
Abstract
Alloxan and streptozotocin are toxic glucose analogues that preferentially accumulate in pancreatic beta cells via the GLUT2 glucose transporter. In the presence of intracellular thiols, especially glutathione, alloxan generates reactive oxygen species (ROS) in a cyclic redox reaction with its reduction product, dialuric acid. Autoxidation of dialuric acid generates superoxide radicals, hydrogen peroxide and, in a final iron-catalysed reaction step, hydroxyl radicals. These hydroxyl radicals are ultimately responsible for the death of the beta cells, which have a particularly low antioxidative defence capacity, and the ensuing state of insulin-dependent ‘alloxan diabetes’. As a thiol reagent, alloxan also selectively inhibits glucose-induced insulin secretion through its ability to inhibit the beta cell glucose sensor glucokinase. Following its uptake into the beta cells, streptozotocin is split into its glucose and methylnitrosourea moiety. Owing to its alkylating properties, the latter modifies biological macromolecules, fragments DNA and destroys the beta cells, causing a state of insulin-dependent diabetes. The targeting of mitochondrial DNA, thereby impairing the signalling function of beta cell mitochondrial metabolism, also explains how streptozotocin is able to inhibit glucose-induced insulin secretion.
Similar content being viewed by others


Introduction
Alloxan and streptozotocin are the most prominent diabetogenic chemicals in diabetes research. Both are cytotoxic glucose analogues. Although their cytotoxicity is achieved via different pathways, their mechanisms of beta cell selective action are identical.
In 1838, Wöhler and Liebig [1] synthesised a pyrimidine derivative, which they later called alloxan [2, 3]. In 1943, alloxan became of interest in diabetes research when Dunn and McLetchie reported that it could induce diabetes in animals [4] as a result of the specific necrosis of the pancreatic beta cells [5–7]. The resulting insulinopenia causes a state of experimental diabetes mellitus called ‘alloxan diabetes’ [4, 8, 9]. The reduction product of alloxan, dialuric acid [1], has also been shown to be diabetogenic in animals [10, 11], and to cause ultrastructural changes identical to those observed in response to alloxan [6].
Streptozotocin is an antimicrobial agent and has also been used as a chemotherapeutic alkylating agent [12–14]. In 1963, Rakieten et al. [15] reported that streptozotocin is diabetogenic. Again, this insulinopenia syndrome, called ‘streptozotocin diabetes’ [13], is caused by the specific necrosis of the pancreatic beta cells and streptozotocin has been the agent of choice for the induction of diabetes mellitus in animals ever since [3, 16].
After decades of research a unifying explanation for the selective toxicity of these two most prominent diabetogenic agents [2, 17–22] can be provided. Since an understanding of the chemical reactivity of these compounds is crucial for understanding their diabetogenicity, this review will provide an integrated view of their chemical properties and biological effects.
Alloxan diabetes and streptozotocin diabetes
Figure 1 shows a schematic diagram of the tetraphasic and triphasic blood glucose responses induced by alloxan and streptozotocin, respectively, when injected [22]. The responses are accompanied by corresponding inverse changes in plasma insulin and sequential ultrastructural changes resulting in necrotic beta cell death.

Phasic blood glucose response to a diabetogenic dose of alloxan (tetraphasic; I–IV) or streptozotocin (triphasic; the first phase does not develop in the case of streptozotocin; II–IV)
A first transient hypoglycaemic phase of up to 30 min starts within minutes of alloxan injection. This short-lived hypoglycaemic response is the result of a transient stimulation of insulin secretion, as documented by an increase in the plasma insulin concentration. The underlying mechanism is a temporarily reduced consumption and increased availability of ATP caused by blockade of glucose phosphorylation through glucokinase inhibition. This initial transient hypoglycaemic phase is not observed in response to streptozotocin injection, since streptozotocin does not inhibit glucokinase. Morphological alterations are minimal during this phase.
The second phase starts with an increase in the blood glucose concentration, 1 h after administration of the toxins, and a decrease in plasma insulin. This first hyperglycaemic phase, which usually lasts 2–4 h, is caused by inhibition of insulin secretion leading to hypoinsulinaemia. During this phase the beta cells show the following morphological characteristics: intracellular vacuolisation, dilation of the rough endoplasmic reticulum, decreased Golgi area, reduced secretory granules and insulin content, and swollen mitochondria.
The third phase, again a hypoglycaemic phase, typically occurs 4–8 h after the injection of the toxins and lasts several hours. It may be so severe that it causes convulsions, and may even be fatal without glucose administration, in particular when liver glycogen stores are depleted through starvation. This severe transitional hypoglycaemia is produced by the flooding of the circulation with insulin as a result of toxin-induced secretory granule and cell membrane rupture. Pancreatectomy prevents this phase. In addition to the morphological changes seen in the first phase, the beta cell nuclei are pyknotic and show no TUNEL-positive staining; these changes are irreversible.
The fourth phase is the permanent diabetic hyperglycaemic phase. Morphologically, complete degranulation and loss of beta cell integrity is seen within 12–48 h. Non-beta cells remain intact, demonstrating the beta cell-selective character of the toxic action. Cell debris originating from the dying beta cells is removed by non-activated scavenger macrophages.
Thus, injections of alloxan and streptozotocin principally induce the same blood glucose and plasma insulin responses and cause an insulin-dependent type 1-like diabetes syndrome. All of the described morphological features of beta cell destruction are characteristic of necrotic cell death [22]. This mechanism is clearly at variance with that which underlies autoimmune type 1 diabetes, both in humans and rodent models of the disease, where beta cell demise is the result of apoptotic cell death without leakage of insulin from ruptured secretory granules [22].
