
Abstract
MicroRNAs (miRNAs) are short non-coding RNAs, highly conserved between species, that are powerful regulators of gene expression. Aberrant expression of miRNAs alters biological processes and pathways linked to human disease. miR-486-5p is a muscle-enriched miRNA localized to the cytoplasm and nucleus, and is highly abundant in human plasma and enriched in small extracellular vesicles. Studies of malignant and non-malignant diseases, including kidney diseases, have found correlations with circulating miR-486-5p levels, supporting its role as a potential biomarker. Pre-clinical studies of non-malignant diseases have identified miR-486-5p targets that regulate major signaling pathways involved in cellular proliferation, migration, angiogenesis, and apoptosis. Validated miR-486-5p targets include phosphatase and tensin homolog (PTEN) and FoXO1, whose suppression activates phosphatidyl inositol-3-kinase (PI3K)/Akt signaling. Targeting of Smad1/2/4 and IGF-1 by miR-486-5p inhibits transforming growth factor (TGF)-β and insulin-like growth factor-1 (IGF-1) signaling, respectively. Other miR-486-5p targets include matrix metalloproteinase-19 (MMP-19), Sp5, histone acetyltransferase 1 (HAT1), and nuclear factor of activated T cells-5 (NFAT5). In this review, we examine the biogenesis, regulation, validated gene targets and biological effects of miR-486-5p in non-malignant diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are short non-coding RNAs, approximately 22–25 nucleotides long that were first identified in Caenorhabditis elegans in 1993 [1]. The human genome alone contains over 1500 miRNAs, although the functional significance of many remains uncertain [2]. miRNAs are highly conserved across species and potently regulate gene expression, as over 60% of human protein-coding genes are conserved targets of miRNAs [3]. The biogenesis of miRNAs is a multistep process that can be divided into canonical and non-canonical pathways, which have been reviewed elsewhere [4]. miRNAs function primarily by post-transcriptional gene silencing via the RNA-induced silencing complex (RISC) pathway in the cytoplasm, wherein the miRNA associates with an Argonaute protein in the RISC, and binds to the 3′ untranslated region (UTR) of its target mRNA [5]. Argonaute proteins associate with a GW182 protein that interacts with poly(A)-binding protein and a cytoplasmic deadenylase complex. Consequently, the miRNA–RISC complex mediates mRNA deadenylation and degradation, although inhibition of mRNA translation may also occur via an unclear mechanism [5]. miRNAs may also bind other regions of target genes, including the 5′UTR and coding sequences, and recent studies have identified a nuclear role as activators or silencers of gene transcription [6, 7]. miRNAs have garnered attention for their potential roles in disease diagnosis, pathogenesis, and therapy, as their expression is dysregulated in both malignant and non-malignant diseases [8,9,10].
miR-486-5p is a muscle-enriched miRNA [11] that circulates at relatively high levels in plasma [12], is enriched within small extracellular vesicles [13], and has been linked to signaling pathways involved in several cancers [9] as well as non-malignant conditions, such as skeletal muscle disorders [14, 15], ischemia–reperfusion injury (IRI) [16,17,18], and organ fibrosis [19,20,21]. Here, we review miR-486-5p biogenesis and regulation of expression, discuss circulating miR-486-5p as a potential biomarker in non-malignant diseases, and the role of exosomal transfer in experimental disease models. We also report major targets of miR-486-5p and affected biological pathways in non-malignant diseases, and highlight the controversies regarding downstream effects on cell and organ function. For information on the role of miR-486-5p in malignant diseases, the reader is referred to a recent comprehensive review [9].
Biogenesis of miR-486-5p
miR-486-5p is highly conserved in mammals, and was first identified as a muscle-enriched miRNA whose expression is downregulated in Duchenne’s muscular dystrophy [22]. miR-486-5p is highly expressed in myoblasts during myogenesis, and in skeletal muscle [14], and is found at high levels in cardiac muscle, with moderate expression in lung, brain, liver, and bladder [11, 23, 24]. miR-486-5p is also a dominant erythroid miRNA, and its expression is upregulated in erythropoiesis [25, 26].
miR-486 is first transcribed from an intron within the ANK1 locus, located on chromosome 8p11.21 (Fig. 1). The ANK1 locus encodes the Ankyrin 1 gene, which is primarily expressed in erythroid cells. The mouse genome contains miR-486a-5p and miR-486b-5p at this locus, both of which consist of the same mature sequence but are transcribed in opposite directions. Similarly, the human genome encodes miR-486-1 and miR-486-2, also consisting of the same mature sequence but transcribed in opposite directions. The biogenesis and expression of miR-486-5p discussed here refers to miR-486a-5p/miR-486-1, as these are both transcribed in the same direction as Ank1. Two miRNAs, miR-486-5p and miR-486-3p [9], are generated from opposite ends of the pre-miRNA hairpin [4]. miRNA strand selection is determined by intrinsic features of the miRNA duplex including the identity and thermodynamic stability of the 5′ nucleotides of each strand, but tissue-specific differences in strand selection (miRNA 5p/3p ratios) also support other regulatory mechanisms involving miRNA processing, duplex remodeling, and degradation [27]. miR-486 processing exhibits Argonaute-2-Slicer dependence, whereby Argonaute-2 catalytic activity is required to generate functional mature miRNA via slicing to remove the star (3p) strand; catalytically inactive Argonaute-2 results in miR-486-3p accumulation, miR-486 duplex arrest, and blunted miR-486-5p activity [25].

miR-486-5p biogenesis a miR-486-5p is transcribed from the last intron of the Ank1 gene, located downstream from alternative exon (39A) of the muscle-specific sAnk1 isoform b genomic organization of miR-486-5p and Ank1 is conserved among mammals c pre-miR-486 requires Dicer cleavage, and also requires Argonaute-2 catalytic activity to remove the 3p star (*) strand, generating functional mature miR-486-5p (adapted from Refs. [11, 25])
miR-486-3p expression and function
The function and expression of the star strand, miR-486-3p, has been less well studied within different tissues. Within the hematopoietic system, miR-486-5p and miR-486-3p are highly expressed in erythroid cells [26]. However, their expression patterns differ in that miR-486-5p increases throughout the differentiation process, whereas miR-486-3p peaks earlier and then declines slightly [26]. Structurally, the pre-miR-486 miRNA/star duplex is perfectly paired and conserved across mammalian species, possibly reflecting conserved activity of miR-486-3p, although functional sensor assays used to validate miRNA activity showed that unlike mature miRNA-486-5p, miR-486-3p failed to repress its sensor [28]. Furthermore, immunoprecipitation of Argonaute proteins revealed that miR-486-3p associated with Argonautes 1 and 3, but functional sensor assays in wild type, Argonaute-2 knockout and Argonaute-2 catalytically inactive mouse embryonic fibroblasts showed that despite its accumulation, miR-486-3p did not exhibit gene regulatory activity [25].
Nonetheless, miR-486-3p target genes have been identified in malignant and non-malignant diseases [9], suggesting a role in gene regulation. In erythroid cells, miR-486-3p targets and downregulates the zinc finger protein BCL11A, associated with increased expression of the γ-globin gene and fetal hemoglobin synthesis [26]. miR-486-3p targets and downregulates the deacetylase Sirtuin 2 (SIRT2), resulting in decreased α-synuclein-induced toxicity in vitro, and suggesting that miR-486-3p may protect against Parkinson’s disease progression [29]. A recent systematic review of 34 studies found that miR-486-3p is among the most frequently altered miRNAs in patients with autism spectrum disorder (ASD) [30]. miR-486-3p was reported to be upregulated in the serum of patients with ASD, and targets and downregulates AT-rich interaction domain 1B (ARID1B) [31], a gene mutated in ASD that also confers an ASD-like phenotype in Arid1b haploinsufficient mice [32]. Although the processing of pre-miR-486 and distribution of miR-486-5p and miR-486-3p within different tissues is not fully understood, these data support miR-486-3p as a functional miRNA with a potential role in human diseases. The remainder of this review will focus on miR-486-5p. For further information on miR-486-3p in human diseases, the reader is referred to a recent review [9].
Regulation of miR-486-5p expression and cellular localization
Although Ank1 mRNA is primarily expressed in erythroid cells, an isoform (sAnk1/Ank1.5) containing a muscle-specific first exon and the last three exons of the Ank1 erythroid gene is produced from an alternate promoter [33] (Fig. 1a, b). The tissue distribution of sAnk1 mirrors that of miR-486-5p, enriched in heart and skeletal muscle [11]. Furthermore, there is increased expression of both sAnk1 mRNA and miR-486-5p during myoblast differentiation [24]. In rat neonatal cardiomyocytes, Small et al.showed that myocardin-related transcription factor A (MRTF-A) induces the expression of both miR-486-5p and sAnk1. The sAnk1 promoter also contains two conserved E boxes for MyoD transcriptional activity, and MyoD activity directly regulates sAnk1 and miR-486-5p expression [24]. These data support the co-regulation of miR-486-5p and sAnk1 and suggest that miR-486-5p is produced from the processing of sAnk1 intronic RNA [11].
The regulation of miR-486-5p expression has derived largely from studies pertaining to malignancies, where its dysregulation is paradoxically associated with either tumor suppression or oncogenesis [9]. Epigenetic modifications of the ANK1 promoter [34] and reduced p53 expression or activity [35] are associated with downregulation of miR-486-5p. The miR-486-5p promoter also contains a binding site for hypoxia-inducible-factor-1α (HIF-1α). HIF-1α overexpression activates the miR-486-5p promoter in HeLa cells, and induces expression of miR-486-5p in prostate cancer cell lines, suggesting that hypoxia stimulates its transcription [36]. The effect of hypoxia on miR-486-5p expression is not limited to malignant cells as hypoxic rat cardiomyocytes also exhibit upregulated miR-486-5p [37]. miR-486-5p expression is also regulated at the post-transcriptional level. For example, long non-coding RNAs (lncRNAs) can directly bind to miR-486-5p, inhibiting its expression and activity in experimental models [21, 38, 39].
Traditionally, miRNA biogenesis is a multi-step process involving nuclear and cytosolic components, with the classical function of mature miRNA being post-transcriptional gene silencing in the cytoplasm via miRISC and the 3′ UTR of the target gene [7]. Studies with high-throughput profiling techniques have identified mature miRNAs that are enriched in cell nuclei [40,41,42], and RISC components including Argonaute-2 and trinucleotide repeat-containing gene 6A protein (TNRC6) have been identified in the nucleus and thus likely play a role in miRNA nuclear transport and activity [43, 44]. Nuclear miRNAs can (1) mediate post-transcriptional gene silencing to downregulate the expression of long non-coding RNAs, (2) interact with pri-miRNAs to inhibit their maturation; (3) activate or suppress gene transcription by interacting with gene promoters in association with Argonaute-2; and (4) induce gene expression by enhancer activation, which may also require Argonaute-2 [7].
