated the addition of ribociclib to the gonadotropin-releasing hormone (GnRH) agonist goserelin with either tamoxifen or a non-steroidal aromatase inhibitor in pre- or perimenopausal women (HR 0.71; 95% CI 0.54–0.95) [54].
The effects of CDK4/6 inhibitors in addition to fulvestrant mainly during the second line of treatment in patients with HR+, HER2− advanced breast cancer were studied in the three large randomized phase III trials; PALOMA-3 (palbociclib), MONALEESA-3 (ribociclib), and MONARCH-2 (abemaciclib) [12, 13, 55]. All phase III trials showed prolongation of the PFS in the CDK4/6 inhibitor group as well as an OS benefit [56,57,58].
Based on the above described results, CDK4/6 inhibitors are registered for first- and second-line treatment in advanced HR+, HER2− breast cancer patients. Their most optimal position during treatment (i.e., first or second line) is a matter of debate (59–61), and is being investigated in the currently ongoing randomized phase III SONIA trial (ClinicalTrials.gov identifier NCT03425838) [61]. Due to the unique study design, this study will also allow for a head-to-head comparison of the three FDA-approved CDK4/6 inhibitors to identify the differences in clinical pharmacology.
4.2 Adjuvant Setting
The positive results in the advanced breast cancer setting raised interest in CDK4/6 inhibitors as potential treatments in the neo-adjuvant and adjuvant setting. For all three CDK4/6 inhibitors, large randomized phase III trials are currently ongoing to investigate the addition of a CDK4/6 inhibitor to standard adjuvant endocrine therapy (NCT03701334) [62, 63]. In the MonarchE trial, the addition of abemaciclib treatment for 2 years in postmenopausal, high risk HR+, HER2− early breast cancer (EBC) patients is explored [62]. After a median follow-up of 15.5 months, 136 of the 2808 patients (4.8%) experienced recurrence of invasive disease in the abemaciclib arm compared with 187 of the 2829 patients (6.6%) in the control arm (HR 0.75; 95% CI 0.60–0.93) [62]. Almost all patients experienced adverse events in the abemaciclib arm (97.9%), with grade ≥ 3 adverse events reported in 45.9% of patients compared with 86.1% and 12.9% in the endocrine monotherapy group, respectively. In total, 463 patients (16.6%) discontinued abemaciclib due to these treatment-related adverse events. These results are in contrast with those of the PALLAS trial, which was terminated early following the interim analysis since no effect of additional palbociclib to adjuvant endocrine therapy was found. After a median follow-up of 23.7 months, 170 of the 2883 patients (5.9%) in the palbociclib arm experienced recurrence of invasive disease compared with 181 of the 2877 patients (6.3%) in the control arm (HR 0.93; 95% CI 0.75–1.14). A remarkably high premature discontinuation rate of 42% was observed in the palbociclib arm of this study, which was attributed to adverse events in 64% of these patients.
These contradictory results in effectiveness raise questions. One important difference between these two trials is the study population. Whereas the PALLAS study included all patients with stage 2–3 EBC, the MonarchE study specifically included patients with high-risk EBC, defined as patients with four or more positive pathologic lymph nodes or patients with one to three pathologic lymph nodes combined with either tumor size ≥ 5 cm, histologic grade 3, or centrally assessed Ki-67 proliferation index of ≥ 20%. Subgroup analyses of only patients with N2–N3 disease and patients with clinically high risk (≥ 4 nodes or 1–3 nodes with either T3–T4 and/or grade 3 disease) disease in the PALLAS trial still did not show a significant effect in recurrence rate with addition of palbociclib, suggesting differences in study population may not be the main cause for the difference in study outcome. However, cross-study comparison should be interpreted with caution. Furthermore, follow-up imaging for the presence of recurrence was not a protocol requirement, but was performed at the discretion of the treating medical oncologist. In combination with the open-label design of the MonarchE trial, this could introduce confounding by indication bias by a likely tendency to more often perform a scan in high-risk EBC patients in the placebo arm compared with the CDK4/6 inhibitor arm. This could lead to more frequently detected recurrences in patients in the placebo arm compared with patients in the abemaciclib arm.
Longer follow-up data of trials are needed to confirm the clinical benefit of CDK4/6 inhibitors in the adjuvant setting, including the impact on OS. Several other studies in the adjuvant setting are still ongoing. The NATALEE trial (NCT03701334) is a study in the adjuvant setting in which ribociclib is added to endocrine treatment for 3 years. Recruitment is ongoing and no results are presented or published yet. The PENELOPE-B trial (NCT01864746) studies the addition of palbociclib to endocrine therapy for 1 year in patients with a high risk of recurrence based on the CPS-EG score, a risk score based on clinical stage before neoadjuvant treatment, pathological stage after neoadjuvant treatment, nuclear grade, and estrogen receptor status. The results are eagerly awaited as, when comparable effectivity is reached with additional palbociclib for 1 year compared with 2 years in the MonarchE trial, this would be advantageous to patients and decrease healthcare costs.