Alloxan: mechanism of action
Alloxan has two distinct pathological effects: it selectively inhibits glucose-induced insulin secretion through specific inhibition of glucokinase, the glucose sensor of the beta cell, and it causes a state of insulin-dependent diabetes through its ability to induce ROS formation, resulting in the selective necrosis of beta cells. These two effects can be assigned to the specific chemical properties of alloxan, the common denominator being selective cellular uptake and accumulation of alloxan by the beta cell.
Beta cell selectivity of alloxan
Alloxan is a very unstable chemical compound [23] with a molecular shape resembling glucose (Fig. 2) [24, 25]. Both alloxan and glucose are hydrophilic and do not penetrate the lipid bilayer of the plasma membrane. The alloxan molecule is structurally so similar to glucose that the GLUT2 glucose transporter in the beta cell plasma membrane accepts this glucomimetic and transports it into the cytosol [25, 26]. Alloxan does not inhibit the function of the transporter [27], and can therefore selectively enter beta cells in an unrestricted manner [28–30]. It is therefore not toxic to insulin-producing cells that do not express this transporter [27, 31]. The half-life of alloxan is short [23, 32]; in aqueous solution it spontaneously decomposes into non-diabetogenic alloxanic acid within minutes [23]. Because of this, it must be taken up and accumulated quickly in the beta cell [28], and is therefore ineffective when blood flow to the pancreas is interrupted for the first few minutes after alloxan injection [33–35]. N-substituted alloxan derivatives with a long carbon side chain, such as butylalloxan (Fig. 2), differ chemically from alloxan in that they are lipophilic [36]. Butylalloxan acts in a similar manner to alloxan and preferentially damages beta cells [6, 11]. But since derivatives such as butylalloxan are lipophilic they can also penetrate plasma membranes that do not express the GLUT2 transporter [27]. Nephrotoxicity is a dominating feature of the toxicity of lipophilic derivatives after systemic administration [11]. This nephrotoxicity is so severe that it causes fatal renal failure in the animals before diabetes can develop [11]. The susceptibility of the kidney to the toxic action of these lipophilic alloxan derivatives is the result of their preferential accumulation in the tubular cells of the kidney, which, like the beta cells, express the GLUT2 glucose transporter.

Chemical formulas of alloxan, dialuric acid, butylalloxan, streptozotocin and methylnitrosourea
Glucokinase inhibition
Selective inhibition of glucose-induced insulin secretion is the major pathophysiological effect of the thiol group reactivity of alloxan [37–39]. Alloxan has a central 5-carbonyl group that reacts very avidly with thiol groups. Glucokinase (hexokinase IV) is the most sensitive thiol enzyme in the beta cell [2, 40], with a half maximal inhibitory concentration in the 1–10 μmol/l range. At higher concentrations, alloxan can inhibit many functionally important enzymes, as well as other proteins and cellular functions [22, 41].
Inhibition of glucokinase reduces glucose oxidation and ATP generation [42], thereby suppressing the ATP signal that triggers insulin secretion [2]. Inhibition of glucokinase is achieved within 1 min of exposure to alloxan.
The inhibition of glucose-induced insulin secretion is preceded by a very transient (1–2 min) stimulation of insulin secretion immediately after exposure to alloxan [43]. This effect can be explained by an initial reduction of ATP consumption resulting from the blockade of glucose phosphorylation by glucokinase [44], which produces a transient increase in ATP in the beta cell and triggers a transient release of insulin.
The inhibition of insulin secretion after exposure to alloxan [43, 45, 46] is restricted to that induced by glucose and its epimer, mannose, both of which induce insulin secretion through interaction with glucokinase [44]. Insulin biosynthesis is also inhibited by alloxan [47, 48], most likely through the same mechanism. The insulin secretory response to other nutrient secretagogues, such as 2-ketoisocaproic acid and leucine, or non-nutrient secretagogues, such as the sulfonylurea drug tolbutamide, remains intact initially because it is not mediated via glucokinase [49], but is lost after a delay of up to 1 h [50] as a consequence of the gradual deterioration of beta cell function.
Thiols such as the tripeptide glutathione (GSH), cysteine and dithiothreitol protect glucokinase against alloxan inhibition because they reduce alloxan to dialuric acid, which is not thiol reactive [23, 51, 52]. However, only dithiols such as dithiothreitol [51, 52] can readily reverse alloxan-induced glucokinase inhibition. They achieve this by reducing functionally essential cysteine residues of the glucokinase enzyme [40], which are oxidised through alloxan action [51, 52].
Likewise, glucose protects against alloxan-induced inhibition of glucose-induced insulin secretion because its binding to the sugar-binding site of glucokinase prevents the oxidation of the functionally essential thiol groups. The non-metabolisable seven carbon sugar mannoheptulose protects glucokinase through the same mechanism, but this alone is not sufficient to prevent alloxan-induced inhibition of insulin secretion. The glucose analogue 3-O-methylglucose, which is not a substrate of glucokinase, does however prevent inhibition. It does this through competitive blockade of alloxan uptake into the beta cell via the GLUT2 glucose transporter.
Thus, the inhibition of glucose-induced insulin secretion by alloxan is the result of the exquisite thiol reactivity of the glucose sensor glucokinase.
Beta cell toxicity and diabetogenicity of alloxan
Alloxan can generate reactive oxygen species (ROS) in a cyclic reaction with its reduction product, dialuric acid (Fig. 3) [53–56], as depicted in the text box ‘Chemical redox cycling reactions between alloxan and dialuric acid, and protective actions of cytoprotective enzymes’ (reactions i–ii). In the beta cells the toxic action of alloxan is initiated by free radicals formed in this redox reaction [53–57]. Autoxidation of dialuric acid generates superoxide radicals (iii–iv) and hydrogen peroxide (iii–iv), and in the Fenton reaction (v), in the presence of a suitable metal catalyst (typically iron) (vi), hydroxyl radicals (v–vii). The autoxidation of dialuric acid involves the intermediate formation of the alloxan radical (i–iv) [54–56].