The cellular localization of mature miR-486-5p is indeed not limited to the cytoplasm. Deep sequencing of the nuclear and cytoplasmic pools of small RNAs from a human nasopharyngeal carcinoma cell line identified 339 nuclear and 324 cytoplasmic miRNAs, the majority of which overlap, including miR-486-5p [42]. The large degree of overlap suggests that miRNAs are imported into the nucleus. Viñas et al. administered intravenous lipid-encapsulated miR-486-5p mimic to mice with bilateral kidney IRI and by in-situ hybridization revealed that miR-486-5p localized to both cytoplasm and nuclei of cortical tubular cells [45]. Thus, it is conceivable that miR-486-5p may exert biological effects in the nucleus via one or more of the identified nuclear roles of miRNAs [7], such as post-transcriptional gene silencing of other non-coding RNAs or regulation of protein-coding gene expression through direct interaction with promoter or enhancer sites.
Circulating miR-486-5p and extracellular vesicles
Circulating miRNAs are stable in plasma and resistant to nuclease digestion [46]. An estimated 90% of circulating miRNAs are present in a non-membrane bound form, associated with the Argonaute-2 ribonucleoprotein complex, whereas a smaller proportion are found within small extracellular vesicles, such as exosomes (50–150 nm diameter) [46, 47]. miR-486-5p is abundant in human plasma [12, 48], where it circulates freely or within exosomes [49]. However, as an erythropoietic miRNA, miR-486-5p is also released by red blood cell hemolysis, thereby increasing miR-486-5p levels in hemolyzed samples. This feature can thereby complicate the interpretation of circulating miR-486-5p levels in biomarker studies [50, 51].
miR-486-5p is differentially expressed in human plasma or serum in a wide range of conditions including, but not limited to solid tumor malignancies [9], sepsis [52], primary muscle diseases (e.g., Duchenne muscular dystrophy [22]), cardiorespiratory diseases (e.g., chronic heart failure [19], cystic fibrosis [53]), diabetic kidney disease [54], osteoarthritis [55], neurological conditions (e.g., vascular dementia [56], Huntington’s disease [57], ASD [58]), and various endocrine disorders (e.g., metabolic syndrome [59], childhood obesity [60], type 2 diabetes mellitus [61], polycystic ovary syndrome [62], and recurrent miscarriage [63]). Accordingly, circulating levels may have utility for diagnosis or prognosis in a variety of diseases (outlined in Supplementary Table 1). Although these studies provide data on clinical associations, further research is required to determine the diagnostic and prognostic potential of circulating miR-486-5p. Of the studies outlined in Supplementary Table 1, the diagnostic accuracy of circulating miR-486-5p was evaluated only in type 2 diabetes [54], vascular dementia [56], and outcomes of embryo transfer with in vitro fertilization (IVF) [63]. Regmi et al.evaluated serum miRNAs in patients with diabetic kidney disease, revealing that miR-486-5p was downregulated, with a receiver operating characteristic area under the curve (ROC-AUC) of 0.853 [54]. In patients with vascular dementia due to cerebral small vessel disease, plasma miR-486-5p had a sensitivity of 75%, specificity of 83%, and ROC–AUC of 90% as a diagnostic marker [56]. In a study of embryo transfer outcomes in IVF, a plasma miRNA signature that included miR-486-5p had a sensitivity of 100% and specificity of 83% for recurrent miscarriage, but sensitivity of only 68.1% and specificity of 54% for successful outcome [63]. Given the variety of non-malignant diseases with dysregulated circulating miR-486-5p levels, its potential as a non-invasive diagnostic and prognostic biomarker warrants further study.
miR-486-5p is enriched within exosomes, which are important mediators of intercellular communication [64] and have shown protective effects in experimental models of organ injury [16, 17, 65, 66]. Indeed, miR-486-5p is among the most abundant miRNAs in both human adipose and bone marrow mesenchymal stem cell (BMSC)-derived exosomes, and over-represented compared to its expression within the parent cells [13]. Furthermore, human cord blood endothelial colony-forming cell (ECFC)-derived exosomes are enriched in miR-486-5p, with levels that are markedly higher compared to ECFC-derived microparticles (100–1000 nm diameter) [16]. Mechanisms involved in selective cargo loading of miRNA and other RNA species into extracellular vesicles remain unclear. However, factors that influence the miRNA profile within extracellular vesicles include the pathophysiological state of the source cell, RNA properties (such as small size, affinity for membrane lipids, and cytoplasmic localization), RNA sequence motifs, post-transcriptional modifications, and associations with RNA binding proteins [64].
Targets of miR-486-5p
Based on 3′UTR sequence homology (http://mirdb.org/cgi-bin/search.cgi), more than 300 predicted miR-486-5p targets have been identified. However, not all predicted targets have been validated by demonstrating a direct interaction between miR-486-5p and the gene transcript 3′ UTR, typically done by luciferase reporter assay. Below, we review key validated targets of miR-486-5p in non-malignant diseases.
Skeletal muscle PTEN and FoXO1
Phosphatase and tensin homolog (PTEN) is a tumor suppressor protein whose expression is tightly regulated at multiple levels [67], and represents the original validated target of miR-486-5p [11]. As a lipid phosphatase, PTEN negatively regulates the phosphatidyl-inositol-3-kinase (PI3K)/Protein kinase B (Akt) signaling pathway by de-dephosphorylating the intracellular messenger and PI3K product phosphatidyl-inositol-triphosphate (PIP3) to PIP2 [68]. Consequently, PTEN inhibits several cellular functions including proliferation, migration, survival, and angiogenesis [68, 69].
FoxO1 belongs to the family of forkhead transcription factors (FoxOs), which are involved in insulin and insulin-like growth factor-1 (IGF-1) signaling, thus affecting cell growth, proliferation, differentiation, apoptosis, oxidative stress, ageing, and metabolism [70]. FoxO1 is a negative regulator of Akt signaling, and its activity is also inhibited via Akt-mediated phosphorylation [71].
Small et al. [11] first identified PTEN and FoxO1 as miR-486-5p targets, by luciferase reporter assay. In mice, an inverse correlation was shown between the expression of miR-486-5p and PTEN in postnatal cardiac growth. Furthermore, miR-486-5p overexpression in cardiomyocytes downregulated endogenous PTEN and Fox01 protein expression and increased p-Akt (at Ser473). These data implicate miR-486-5p as a promoter of cardiac muscle growth by targeting and downregulating PTEN and Fox01, thereby activating PI3K/Akt signaling [11].
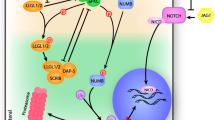
PTEN and PI3K/Akt signaling has also been implicated in primary muscle disorders, including Duchenne muscular dystrophy (DMD), caused by mutations in the dystrophin gene [72]. miR-486-5p expression is downregulated in skeletal muscle of patients with DMD compared to healthy controls [22] and in dystrophin-deficient muscles in mice [23]. Dystrophin-deficient animals have increased PTEN expression and decreased Akt phosphorylation within dystrophin-deficient muscle tissue [73], and modulation of Akt signaling improves muscle function in these models [74]. At the cellular level, miR-486-5p inhibition is detrimental to myoblasts, resulting in reduced myotube formation, failed migration, increased caspase-3/7 levels and enhanced apoptosis [14]. In human myotubes that overexpress DOCK3 (a validated target of miR-486-5p in skeletal muscle), PTEN expression is increased, associated with reduced Akt phosphorylation, higher expression of activated caspases-3/7 and increased myotube apoptosis [23]. These results provide strong evidence that miR-486-5p modulates these pathways via targeting PTEN and DOCK3 (Fig. 2).

miR-486-5p can activate or suppress PI3K/Akt signaling via its targets. miR-486-5p targets PTEN, DOCK3, and FoxO1 to activate PI3K/Akt signaling, and confers protective effects in skeletal muscle disorders, and kidney and cardiac IR injury (top). miR-486-5p targets IGF-1 to inhibit PI3K/Akt signaling, with possible negative consequences in cyanotic congenital heart disease and pre-eclampsia. CHD congenital heart disease, DMD Duchenne muscular dystrophy, IGF-1 insulin-like growth factor 1, IR ischemia reperfusion, PI3K phosphatidylinositol-3-kinase, PTEN phosphatase and tensin homolog (created with BioRender.com)
The dynamic expression of miR-486-5p in normal muscle regeneration is critical for its regenerative effect [14]. A comparison of wild type and dystrophin-deficient mice subjected to cardiotoxin-induced tibialis anterior muscle injury revealed that miR-486-5p expression is transiently upregulated after injury, with normal muscle regeneration, while dystrophin-deficient mice have reduced miR-486-5p expression following injury and impaired muscle regeneration. The authors also evaluated muscle regeneration in transgenic mice that overexpress miR-486-5p exclusively in heart and skeletal muscle. Transgenic mice with muscle-specific miR-486-5p overexpression are viable, with no significant phenotypic differences from wild-type controls at 6 months of age, other than slight weight gain. However, when subjected to cardiotoxin-induced skeletal muscle injury, the transgenic mice displayed slowed muscle regeneration and altered architecture, with increased multinucleated myofibers and a higher proliferative index [14]. Post-skeletal muscle injury, mice with muscle-specific miR-486-5p overexpression had decreased expression of target genes PTEN and FoxO1 (compared to wild type mice) along with downregulation of cyclin-dependent kinase inhibitors p21and p27 [14], which are FoxO1 targets [75]. These findings suggest that maintaining high miR-486-5p expression (rather than dynamic miR-486-5p expression) influences muscle satellite cell kinetics and fusion associated with targeting of PTEN and FoxO1, resulting in delayed and abnormal skeletal muscle regeneration [14].
In addition to myogenic differentiation and DMD, miR-486-5p protects against muscle wasting through targets PTEN and FoxO1 [15, 66]. In experimental chronic kidney disease (CKD), the catabolic environment increases glucocorticoid production and suppresses insulin signaling, resulting in decreased PI3K/Akt activity [76, 77]. Suppression of Akt signaling induces FoxO1 dephosphorylation, which translocates to the nucleus and activates expression of E3 ubiquitin ligases responsible for muscle proteolysis [77]. In mice with muscle-specific gene deletion of FoXO1, Xu et al. reported protection against CKD-induced muscle wasting [15]. In vivo, muscle miR-486-5p levels were decreased in CKD mice, and electroporation of miR-486-5p mimic into muscle improved mass, and decreased expression of ubiquitin E3 ligases, FoxO1 and PTEN [15]. In dexamethasone-treated cultured myotubes, Li et al.demonstrated that BMSC-derived exosomes (enriched in miR-486-5p) improved myotube quantity, reduced expression of muscle atrophy markers, and downregulated the nuclear translocation of FoxO1, whereas these effects were blunted using exosomes treated with miR-486-5p inhibitor [66]. Accordingly, these data suggest that miR-486-5p is a potential therapeutic strategy for disorders of muscle catabolism via its targeting of PTEN and FoxO1.