4.3 Neoadjuvant Setting
The addition of CDK4/6 inhibitors in the neoadjuvant setting could potentially increase antiproliferative effects on the primary tumor before surgery takes place and ultimately result in an improved OS. Although no long-term clinical outcomes have been published yet in the neoadjuvant setting, several studies have shown an increase in cell cycle arrest with the addition of a CDK4/6 inhibitor to neoadjuvant endocrine treatment. In the NeoPalAna trial, a complete cell cycle arrest, as determined by the Ki67 index, was reached in a significantly higher proportion of the patients who received palbociclib and anastrazole compared with patients who received anastrazole monotherapy [64]. The addition of ribociclib to letrozole during six cycles in the neoadjuvant setting was explored in the FELINE trial. It showed a higher number of patients with a complete cell cycle arrest after 2 weeks of treatment in the ribociclib group compared with the placebo group, although at surgery no difference in proportion of patients with complete cell cycle arrest was found [65]. The NeoMonarch trial showed a reduction in Ki67 when abemaciclib was added to anastrazole compared with anastrazole monotherapy after 2 weeks of treatment [66]. In all studies, the number patients with a complete cell cycle arrest decreased after discontinuation of the CDK4/6 inhibitor, which indicates the importance of continuation of CDK4/6 inhibition until surgery takes place.
5 Tolerability
The main side effects of CDK4/6 inhibitors are bone marrow suppression—such as neutropenia, anemia, and thrombopenia—and gastrointestinal toxicities. However, there seem to be some distinct differences between the three drugs. Interestingly, despite these differences in toxicity profiles, all three CDK4/6 inhibitors showed comparable EORTC QLQ-C30 quality-of-life scores [67,68,69]. Approximately 40–50% of patients treated in phase III trials needed a dose reduction in cycle 1 or 2 (Table 2). Importantly, permanent discontinuation was reported more often with abemaciclib treatment (20%) compared with palbociclib and ribociclib (both 7.5%) [8,9,10]. The most common reasons for dose modification were myelosuppression for palbociclib and ribociclib and diarrhea for abemaciclib.
5.1 Neutropenia
Grade 3–4 neutropenia—scored by the common terminology criteria for adverse events (CTCAE)—was the most reported adverse event for palbociclib (66%) and ribociclib (60%) and was the second most reported adverse event in patients treated with abemaciclib (22%). Neutropenia is usually observed in the first cycles of both palbociclib and ribociclib. Median time from first dose to onset of first episode of neutropenia grade ≥ 3 is 28.0 (12–854) days with a median duration of 7 days [70]. Also, for abemaciclib, neutropenia often occurs in the first cycle, however in some cases a more delayed onset of neutropenia was observed in cycle 2 or later [10, 32, 71, 72]. Febrile neutropenia was observed at low frequency in patients treated with a CDK4/6 inhibitor (2% for both palbociclib and ribociclib and < 1% for abemaciclib) [8, 10, 13]. A pharmacokinetic and pharmacodynamic model suggested that higher palbociclib exposure was associated with lower absolute neutrophil counts [73]. However, in general, no clear correlation between race and toxicity was found in all phase III trials [8,9,10,11, 13, 58]. Therefore, more research is needed to identify pharmacokinetic targets for the three CDK4/6 inhibitors.
5.2 Gastrointestinal Toxicity
Grade 3 diarrhea (i.e., an increase of seven or more stools a day) was more frequently reported in patients treated with abemaciclib (9%) compared with palbociclib and ribociclib (both 1%) (Table 2) [8,9,10, 52, 74, 75]. Median time to onset of diarrhea was 6 days with a median duration of 6–8 days. The highest rate of diarrhea was noticed in the first cycle and decreased in following cycles [76]. Prevention of diarrhea is important, since this side effect can negatively impact drug absorption. A population pharmacokinetic and pharmacodynamic model demonstrated that administration of abemaciclib 200 mg twice daily resulted in proportionally less drug absorption than administration of 150 mg twice daily [77]. An explanation for this phenomenon is the increasing incidence of gastrointestinal toxicity and diarrhea at higher abemaciclib doses, resulting in decreased absorption as a consequence of disrupted intestinal endothelial cells [39, 77]. In most patients, this side effect does not lead to drug discontinuation. If there are no signs of infections, diarrhea can be treated with dietary modifications, hydration, and loperamide or other antidiarrheal agents. After recovery, a dose reduction can be considered [31].
5.3 QTc Prolongation
Following ribociclib administration, the QTc interval might be prolonged, whereas the other two drugs are not associated with a clinically relevant QTc time prolongation (> 20 ms) at the recommended dosing schedules [31, 79]. Therefore, QTc prolongation seems to be rather drug specific, instead of a class effect of CDK4/6 inhibitors [80]. QTc prolongation (on average 22.9 ms) was mainly observed in the first cycle of ribociclib in combination with endocrine therapy. In the MONALEESA-2 trial, 11 patients (3.3%) were diagnosed with a QTc prolongation up to > 480 ms in combination with letrozole. In the MONALEESA-7 trial, a higher incidence of QTc prolongation (> 60 ms from baseline) was reported in patients receiving ribociclib in combination with tamoxifen (16%) compared with the combination of ribociclib and a non-steroidal aromatase inhibitor (7%) [81]. Based on this, concomitant intake of ribociclib and drugs with a known potential for QTc prolongation—such as anti-arrhythmic drugs—should be avoided or closely monitored [80].