Redox cycling reactions between alloxan and dialuric acid. A, alloxan; \( {\text{AH}}^{{\text{ $ \bullet $ }}} \), alloxan radical; AH2, dialuric acid; \( {\text{GS}}^{{\text{ $ \bullet $ }}} \), glutathione radical; GSSG, oxidised glutathione; \( {\text{OH}}^{{\text{ $ \bullet $ }}} \), hydroxyl radical; \( {\text{O}}^{{{\text{ $ \bullet $ - }}}}_{{\text{2}}} \), superoxide radical
The reduction of alloxan to dialuric acid in the cell requires the presence of a suitable thiol, typically the tripeptide glutathione (GSH) to generate the redox cycling partner, dialuric acid, and oxidised glutathione (viii) [56, 58]. The triketone structure of alloxan is vitally important for this two-step reaction with glutathione [59], which generates the alloxan radical as an intermediate product (ix–x). Other thiols such as cysteine, which are present at lower concentrations in the cell, dithiols and ascorbic acid are also suitable reducing agents and may therefore contribute to alloxan reduction [60]. Alloxan can also generate ROS by reacting with thiol groups on proteins such as enzymes [52, 61] and albumin [62]. During each typical redox cycle a small amount of ‘Compound 305’, an alloxan–GSH adduct [23, 32, 56, 63], is formed. The intracellular concentration of Compound 305 increases in a time-dependent manner, which gradually decreases the amount of reduced GSH available in the cell for redox cycling, thus producing a lower pro-oxidative ratio between alloxan and GSH, rather than a higher antioxidative ratio [54–56].
Paradoxically the thiols cysteine and GSH have long been reported to protect rats against the development of alloxan diabetes when injected together with alloxan [64, 65]. This observation can now be explained in light of the established molecular mechanism of alloxan action. When concentrations of reducing agents in the blood stream or in the extracellular space are significantly increased through injection of a thiol, more alloxan is reduced extracellularly so that less is available for intracellular accumulation. Normally the capacity for alloxan reduction, redox cycling and the generation of ROS in the circulation [62] is not sufficient to prevent the alloxan molecule from reaching and entering the beta cell.
The major oxidation pathway of dialuric acid, a chain reaction dependent upon superoxide radicals, is inhibited by superoxide dismutase (SOD; xi). In the presence of SOD, an autocatalytic process involving the interaction between dialuric acid and alloxan becomes important [54], while in the presence of a transition metal, a third oxidation mechanism, dependent upon hydrogen peroxide, has been identified [54]. This latter step is inhibited by the hydrogen peroxide inactivating enzyme catalase [54] (xii; text box: Chemical redox cycling reactions between alloxan and dialuric acid, and protective actions of cytoprotective enzymes). The other hydrogen peroxide inactivating enzyme, glutathione peroxidase, can principally act in a similar manner. But this enzyme requires GSH, which is oxidised in this reaction (xiii).
When kept in the oxidised form, alloxan does not generate ROS [60]. Thus, alloxan is not cytotoxic in the absence of thiols such as GSH or when restricted to the extracellular space [60]. Thiols in the plasma membrane, with which alloxan could interact and generate ROS in a redox cycle, are apparently not present or not accessible to a sufficient extent to allow the generation of ROS and damage the cells [60].
The reduction product dialuric acid is also not toxic when kept in the reduced form [54, 56, 60]. However, in contrast to alloxan, dialuric acid autoxidises spontaneously in the presence of O2, thus generating cytotoxic ROS in the absence of thiols [54, 55]. As a result of subsequent redox cycling it can also induce diabetes [10, 11] and cause beta cell lesions as alloxan [6].
The antioxidative enzyme SOD has a cytoprotective effect against both alloxan and dialuric acid in the presence of GSH by virtue of its ability to scavenge superoxide radicals, which are generated in the O2-dependent chain reaction between dialuric acid and alloxan. The resultant suppression of dialuric acid autoxidation prevents the generation of further ROS [54, 56], although increasing the concentrations of the toxins can reinstate the toxic action of both compounds. This is due to an autocatalytic reaction between the oxidised and reduced pyrimidine, which generates ROS even when the chain reaction is suppressed by SOD [54, 56].
Apparently, the superoxide radical is not the species responsible for the cytotoxicity of alloxan and dialuric acid. Several lines of evidence point to hydroxyl radicals as the principal culprit.
First, the hydrogen peroxide-inactivating enzyme catalase provides significantly better protection against the toxic effects of alloxan and dialuric acid on insulin-producing cells than the superoxide radical-inactivating enzyme SOD, though catalase does not prevent redox cycling and therefore superoxide radical formation [54, 56].
Second, since intracellular GSH concentrations are in the same millimolar concentration range in insulin-producing cells as in liver cells, and since both cell types express the GLUT2 glucose transporter, a difference in the intracellular GSH concentration cannot be responsible for the much greater susceptibility in vivo of insulin-producing cells compared with liver cells to the toxicity of alloxan [17]. However, liver cells are much better endowed with the hydrogen peroxide-inactivating enzyme catalase compared with insulin-producing cells [66, 67]. When the low intracellular levels of catalase are raised in insulin-producing cells through overexpression of the gene for this enzyme, these cells are protected equally well [60].
Third, a multitude of metal chelators, iron chelators in particular, and hydroxyl scavengers have been tested for their protective action both in vitro and in vivo, but the results obtained have not been unequivocal [18, 22, 66, 68–70]. Given the extremely short half-life and the extraordinary chemical reactivity of the hydroxyl radical, it is not surprising that complete protection against alloxan toxicity cannot be achieved. Hydroxyl radicals interact with biological targets before they can be inactivated through hydroxyl radical scavengers. In the same way, it is not surprising that it has been extremely difficult to efficiently suppress metal-catalysed hydroxyl radical formation through chelators [69–72], since it is crucial that metal chelation occurs before hydroxyl radical formation is initiated. Owing to the chemical properties of the scavengers and chelators, this cannot be completely achieved unless experimental conditions are optimised. In such a setting the toxicity of alloxan and dialuric acid to insulin-producing cells in vitro is suppressed by the iron chelator desferrioxamine [60], which prevents the generation of the very toxic hydroxyl radicals in the iron-catalysed Fenton reaction [54, 56] (Fig. 3).