Targeting of PTEN and PI3K/Akt signaling in cardiac ischemia
Acute myocardial infarction is associated with IRI [78] and coronary microembolization [79], which lead to myocardial cell apoptosis. In a swine model of coronary microembolization, activation of the PTEN/Akt pathway contributes to cardiomyocyte apoptosis [80]. In rats with coronary microembolization, ventricular miR-486-5p expression is downregulated, and its overexpression activates PI3K/Akt signaling by reducing PTEN expression, resulting in decreased cardiomyocyte apoptosis, reduced microinfarct area and improved cardiac function [81]. In a preclinical study of myocardial IRI, hypoxia/re-oxygenation triggers cardiomyocyte apoptosis with increased PTEN expression and reduced miR-486-5p expression [17]. Exosomes derived from rat BMSCs overexpressing miR-486-5p have a pronounced inhibitory effect on PTEN expression, leading to Akt activation, apoptosis reduction and significant reduction in myocardial infarct size compared to unaltered exosomes [17].
Bei et al. demonstrated that endogenous miR-486-5p was downregulated in cardiac tissue from mice subjected to IRI [18]. Injection of adeno-associated virus 9 (AAV9)-expressing miR-486-5p to mice subjected to cardiac IRI significantly reduced infarct size and cardiomyocyte apoptosis at 24 h, preserved cardiac function and reduced cardiac fibrosis at 3 weeks. Long term cardiac remodeling and dysfunction (at 6 weeks) was also prevented [18]. At 3 weeks after IRI, there was decreased expression of PTEN and FoxO1 in cardiac tissue of mice treated with miR-486-5p. Moreover, cardiomyocyte-specific expression of miR-486-5p suppressed apoptosis, attenuated cardiac dysfunction and fibrosis at 3 weeks, and downregulated PTEN and FoxO1 expression. On the other hand, miR-486-5p inhibition by miR-486-5p sponge AAV9 in cardiac IRI did not further increase infarct size at 24 h or worsen cardiac dysfunction at 3 weeks [18]. Of interest, swimming exercise upregulated miR-486-5p expression in isolated cardiomyocytes from mouse heart tissue, downregulated cardiac PTEN and FoxO1 expression, and protected against cardiac IRI [18]. Furthermore, miR-486-5p knock out mice subjected to exercise prior to cardiac IRI had significantly increased infarct size at 24 h compared to wild type controls [18]. miR-486-5p mimic suppressed apoptosis in cultured rat neonatal and human induced pluripotent stem cell-derived (hiPSC) cardiomyocytes, along with decreased expression of PTEN and FoxO1 and activation of Akt signaling. Finally, while anti-miR-486-5p increased apoptosis in rat cardiomyocytes, silencing either PTEN or FoxO1 in anti-miR-486-5p-transfected cardiomyocytes attenuated the pro-apoptotic effect of miR-486-5p inhibition [18].
These compelling results suggest that exosomal transfer or direct administration of miR-486-5p protects against myocardial IRI by targeting PTEN and FoxO1, thereby activating Akt signaling and suppressing cardiomyocyte apoptosis (Fig. 2). Furthermore, these data also support an important role of miR-486-5p for the cardioprotective benefit of exercise in cardiac IRI.
Matrix metalloproteinase-19 (MMP-19) and promotion of angiogenesis in myocardial infarction
Besides targeting PTEN/PI3K/Akt signaling, miR-486-5p may protect against myocardial ischemic injury via other pathways. De novo angiogenesis may protect injured cardiomyocytes, prevent adverse cardiac remodeling and thereby reduce the risk of heart failure after myocardial infarction [82]. In BM-MSCs under hypoxia, the expression of miR-486-5p and the pro-angiogenic vascular endothelial growth factor (VEGF) is upregulated [83]. miR-486-5p overexpression increases VEGF mRNA, and miR-486-5p inhibition downregulates VEGF mRNA and secretion, suggesting that miR-486-5p confers a proangiogenic effect [83].
Matrix metalloproteinase 19 (Mmp-19) is an inhibitor of angiogenesis whose expression is downregulated in invasive carcinomas [84]. Mmp-19-deficient mice display earlier onset tumor angiogenesis [85] and Mmp-19 reduces endothelial cell angiogenesis by proteolytic cleavage of plasminogen, generating angiostatin-like fragments as endogenous angiogenesis inhibitors [86]. Li et al.studied the effect of exosomal miR-486-5p on angiogenesis after myocardial infarction in mice and non-human primates [65]. In mice with myocardial infarction, intra-myocardial administration of hypoxia-preconditioned MSC-derived exosomes, enriched in miR-486-5p, reduced infarct size, improved left ventricular ejection fraction and increased vascular density after 4 weeks. Exosomes also increased vessel sprouting from isolated aortic rings, further supporting their pro-angiogenic effect. Hypoxia-preconditioned exosomes from MSCs treated with anti-miR-486-5p failed to promote myocardial repair and angiogenesis, suggesting that their protective effect is mediated in part by miR-486-5p. RNA sequencing of mouse myocardial tissues identified Mmp-19 as significantly downregulated in the exosome-treated hearts. Mmp-19 is highly expressed in cardiac fibroblasts, and is a direct target of miR-486-5p by luciferase reporter assay. Furthermore, pathway analysis revealed that VEGF signaling is upregulated in myocardial tissue from exosome-treated mice, and levels of uncleaved VEGF-A are higher in fibroblasts overexpressing miR-486-5p or with silencing of Mmp-19 [65], thus linking miR-486-5p to angiogenesis through its target Mmp-19.
Further studies in a non-human primate model of myocardial infarction revealed that hypoxia-preconditioned exosomes (enriched in miR-486-5p) promote cardiac angiogenesis, reduce infarct size, and improve cardiac function [65]. After 17 month follow-up, increased vascular and arterial density was found within exosome-treated hearts, indicating long-lasting cardiac angiogenesis. Intramyocardial injection of miR-486-5p-overexpressing exosomes improves cardiac function, reduces infarct size, and increases vascular density [65]. These results suggest that miR-486-5p promotes angiogenesis and functional myocardial recovery in two pre-clinical models of myocardial infarction. The pro-angiogenic effect may occur via a paracrine mechanism from fibroblasts that targets Mmp-19 expression, resulting in decreased cleavage of extracellular VEGF-A.
Targeting of the PTEN/Akt pathway in kidney ischemic injury
Acute kidney injury (AKI) refers to a rapid decline in kidney function and is a common complication of hospitalization, affecting up to 20% of patients with a higher prevalence in critical care settings [87]. In-hospital mortality rises with increasing severity of AKI with a rate for severe AKI up to 50%[88]. Patients who recover from AKI are at risk of adverse outcomes including new or progressive CKD, and kidney failure [89]. Yet, no effective treatments for AKI exist and preventative measures are limited [90].
The role of miR-486-5p has been evaluated in the context of kidney IRI, an experimental model of human ischemic AKI characterized by tubular cell damage and apoptosis/necrosis, as well as endothelial cell dysfunction and loss [91]. Human cord blood ECFC-derived exosomes, highly enriched in miR-486-5p, suppress apoptosis of cultured human endothelial cells subjected to hypoxia/reoxygenation by transfer of miR-486-5p, targeting PTEN and activating Akt signaling [16, 92]. In mice with kidney IRI, ECFC-derived exosomes decrease kidney PTEN expression (and activate Akt), associated with reduced apoptosis, histologic injury and improved kidney function [16]. These data suggest that ECFC-derived exosomes protect against kidney IR injury via transfer of miR-486-5p.
A recent study by Viñas et al. examined the effects of direct intravenous administration of lipid-encapsulated miR-486-5p mimic to mice with ischemic AKI, and evaluated the transcriptome of kidney proximal tubules and endothelial cells [45]. miR-486-5p mimic significantly improved kidney function and decreased histological injury and apoptosis in mice subjected to kidney IRI, and prevented the proximal tubular activation of genes commonly associated with ischemic injury. Kidney PTEN protein expression was decreased by miR-486-5p, associated with activation of Akt.
Importantly, the protective effects of miR-486-5p in ischemic AKI may involve other distinct miR-486-5p targets besides PTEN. In particular, distinct proximal tubular genes associated with apoptosis and the tumor necrosis family (TNF) pathway are significantly downregulated by miR-486-5p 24 h after IRI [45]. Furthermore, RNA sequencing of kidney proximal tubular cells and endothelial cells revealed only few known miR-486-5p targets downregulated by mimic 24 h after IRI. Notably, in-situ hybridization revealed that miR-486-5p localizes to the cytoplasm and nucleus of kidney cortical tubular cells [45], raising the possibility that miR-486-5p may directly regulate gene transcription at the nuclear level, in addition to its effects via 3′UTR targeting.
Targeting of NFAT5 in chronic kidney disease: diabetic nephropathy
CKD is defined by the presence of abnormalities of kidney structure or function for at least 3 months [93]. Diabetes mellitus is the leading cause of CKD world-wide, and diabetic nephropathy (DN) affects approximately 30% of patients with diabetes [94]. The pathophysiology of DN involves hemodynamic and metabolic factors including increased intraglomerular pressure and hyperfiltration, and the production of advanced glycation end-products. This promotes the generation of growth factors and hormones, such as transforming growth factor (TGF)-β and angiotensin II, reactive oxygen species, and inflammatory mediators, all of which contribute to kidney histological changes including glomerular basement membrane thickening, extracellular matrix (ECM) deposition within the mesangium, proliferative changes, and ultimately, tubulointerstitial fibrosis and glomerulosclerosis [94, 95].
The nuclear factor of activated T-cells (NFAT), the substrate for calcineurin, represents a family of calcium-dependent transcription factors. NFAT5 is ubiquitously expressed in all tissue types, and is an important gene regulator in organs with high hyperosmotic pressure risk, such as kidneys, heart, and brain [96]. However, NFAT5 can have a pathogenic role in disease as it regulates the expression of pro-inflammatory cytokines [96], increases nuclear factor (NF)-κB activity [97], and increases the expression of genes involved in vascular smooth muscle cell and macrophage migration, platelet activation, and angiogenesis [96]. In experimental DN, the NFAT family of transcription factors is required for ECM protein accumulation and glomerular hypertrophy [98], and NFAT inhibition reduces podocyte injury [99] and renal fibrosis [100].
In this context, Duan et al. demonstrated that NFAT5 is a validated target of miR-486-5p in DN [21]. In addition, miR-486-5p is a downstream target of lnc-ISG20 RNA: lnc-ISG20 expression increases in kidneys of diabetic mice and in mesangial cells cultured in high glucose, while miR-486-5p decreases; NFAT5 expression also increases in both models [21]. NFAT5-induced Akt phosphorylation promotes fibrosis by increasing the expression of collagen, fibronectin and TGF-β in mesangial cells cultured in high glucose. In mice with DN, lnc-ISG20 overexpression promotes kidney fibrosis via Akt phosphorylation, while NFAT5 knockdown prevents fibrosis [21]. Thus, lnc-ISG20 downregulates miR-486-5p expression, resulting in increased NFAT5 expression, Akt activation, and increased expression of pro-fibrotic genes [21]. These data support an anti-fibrotic role of miR-486-5p in diabetic nephropathy.
