
Abstract
The transforming growth factor beta (TGF-β) is a crucial cytokine that get increasing concern in recent years to treat human diseases. This signal controls multiple cellular responses during embryonic development and tissue homeostasis through canonical and/or noncanonical signaling pathways. Dysregulated TGF-β signal plays an essential role in contributing to fibrosis via promoting the extracellular matrix deposition, and tumor progression via inducing the epithelial-to-mesenchymal transition, immunosuppression, and neovascularization at the advanced stage of cancer. Besides, the dysregulation of TGF-beta signal also involves in other human diseases including anemia, inflammatory disease, wound healing and cardiovascular disease et al. Therefore, this signal is proposed to be a promising therapeutic target in these diseases. Recently, multiple strategies targeting TGF-β signals including neutralizing antibodies, ligand traps, small-molecule receptor kinase inhibitors targeting ligand–receptor signaling pathways, antisense oligonucleotides to disrupt the production of TGF-β at the transcriptional level, and vaccine are under evaluation of safety and efficacy for the forementioned diseases in clinical trials. Here, in this review, we firstly summarized the biology and function of TGF-β in physiological and pathological conditions, elaborated TGF-β associated signal transduction. And then, we analyzed the current advances in preclinical studies and clinical strategies targeting TGF-β signal transduction to treat diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytokine-based targeting presents a promising therapy in many disorders, including cancer, inflammatory or infectious diseases, and fibrotic diseases [1,2,3]. One multifunctional polypeptide cytokine, transforming growth factor beta (TGF-β), becomes a potential therapeutic target for its bio function in regulating the growth and differentiation of cells. This cytokine belongs to the TGF-β superfamily, which comprises many proteins, including growth differentiation factors and activins. The structurally-related TGF-β1, TGF-β2, and TGF-β3 cytokines are three isoforms of the TGF-β family [4, 5]. If not otherwise specified, TGF-β in the following statement will stand for TGF-β1. Generally, latent TGF-β is stored in the multiple extracellular matrix. Under enzymatic and non-enzymatic action, latent TGF transforms into activated TGF. Only activated TGF can bind to the TGF receptor complex and induce canonical and noncanonical pathways of TGF-β signal transduction.
Although scientists have proposed the critical role of TGF-β signaling pathways in fibrosis, tumorigenesis, and regulating immune responses, developing TGF-β-targeted therapeutic drugs is a great obstacle for the dual role and paradoxical effects on fibrosis and immune systems regulation in the occurrence and development of disease. TGF-β is critical in regulating tissue homeostasis and renewal in physiological conditions. In pathological conditions, TGF-β signaling plays a critical role in regulating inflammatory progression and wound healing [6,7,8]. Moreover, TGF-β signaling also contributes to fibrosis by inducing extracellular matrix deposition [4, 9]. Dysregulated TGF-β regulates both adaptive immunity and innate immunity during tumorigenesis. At early tumorigenesis, TGF-β becomes cytostatic, apoptotic, and tumor suppressive and acts as a tumor suppressor by inhibiting excessive inflammation and inducing tolerance. While during advanced cancer, TGF-β is necessary to promote tumor tolerance, inflammation suppression, T cell exclusion, epithelial-mesenchymal transition, migration, invasion, and progression [10].
Regulation of TGF-β signal transduction occurs at several levels, including the production of TGF-β ligands, ligand-receptor interactions, downstream signal cascades after kinase receptor activation, and transcriptional disruption. These crucial roles and promising therapeutic potential of TGF-β in the diseases mentioned above, the specific mechanisms of TGF-β driving these diseases and therapies based on TGF-β signal transduction provide targeted therapeutic strategies. Recently, many TGF-β-targeted drugs are under preclinical and clinical trials. Neutralizing antibodies, TGF-β ligand traps, small-molecule receptor kinase inhibitors, antisense oligonucleotides, and vaccine-based therapy are the main targeted strategies of TGF-β [11,12,13,14,15]. However, most of them are in phase 1/2 clinical trials. In this review, we elaborated on the biology and function of TGF-β, and summarized the recent advances in TGF-β associated targeted therapy.
The biology of TGF-β
The production and activation of TGF-β
TGF-β usually exists extracellularly as heterodimers or homodimers [16]. Generally, TGF-βs have three mammalian genome-encoded isoforms: TGF-β1, TGF-β2, and TGF-β3 [4]. Each isoform is synthesized in the rough endoplasmic reticulum as a precursor molecule that consists of an N-terminal signal peptide, the latency-associated polypeptide (LAP), and a mature polypeptide at the C-terminal [17]. When the signal peptide is removed, the precursor is elaborated through proteolytic cleavage, thereby separating the N-terminal prodomain from the C-terminal mature polypeptide. Among the three isoforms, the TGF-β1 homodimer is the most widely studied subtype and was the first purified protein, which is characterized by complementary DNA cloning [18]. For cell origination, TGF-β1 is originally purified from platelets [19]. In addition, tumor cells, tumor-associated macrophages and stromal cancer-associated fibroblasts in the tumor microenvironment also express TGF-β1 in a heterogeneous manner, not necessarily the expression of TGF-β2 or TGF-β3 [17, 20, 21].