6 Future Perspectives
6.1 Refining CDK4/6 Inhibitor Therapy by Using Biomarkers
Despite the fact that many HR+, HER2− advanced BC patients respond well to CDK4/6 inhibitors in the first line of treatment, in subsequent lines the response period is evidently shorter and in the end all patients will develop resistance to CDK4/6 inhibitors. Consequently, predictive and prognostic biomarkers are needed to further refine CDK4/6 inhibitor treatment of advanced HR+/HER2− BC patients. In the large, randomized, controlled, phase III SONIA trial (NCT03425838), the optimal position of CDK4/6 inhibitors in HR+, HER2− advanced breast cancer in the first- or in second-line treatment setting will be determined. Important secondary aims of this phase III trial are to investigate the possible associations between pharmacokinetics, pharmacodynamics, and circulating tumor DNA (ctDNA) markers for each CDK4/6 inhibitor in relationship to clinical outcome [61]. The identification of biomarkers will be of importance both for selection of responsive patients and the optimization of the therapeutic response within a patient.
6.1.1 Biomarkers Reflecting Sensitivity or Resistance to CDK4/6 Inhibitors
Biomarkers reflecting increased activity of the CDK-RB1-E2F pathway in tumor cells could help to identify patients who are sensitive to CDK4/6 inhibitor treatment. However, within the PALOMA-1 trial, the use of CCND1 amplification or p16 loss as additional biomarkers for patient selection did not result in an improved PFS compared with all ER+/HER2− patients [84]. Vice versa, identification of resistance mechanisms may yield biomarkers to identify tumors resistant to CDK4/6 inhibitors. Whole exome sequencing of CDK4/6 inhibitor-exposed breast cancer tissues identified eight potential resistance mechanisms, including RB1 loss, activating alterations in AKT1, RAS, AURKA, CCNE2, ERBB2, and FGFR2, and loss of ESR1 expression, together explaining 66% of the observed resistance [85]. In the PALOMA-3 trial, RB1 mutations were acquired in 5% of patients after treatment with palbociclib, RB1 deletions were observed more frequently but no evidence was found for selection of RB1 deletion during treatment [87, 88]. Next to RB1, particularly the E-type cyclins, encoded by the CCNE1 and CCNE2 genes, appear to represent promising markers of resistance. High cyclin E expression was observed in preclinical models with acquired resistance to CDK4/6 inhibitors [88] and gene expression analyses of breast cancer tissues from the PALOMA-3 study showed that low expression of CCNE1 before treatment start was associated with a longer PFS [89]. In the NeoPalAna trial described above, expression of CCNE1, CCND3, and CDKN2D genes remained elevated during treatment with CDK4/6 inhibitors only in non-responding patients [64]. Large clinical trials have to confirm this potential value of cyclin E proteins for identification of CDK4/6-resistant patients.
6.1.2 Biomarkers in Liquid Biopsies
Liquid biopsies, usually referring to detection of tumor-specific DNA fragments in a patient’s blood, represent a promising means for real-time monitoring of treatment response. Targeted next-generation sequencing analyses on blood-derived cell-free DNA (cfDNA) from baseline samples were performed in the PALOMA-3 trial and showed that copy number gains of FGFR1, CDK4, MYC, CCNE1, and MCL1 as well as p53 mutations were associated with a decreased PFS in patients treated with fulvestrant and palbociclib. However, in multivariable analysis, FGFR1 gain, TP53 mutations, and estimated ctDNA tumor fraction were significantly associated with PFS, irrespective of treatment arm. Results of this study demonstrate that no specific alteration at baseline was associated with PFS specifically in the palbociclib group [85].
In the same cohort, PIK3CA and ESR1 mutations were determined at baseline and after 2 weeks of treatment only in the palbociclib arm. Interestingly, a decrease in the level of detected mutant PIK3CA molecules, but not ESR1 mutations, within the first 2 weeks of CDK4/6 inhibitor treatment was associated with a longer PFS. Although this phenomenon is probably not CDK4/6-inhibitor specific and is likely to reflect ctDNA tumor fraction (i.e., the amount of tumor-derived DNA in the blood), it could provide a potential tool for early monitoring of treatment response irrespective of treatment [86].
6.1.3 Therapeutic Drug Monitoring (TDM)
For many drugs, it has been shown that optimization of drug dosage by plasma concentration within a predefined therapeutic window—also known as therapeutic drug monitoring (TDM)—improves clinical outcome or reduces toxicity [92,93,94]. For CDK4/6 inhibitors, this represents an interesting concept as well. However, at present a minimal effective concentration has not been established for any of these three agents. On the other hand, the correlation between palbociclib exposure and toxicity suggests that we can improve the tolerability by dose reductions [42, 73]. Further research is needed to investigate whether the current practice of toxicity-guided dose adaptations can be further improved by implementing TDM.
6.2 Widening the Application of CDK4/6 Inhibitors
6.2.1 HER2+ Breast Cancer
Several studies have shown that a crosstalk exists between HER2 signaling and estrogen receptor pathways in a bidirectional way, which make CDK4/6 inhibitors an attractive therapeutic option in this breast cancer subtype [95]. In a preclinical study it has been shown that the CDK4/6 pathway could mediate resistance against HER2− targeted therapies [96]. A phase II randomized trial explored the safety and efficacy of the combination of abemaciclib plus fulvestrant with trastuzumab in HR+, HER2+ breast cancer patients [97]. Patients were included after at least two lines of therapy in the advanced setting. The experimental combination of trastuzumab plus abemaciclib and fulvestrant in comparison with trastuzumab plus standard-of-care chemotherapy of physician’s choice showed a PFS of 8.3 months versus 5.7 months in the standard-of-care arm (HR 0.67; 95% CI 0.45–1.00). This study confirms preclinical data and is promising since a significant increase in PFS is present, even after two or more lines of treatment. However, results of phase III studies are needed to confirm this effectiveness.