Taken together, these data provide convincing evidence that the hydroxyl radical [56, 60, 73], rather than the superoxide radical, is the ultimate toxic ROS species, and that its formation is prevented by the destruction of hydrogen peroxide by catalase [54, 56].
Optimal protection against the cytotoxic action of alloxan and dialuric acid is provided only by a combination of SOD plus catalase, which completely prevents redox cycling between alloxan and dialuric acid, and thus the generation of all ROS species in this reaction pathway.
Glucose also provides complete protection against all toxic effects of alloxan both in vivo and in vitro [2]. This universal protection is achieved through the prevention of glucokinase inhibition and the preservation of the antioxidative defence mechanisms of the beta cell [22]. The non-metabolisable glucose analogue 3-O-methylglucose also provides protection, but does this exclusively through the prevention of alloxan uptake into the beta cell via the GLUT2 glucose transporter [22].
Thus, it can be concluded that the pancreatic beta cell toxicity and the resultant diabetogenicity of alloxan are due to redox cycling and the generation of toxic ROS.
Streptozotocin: mechanism of action
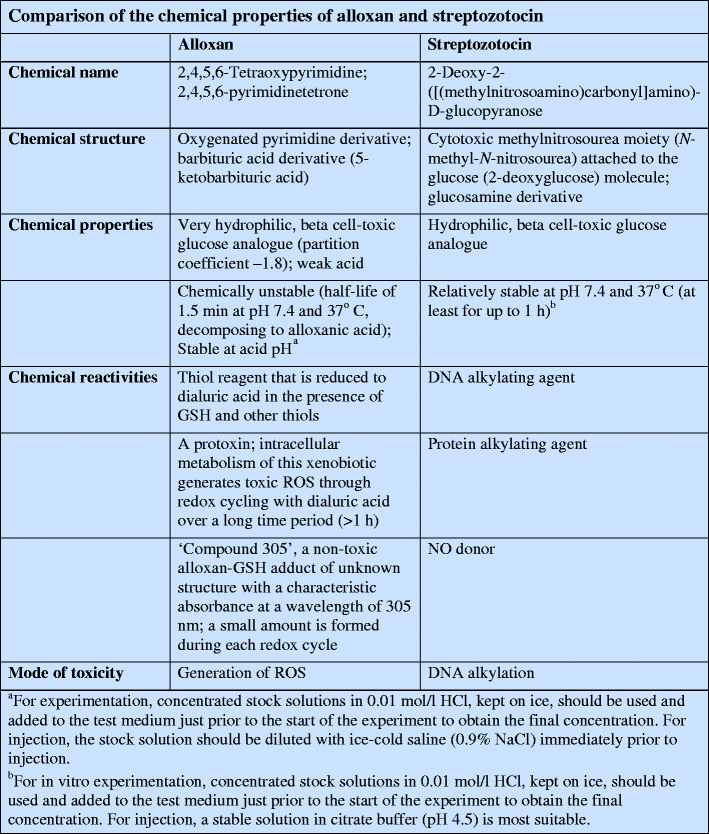
Streptozotocin (Fig. 2) inhibits insulin secretion and causes a state of insulin-dependent diabetes mellitus. Both effects can be attributed to its specific chemical properties, namely its alkylating potency (see text box: Comparison of the chemical properties of alloxan and streptozotocin). As with alloxan, its beta cell specificity is mainly the result of selective cellular uptake and accumulation.
Beta cell selectivity of streptozotocin
Streptozotocin is a nitrosourea analogue in which the N-methyl-N-nitrosourea (MNU) moiety (Fig. 2) is linked to the carbon-2 of a hexose. The toxic action of streptozotocin and chemically related alkylating compounds requires their uptake into the cells. Nitrosoureas are usually lipophilic and tissue uptake through the plasma membrane is rapid; however, as a result of the hexose substitution, streptozotocin is less lipophilic. Streptozotocin is selectively accumulated in pancreatic beta cells via the low-affinity GLUT2 glucose transporter in the plasma membrane [74, 75]. Thus, insulin-producing cells that do not express this glucose transporter are resistant to streptozotocin [76–78]. This observation also explains the greater toxicity of streptozotocin compared with N-methyl-N-nitrosourea in cells that express GLUT2, even though both substances alkylate DNA to a similar extent [77, 79, 80]. The importance of the GLUT2 glucose transporter in this process is also shown by the observation that streptozotocin damages other organs expressing this transporter, particularly kidney and liver [17, 19].
Beta cell toxicity of streptozotocin
It is generally assumed that the toxicity of streptozotocin is dependent upon the DNA alkylating activity of its methylnitrosourea moiety [79–83], especially at the O6 position of guanine [22]. The transfer of the methyl group from streptozotocin to the DNA molecule causes damage, which along a defined chain of events [84], results in the fragmentation of the DNA [85]. Protein glycosylation may be an additional damaging factor [41]. In the attempt to repair DNA, poly(ADP-ribose) polymerase (PARP) is overstimulated. This diminishes cellular NAD+, and subsequently ATP, stores [82, 85–87]. The depletion of the cellular energy stores ultimately results in beta cell necrosis. Although streptozotocin also methylates proteins [80, 81], DNA methylation is ultimately responsible for beta cell death, but it is likely that protein methylation contributes to the functional defects of the beta cells after exposure to streptozotocin.