Of note, the effect of glucose on miR-486-5p expression may be cell-specific. Although high glucose downregulates miR-486-5p expression in mesangial cells [21], exposure of human adipose tissue-derived MSCs to high glucose upregulates miR-486-5p expression, which inhibits cellular proliferation by targeting and downregulating SIRT1 [101], a deacetylase that regulates gene expression and is implicated in insulin sensitivity in type 2 diabetes [102]. SIRT1 expression is downregulated in adipose tissue of patients with diabetes [102], and in endothelial progenitor cells exposed to high glucose [103]. Bouchareychas et al. showed that miR-486-5p is upregulated in monocytes from hyperglycemic mice, in exosomes derived from BM-derived macrophages exposed to hyperglycemia, and in plasma-derived exosomes from patients with diabetes and peripheral arterial disease [104]. Thus, although miR-486-5p may protect against kidney fibrosis by targeting NFAT5 in DN, its upregulation in other cell types in the context of hyperglycemia may have adverse consequences.
Targeting of IGF-1
Insulin-like growth factor-1 (IGF-1) is a polypeptide trophic factor whose biological actions are mediated by the IGF-1 receptor, a tyrosine kinase that phosphorylates intracellular proteins to activate multiple signaling pathways, including PI3K/Akt and mitogen-activated protein kinase (MAPK) [105]. Consequently, IGF-1 is involved in several cellular functions including survival, growth, and differentiation. Targeting of IGF-1 by miR-486-5p in malignant diseases has been reported to reduce cancer cell growth and migration, and stimulate apoptosis [35, 106].
Studies of miR-486-5p and IGF-1 signaling in non-malignant diseases have been limited to in vitro models, and reveal conflicting effects on proliferation, migration, and apoptosis. IGF-1 is an essential regulator of cardiac structure and homeostasis [107] and reduced serum levels of IGF-1 are found in patients with cyanotic congenital heart disease, who experience chronic hypoxemia [108]. Erythrocyte levels of miR-486-5p levels are increased in pediatric patients with cyanotic congenital heart disease compared to healthy controls [109]. Fan et al. showed that miR-486-5p expression is upregulated in rat cardiomyocytes exposed to hypoxia, and demonstrated IGF-1 is a direct target of miR-486-5p by luciferase reporter assay. Downregulation of miR-486-5p increases IGF-1 expression and cell viability, and suppresses hypoxia-induced cardiomyocyte apoptosis, whereas silencing of IGF-1 has the opposite effect [37]. The data suggest that miR-486-5p may have a pathological role in cyanotic congenital heart disease by targeting IGF-1, thereby promoting cardiomyocyte apoptosis (Fig. 2).
The role of miR-486-5p and IGF-1 targeting has also been studied in pre-eclampsia, a serious pregnancy-specific complication mediated by placental dysfunction. Human placental microvascular endothelial cell-derived exosomes are enriched in miR-486-5p [110] and levels are further upregulated following exposure to hypoxia/reoxygenation. Administration of hypoxia-exposed exosomes to trophoblast cells results in transfer of miR-486-5p, associated with decreased cell viability, proliferation, migration and invasion, via targeting of IGF-1 [110]. These data suggest that miR-486-5p may have a pathological role in the development and progression of pre-eclampsia by causing trophoblast dysfunction. Similarly, an in vitro study in hypertrophic scar fibroblasts showed that miR-486-5p overexpression inhibits cell viability, migration and expression of collagens and promotes apoptosis by targeting IGF-1 [111]. Interestingly, miR-486-5p has opposing biological effects in this model, suppressing PI3K/Akt signaling through IGF-1, in contrast to skeletal muscle [15, 23] and models of cardiac IRI [17, 81] Thus, cell-specific factors influence the regulation of miR-486-5p expression and its affected target genes, which ultimately determine biological effects. Indeed, in malignant diseases miR-486-5p has been reported to have both oncogenic and tumor suppressor roles [9].
Targeting SMAD1/2/4 and inhibition of TGF-β signaling
TGF-β belongs to a superfamily of related growth factors and comprises three isoforms in mammals (TGF-β1, TGF-β2, TGF-β3), all of which bind the TGF-β receptor 2 (TGFR2) to recruit TGFR1 and activate signaling [112]. TGF-β signaling is the primary driver of tissue fibrosis, but also has effects on cell proliferation, differentiation, apoptosis, and immunity [112]. TGF-β signaling activates Smad-based pathways by phosphorylation and activation of Smad2/3 by the TGFR1, resulting in nuclear translocation of the Smad complex and transcription of pro-fibrotic genes, such as smooth muscle actin (α-SMA), collagens, and fibronectin [112]. TGF-β also interacts with non-Smad-based signaling including the MAPK and PTEN/PI3K/Akt pathways. Ultimately, TGF-β signaling through Smad and non-Smad-based pathways induces fibrosis via myofibroblast activation, excessive ECM production, and inhibition of ECM degradation [112].
In mice with pulmonary fibrosis, lung miR-486-5p levels diminish, and decreased levels are found in the serum and lung tissue of patients with silicosis and idiopathic pulmonary fibrosis [20]. Administration of miR-486-5p attenuates pulmonary fibrosis in mice exposed to silica or bleomycin [20]. In cultured mouse fibroblasts miR-486-5p directly targets Smad2, inhibits TFG-β-induced expression of pro-fibrotic genes and reduces fibroblast proliferation [20]. In human hypertrophic scar fibroblasts, miR-486-5p targets Smad2 to inhibit proliferation and induce apoptosis [113]. These data support an anti-fibrotic role for miR-486-5p in models of pulmonary fibrosis and hypertrophic scar.
Epithelial–mesenchymal transition (EMT) is another TGF-β-mediated mechanism that contributes to fibrosis [112], and is implicated in development of posterior capsular opacification, (also known as secondary cataract), a complication of cataract surgery. In vitro studies using cultured human lens epithelial cells reported that miR-486-5p expression is downregulated in TGF-β2-induced lens cells, and overexpression of miR-486-5p reduces proliferation, migration, and EMT by targeting Smad2 and Smad4, suggesting that miR-486-5p may prevent the progression of cataracts [114, 115].
Heart failure, a common end-stage manifestation of cardiac diseases, is associated with cardiac remodeling, characterized by cardiomyocyte hypertrophy, apoptosis, cardiac fibroblast activation and ECM deposition [116]. Serum IgE levels are elevated in patients with heart failure, and the IgE receptor (FCεR1) is upregulated in heart tissue from these patients [117]. Zhao et al. showed that blocking the IgE-FCεR1 pathway alleviates pathological cardiac remodeling in two distinct mouse models of heart failure [117]. Treatment of rat cardiac fibroblasts with IgE results in fibroblast activation, matrix protein production, and upregulation of TGF-β signaling in an FCεR1-dependent manner [117].
Zhao et al. [19] subsequently reported that miR-486-5p is downregulated (and its predicted target Smad1 is upregulated) in mouse cardiac fibroblasts treated with IgE in an FCεR1-dependent manner, while the expression of collagens and α-SMA is also increased. Luciferase reporter assay revealed that Smad1 is a direct target of miR-486-5p, while miR-486-5p mimic decreases expression of Smad1/p-Smad1 in primary mouse cardiac fibroblasts. Smad1 also promotes activation and collagen expression in cardiac fibroblasts treated with IgE. These data support a role for the miR-486-5p/Smad1 pathway in IgE-induced collagen expression, and suggest that miR-486-5p protects against cardiac fibrosis by targeting and downregulating Smad1.
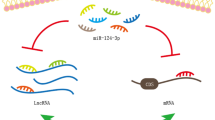
Although targeting of TGF-β/Smad signaling by miR-486-5p may protect against select fibrotic diseases, other processes may be negatively impacted. Shi et al. reported that patients with osteoarthritis (the most common form of arthritis caused by degeneration of joint cartilage) exhibit low levels of Smad2 and elevated miR-486-5p levels in cartilage tissue compared to control patients. In cultured human chondrocytes, miR-486-5p targets Smad2 and decreases cell proliferation and type II collagen expression. These data suggest that miR-486-5p may promote osteoarthritis progression by targeting Smad2 [118] (Fig. 3). However, using collagen-induced arthritic mice as a model for the inflammatory autoimmune disease rheumatoid arthritis, Chen et al.demonstrated that exosomes containing miR-486-5p alleviate disease severity by decreasing the expression of Tob1, an antiproliferative protein that interacts with Smad family proteins [119], inducing osteoblast differentiation [120]. Thus the role of miR-486-5p in arthritis is complex and may differ based on arthritis etiology.

miR-486-5p targets Smad-dependent TGF-β signaling. miR-486-5p confers protective effects in models of cardiac and pulmonary fibrosis, hypertrophic scar, and cataract progression but may have a role in osteoarthritis pathogenesis. EMT epithelial-to-mesenchymal transition, NFAT-5 nuclear factor of activated T cells-5, OA osteoarthritis, α-SMA α-smooth muscle actin, TGF-β transforming growth factor β (created with BioRender.com)
Targeting Sp5/Wnt signaling
Successful wound healing requires cellular proliferation, migration, and angiogenesis [121]. The Wnt/β-catenin pathway involves evolutionary conserved signaling that is essential for cell fate and organization during embryogenesis, and plays a role in adult tissue homeostasis and regeneration [122]. The Sp5 transcription factor is a Wnt target gene that negatively regulates Wnt signaling by transcriptional repression of downstream genes, such as p21 and cyclin-D2, both of which regulate cellular proliferation, and the latter has also been implicated in endothelial cell repair [123].
Lu et al.investigated the role of adipose-derived stem cell-secreted extracellular vesicles, enriched in miR-486-5p, on cutaneous wound healing and the validated target Sp5 [121]. In vitro experiments revealed that vesicle transfer of miR-486-5p enhances proliferation and migration of human skin fibroblasts, and stimulates proliferation, migration, and angiogenesis of human microvascular endothelial cells by inhibiting the expression of Sp5, thereby increasing cyclin-D2 expression. Thus, the relationship between miR-486-5p targeting of the transcriptional repressor Sp5 and endothelial repair warrants further exploration in other models of organ injury and recovery that require de novo angiogenesis.
miR-486-5p targets Histone acetyltransferase 1
Not all pre-clinical data demonstrate protective effects of miR-486-5p in disease. In this regard, histone acetylation is an epigenetic modification that changes chromatin structure and affects DNA replication, repair, and activation of gene transcription [124]. Histone acetyltransferase 1 (HAT1) partially localizes to the cytoplasm and deacetylates newly synthesized histone H4 on lysines 5 and 12 (H4K5, H4K12) [125]. HAT1 is involved in several biological processes including chromatin assembly, DNA replication, DNA repair, cell proliferation and glucose metabolism [124].