6.2.2 Triple-Negative Breast Cancer
Triple-negative breast cancer (TNBC) is a heterogeneous and aggressive type of breast cancer with limited therapeutic options. Currently, CDK4/6 inhibitors do not play a role in the treatment since efficacy is expected to be limited. Several preclinical and clinical studies are examining a role for CDK4/6 inhibitors in TNBC with diverse biological rationales and hypotheses. TNBC can be divided into subgroups by gene expression profiling: luminal-AR, mesenchymal, basal-like immune-suppressed, and basal-like immune-activated [98]. Experiments in TNBC cell lines have shown that these subtypes differ in their sensitivity to CDK4/6 inhibition, with the luminal-AR subtype being more sensitive to CDK4/6 inhibitors, whereas the basal-like subtype is highly insensitive. These results indicate that patients with TNBC should be selected based on their subgroup for treatment with CDK4/6 inhibitors [99].
Research is ongoing into newly developed CDK4/6 inhibitors in combination with other drug modalities in breast cancer. A phase II trial explored the benefit of an addition of trilaciclib, a novel CDK4/6 inhibitor, to chemotherapy (gemcitabine and carboplatin) in patients with metastatic TNBC. An improvement of antitumor effect and in the tolerability of chemotherapy by myelopreservation was observed in patients who received additional trilaciclib [100]. One of the hypotheses to explain the observed effect is the enhancement of antitumor immunity by trilaciclib [101], an effect of CDK4/6 inhibitors that has recently raised more interest. Based on the foregoing information, phase III trials in which TNBC patients are selected based on their underlying subtype are needed to further explore the potential effects of adding CDK4/6 inhibitors to chemotherapy.
6.2.3 CDK4/6 Inhibitors and Chemotherapy
CDK4/6 inhibitors and chemotherapy target different phases of the cell cycle. CDK4/6 inhibitors induce cell cycle arrest in the G0–G1 phase, preventing cells from entering subsequent phases of the cell cycle. This antagonizes the effect of classic chemotherapeutics, of which the main mode of action takes place in these subsequent phases. Several preclinical studies in breast cancer explored the effect of treatment with CDK4/6 inhibitors followed by chemotherapy. Monotherapy was superior to the combination of these therapies [102, 103]. Interestingly, a preclinical study in pancreatic cancer showed that reversing the order of treatments by giving CDK4/6 inhibitors after chemotherapy could increase the effect of chemotherapy by repressive effects on homologous recombination proteins [104]. Based on these studies, sequence and timing of therapies seems to be essential to reach optimal antitumor effects. Further preclinical investigations potentially followed by clinical studies in breast cancer are warranted to explore the implications for future clinical use of CDK4/6 inhibitors after DNA-damaging therapies.
6.2.4 HR+, HER2− Breast Cancer
Next to CDK4/6-RB1 signaling, PI3K/AKT/mTOR signaling is an important growth stimulatory pathway in HR+, HER2− metastatic breast cancer. One of the described mechanisms of resistance to CDK4/6 inhibition is the PI3K-dependent activation of the cyclin D1/CDK2 complex. This complex could phosphorylate pRb leading to S-phase entry independent of CDK4/6. Blocking the activation of cyclin D1 by inhibiting PIK3CA could be a strategy to overcome this resistance, which was effective in cell line experiments [88]. Similarly, mTORC1/2 inhibitors have shown potency to restore sensitivity to CDK4/6 inhibitors in resistant ER+ breast cancer cell lines [105]. Combining fulvestrant, a CDK4/6 inhibitor, and a PIK3CA inhibitor, as was explored in breast cancer patients with advanced disease in a phase I trial, results in promising efficacy and safety. A retrospective clinical trial found a limited effect of palbociclib when it was given after progression on everolimus, which suggests a role for mTOR inhibitors during or after CDK4/6 inhibitor treatment [106]. These studies imply that additional treatments that block the PI3K/AKT/mTOR signaling pathway could be of value in overcoming resistance and prolonging the anti-tumor effects of CDK4/6 inhibitors. Sequence and timing will be essential and biomarkers are needed to determine the optimal sequence and timing of treatment on the individual patient level.
6.2.5 Other Tumor Types
Since aberrations in the CDK-RB1-E2F pathway are frequently observed in many types of tumors, it is expected that CDK4/6 inhibitors have a wider applicability than in breast cancer alone. Amplification of cyclin D (CCND1), for example, is described in non-small lung cancer, melanoma, and endometrial cancer, among others. Several clinical trials in these tumor types are ongoing and the first encouraging results have been presented [39]. A recently performed phase II trial in patients with HR+ recurrent or advanced endometrial cancer showed a prolongation in PFS with the addition of palbociclib to letrozole [107].
In addition, several preclinical studies suggest other potential mechanisms of action of CDK4/6 inhibitors that could be of interest for the application in other types of tumors as well. For example, the ability of CDK4/6 inhibitors to increase the capacity of tumor cells to express antigens and enhance T cell infiltration was shown in vitro [108]. This suggests CDK4/6 in combination with immune checkpoint blockades could be of interest in specific tumor types. However, possible applications first need to be explored further in clinical trials.