The mature polypeptides form mature homodimeric and heterodimeric complexes through disulfide-linked dimerization [22]. The latent TGF-β complex is associated with TGF-β binding protein through disulfide bonding to the large latent complex (Fig. 1). The large latent complex is relevant to the extracellular matrix or to the glycoprotein-A repetitions predominant (GARP, also known as leucine-rich repeat containing 32, LRRC32) on the cell surface [23, 24]. Further activation of latent TGF-β is required to release mature TGF-β, which binds to TGF-β receptors on adjacent cells [24]. Besides, TGF-β is supposed to act in a cell- and context-dependent manner [25]. Moreover, latent TGF-β is associated with GARP on mesenchymal stromal cells, platelets, and Tregs, thereby promoting GARP to manage the preservation of these TGF-β complexes [26, 27]. GARP is expressed on fibroblasts, megakaryocytes, and endothelial cells, raising the possibility that GARP plays a broad role in TGF-β1 latency. In addition to GARP, another GARP-related protein, LRRC33, is associated with latent TGF-β and regulates TGF-β activation [23, 28]. Besides, LRRC15, expressed on stromal fibroblasts in advanced tumors, plays similar roles in the cell-associated preservation of TGF-β [29, 30].

Schematic diagram and activation of latent TGF-β. The pro-TGF-β precursor consists of an N-terminal peptide with latency-associated peptide (LAP) and a mature C-terminal fragment. The pro-TGF-β precursor is cleaved by the convertase furin, and then the LAP dimer binds to mature TGF-β and forms the small latent complex (SLC). Proteases including plasmin, cathepsin and matrix metalloproteinase 9/14 (MMP9/14)) can cleave LAP and release active TGF-β in the extracellular matrix (ECM). SLC binds to latent TGF-β-binding protein (LTBP) with ECM proteins, including fibronectin and fibrillin, mediating the release of active TGF-β via interaction with αβ-integrin. TGF-β can also be activated through SLC anchoring to glycoprotein A repetition predominant protein (GARP)
The canonical and noncanonical pathways of TGF-β signal transduction
TGF-β signal transduction depends on canonical and noncanonical pathways (Fig. 2). For the canonical pathway, TGF-β ligands primarily bind to the TGF-β type III receptor (TGF-βRIII, also called betaglycan), which has a high expression level on many cell types. Among the three isoforms of TGF-β, TGF-β2 primarily depends on TGF-βRIII for signaling compared with the other two isoforms [31]. The receptor complex of TGF-β is a tetramer composed of two paired serine or threonine protein kinases, TGF-βRIs and TGF-βRIIs [32]. After binding to TGF-βRIII, TGF-βRIII presents TGF-β to the TGF-βRI/TGF-βRII complex, which has a high affinity for TGF-β [33]. In addition, TGF-β binding to TGF-β can recruit and phosphorylate TGF-βRI, which is a requirement for signal transduction [34]. TGF-βRI phosphorylates SMAD2 on a carboxyl-terminal fragment which contains three serine residues specially at positions 465 and 467 [35]. Then, the phosphorylated SMAD2/3 separates immediately from TGF-βRI and aggregates with SMAD4 to form a heteromeric complex. The formative SMAD2/3-SMAD4 complex translocate into the cell nucleus and activates or restrains target gene expression [36]. TGF-β induced SMAD7 to encode a negative regulator of TGF-β/SMAD signals, which is associated with TGF-βRI, thereby blocking SMAD2 phosphorylation and activation. Moreover, SMAD7 antagonizes TGF-β signals by affecting the formation of the SMAD-DNA complex in the nucleus [37] and inhibits the formation and translocation of the SMAD2-SMAD4 complex [38, 39]. SMAD7 is demonstrated to form complex with SMAD2/3 to mitigate signaling. SMAD7 affects the TGF-β signaling cascades by deactivation of SMAD2/3 and non-SMAD pathways, without any influences on TGF-β receptor activity. SMAD7 is demonstrated to induce myofibroblasts as an endogenous TGF-β-related negative feedback mechanism which inhibits postinfarction fibrosis by restraining TGF-β-independent fibrogenic functions [40]. Overexpression of SMAD7 is associated with inflammatory diseases and is regarded as an inhibitor of TGF-β1 activity [41]. SMAD7 recruits E3 ubiquitin ligases, including tripartite motif-containing protein 31 (TRIM31), SMAD ubiquitination regulatory factor 1/2 (Smurf1/2), and neural precursor cell expressed developmentally downregulated 4–2 (NEDD4–2) to TGF-βRI, promoting proteasomal or lysosomal degradation [42,43,44]. TGF-βRI ubiquitination is reversed by deubiquitinating enzymes, such as ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) and ubiquitin-specific protease 4 (USP4) [45, 46].