Liu et al. investigated the relationship between miR-486-5p and ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux in macrophage-derived foam cells [126]. ABCA1 expression and atherogenesis are regulated by epigenetic modifications [127], but ABCA1 is not a predicted target of miR-486-5p. However, in this study miR-486-5p directly targeted HAT1 by luciferase reporter assay. Treatment of macrophage-derived foam cells with miR-486-5p mimic decreases HAT1 expression, decreases H4K5/H4K12 acetylation, and blocks cholesterol efflux. HAT1 overexpression increases ABCA1 expression, while miR-486-5p mimic inhibits ABCA1 expression [126]. Further supporting the role of miR-486-5p in atherosclerosis, Apoe−−/e−− mice treated with exosomes from BM-derived macrophages (enriched in miR-486-5p) develop atherosclerotic lesions with macrophage foam cells [104]. Together, these studies suggest that macrophage miR-486-5p may promote atherosclerosis.
The relationship between miR-486-5p and HAT1 has also been evaluated in the context of chronic obstructive pulmonary disease (COPD). Zhang et al. reported that miR-486-5p is upregulated in lung tissues of patients with COPD compared to smokers without COPD, and in alveolar macrophages and peripheral monocytes of COPD patients and smokers compared to healthy non-smokers [128]. Rat pulmonary macrophages exposed to cigarette smoking extract upregulate endogenous miR-486-5p and toll-like receptor 4 (TLR4), a known inflammatory trigger, and downregulate HAT1, a direct miR-486-5p target. miR-486-5p negatively regulates HAT1 expression in rat pulmonary macrophages, and HAT1 suppression increases expression of TLR4 and inflammatory cytokines [128]. miR-486-5p may therefore play a pathological role in COPD by regulating TLR-4 triggered inflammation via its target HAT1.
miR-486-5p targets in development
In skeletal muscle development, the transcription factor Pax7 is expressed in muscle satellite cells, and is downregulated in activated satellite cells for differentiation [129]. In C2C12 myoblasts, Dey et al.showed that miR-486-5p expression is upregulated during myoblast differentiation [24]. miR-486-5p overexpression accelerates myoblast differentiation, while miR-486-5p inhibition delays differentiation with persistent expression of Pax7 protein, which is a target of miR-486-5p. Thus, miR-486-5p promotes myogenesis by targeting and downregulating Pax7, a transcription factor required for satellite cell biogenesis and survival [24].
In hematopoiesis, miR-486-5p expression is upregulated throughout erythroid differentiation [25, 26]. miR-486-5p overexpression in cord blood CD34+ cells (megakaryocyte–erythroid progenitors) enhances cell growth, erythroid differentiation and cell survival, while miR-486-5p inhibition suppresses these processes [130]. miR-486-5p inhibition also upregulates PTEN and FoxO1 protein expression, decreases p-Akt, and promotes apoptosis and cell growth inhibition. FoxO1 knockdown rescued the effects of miR-486-5p inhibition but did not influence erythroid differentiation. Thus contributing targets of miR-486-5p other than FoxO1 remain to be uncovered [130]. The role of miR-486-5p in erythropoiesis has been further characterized in homozygous miR-486-5p knockout mice [25]. The mice are viable, but display defects within the erythroid lineage in bone marrow and spleen, with accumulation of early stage erythroblasts, and a reduced proportion of mature erythrocytes. When subjected to oxidative stress, peripheral blood from miR-486-5p knockout mice shows a greater reduction of red blood cells and increased proportion of reticulocytes [25].
miR-486-5p is also implicated in neurogenesis. Dori et al. isolated discrete populations of neural progenitor cells (proliferative progenitors that expand the stem cell pool and differentiative progenitors that generate neurons) and profiled global miRNA expression during cortical development [131]. miR-486-5p expression is transiently downregulated in differentiative progenitors compared with proliferative progenitors and neurons, termed an ‘off-switch’ transcript. In utero delivery of a locked nucleic acid miR-486-5p inhibitor to mice at embryonic day 13.5 (the developmental stage, where cortical progenitor cells are mostly proliferative progenitors) increases the proportion of cortical progenitor cells at the expense of neurons without affecting neuron migration or survival [131]. These findings suggest that miR-486-5p regulates cell fate in neurogenesis, but the precise targets of miR-486-5p in cortical development remain unknown. Thus, understanding the mechanisms by which miR-486-5p regulates developmental processes could provide additional insight into its role in disease states and identify new targets and pathways for further study.
Conclusions
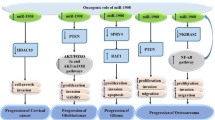
miR-486-5p is implicated in several non-malignant diseases as an important regulator of critical signaling pathways including PTEN/Akt, IGF-1, MMP-19/VEGF, Smad-dependent TGF-β, and Sp5/Wnt/β-catenin, affecting biological processes including apoptosis, cellular migration and proliferation, angiogenesis, and fibrosis (summarized in Fig. 4). miR-486-5p also regulates epigenetic modifications by targeting and downregulating HAT1. However, varying biological effects of miR-486-5p have been reported depending on the targeted genes, which may also be specific to cell type and injury model. In addition to its validated 3′UTR targets that are downregulated by post-transcriptional gene silencing via the cytoplasmic RISC pathway, miR-486-5p also localizes to the nucleus, and thus may affect gene expression beyond classical post-transcriptional gene regulation.

Overview of miR-486-5p target genes, affected signaling pathways, cellular processes, and possible biological effects. miR-486-5p has therapeutic potential in non-malignant diseases by modulating signaling pathways that control critical cellular processes that are involved in tissue regeneration, recovery of organ function, prevention of adverse long-term consequences after organ injury, and organ fibrosis. ABCA1 ATP binding cassette subfamily A member 1, COPD chronic obstructive pulmonary disease, HAT-1 histone acetyltransferase 1, IGF-1 insulin-like growth factor-1, Mmp-19 matrix metalloproteinase-19, NFAT5 nuclear factor of activated T cells-5, OA osteoarthritis, PI3K phosphatidylinositol-3-kinase, PTEN phosphatase and tensin homolog, TGF-ß transforming growth factor-ß, TLR-4 toll-like receptor-4, VEGFA vascular endothelial growth factor-A (created with BioRender.com)
Pre-clinical studies support miR-486-5p as a promising therapy for several non-malignant diseases, including cardiac and kidney disorders, due to its proliferative, pro-angiogenic, anti-apoptotic, and anti-fibrotic effects. miR-486-5p protects against cardiac ischemic injury and targets PTEN, suppressing cardiomyocyte apoptosis in cardiac IRI [17, 81], improves cardiac function and promotes angiogenesis after myocardial infarction associated with targeting Mmp-19 in cardiac fibroblasts [65], and reduces IgE-mediated cardiac fibrosis by targeting Smad1 [19]. miR-486-5p also protects against ischemic kidney injury and targets PTEN [16, 45], associated with decreased expression of select genes involved in apoptosis and the TNF inflammatory pathway, and may confer a protective anti-fibrotic role in diabetic kidney disease by targeting NFAT5 [21]. Although there are no human clinical trials involving miR-486-5p therapy to date, clinical trials are underway for other miRNAs [132]. For instance, a phase I trial of intravenous liposomal miR-34a mimic administered to patients with advanced solid tumors was found to target tumors but unfortunately led to serious immune-mediated adverse events, resulting in early trial termination [133]. In Alport nephropathy—a hereditary nephropathy—Phase 1 trials involving use of the miR-21 inhibitor RG-012 have been completed and will move to Phase 2 [132]. Accordingly, miR-486-5p has potential as a bio-therapeutic agent in several non-malignant diseases. Nonetheless, studies addressing the safety profile and longer term effects of miR-486-5p delivery are warranted, since miR-486-5p affects cell cycle kinetics [14], its expression is tightly regulated in growth and development [11], and dysregulated expression has been associated with malignancy potential [9].
Data availability
Not applicable.