Inhibitors of poly ADP-ribosylation suppress the process of DNA methylation. Thus, injection of nicotinamide and other PARP inhibitors in parallel with, or prior to the administration of streptozotocin is well known to protect beta cells against the toxic action of streptozotocin and to prevent the development of a diabetic state [13]. Also, mice deficient in PARP are resistant to beta cell death mediated by streptozotocin, in spite of DNA fragmentation. The absence of PARP prevents the depletion of the cofactor NAD+ and the subsequent loss of ATP [84, 88–90] and thus cell death.
The role of alkylation in beta cell damage has also been examined by the use of ethylating agents, which are less toxic than their methylating counterparts, on account of O6-ethylguanine being less toxic than O6-methylguanine [91]. The fact that N-ethyl-N-nitrosourea and ethyl methanesulphonate are significantly less toxic to insulin-producing cells than MNU and methyl methanesulphonate [77, 91] has been taken as support for the notion that in insulin-producing cells, as in other cell types, the mechanism of toxic action is due to alkylation, with methylation of DNA bases being more toxic than ethylation [22, 81].
An alternative hypothesis proposes that part of the diabetogenic effect of streptozotocin may relate not to its alkylating ability but to its potential to act as an intracellular nitric oxide (NO) donor [92]. Both streptozotocin and MNU contain a nitroso group (Fig. 2) and can liberate NO. In fact, streptozotocin has been shown to increase the activity of guanylyl cyclase and the formation of cGMP, which are characteristic effects of NO. However, the alkylating agent methyl methanesulphonate is the most toxic compound, though unlike MNU, it is not a NO donor [91], indicating that NO is not an indispensable prerequisite for the toxic action of the family of alkylating agents that streptozotocin belongs to.
Finally, some minor generation of ROS, including superoxide and hydroxyl radicals originating from hydrogen peroxide dismutation during hypoxanthine metabolism [93], may accompany the effect of streptozotocin and accelerate the process of beta cell destruction but ROS do not play a crucial role [22].
Inhibition of insulin secretion by streptozotocin
The effects of streptozotocin on glucose and insulin homeostasis reflect the toxin-induced abnormalities in beta cell function. Initially, insulin biosynthesis, glucose-induced insulin secretion and glucose metabolism (both glucose oxidation and oxygen consumption) are all affected [93–95]. On the other hand, streptozotocin has no immediate, direct inhibitory effect upon glucose transport [77] or upon glucose phosphorylation by glucokinase [39]. However, at later stages of functional beta cell impairment, deficiencies in terms of gene expression and protein production lead to the deterioration of both glucose transport and metabolism [96].
Even before the negative effects of mitochondrial DNA and protein alkylation and glycosylation become evident, streptozotocin-induced depletion of NAD+ may result in the inhibition of insulin biosynthesis and secretion [82, 85, 94]. Later, inhibition of glucose-induced and amino acid-induced insulin secretion [97] as a result of mitochondrial enzyme dysfunction [98] and damage to the mitochondrial genome [99] become apparent. This impairment is more marked for nutrient- than for non-nutrient-induced insulin secretion. This interpretation has been confirmed through studies which have shown that pre-treatment of isolated pancreatic islets with the poly(ADP-ribose) polymerase (PARP) inhibitor nicotinamide prevents early inhibition of beta cell function during the first day after streptozotocin exposure, while long-term inhibition of insulin secretion 6 days after streptozotocin exposure was not counteracted by nicotinamide [100].
Conclusion
Both alloxan and streptozotocin induce insulin deficiency. While the mechanisms of beta cell-selective action through uptake via the GLUT2 glucose transporter and beta cell death via necrosis are identical, ROS in the case of alloxan and DNA alkylation in the case of streptozotocin mediate the toxic action of these glucose analogues (Fig. 4). This also explains why a lack of significant expression of this glucose transporter isoform, such as in human beta cells, provides insensitivity to the toxins [22, 101]. On the other hand, expression of this glucose transporter in other organs like in tubular cells and hepatocytes explains why the toxins can cause damage to kidney and liver [22].

Schematic representation of the toxic effects of the glucose analogues alloxan and streptozotocin in beta cells, which produce chemical diabetes
Due to its chemical properties, in particular the greater stability (see text box: Comparison of the chemical properties of alloxan and streptozotocin), streptozotocin is the agent of choice for reproducible induction of a diabetic metabolic state in experimental animals. Alloxan, on the other hand, as a model compound of ROS mediated beta cell toxicity, is the agent with the greater impact upon the understanding of ROS mediated mechanisms of beta cell death in type 1 and type 2 diabetes mellitus.