6.3 New Types of CDK4/6 Inhibitors
Research is ongoing into the novel CDK4/6 inhibitors lerociclib (NCT03455829; NCT02983071) and trilaciclib (NCT02978716; NCT03041311; NCT02514447) in combination with other targeted therapies or chemotherapy in lung and breast cancer. Preclinical work showed a different pharmacokinetic and pharmacodynamic profile for lerociclib compared with the other CDK4/6 inhibitors, since lerociclib accumulated in xenograft tumors but not in plasma [109]. This resulted in less inhibition of myeloid progenitor cells. A recently performed phase I/II trial of lerociclib in patients with HR+, HER2− metastatic breast cancer showed the potential clinical benefits of lerociclib since a low rate of both neutropenia grade 3 or 4 and gastrointestinal adverse events was seen compared with the other CDK4/6 inhibitors [110]. These results await confirmation in phase III trials. Trilaciclib differs from the clinically available CDK4/6 inhibitors since it is administered intravenously and is developed to be combined with chemotherapy, mainly to reduced side effects. As earlier described, the first phase I/II trial showed promising results in terms of efficacy.
7 Conclusions
CDK4/6 inhibitors—palbociclib, ribociclib, and abemaciclib—play an eminent role in the treatment of advanced breast cancer. The pharmacokinetics, pharmacodynamics, and efficacy of the three CDK4/6 inhibitors seem to be comparable, although there are also interesting differences such as ability for brain penetration, side effects, and dosing schedules. These differences between the three CDK4/6 inhibitors can be used to optimize selection of treatment for individual patients. Further research is needed to investigate the optimal treatment sequence of CDK4/6 inhibitors in different breast cancer settings and different subtypes and to develop (pharmacodynamic) biomarkers for selecting patients, predicting response, and to optimize the treatment schedule.
References
Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LAG, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5):dju055.
Parise CA, Bauer KR, Brown MM, Caggiano V. Breast cancer subtypes as defined by the estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) among women with invasive breast cancer in California, 1999–2004. Breast J. 2009;15(6):593–602.
Lobbezoo DJA, van Kampen RJW, Voogd AC, Dercksen MW, van den Berkmortel F, Smilde TJ, et al. Prognosis of metastatic breast cancer subtypes: the hormone receptor/HER2-positive subtype is associated with the most favorable outcome. Breast Cancer Res Treat. 2013;141(3):507–14.
Silberholz J, Bertsimas D, Vahdat L. Clinical benefit, toxicity and cost of metastatic breast cancer therapies: systematic review and meta-analysis. Breast Cancer Res Treat. 2019;176(3):535–43.
Giuliano M, Schettini F, Rognoni C, Milani M, Jerusalem G, Bachelot T, et al. Endocrine treatment versus chemotherapy in postmenopausal women with hormone receptor-positive, HER2-negative, metastatic breast cancer: a systematic review and network meta-analysis. Lancet Oncol. 2019;20(10):1360–9.
Aggelis V, Johnston SRD. Advances in endocrine-based therapies for estrogen receptor-positive metastatic breast cancer. Drugs. 2019;79(17):1849–66.
Milani A, Geuna E, Mittica G, Valabrega G. Overcoming endocrine resistance in metastatic breast cancer: current evidence and future directions. World J Clin Oncol. 2014;5(5):990–1001.
Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–36.
Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–48.
Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638–46.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–19.
Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–84.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im S-A, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465–72.
Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34(1):9–20.
O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–30.
Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–87.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 Inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. 2018;28(11):911–25.
Lange CA, Yee D. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr Relat Cancer. 2011;18(4):C19-24.
Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910–7.
Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, et al. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. 2020;21(6):1960. https://doi.org/10.3390/ijms21061960.
Parylo S, Vennepureddy A, Dhar V, Patibandla P, Sokoloff A. Role of cyclin-dependent kinase 4/6 inhibitors in the current and future eras of cancer treatment. J Oncol Pharm Pract Off Publ Int Soc Oncol Pharm Pract. 2019;25(1):110–29.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
McDonald ER, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell. 2017;170(3):577-592.e10.
Mohammedi L, Doula FD, Mesli F, Senhadji R. Cyclin D1 overexpression in Algerian breast cancer women: correlation with CCND1 amplification and clinicopathological parameters. Afr Health Sci. 2019;19(2):2140–6.
Li H, Zheng B. Overexpression of the ubiquitin-specific peptidase 9 X-Linked (USP9X) gene is associated with upregulation of cyclin D1 (CCND1) and downregulation of cyclin-dependent inhibitor kinase 1A (CDKN1A) in breast cancer tissue and cell lines. Med Sci Monit Int Med J Exp Clin Res. 2019;6(25):4207–16.
Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol. 2018;10:1758835918786451.
Abraham J, Coleman R, Elias A, Holmes FA, Kalinsky K, Kittaneh M, et al. Use of cyclin-dependent kinase (CDK) 4/6 inhibitors for hormone receptor-positive, human epidermal growth factor receptor 2-negative, metastatic breast cancer: a roundtable discussion by The Breast Cancer Therapy Expert Group (BCTEG). Breast Cancer Res Treat. 2018;171(1):11–20.
Johnson J, Thijssen B, McDermott U, Garnett M, Wessels LFA, Bernards R. Targeting the RB-E2F pathway in breast cancer. Oncogene. 2016;35(37):4829–35.
Scott SC, Lee SS, Abraham J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin Oncol. 2017;44(6):385–94.
Summary of Product Characteristics abemaciclib. Food and Drug Administration. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm578081.htm. Accessed 15 Nov 2020.