References
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854. https://doi.org/10.1016/0092-8674(93)90529-y
Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488. https://doi.org/10.1038/nrm3611
Friedman RC, Farh KK-H, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. https://doi.org/10.1101/gr.082701.108
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol 9:402. https://doi.org/10.3389/fendo.2018.00402
Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet 16:421–433. https://doi.org/10.1038/nrg3965
Catalanotto C, Cogoni C, Zardo G (2016) MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci 17:1712. https://doi.org/10.3390/ijms17101712
Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y (2018) Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol Cancer 17:64. https://doi.org/10.1186/s12943-018-0765-5
Brandenburger T, Lorenzen JM (2020) Diagnostic and therapeutic potential of microRNAs in acute kidney injury. Front Pharmacol 11:657. https://doi.org/10.3389/fphar.2020.00657
ElKhouly AM, Youness RA, Gad MZ (2020) MicroRNA-486-5p and microRNA-486-3p: multifaceted pleiotropic mediators in oncological and non-oncological conditions. Non-coding RNA Res 5:11–21. https://doi.org/10.1016/j.ncrna.2020.01.001
Gadde S, Rayner KJ (2016) Nanomedicine meets microRNA: Current advances in RNA- based nanotherapies for atherosclerosis. Arterioscler Thromb Vasc Biol 36:e73–e79. https://doi.org/10.1161/ATVBAHA.116.307481
Small EM, O’Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, Richardson JA, Olson EN (2010) Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. PNAS 107:4218–4223. https://doi.org/10.1073/pnas.1000300107
Bayés-Genis A, Lanfear DE, Ronde MWJD, Lupón J, Leenders JJ, Liu Z, Zuithoff NPA, Eijkemans MJC, Zamora E, Antonio MD et al (2018) Prognostic value of circulating microRNAs on heart failure-related morbidity and mortality in two large diverse cohorts of general heart failure patients. Eur J Heart Fail 20:67–75. https://doi.org/10.1002/ejhf.984
Baglio SR, Rooijers K, Koppers-Lalic D, Verweij FJ, Lanzón MPR, Zini N, Naaijkens B, Perut F, Niessen HWM, Baldini N et al (2015) Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res Ther 6:127. https://doi.org/10.1186/s13287-015-0116-z
Alexander MS, Casar JC, Motohashi N, Myers JA, Eisenberg I, Gonzalez RT, Estrella EA, Kang PB, Kawahara G, Kunkel LM (2011) Regulation of DMD pathology by an ankyrin-encoded miRNA. Skelet Muscle 1:27. https://doi.org/10.1186/2044-5040-1-27
Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z (2012) Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int 82:401–411. https://doi.org/10.1038/ki.2012.84
Viñas JL, Burger D, Zimpelmann J, Haneef R, Knoll W, Campbell P, Gutsol A, Carter A, Allan DS, Burns KD (2016) Transfer of microRNA-486-5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int 90:1238–1250. https://doi.org/10.1016/j.kint.2016.07.015
Sun X-H, Wang X, Zhang Y, Hui J (2019) Exosomes of bone-marrow stromal cells inhibit cardiomyocyte apoptosis under ischemic and hypoxic conditions via miR-486-5p targeting the PTEN/ PI3K/AKT signaling pathway. Thromb Res 177:23–32. https://doi.org/10.1016/j.thromres.2019.02.002
Bei Y, Lu D, Bär C, Chatterjee S, Costa A, Riedel I, Mooren FC, Zhu Y, Huang Z, Wei M et al (2022) miR-486 attenuates cardiac ischemia/reperfusion injury and mediates the beneficial effect of exercise for myocardial protection. Mol Ther 30:1675–1691. https://doi.org/10.1016/j.ymthe.2022.01.031
Zhao H, Yang H, Geng C, Chen Y, Tang Y, Li Z, Pang J, Shu T, Nie Y, Liu Y et al (2021) Elevated IgE promotes cardiac fibrosis by suppressing miR-486a-5p. Theranostics 11:7600–7615. https://doi.org/10.7150/thno.47845
Ji X, Wu B, Fan J, Han R, Luo C, Wang T, Yang J, Han L, Zhu B, Wei D et al (2015) The anti-fibrotic effects and mechanisms of MicroRNA-486-5p in pulmonary fibrosis. Sci Rep 5:14131. https://doi.org/10.1038/srep14131
Duan Y-R, Chen B-P, Chen F, Yang S-X, Zhu C-Y, Ma Y-L, Li Y, Shi J (2021) LncRNA lnc-ISG20 promotes renal fibrosis in diabetic nephropathy by inducing AKT phosphorylation through miR-486-5p/NFAT5. J Cell Mol Med 25:4922–4937. https://doi.org/10.1111/jcmm.16280
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S et al (2007) Distinctive patterns of microRNA expression in primary muscular disorders. PNAS 104:17016–17021. https://doi.org/10.1073/pnas.0708115104
Alexander MS, Casar JC, Motohashi N, Vieira NSM, Eisenberg I, Marshall JL, Gasperini MJ, Lek A, Myers JA, Estrella EA et al (2014) MicroRNA-486-dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy-associated symptoms. J Clin Investig 124:2651–2667. https://doi.org/10.1172/JCI73579
Dey BK, Gagan J, Dutta A (2011) miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol 31:203–214. https://doi.org/10.1128/MCB.01009-10
Jee D, Yang J-S, Park S-M, Farmer DJT, Wen J, Chou T, Chow A, McManus MT, Kharas MG, Lai EC (2018) Dual strategies for argonaute2-mediated biogenesis of erythroid miRNAs underlie conserved requirements for slicing in mammals. Mol Cell 69:265–278. https://doi.org/10.1016/j.molcel.2017.12.027
Lulli V, Romania P, Morsilli O, Cianciulli P, Gabbianelli M, Testa U, Giuliani A, Marziali G (2013) MicroRNA-486-3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS ONE 8:e60436. https://doi.org/10.1371/journal.pone.0060436
Medley JC, Panzade G, Zinovyeva AY (2021) microRNA strand selection: unwinding the rules. WIREs RNA 12:e1627. https://doi.org/10.1002/wrna.1627
Yang J-S, Phillips MD, Betel D, U P, Ventura A, Siepel AC, Chen KC, Lai EC (2011) Widespread regulatory activity of vertebrate microRNA* species. RNA 17:312–326. https://doi.org/10.1261/rna.2537911
Wang Y, Cai Y, Huang H, Chen X, Chen X, Chen X, Mai H, Li X, Zhao J, Yang J et al (2018) miR-486-3p influences the neurotoxicity of a-synuclein by targeting the SIRT2 gene and the polymorphisms at target sites contributing to Parkinson’s disease. Cell Physiol Biochem 51:2732–2745. https://doi.org/10.1159/000495963
Huang Z-X, Chen Y, Guo H-R, Chen G-F (2021) Systematic review and bioinformatic analysis of microRNA expression in autism spectrum disorder identifies pathways associated with cancer, metabolism, cell signaling, and cell adhesion. Front Psychiatry 12:630876. https://doi.org/10.3389/fpsyt.2021.630876
Yu D, Jiao X, Cao T, Huang F (2018) Serum miRNA expression profiling reveals miR-486-3p may play a significant role in the development of autism by targeting ARID1B. NeuroReport 29:1431–1436. https://doi.org/10.1097/WNR.0000000000001107
Shibutani M, Horii T, Shoji H, Morita S, Kimura M, Terawaki N, Miyakawa T, Hatada I (2017) Arid1b haploinsufficiency causes abnormal brain gene expression and autism-related behaviors in mice. Int J Mol Sci 18:1872. https://doi.org/10.3390/ijms18091872
Gallagher PG, Forget BG (1998) An alternate promoter directs expression of a truncated, muscle-specific isoform of the human ankyrin 1 gene*. J Biol Chem 273:1339–1348. https://doi.org/10.1074/jbc.273.3.1339
Tessema M, Yingling CM, Picchi MA, Wu G, Ryba T, Lin Y, Bungum AO, Edell ES, Spira A, Belinsky SA (2017) ANK1 methylation regulates expression of microRNA-486-5p and discriminates lung tumors by histology and smoking status. Cancer Lett 410:191–200. https://doi.org/10.1016/j.canlet.2017.09.038
Peng Y, Dai Y, Hitchcock C, Yang X, Kassis ES, Liu L, Luo Z, Sun H-L, Cui R, Wei H et al (2013) Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. PNAS 110:15043–15048. https://doi.org/10.1073/pnas.1307107110
Yang Y, Ji C, Guo S, Su X, Zhao X, Zhang S, Liu G, Qiu X, Zhang Q, Guo H et al (2017) The miR-486-5p plays a causative role in prostate cancer through negative regulation of multiple tumor suppressor pathways. Oncotarget 8:72835–72846. https://doi.org/10.18632/oncotarget.20427
Fan J, Shi S, Qiu Y, Zheng Z, Yu L (2019) MicroRNA-486-5p down-regulation protects cardiomyocytes against hypoxia-induced cell injury by targeting IGF-1. Int J Clin Exp Pathol 12:2544–2551
Xiong F, Wei W-P, Liu Y-B, Wang Y, Zhang H-Y, Liu R (2021) Long noncoding RNA XIST enhances cerebral ischemia-reperfusion injury by regulating miR-486-5p and GAB2. Eur Rev Med Pharmacol Sci 25:2013–2020. https://doi.org/10.26355/eurrev_202102_25103
Li D, Wu L, Knox B, Chen S, Tolleson WH, Liu F, Yu D, Guo L, Tong W, Ning B (2020) Long noncoding RNA LINC00844-mediated molecular network regulates expression of drug metabolizing enzymes and nuclear receptors in human liver cells. Arch Toxicol 94:1637–1653. https://doi.org/10.1007/s00204-020-02706-5
Park CW, Zeng Y, Zhang X, Subramanian S, Steer CJ (2010) Mature microRNAs identified in highly purified nuclei from HCT116 colon cancer cells. RNA Biol 7:606–614. https://doi.org/10.4161/rna.7.5.13215
Chen B, Zhang B, Luo H, Yuan J, Skogerbø G, Chen R (2012) Distinct microRNA subcellular size and expression patterns in human cancer cells. Int J Cell Biol 2012:672462. https://doi.org/10.1155/2012/672462
Liao J-Y, Ma L-M, Guo Y-H, Zhang Y-C, Zhou H, Shao P, Chen Y-Q, Qu L-H (2010) Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 39 trailers. PLoS ONE 5:e10563. https://doi.org/10.1371/journal.pone.0010563
Nishi K, Nishi A, Nagasawa T, Ui-Tei K (2013) Human TNRC6A is an argonaute-navigator protein for microRNA-mediated gene silencing in the nucleus. RNA 19:17–35. https://doi.org/10.1261/rna.034769.112
Kalantari R, Hicks JA, Li L, Gagnon KT, Sridhara V, Lemoff A, Mirzaei H, Corey DR (2016) Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA 22:1085–1098. https://doi.org/10.1261/rna.056499.116
Viñas JL, Spence M, Porter CJ, Douvris A, Guts A, Zimpelmann JA, Campbell PA, Burns KD (2021) micro-RNA-486-5p protects against kidney ischemic injury and modifies the apoptotic transcriptome in proximal tubules. Kidney Int 100:597–612. https://doi.org/10.1016/j.kint.2021.05.034
Turchinovich A, Weiz L, Langheinz A, Burwinkel B (2011) Characterization of extracellular circulating microRNA. Nucleic Acids Res 39:7223–7233. https://doi.org/10.1093/nar/gkr254
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL et al (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. PNAS 108:5003–5008. https://doi.org/10.1073/pnas.1019055108