Abbreviations
- GSH:
-
glutathione
- MNU:
-
N-methyl-N-nitrosourea
- NO:
-
nitric oxide
- PARP:
-
poly(ADP-ribose) polymerase
- ROS:
-
reactive oxygen species
- SOD:
-
superoxide dismutase
References
Wöhler F, Liebig J (1838) Untersuchungen über die Natur der Harnsäure. [Investigations on the nature of uric acid]. Ann Pharm 26:241–340 (article in German)
Lenzen S, Panten U (1988) Alloxan: history and mechanism of action. Diabetologia 31:337–342
Lenzen S, Tiedge M, Jörns A, Munday R (1996) Alloxan derivatives as a tool for the elucidation of the mechanism of the diabetogenic action of alloxan. In: Shafrir E (ed) Lessons from animal diabetes. Birkhäuser, Boston, pp 113–122
Dunn JS, McLetchie NGB (1943) Experimental alloxan diabetes in the rat. Lancet 245:384–387
Dunn JS, Sheehan HL, McLetchie NGB (1943) Necrosis of islets of Langerhans produced experimentally. Lancet 244:484–487
Jörns A, Munday R, Tiedge M, Lenzen S (1997) Comparative toxicity of alloxan, N-alkylalloxans and ninhydrin to isolated pancreatic islets in vitro. J Endocrinol 155:283–293
Peschke E, Ebelt H, Brömme HJ, Peschke D (2000) ‘Classical’ and ‘new’ diabetogens—comparison of their effects on isolated rat pancreatic islets in vitro. Cell Mol Life Sci 57:158–164
Goldner MG, Gomori G (1943) Alloxan diabetes in the dog. Endocrinology 33:297–308
McLetchie NGB (1982) Alloxan diabetes: the sorcerer and his apprentice. Diabetologia 23:72–75
Bailey CC, Bailey OT, Leech RS (1946) Diabetes mellitus in rabbits injected with dialuric acid. Proc Soc Exp Biol Med 63:502–505
Brückmann G, Wertheimer E (1947) Alloxan studies: the action of alloxan homologues and related compounds. J Biol Chem 168:241–256
White FR (1963) Streptozotocin. Cancer Chemother Rep 30:49–53
Schein PS, Cooney DA, Vernon ML (1967) The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer Res 27:2324–2332
Schein PS, O’Connell MJ, Blom J et al (1974) Clinical antitumor activity and toxicity of streptozotocin (NSC-85998). Cancer 34:993–1000
Rakieten N, Rakieten ML, Nadkarni MV (1963) Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother Rep 29:91–98
Arison RN, Ciaccio EI, Glitzer MS, Cassaro JA, Pruss MP (1967) Light and electron microscopy of lesions in rats rendered diabetic with streptozotocin. Diabetes 16:51–56
Rerup CC (1970) Drugs producing diabetes through damage of the insulin secreting cells. Pharmacol Rev 22:485–518
Cooperstein SJ, Watkins D (1981) Action of toxic drugs on islet cells. In: Cooperstein SJ, Watkins D (eds) The islet of Langerhans. Academic, New York, pp 387–425
Weiss RB (1982) Streptozocin: a review of its pharmacology, efficacy, and toxicity. Cancer Treat Rep 66:427–438
Preston AM (1985) Modification of streptozotocin-induced diabetes by protective agents. Nutr Res 5:435–446
Szkudelski T (2001) The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 50:537–546
Lenzen S (2007) Alloxan and streptozotocin diabetes. In: Peschke E (ed) Endokrinologie III Vorträge im Rahmen des Projektes ‘Zeitstrukturen endokriner Systeme’. [Endocrinology III lectures within the ‘time structures of endocrine systems’ project framework]. Abhandlung der Sächs. Akad. Wiss., Math-naturwiss Klasse, Verlag der Sächsischen Akademie der Wissenschaften, Leipzig, commissioned by S. Hirzel Verlag, Stuttgart/Leipzig, pp 119–138
Lenzen S, Munday R (1991) Thiol-group reactivity, hydrophilicity and stability of alloxan, its reduction products and its N-methyl derivatives and a comparison with ninhydrin. Biochem Pharmacol 42:1385–1391
Weaver DC, Barry CD, McDaniel ML, Marshall GR, Lacy PE (1979) Molecular requirements for recognition at glucoreceptor for insulin release. Mol Pharmacol 16:361–368
Gorus FK, Malaisse WJ, Pipeleers DG (1982) Selective uptake of alloxan by pancreatic B-cells. Biochem J 208:513–515
Weaver DC, McDaniel ML, Lacy PE (1978) Alloxan uptake by isolated rat islets of Langerhans. Endocrinology 102:1847–1855
Elsner M, Tiedge M, Guldbakke B, Munday R, Lenzen S (2002) Importance of the GLUT2 glucose transporter for pancreatic beta cell toxicity of alloxan. Diabetologia 45:1542–1549
Hammarström L, Hellman B, Ullberg S (1967) On the accumulation of alloxan in the pancreatic beta-cells. Diabetologia 3:340–344
Boquist L, Nelson L, Lorentzon R (1983) Uptake of labeled alloxan in mouse organs and mitochondria in vivo and in vitro. Endocrinology 113:943–948
Malaisse WJ, Doherty M, Ladriere L, Malaisse-Lagae F (2001) Pancreatic uptake of [2-14C]alloxan. Int J Mol Med 7:311–315
Bloch KO, Zemel R, Bloch OV, Grief H, Vardi P (2000) Streptozotocin and alloxan-based selection improves toxin resistance of insulin-producing RINm cells. Int J Exp Diabetes Res 1:211–219
Patterson JW, Lazarow A, Levey S (1949) Alloxan and dialuric acid: their stabilities and ultraviolet absorption spectra. J Biol Chem 177:187–196
Gomori G, Goldner MG (1945) Acute nature of alloxan damage. Proc Soc Exp Biol (NY) 58:232–233
Bailey CC, Collins-Williams J, Le Compte PM (1950) Effect of alloxan in rabbits with temporary occlusion of the arteries to the pancreas. Proc Soc Exp Biol 71:580–583
Hellman B, Diderholm H (1955) The diabetogenic effect of alloxan after elimination of extra-pancreatic factors. Acta Endocrinol (Copenh) 20:81–87
Munday R, Ludwig K, Lenzen S (1993) The relationship between the physicochemical properties and the biological effects of alloxan and several N-alkyl substituted alloxan derivatives. J Endocrinol 139:153–163
Hara H, Miwa I, Okuda J (1986) Inhibition of rat liver glucokinase by alloxan and ninhydrin. Chem Pharm Bull (Tokyo) 34:4731–4737