Summary of Product Characteristics palbociclib. Food and Drug Administration. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm549978.htm. Accessed 15 Nov 2020.
Summary of Product Characteristics ribociclib. Food and Drug Administration. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Accessed 15 Nov 2020.
Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig New Drugs. 2014;32(5):825–37.
Tate SC, Cai S, Ajamie RT, Burke T, Beckmann RP, Chan EM, et al. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin Cancer Res. 2014;20(14):3763–74.
O’Brien N, Conklin D, Beckmann R, Luo T, Chau K, Thomas J, et al. Preclinical activity of abemaciclib alone or in combination with antimitotic and targeted therapies in breast cancer. Mol Cancer Ther. 2018;17(5):897–907.
Torres-Guzmán R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget. 2017;8(41):69493–507.
Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab Dispos Biol Fate Chem. 2015;43(9):1360–71.
Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740–53.
de Gooijer MC, Zhang P, Thota N, Mayayo-Peralta I, Buil LCM, Beijnen JH, et al. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Investig New Drugs. 2015;33(5):1012–9.
Martínez-Chávez A, van Hoppe S, Rosing H, Lebre MC, Tibben M, Beijnen JH, et al. P-glycoprotein limits ribociclib brain exposure and CYP3A4 restricts its oral bioavailability. Mol Pharm. 2019;16(9):3842–52.
Yu Y, Loi C-M, Hoffman J, Wang D. Physiologically based pharmacokinetic modeling of palbociclib. J Clin Pharmacol. 2017;57(2):173–84.
Ladumor MK, Bhatt DK, Gaedigk A, Sharma S, Thakur A, Pearce RE, et al. Ontogeny of hepatic sulfotransferases and prediction of age-dependent fractional contribution of sulfation in acetaminophen metabolism. Drug Metab Dispos Biol Fate Chem. 2019;47(8):818–31.
Kozyra M, Ingelman-Sundberg M, Lauschke VM. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med Off J Am Coll Med Genet. 2017;19(1):20–9.
Im S-A, Mukai H, Park IH, Masuda N, Shimizu C, Kim S-B, et al. Palbociclib plus letrozole as first-line therapy in postmenopausal asian women with metastatic breast cancer: results from the phase III, randomized PALOMA-2 study. J Glob Oncol. 2019;5:1–19.
van Dyk M, Marshall J-C, Sorich MJ, Wood LS, Rowland A. Assessment of inter-racial variability in CYP3A4 activity and inducibility among healthy adult males of Caucasian and South Asian ancestries. Eur J Clin Pharmacol. 2018;74(7):913–20.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66.
Laurenti E, Frelin C, Xie S, Ferrari R, Dunant CF, Zandi S, et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 2015;16(3):302–13.
Lallena MJ, Boehnke K, Torres R, Hermoso A, Amat J, Calsina B, et al. Abstract 3101: In-vitro characterization of Abemaciclib pharmacology in ER+ breast cancer cell lines. Cancer Res. 2015;75(15 Supplement):3101–3101.
Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther. 2016;15(10):2273–81.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–46.
Shohdy KS, Lasheen S, Kassem L, Abdel-Rahman O. Gastrointestinal adverse effects of cyclin-dependent kinase 4 and 6 inhibitors in breast cancer patients: a systematic review and meta-analysis. Ther Adv Drug Saf. 2017;8(11):337–47.
Chong Q-Y, Kok Z-H, Bui N-L-C, Xiang X, Wong AL-A, Yong W-P, et al. A unique CDK4/6 inhibitor: current and future therapeutic strategies of abemaciclib. Pharmacol Res. 2020;156:104686.
Im S-A, Lu Y-S, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. 2019;381(4):307–16.
Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–39.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im S-A, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–36.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im S-A, et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl J Med. 2020;382(6):514–24.
Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. The effect of abemaciclib plus fulvestrant on overall survival in hormone receptor-positive, ERBB2-negative breast cancer that progressed on endocrine therapy-MONARCH 2: a randomized clinical trial. JAMA Oncol. 2019;6(1):116–24.
Gao JJ, Cheng J, Bloomquist E, Sanchez J, Wedam SB, Singh H, et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2020;21(2):250–60.
Azim HA, Dawood S, El-Saghir N, Kassem L, Azim HA. Understanding the benefits and challenges of first-line cyclin-dependent kinases 4 and 6 inhibitors in advanced breast cancer among postmenopausal women. Breast J. 2020;26(4):630–42.
van Ommen-Nijhof A, Konings IR, van Zeijl CJJ, Uyl-de Groot CA, van der Noort V, Jager A, et al. Selecting the optimal position of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer - the SONIA study: study protocol for a randomized controlled trial. BMC Cancer. 2018;18(1):1146.
Johnston SRD, Harbeck N, Hegg R, Toi M, Martin M, Shao ZM, et al. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). J Clin Oncol Off J Am Soc Clin Oncol. 2020;JCO2002514.
Mayer EL, Gnant MI, DeMichele A, Martin M, Burstein H, Prat A, et al. LBA12 PALLAS: a randomized phase III trial of adjuvant palbociclib with endocrine therapy versus endocrine therapy alone for HR+/HER2- early breast cancer. Ann Oncol. 2020;1(31):S1145.
Ma CX, Gao F, Luo J, Northfelt DW, Goetz M, Forero A, et al. NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor-positive breast cancer. Clin Cancer Res. 2017;23(15):4055–65.