LaBelle J, Bowser M, Brown A, Farnam L, Kho A, Li J, McGeachie M, Chase R, Piehl S, Allen K et al (2021) Commercially available blocking oligonucleotides effectively suppress unwanted hemolysis-related miRNAs in a large whole-blood RNA cohort. J Mol Diagn 23:671–682. https://doi.org/10.1016/j.jmoldx.2021.03.006
Wu Q, Yu L, Lin X, Zheng Q, Zhang S, Chen D, Pan X, Huang Y (2020) Combination of serum miRNAs with serum exosomal miRNAs in early diagnosis for non-small-cell lung cancer. Cancer Manag Res 12:485–495. https://doi.org/10.2147/CMAR.S232383
Pritchard CC, Kroh E, Wood B, Arroyo JD, Dougherty KJ, Miyaji MM, Tait JF, Tewari M (2012) Blood cell origin of circulating microRNAs: a cautionary note for cancer biomarker studies. Cancer Prev Res (Phila) 5:492–497. https://doi.org/10.1158/1940-6207.CAPR-11-0370
Shkurnikov MY, Knyazev EN, Fomicheva KA, Mikhailenko DS, Nyushko KM, Saribekyan EK, Samatov TR, Alekseev BY (2016) Analysis of Plasma microRNA Associated with Hemolysis. Bull Exp Biol Med 160:748–750. https://doi.org/10.1007/s10517-016-3300-y
Sun B, Guo S (2021) miR-486-5p serves as a diagnostic biomarker for sepsis and its predictive value for clinical outcomes. J Inflamm Res 14:3687–3695. https://doi.org/10.2147/JIR.S323433
Ideozu JE, Zhang X, Rangaraj V, McColley S, Levy H (2019) Microarray profiling identifies extracellular circulating miRNAs dysregulated in cystic fibrosis. Sci Rep 9:15483. https://doi.org/10.1038/s41598-019-51890-7
Regmi A, Liu G, Zhong X, Hu S, Ma R, Zafar LGMI, Chen L (2019) Evaluation of serum microRNAs in patients with diabetic kidney disease: a nested case-controlled study and bioinformatic analysis. Med Sci Monit 25:1699–1708. https://doi.org/10.12659/MSM.913265
Kong R, Gao J, Si Y, Zhao D (2017) Combination of circulating miR-19b-3p, miR-122-5p and miR-486-5p expressions correlates with risk and disease severity of knee osteoarthritis. Am J Transl Res 9:2852–2864
Prabhakar P, Chandra SR, Christopher R (2017) Circulating microRNAs as potential biomarkers for the identification of vascular dementia due to cerebral small vessel disease. Age Ageing 46:861–864. https://doi.org/10.1093/ageing/afx090
Hoss AG, Lagomarsino VN, Frank S, Hadzi TC, Myers RH, Latourelle JC (2015) Study of plasma-derived miRNAs mimic differences in Huntington’s disease brain. Mov Disord 30:1961–1964. https://doi.org/10.1002/mds.26457
Ghahramani-Seno MM, Hu P, Gwadry FG, Pinto D, Marshall CR, Casallo G, Scherer SW (2011) Gene and miRNA expression profiles in autism spectrum disorders. Brain Res 1380:85–97. https://doi.org/10.1016/j.brainres.2010.09.046
Zaki MB, Abulsoud AI, Elsisi AM, Doghish AS, Mansour OAE, Amin AI, Elrebehy MA, Mohamed MY, Goda MA (2019) Potential role of circulating microRNAs (486–5p, 497, 509–5p and 605) in metabolic syndrome Egyptian male patients. Diabetes Metab Syndr Obes 12:601–611. https://doi.org/10.2147/DMSO.S187422
Prats-Puig A, Ortega FJ, Mercader JM, Moreno-Navarrete JM, Moreno M, Bonet N, Ricart W, López-Bermejo A, Fernández-Real JM (2013) Changes in circulating microRNAs are associated with childhood obesity. J Clin Endocrinol Metab 98:E1655-1660. https://doi.org/10.1210/jc.2013-1496
Matsha TE, Kengne AP, Hector S, Mbu DL, Yako YY, Erasmus RT (2018) MicroRNA profiling and their pathways in South African individuals with prediabetes and newly diagnosed type 2 diabetes mellitus. Oncotarget 9:30485–30498. https://doi.org/10.18632/oncotarget.25271
Butler AE, Ramachandran V, Sathyapalan T, David R, Gooderham NJ, Benurwar M, Dargham SR, Hayat S, Najafi-Shoushtar SH, Atkin SL (2020) microRNA expression in women with and without polycystic ovarian syndrome matched for body mass index. Front Endocrinol (Lausanne) 11:206. https://doi.org/10.3389/fendo.2020.00206
Yang Q, Gu W-W, Gu Y, Yan N-N, Mao Y-Y, Zhen X-X, Wang J-M, Yang J, Shi H-J, Zhang X et al (2018) Association of the peripheral blood levels of circulating microRNAs with both recurrent miscarriage and the outcomes of embryo transfer in an in vitro fertilization process. J Transl Med 16:186. https://doi.org/10.1186/s12967-018-1556-x
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO (2020) RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 21:585–606. https://doi.org/10.1038/s41580-020-0251-y
Li Q, Xu Y, Lv K, Wang Y, Zhong Z, Xiao C, Zhu K, Ni C, Wang K, Kong M et al (2021) Small extracellular vesicles containing miR-486–5p promote angiogenesis after myocardial infarction in mice and nonhuman primates. Sci Transl Med 13:eabb0202. https://doi.org/10.1126/scitranslmed.abb0202
Li Z, Liu C, Li S, Li T, Li Y, Wang N, Bao X, Xue P, Liu S (2021) BMSC-derived exosomes inhibit dexamethasone-induced muscle atrophy via the miR-486-5p/FoxO1 axis. Front Endocrinol 12:681267. https://doi.org/10.3389/fendo.2021.681267
Brito MB, Goulielmaki E, Papakonstanti EA (2015) Focus on PTEN regulation. Front Oncol 5:166. https://doi.org/10.3389/fonc.2015.00166
Ghafouri-Fard S, Abak A, Shoorei H, Mohaqiq M, Majidpoor J, Sayad A, Taheri M (2021) Regulatory role of microRNAs on PTEN signaling. Biomed Pharmacother 113:110986. https://doi.org/10.1016/j.biopha.2020.110986
Gomez-Manzano C, Fueyo J, Jiang H, Glass TL, Lee H-Y, Hu M, Liu J-L, Jasti SL, Liu T-J, Conrad CA et al (2003) Mechanisms underlying PTEN regulation of vascular endothelial growth factor and angiogenesis. Ann Neurol 53:109–117. https://doi.org/10.1002/ana.10396
Lee S, Dong HH (2017) FoxO integration of insulin signaling with glucose and lipid metabolism. J Endocrinol 233:R67–R79. https://doi.org/10.1530/JOE-17-0002
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochem Biophys Acta 1813:1938–1945. https://doi.org/10.1016/j.bbamcr.2011.06.002
Emery AE (2002) The muscular dystrophies. Lancet 359:687–695. https://doi.org/10.1016/S0140-6736(02)07815-7
Feron M, Guevel L, Rouger K, Dubreil L, Arnaud M-C, Ledevin M, Megeney LA, Cherel Y, Sakanyan V (2009) PTEN contributes to profound PI3K/Akt signaling pathway deregulation in dystrophin-deficient dog muscle. Am J Pathol 174:1459–1470. https://doi.org/10.2353/ajpath.2009.080460
Kim MH, Kay DI, Rudra RT, Chen BM, Hsu N, Izumiya Y, Martinez L, Spencer MJ, Walsh K, Grinnell AD et al (2011) Myogenic Akt signaling attenuates muscular degeneration, promotes myofiber regeneration and improves muscle function in dystrophin-deficient mdx mice. Hum Mol Genet 20:1324–1338. https://doi.org/10.1093/hmg/ddr015
Lees SJ, Childs TE, Booth FW (2008) Age-dependent FOXO regulation of p27Kip1 expression via a conserved binding motif in rat muscle precursor cells. Am J Physiol Cell Physiol 295:C1238-1246. https://doi.org/10.1152/ajpcell.00349.2008
Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE (2006) Chronic kidney disease causes defects in signaling through the insulin receptor substrate phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol 17:1388–1394. https://doi.org/10.1681/ASN.2004100842
Robinson KA, Baker LA, Graham-Brown MPM, Watson EL (2020) Skeletal muscle wasting in chronic kidney disease: the emerging role of microRNAs. Nephrol Dial Transplant 35:1469–1478. https://doi.org/10.1093/ndt/gfz193
Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T (2012) Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth 16:123–132. https://doi.org/10.1177/1089253211436350
Bekkers SC, Yazdani SK, Virmani R, Waltenberger J (2010) Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J Am Coll Cardiol 55:1649–1660. https://doi.org/10.1016/j.jacc.2009.12.037
Wang J, Che H, Su Q, You Zhou M, Liu T, Li L (2016) The PTEN/Akt signaling pathway mediates myocardial apoptosis in swine after coronary microembolization. J Cardiovasc Pharmacol Ther 21:471–477. https://doi.org/10.1177/1074248415624158
Zhu H-H, Wang X-T, Sun Y-H, He W-K, Liang J-B, Mo B-H, Li L (2019) MicroRNA-486-5p targeting PTEN protects against coronary microembolization-induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur J Pharmacol 855:244–251. https://doi.org/10.1016/j.ejphar.2019.03.045
Gogiraju R, Bochenek ML, Schäfer K (2019) Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front Cardiovasc Med 6:20. https://doi.org/10.3389/fcvm.2019.00020
Shi X-F, Wang H, Xiao F-J, Yin Y, Xu Q-Q, Ge R-L, Wang L-S (2016) MiRNA-486 regulates angiogenic activity and survival of mesenchymal stem cells under hypoxia through modulating Akt signal. Biochem Biophys Res Commun 470:670–677. https://doi.org/10.1016/j.bbrc.2016.01.084
Velinov N, Aebersold D, Haeni N, Hlushchuk R, Weinstein F, Sedlacek R, Djonov V (2007) Matrix metalloproteinase-19 is a predictive marker for tumor invasiveness in patients with oropharyngeal squamous cell carcinoma. Int J Biol Markers 22:265–273. https://doi.org/10.5301/JBM.2008.2632
Jost M, Folgueras AR, Frérart F, Pendas AM, Blacher S, Houard X, Berndt S, Munaut C, Cataldo D, Alvarez J et al (2006) Earlier onset of tumoral angiogenesis in matrix metalloproteinase-19-deficient mice. Cancer Res 66:5234–5241. https://doi.org/10.1158/0008-5472.CAN-05-4315
Brauer R, Beck IM, Roderfeld M, Roeb E, Sedlacek R (2011) Matrix metalloproteinase-19 inhibits growth of endothelial cells by generating angiostatin-like fragments from plasminogen. BMC Biochem 12:38. https://doi.org/10.1186/1471-2091-12-38
Wang HE, Muntner P, Chertow GM, Warnock DG (2012) Acute kidney injury and mortality in hospitalized patients. Am J Nephrol 35:349–355. https://doi.org/10.1159/000337487
Uchino S, Bellomo R, Goldsmith D, Bates S, Ronco C (2006) An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit Care Med 34:1913–1917. https://doi.org/10.1097/01.CCM.0000224227.70642.4F
See EJ, Jayasinghe K, Glassford N, Bailey M, Johnson DW, Polkinghorne KR, Toussaint ND, Bellomo R (2019) Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int 95:160–172. https://doi.org/10.1016/j.kint.2018.08.036
Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group (2012) KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2:19–36. https://doi.org/10.1159/000339789
Bonventre JV, Yang L (2011) Cellular pathophysiology of ischemic acute kidney injury. J Clin Investig 121:4210–4221. https://doi.org/10.1172/JCI45161