Meglasson MD, Burch PT, Berner DK, Najafi H, Matschinsky FM (1986) Identification of glucokinase as an alloxan-sensitive glucose sensor of the pancreatic beta-cell. Diabetes 35:1163–1173
Lenzen S, Tiedge M, Panten U (1987) Glucokinase in pancreatic B-cells and its inhibition by alloxan. Acta Endocrinol (Copenh) 115:21–29
Tiedge M, Richter T, Lenzen S (2000) Importance of cysteine residues for the stability and catalytic activity of human pancreatic beta cell glucokinase. Arch Biochem Biophys 375:251–260
Konrad RJ, Kudlow JE (2002) The role of O-linked protein glycosylation in beta-cell dysfunction. Int J Mol Med 10:535–539
Gunnarsson R, Hellerström C (1973) Acute effects of alloxan on the metabolism and insulin secretion of the pancreatic B-cell. Horm Metab Res 5:404–409
Weaver DC, McDaniel ML, Naber SP, Barry CD, Lacy PE (1978) Alloxan stimulation and inhibition of insulin release from isolated rat islets of Langerhans. Diabetes 27:1205–1214
Lenzen S, Panten U (1988) Signal recognition by pancreatic B-cells. Biochem Pharmacol 37:371–378
Ishibashi F, Onari K, Sato T, Kawate R (1978) Studies on the mechanism of alloxan inhibition of glucose-induced insulin release. Hiroshima J Med Sci 27:211–219
Miwa I, Hara H, Okuda J (1986) Parallel inhibition of islet glucokinase and glucose-stimulated insulin secretion by either alloxan or ninhydrin. J Clin Biochem Nutr 1:237–245
Jain K, Logothetopoulos J (1976) Proinsulin biosynthesis by pancreatic islets of the rat and the study of alloxan cytotoxicity in vitro. Biochim Biophys Acta 435:145–151
Niki A, Niki H, Miwa I, Lin BJ (1976) Interaction of alloxan and anomers of d-glucose on glucose-induced insulin secretion and biosynthesis in vitro. Diabetes 25:574–579
Lenzen S, Brand FH, Panten U (1988) Structural requirements of alloxan and ninhydrin for glucokinase inhibition and of glucose for protection against inhibition. Br J Pharmacol 95:851–859
Borg LA (1981) Effects of alloxan on the islets of Langerhans: inhibition of leucine metabolism and insulin secretion. Biochim Biophys Acta 677:257–262
Lenzen S, Freytag S, Panten U (1988) Inhibition of glucokinase by alloxan through interaction with SH groups in the sugar-binding site of the enzyme. Mol Pharmacol 34:395–400
Lenzen S, Mirzaie-Petri M (1991) Inhibition of glucokinase and hexokinase from pancreatic B-cells and liver by alloxan, alloxantin, dialuric acid, and t-butylhydroperoxide. Biomed Res 12:297–307
Cohen G, Heikkila RE (1974) The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J Biol Chem 249:2447–2452
Munday R (1988) Dialuric acid autoxidation. Effects of transition metals on the reaction rate and on the generation of ‘active oxygen’ species. Biochem Pharmacol 37:409–413
Winterbourn CC, Cowden WB, Sutton HC (1989) Auto-oxidation of dialuric acid, divicine and isouramil. Superoxide dependent and independent mechanisms. Biochem Pharmacol 38:611–618
Winterbourn CC, Munday R (1989) Glutathione-mediated redox cycling of alloxan. Mechanisms of superoxide dismutase inhibition and of metal-catalyzed \( OH^{ \bullet } \) formation. Biochem Pharmacol 38:271–277
Oberley LW (1988) Free radicals and diabetes. Free Radic Biol Med 5:113–124
Brömme HJ, Mörke W, Peschke D, Ebelt H (2000) Scavenging effect of melatonin on hydroxyl radicals generated by alloxan. J Pineal Res 29:201–208
Elsner M, Gurgul-Convey E, Lenzen S (2007) Relation between triketone structure, generation of reactive oxygen species and selective toxicity of the diabetogenic agent alloxan. Antioxid Redox Signal (in press)
Elsner M, Gurgul-Convey E, Lenzen S (2006) Relative importance of cellular uptake and reactive oxygen species for the toxicity of alloxan and dialuric acid to insulin-producing cells. Free Radic Biol Med 41:825–834
Lenzen S, Mirzaie-Petri M (1992) Inhibition of aconitase by alloxan and the differential modes of protection of glucose, 3-O-methylglucose, and mannoheptulose. Naunyn Schmiedebergs Arch Pharmacol 346:532–536
Sakurai K, Miura T (1989) Generation of free radicals by alloxan in the presence of bovine serum albumin: a role of protein sulfhydryl groups in alloxan cytotoxicity. Biochem Int 19:405–412
Brömme HJ, Mörke W, Weinandy R, Peschke D, Peschke E (2002) Formation of compound 305 requires the simultaneous generation of both alloxan and GSH radicals. Horm Metab Res 34:62–66
Lazarow A, Patterson JW, Levey S (1948) The mechanism of cysteine and glutathione protection against alloxan diabetes. Science 108:308–309
Sen PB, Bhattacharya G (1952) Reversal of the diabetogenic action of alloxan by sulfhydryl compounds. Science 115:41–43
Grankvist K, Marklund S, Sehlin J, Taljedal IB (1979) Superoxide dismutase, catalase and scavengers of hydroxyl radical protect against the toxic action of alloxan on pancreatic islet cells in vitro. Biochem J 182:17–25
Tiedge M, Lortz S, Drinkgern J, Lenzen S (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46:1733–1742
Tibaldi J, Benjamin J, Cabbat FS, Heikkila RE (1979) Protection against alloxan-induced diabetes by various urea derivatives: relationship between protective effects and reactivity with the hydroxyl radical. J Pharmacol Exp Ther 211:415–418
Fischer LJ, Harman AW (1982) Oxygen free radicals and the diabetogenic action of alloxan. In: Autor AP (ed) Pathology of oxygen. Academic, New York, pp 261–275
Jörns A, Tiedge M, Lenzen S, Munday R (1999) Effect of superoxide dismutase, catalase, chelating agents, and free radical scavengers on the toxicity of alloxan to isolated pancreatic islets in vitro. Free Radic Biol Med 26:1300–1304
Grankvist K, Marklund S, Täljedal IB (1979) Influence of trace metals on alloxan cytotoxicity in pancreatic islets. FEBS Lett 105:15–18