Letrozole + ribociclib versus letrozole + placebo as neoadjuvant therapy for ER+ breast cancer (FELINE trial). https://doi.org/10.1200/JCO.2020.38.15_suppl.505.
Hurvitz SA, Martin M, Press MF, Chan D, Fernandez-Abad M, Petru E, et al. Potent cell-cycle inhibition and upregulation of immune response with abemaciclib and anastrozole in neoMONARCH, phase II neoadjuvant study in HR+/HER2- breast cancer. Clin Cancer Res. 2020;26(3):566–80.
Kaufman PA, Toi M, Neven P, Sohn J, Grischke E-M, Andre V, et al. Health-related quality of life in MONARCH 2: abemaciclib plus fulvestrant in hormone receptor-positive, HER2-negative advanced breast cancer after endocrine therapy. Oncologist. 2020;25(2):e243–51.
Harbeck N, Iyer S, Turner N, Cristofanilli M, Ro J, André F, et al. Quality of life with palbociclib plus fulvestrant in previously treated hormone receptor-positive, HER2-negative metastatic breast cancer: patient-reported outcomes from the PALOMA-3 trial. Ann Oncol. 2016;27(6):1047–54.
Verma S, O’Shaughnessy J, Burris HA, Campone M, Alba E, Chandiwana D, et al. Health-related quality of life of postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer treated with ribociclib + letrozole: results from MONALEESA-2. Breast Cancer Res Treat. 2018;170(3):535–45.
Diéras V, Harbeck N, Joy AA, Gelmon K, Ettl J, Verma S, et al. Palbociclib with letrozole in postmenopausal women with ER+/HER2- advanced breast cancer: hematologic safety analysis of the randomized PALOMA-2 Trial. Oncologist. 2019;24(12):1514–25.
Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2016;22(23):5696–705.
Verma S, Bartlett CH, Schnell P, DeMichele AM, Loi S, Ro J, et al. Palbociclib in combination with fulvestrant in women with hormone receptor-positive/HER2-negative advanced metastatic breast cancer: detailed safety analysis from a multicenter, randomized, placebo-Controlled, phase III study (PALOMA-3). Oncologist. 2016;21(10):1165–75.
Sun W, O’Dwyer PJ, Finn RS, Ruiz-Garcia A, Shapiro GI, Schwartz GK, et al. Characterization of neutropenia in advanced cancer patients following palbociclib treatment using a population pharmacokinetic-pharmacodynamic modeling and simulation approach. J Clin Pharmacol. 2017;57(9):1159–73.
Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet. 2020;395(10226):817–27.
Spring LM, Zangardi ML, Moy B, Bardia A. Clinical management of potential toxicities and drug interactions related to cyclin-dependent kinase 4/6 inhibitors in breast cancer: practical considerations and recommendations. Oncologist. 2017;22(9):1039–48.
Rugo HS, Huober J, García-Sáenz JA, Masuda N, Sohn JH, Andre VAM, et al. Management of abemaciclib-associated adverse events in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: safety analysis of MONARCH 2 and MONARCH 3. Oncologist. 2020.
Tate SC, Sykes AK, Kulanthaivel P, Chan EM, Turner PK, Cronier DM. A population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a phase I clinical trial in cancer patients. Clin Pharmacokinet. 2018;57(3):335–44.
Johnston S, Martin M, Di Leo A, Im S-A, Awada A, Forrester T, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. 2019;5:5.
Durairaj C, Ruiz-Garcia A, Gauthier ER, Huang X, Lu DR, Hoffman JT, et al. Palbociclib has no clinically relevant effect on the QTc interval in patients with advanced breast cancer. Anticancer Drugs. 2018;29(3):271–80.
Kloth JSL, Pagani A, Verboom MC, Malovini A, Napolitano C, Kruit WHJ, et al. Incidence and relevance of QTc-interval prolongation caused by tyrosine kinase inhibitors. Br J Cancer. 2015;112(6):1011–6.
Tripathy D, Im S-A, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018;19(7):904–15.
Duan J, Tao J, Zhai M, Li C, Zhou N, Lv J, et al. Anticancer drugs-related QTc prolongation, torsade de pointes and sudden death: current evidence and future research perspectives. Oncotarget. 2018;9(39):25738–49.
Hussaarts KGAM, Berger FA, Binkhorst L, Oomen-de Hoop E, van Leeuwen RWF, van Alphen RJ, et al. The risk of QTc-interval prolongation in breast cancer patients treated with tamoxifen in combination with serotonin reuptake inhibitors. Pharm Res. 2019;37(1):7.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov. 2020;10(8):1174–93.
Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018;29(3):640–5.
O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8(11):1390–403.
Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–13.
Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2019;37(14):1169–78.
O’Leary B, Cutts RJ, Huang X, Hrebien S, Liu Y, André F, et al. Circulating tumor DNA markers for early progression on fulvestrant with or without palbociclib in ER+ advanced breast cancer. J Natl Cancer Inst. 2020.
O’Leary B, Hrebien S, Morden JP, Beaney M, Fribbens C, Huang X, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9(1):896.
Groenland SL, van Nuland M, Verheijen RB, Schellens JHM, Beijnen JH, Huitema ADR, et al. Therapeutic drug monitoring of oral anti-hormonal drugs in oncology. Clin Pharmacokinet. 2018.