Viñas JL, Spence M, Gutsol A, Knoll W, Burger D, Zimpelmann J, Allan DS, Burns KD (2018) Receptor-ligand interaction mediates targeting of endothelial colony forming cell-derived exosomes to the kidney after ischemic injury. Sci Rep 8:16320. https://doi.org/10.1038/s41598-018-34557-7
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group (2013) KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 3:1–150
Umanath K, Lewis JB (2018) Update on diabetic nephropathy: core curriculum 2018. Am J Kidney Dis 71:884–895. https://doi.org/10.1053/j.ajkd.2017.10.026
Kato M, Natarajan R (2014) Diabetic nephropathy—emerging epigenetic mechanisms. Nat Rev Nephrol 10:517–530. https://doi.org/10.1038/nrneph.2014.116
Cen L, Xing F, Xu L, Cao Y (2020) Potential role of gene regulator NFAT5 in the pathogenesis of diabetes mellitus. J Diabetes Res. https://doi.org/10.1155/2020/6927429
Zhai S, Li M, Sun B, Han Y (2019) Amelioration of lipopolysaccharide-induced nephrotic proteinuria by NFAT5 depletion involves suppressed NF-κB activity. Inflammation 42:1326–1335. https://doi.org/10.1007/s10753-019-00993-4
Gooch JL, Barnes JL, Garcia S, Abboud HE (2003) Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol 284:F144–F154. https://doi.org/10.1152/ajprenal.00158.2002
Zhang L, Li R, Shi W, Liang X, Liu S, Ye Z, Yu C, Chen Y, Zhang B, Wang W et al (2013) NFAT2 inhibitor ameliorates diabetic nephropathy and podocyte injury in db/db mice. Br J Pharmacol 170:426–439. https://doi.org/10.1111/bph.12292
Lu A, Pallero MA, Owusu BY, Borovjagin AV, Lei W, Sanders PW, Murphy-Ullrich JE (2020) Calreticulin is important for the development of renal fibrosis and dysfunction in diabetic nephropathy. Matrix Biol Plus 8:100034. https://doi.org/10.1016/j.mbplus.2020.100034
Kim YJ, Hwang SH, Lee SY, Shin KK, Cho HH, Bae YC, Jung JS (2012) miR-486-5p induces replicative senescence of human adipose tissue-derived mesenchymal stem cells and its expression is controlled by high glucose. Stem Cells Dev 21:1749–1760. https://doi.org/10.1089/scd.2011.0429
Rutanen J, Yaluri N, Modi S, Pihlajamäki J, Vänttinen M, Itkonen P, Kainulainen S, Yamamoto H, Lagouge M, Sinclair DA et al (2010) SIRT1 mRNA expression may be associated with energy expenditure and insulin sensitivity. Diabetes 59:829–835. https://doi.org/10.2337/db09-1191
Balestrieri ML, Rienzo M, Felice F, Rossiello R, Grimaldi V, Milone L, Casamassimi A, Servillo L, Farzati B, Giovane A et al (2008) High glucose downregulates endothelial progenitor cell number via SIRT1. Biochim Biophys Acta 1784:936–945. https://doi.org/10.1016/j.bbapap.2008.03.004
Bouchareychas L, Duong P, Phu TA, Alsop E, Meechoovet B, Reiman R, Ng M, Yamamoto R, Nakauchi H, Gasper WJ et al (2021) High glucose macrophage exosomes enhance atherosclerosis by driving cellular proliferation & hematopoiesis. Science 24:102847. https://doi.org/10.1016/j.isci.2021.102847
Zheng W-H, Quirion R (2006) Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci 7:51. https://doi.org/10.1186/1471-2202-7-51
Youness RA, El-Tayebi HM, Assal RA, Hosny K, Esmat G, Abdelaziz AI (2016) MicroRNA-486-5p enhances hepatocellular carcinoma tumor suppression through repression of IGF-1R and its downstream mTOR, STAT3 and c-Myc. Oncol Lett 12:2567–2573. https://doi.org/10.3892/ol.2016.4914
Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, Kahn CR (2007) Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol Cell Biol 27:1649–1664. https://doi.org/10.1128/MCB.01110-06
Dündar B, Akçoral A, Saylam G, Unal N, Meşe T, Hüdaoğlu S, Büyükgebiz B, Böber E, Büyükgebiz A (2000) Chronic hypoxemia leads to reduced serum IGF-1 levels in cyanotic congenital heart disease. J Pediatr Endocrinol Metab 13:431–436. https://doi.org/10.1515/jpem.2000.13.4.431
Mukai N, Nakayama Y, Murakami S, Tanahashi T, Sessler DI, Ishii S, Ogawa S, Tokuhira N, Mizobe T, Sawa T et al (2018) Potential contribution of erythrocyte microRNA to secondary erythrocytosis and thrombocytopenia in congenital heart disease. Pediatr Res 83:867–873. https://doi.org/10.1038/pr.2017.327
Ma R, Liang Z, Shi X, Xu L, Li X, Wu J, Zhao L, Liu G (2021) Exosomal miR-486-5p derived from human placental microvascular endothelial cells regulates proliferation and invasion of trophoblasts via targeting IGF1. Hum Cell 34:1310–1323. https://doi.org/10.1007/s13577-021-00543-x
Xiao Y (2020) MiR-486-5p inhibits the hyperproliferation and production of collagen in hypertrophic scar fibroblasts via IGF1/PI3K/AKT pathway. J Dermatol Treat. https://doi.org/10.1080/09546634.2020.1728210
Meng X-M, Nikolic-Paterson DJ, Lan HY (2016) TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338. https://doi.org/10.1038/nrneph.2016.48
Shi Y, Wang L, Yu P, Liu Y, Chen W (2019) MicroRNA-486-5p inhibits the growth of human hypertrophic scar fibroblasts by regulating Smad2 expression. Mol Med Rep 19:5203–5210. https://doi.org/10.3892/mmr.2019.10186
Liu B, Sun J, Lei X, Zhu Z, Pei C, Qin L (2017) MicroRNA-486-5p suppresses TGF-ß2-induced proliferation, invasion and epithelial mesenchymal transition of lens epithelial cells by targeting Smad2. J Biosci 42:575–584. https://doi.org/10.1007/s12038-017-9709-2
Wang H, Zheng G (2020) LncRNA NEAT1 promotes proliferation, migration, invasion and epithelial-mesenchymal transition process in TGF-β2-stimulated lens epithelial cells through regulating the miR-486-5p SMAD4 axis. Cancer Cell Int 20:529. https://doi.org/10.1186/s12935-020-01619-8
Piek A, de-Boer RA, Silljé HHW (2016) The fibrosis-cell death axis in heart failure. Heart Fail Rev 21:199–211. https://doi.org/10.1007/s10741-016-9536-9
Zhao H, Yang H, Geng C, Chen Y, Pang J, Shu T, Zhao M, Tang Y, Li Z, Li B et al (2021) Role of IgE-FcεR1 in pathological cardiac remodeling and dysfunction. Circulation 143:1014–1030. https://doi.org/10.1161/CIRCULATIONAHA.120.047852
Shi J, Guo K, Su S, Li J, Li C (2018) miR-486-5p is upregulated in osteoarthritis and inhibits chondrocyte proliferation and migration by suppressing SMAD2. Mol Med Rep 18:502–508. https://doi.org/10.3892/mmr.2018.8931
Tzachanis D, Freeman GJ, Hirano N, Puijenbroek AAV, Delfs MW, Berezovskaya A, Nadler LM, Boussiotis VA (2001) Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat Immunol 2:1174–1182. https://doi.org/10.1038/ni730
Chen J, Liu M, Luo X, Peng L, Zhao Z, Hea C, He Y (2020) Exosomal miRNA-486-5p derived from rheumatoid arthritis fibroblast-like synoviocytes induces osteoblast differentiation through the Tob1/BMP/Smad pathway. Biomater Sci 8:3430. https://doi.org/10.1039/c9bm01761e
Lu Y, Wen H, Huang J, Liao P, Liao H, Tu J, Zeng Y (2020) Extracellular vesicle-enclosed miR-486-5p mediates wound healing with adipose-derived stem cells by promoting angiogenesis. J Cell Mol Med 00:1–15. https://doi.org/10.1111/jcmm.15387
Schunk SJ, Floege JR, Fliser D, Speer T (2021) WNT-β-catenin signalling—a versatile player in kidney injury and repair. Nat Rev Nephrol 17:172. https://doi.org/10.1038/s41581-020-00343-w
Huang R, Hu Z, Cao Y, Li H, Zhang H, Su W, Xu Y, Liang L, Melgiri ND, Jiang L (2019) MiR-652-3p inhibition enhances endothelial repair and reduces atherosclerosis by promoting Cyclin D2 expression. EBioMedicine 40:685–694. https://doi.org/10.1016/j.ebiom.2019.01.032
Poziello A, Nebbioso A, Stunnenberg HG, Martens JHA, Carafa V, Altuccia L (2021) Recent insights into histone acetyltransferase-1: biological function and involvement in pathogenesis. Epigenetics 16:838–850. https://doi.org/10.1080/15592294.2020.1827723
Parthun MR (2012) Histone acetyltransferase 1: more than just an enzyme? Biochim Biophys Acta 1819:256–263. https://doi.org/10.1016/j.bbagrm.2011.07.006
Liu D, Zhang M, Xie W, Lan G, Cheng H-P, Gong D, Huang C, Lv Y-C, Yao F, Tan Y-L et al (2016) MiR-486 regulates cholesterol efflux by targeting HAT1. Biochem Biophys Res Commun 472:418–424. https://doi.org/10.1016/j.bbrc.2015.11.128
Cao Q, Rong S, Repa JJ, Clair RS, Parks JS, Mishra N (2014) Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol 34:1871–1879. https://doi.org/10.1161/ATVBAHA.114.303393
Zhang J, Xu Z, Kong L, Gao H, Zhang Y, Zheng Y, Wan Y (2020) miRNA-486-5p promotes COPD progression by targeting HAT1 to regulate the TLR4-triggered inflammatory response of alveolar macrophages. Int J Chron Obstruct Pulmon Dis 15:2991–3001. https://doi.org/10.2147/COPD.S280614
Zammit PS, Relaix F, Nagata Y, Ruiz APR, Collins CA, Partridge TA, Beauchamp JR (2006) Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci 119:1824–1832. https://doi.org/10.1242/jcs.02908
Wang L-S, Li L, Li L, Chu S, Shiang K-D, Li M, Sun H-Y, Xu J, Xiao F-J, Sun G et al (2015) MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 125:1302–1313. https://doi.org/10.1182/blood-2014-06-581926
Dori M, Cavalli D, Lesche M, Massalini S, Alieh LHA, Toledo BCD, Khudayberdiev S, Schratt G, Dahl A, Calegari F (2020) MicroRNA profiling of mouse cortical progenitors and neurons reveals miR-486-5p as a regulator of neurogenesis. Development 147:190520. https://doi.org/10.1242/dev.190520
Hanna J, Hossain GS, Kocerha J (2019) The Potential for microRNA therapeutics and clinical research. Front Genet 10:478. https://doi.org/10.3389/fgene.2019.00478
Hong DS, Kang Y-K, Borad M, Sachdev J, Ejadi S, Lim HY, Brenner AJ, Park K, Lee J-L, Ki T-Y et al (2020) Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer 122:1630–1637. https://doi.org/10.1038/s41416-020-0802-1
Acknowledgements
The generous research support of the Jones Family Foundation (of The Ottawa Hospital Foundation) is gratefully acknowledged.
Funding
Supported by a Grant to KDB from the Canadian Institutes of Health Research (HDK-388364).
Author information
Authors and Affiliations
Contributions
KDB and JV conceptualized the study. AD, JV, and KDB drafted the initial manuscript. AD, JV, and KDB all contributed to the literature search, manuscript review and editing, and approved the final version. KDB is the guarantor of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Douvris, A., Viñas, J. & Burns, K.D. miRNA-486-5p: signaling targets and role in non-malignant disease. Cell. Mol. Life Sci. 79, 376 (2022). https://doi.org/10.1007/s00018-022-04406-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04406-y