Heikkila RE, Cabbat FS (1982) The prevention of alloxan-induced diabetes in mice by the iron-chelator detapac: suggestion of a role for iron in the cytotoxic process. Experientia 38:378–379
Heikkila RE, Winston B, Cohen G (1976) Alloxan-induced diabetes-evidence for hydroxyl radical as a cytotoxic intermediate. Biochem Pharmacol 25:1085–1092
Tjälve H, Wilander E, Johansson EB (1976) Distribution of labelled streptozotocin in mice: uptake and retention in pancreatic islets. J Endocrinol 69:455–456
Karunanayake EH, Baker JR, Christian RA, Hearse DJ, Mellows G (1976) Autoradiographic study of the distribution and cellular uptake of (14C)-streptozotocin in the rat. Diabetologia 12:123–128
Ledoux SP, Wilson GL (1984) Effects of streptozotocin on a clonal isolate of rat insulinoma cells. Biochim Biophys Acta 804:387–392
Elsner M, Guldbakke B, Tiedge M, Munday R, Lenzen S (2000) Relative importance of transport and alkylation for pancreatic beta-cell toxicity of streptozotocin. Diabetologia 43:1528–1533
Schnedl WJ, Ferber S, Johnson JH, Newgard CB (1994) STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 43:1326–1333
Ledoux SP, Woodley SE, Patton NJ, Wilson GL (1986) Mechanisms of nitrosourea-induced beta-cell damage. Alterations in DNA. Diabetes 35:866–872
Wilson GL, Hartig PC, Patton NJ, LeDoux SP (1988) Mechanisms of nitrosourea-induced beta-cell damage. Activation of poly(ADP-ribose) synthetase and cellular distribution. Diabetes 37:213–216
Bennett RA, Pegg AE (1981) Alkylation of DNA in rat tissues following administration of streptozotocin. Cancer Res 41:2786–2790
Uchigata Y, Yamamoto H, Kawamura A, Okamoto H (1982) Protection by superoxide dismutase, catalase, and poly(ADP-ribose) synthetase inhibitors against alloxan- and streptozotocin-induced islet DNA strand breaks and against the inhibition of proinsulin synthesis. J Biol Chem 257:6084–6088
Murata M, Takahashi A, Saito I, Kawanishi S (1999) Site-specific DNA methylation and apoptosis: induction by diabetogenic streptozotocin. Biochem Pharmacol 57:881–887
Pieper AA, Verma A, Zhang J, Snyder SH (1999) Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20:171–181
Yamamoto H, Uchigata Y, Okamoto H (1981) Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature 294:284–286
Schein PS, Loftus S (1968) Streptozotocin: depression of mouse liver pyridine nucleotides. Cancer Res 28:1501–1506
Sandler S, Swenne I (1983) Streptozotocin, but not alloxan, induces DNA repair synthesis in mouse pancreatic islets in vitro. Diabetologia 25:444–447
Pieper AA, Brat DJ, Krug DK et al (1999) Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96:3059–3064
Burkart V, Wang ZQ, Radons J et al (1999) Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med 5:314–319
Masutani M, Suzuki H, Kamada N et al (1999) Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96:2301–2304
Delaney CA, Dunger A, Di Matteo M, Cunningham JM, Green MH, Green IC (1995) Comparison of inhibition of glucose-stimulated insulin secretion in rat islets of Langerhans by streptozotocin and methyl and ethyl nitrosoureas and methanesulphonates. Lack of correlation with nitric oxide-releasing or O6-alkylating ability. Biochem Pharmacol 50:2015–2020
Turk J, Corbett JA, Ramanadham S, Bohrer A, McDaniel ML (1993) Biochemical evidence for nitric oxide formation from streptozotocin in isolated pancreatic islets. Biochem Biophys Res Commun 197:1458–1464
Nukatsuka M, Yoshimura Y, Nishida M, Kawada J (1990) Allopurinol protects pancreatic beta cells from the cytotoxic effect of streptozotocin: in vitro study. J Pharmacobiodyn 13:259–262
Strandell E, Eizirik DL, Korsgren O, Sandler S (1988) Functional characteristics of cultured mouse pancreatic islets following exposure to different streptozotocin concentrations. Mol Cell Endocrinol 59:83–91
Bedoya FJ, Solano F, Lucas M (1996) N-Monomethyl-arginine and nicotinamide prevent streptozotocin-induced double strand DNA break formation in pancreatic rat islets. Experientia 52:344–347
Wang Z, Gleichmann H (1998) GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 47:50–56
Eizirik DL, Sandler S, Sener A, Malaisse WJ (1988) Defective catabolism of d-glucose and l-glutamine in mouse pancreatic islets maintained in culture after streptozotocin exposure. Endocrinology 123:1001–1007
Rasschaert J, Eizirik DL, Malaisse WJ (1992) Long term in vitro effects of streptozotocin, interleukin-1, and high glucose concentration on the activity of mitochondrial dehydrogenases and the secretion of insulin in pancreatic islets. Endocrinology 130:3522–3528
Eizirik DL, Sandler S, Ahnström G, Welsh M (1991) Exposure of pancreatic islets to different alkylating agents decreases mitochondrial DNA content but only streptozotocin induces long-lasting functional impairment of B-cells. Biochem Pharmacol 42:2275–2282
Strandell E, Eizirik DL, Sandler S (1989) Survival and B-cell function of mouse pancreatic islets maintained in culture after concomitant exposure to streptozotocin and nicotinamide. Exp Clin Endocrinol 93:219–224
Elsner M, Tiedge M, Lenzen S (2003) Mechanism underlying resistance of human pancreatic beta cells against toxicity of streptozotocin and alloxan. Diabetologia 46:1713–1714
Acknowledgements
The author is most grateful to Frits Holleman for excellent editorial support.
Duality of interest
The author states no duality of interest.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 51, 216–226 (2008). https://doi.org/10.1007/s00125-007-0886-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0886-7