Groenland SL, van Eerden RAG, Verheijen RB, Koolen SLW, Moes DJAR, Desar IME, et al. Therapeutic drug monitoring of oral anticancer drugs: the Dutch pharmacology oncology group-therapeutic drug monitoring protocol for a prospective study. Ther Drug Monit. 2019;41(5):561–7.
Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102(5):765–76.
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96(12):926–35.
Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29(3):255–69.
Tolaney SM, Wardley AM, Zambelli S, Hilton JF, Troso-Sandoval TA, Ricci F, et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(6):763–75.
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SAW, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–98.
Asghar US, Barr AR, Cutts R, Beaney M, Babina I, Sampath D, et al. Single-cell dynamics determines response to cdk4/6 inhibition in triple-negative breast cancer. Clin Cancer Res. 2017;23(18):5561–72.
Weiss JM, Csoszi T, Maglakelidze M, Hoyer RJ, Beck JT, Domine Gomez M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30(10):1613–21.
Tan AR, Wright GS, Thummala AR, Danso MA, Popovic L, Pluard TJ, et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2019;20(11):1587–601.
Jin D, Tran N, Thomas N, Tran DD. Combining CDK4/6 inhibitors ribociclib and palbociclib with cytotoxic agents does not enhance cytotoxicity. PLoS ONE. 2019;14(10):e0223555.
McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle Georget Tex. 2012;11(14):2747–55.
Salvador-Barbero B, Álvarez-Fernández M, Zapatero-Solana E, El Bakkali A, Menéndez MDC, López-Casas PP, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37(3):340-353.e6.
Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, Carnevalli LS, et al. Combined inhibition of mTOR and CDK4/6 Is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol Cancer Ther. 2018;17(5):908–20.
Dhakal A, Matthews CM, Levine EG, Salerno KE, Zhang F, Takabe K, et al. Efficacy of palbociclib combinations in hormone receptor-positive metastatic breast cancer patients after prior everolimus treatment. Clin Breast Cancer. 2018;18(6):e1401–5.
Mirza MR, Bjørge L, Marmé F, Christensen RD, Gil-Martin M, Auranen A, et al. LBA28 A randomised double-blind placebo-controlled phase II trial of palbociclib combined with letrozole (L) in patients (pts) with oestrogen receptor-positive (ER+) advanced/recurrent endometrial cancer (EC): NSGO-PALEO / ENGOT-EN3 trial. Ann Oncol. 2020;1(31):S1160.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–5.
Bisi JE, Sorrentino JA, Jordan JL, Darr DD, Roberts PJ, Tavares FX, et al. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget. 2017;8(26):42343–58.
Krastev B, Rai R, Bulat I, Maglakelidze M, Murias C, Arkenau H-T, et al. 278MO cfDNA analysis from phase I/II study of lerociclib (G1T38), a continuously dosed oral CDK4/6 inhibitor, with fulvestrant in HR+/HER2- advanced breast cancer patients. Ann Oncol. 2020;1(31):S351–2.
Flaherty KT, LoRusso PM, DeMichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568–76.
Choo JR-E, Lee S-C. CDK4-6 inhibitors in breast cancer: current status and future development. Expert Opin Drug Metab Toxicol. 2018;14(11):1123–38.
Ruiz-Garcia A, Plotka A, O’Gorman M, Wang DD. Effect of food on the bioavailability of palbociclib. Cancer Chemother Pharmacol. 2017;79(3):527–33.
Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011;104(12):1862–8.
Samant TS, Dhuria S, Lu Y, Laisney M, Yang S, Grandeury A, et al. Ribociclib bioavailability is not affected by gastric pH changes or food intake: in silico and clinical evaluations. Clin Pharmacol Ther. 2018;104(2):374–83.
Posada MM, Morse BL, Turner PK, Kulanthaivel P, Hall SD, Dickinson GL. Predicting clinical effects of CYP3A4 modulators on abemaciclib and active metabolites exposure using physiologically based pharmacokinetic modeling. J Clin Pharmacol. 2020;60(7):915–30.
Fujiwara Y, Tamura K, Kondo S, Tanabe Y, Iwasa S, Shimomura A, et al. Phase 1 study of abemaciclib, an inhibitor of CDK 4 and 6, as a single agent for Japanese patients with advanced cancer. Cancer Chemother Pharmacol. 2016;78(2):281–8.
Thakkar D, Kate AS. Update on metabolism of abemaciclib: In silico, in vitro, and in vivo metabolite identification and characterization using high resolution mass spectrometry. Drug Test Anal. 2020;12(3):331–42.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38.
Slamon DJ, Fasching PA, Patel R, Verma S, Hurvitz SA, Chia SKL, et al. NATALEE: Phase III study of ribociclib (RIBO) + endocrine therapy (ET) as adjuvant treatment in hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2–) early breast cancer (EBC). J Clin Oncol. 2019;37(15_suppl):TPS597–TPS597.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
FundIng
No external funding was used in the preparation of this manuscript.
Conflict of interest
Stijn L.W. Koolen has received a research grant from Novartis. C. Louwrens Braal, Elisabeth M. Jongbloed, Saskia M. Wilting, Agnes Jager and Ron H.J. Mathijssen declare they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
All authors participated in data analysis and interpretation, creation of tables, and writing and editing of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Braal, C.L., Jongbloed, E.M., Wilting, S.M. et al. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 81, 317–331 (2021). https://doi.org/10.1007/s40265-020-01461-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01461-2