TGF-β induced canonical and noncanonical signaling pathways. TGF-β is presented to TGF-βRII, which phosphorylates TGF-βRI to initiate the subsequent TGF-β pathway. In the canonical pathway, TGF-βRI phosphorylates SMAD2/3 to form the SMAD2/3-SMAD4 complex. SMAD2/3, SMAD4 and transcription factor (TF) complexes are transferred to the cell nucleus, modulating the expression of target genes. SMAD7, one of the target genes, regulates the duration and intensity of TGF-β with a negative feedback loop. In the noncanonical pathway, TGF-β can activate PI3K, RHO, PAR6, RAS, TRAF4/6, JNK, P38, NF-κB and ERK signaling
In addition to the canonical signal transduction of TGF-β, the noncanonical signaling pathways play vital roles in diseases [47]. TGF-βRI is demonstrated to activate RHO small GTPases, which control the activity of LIM kinase (LIMK) and phosphorylate cofilin, thereby reorganizing the actin cytoskeleton and participating in cell adhesion and proliferation [48, 49]. TGF-βRII phosphorylates the cell polarity regulator PAR6 and is associated with tight junctions and epithelial-to-mesenchymal transition (EMT) [50, 51]. TGF-β is demonstrated to activate the c-Jun N-terminal kinase (JNK) and P38/mitogen-activated protein kinase (MAPK)/nuclear factor kappa-B (NF-κB) pathways, which are downstream of tumor necrosis factor-associated factor 4/6 (TRAF4/6) [52,53,54]. PI3K/AKT pathway is also activated as the downstream signal transduction of noncanonical TGF-β signals [55, 56]. In addition, TGF-β induced the phosphorylation of Src homology domain 2-containing protein and then activated the rat sarcoma signal (RAS), rapid accelerated fibrosarcoma signal (RAF), MAPK, and extracellular signal regulated kinase (ERK) pathways [57,58,59]. RAS-responsive element-binding protein 1 (RREB1) is supposed to provide a connection between RAS and TGF-β signals that coordinate the initiation of fibrogenic EMT [60]. There is crosstalk between the canonical and noncanonical signals induced by TGF-β, which is regulated by receptor tyrosine kinases. TGF-β activated the above pathways by influencing the expression of platelet-derived growth factor (PDGF) in a paracrine or autocrine manner [61].
The function of TGF-β signals in disease
TGF-β and the tumor microenvironment (TME)
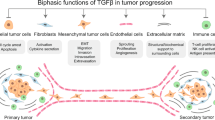
TGF-β signals play critical roles in the regulation of the TME, which has a complex impact on the progression of cancers (Fig. 3). TGF-β may be used as a biomarker in cancer [62]. The TME contains various types of immune cells, such as tumor-associated macrophages (TAMs), neutrophils, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), T cells and B cells, and nonimmune cells, including cancer-associated fibroblasts (CAFs) and stromal cells, as well as a wide range of cytokines [63,64,65,66]. Some immunosuppressive cells, such as TAMs and MDSCs, accumulate early in the TME during tumor growth and suppress the T-cell responses that maintain an immunosuppressive environment [67, 68]. In turn, immune cells and stromal cells in the TME are the primary sources of cytokines, including TGF-β [69]. In fact, TGF-β plays a dual role during tumor progression, functioning as a tumor suppressor in the early stage of cancer and as a tumor promoter in the late stages of cancer, such as breast cancer, hepatocellular carcinoma, lung cancer and pancreatic cancer [70,71,72,73]. Generally, TGF-β inhibits the proliferation of immunosuppressive myeloid cells, especially in early-stage cancers [74, 75]. In advanced tumors, TGF-β produced by myeloid cells suppresses antitumor immunity and then promotes tumor metastasis [76, 77].

The functions of TGF-β in the TME. Tumor cells, endothelial cells, mesenchymal stem cells, cancer-associated fibroblasts, and macrophages can induce the production and secretion of TGF-β in the TME. TGF-β suppresses tumor immune responses by modulating the multiple functions of immune cells in the TME, as depicted
TGF-β inhibited naive CD4+ T cells from differentiating into other effector subtypes, such as Tregs, thereby suppressing the antitumor immune response [78]. Depletion of TGF-βRII in CD4+ T cells inhibited tumor progression, which resulted in tumor cell death in distant avascular regions due to vascular remodeling [79]. DCs are antigen-presenting cells that deliver tumor antigens to natural killer cells (NK cells) and T cells, inducing antitumor cytotoxic effects [80, 81]. TGF-β blocks cytotoxic CD8+ T-cell activation and maturation by inhibiting DC tumor antigen presentation. In addition, TGF-β also inhibited the proliferation and function of CD8+ T cells by reducing the secretion of interferon-γ (IFN-γ) and interleukin-2 (IL-2) [82, 83]. TGF-β promoted the expression of antigen-induced programmed death 1 (PD-1) on CD8+ T cells, leading to the exhaustion of T cells [84]. TGF-β signals maintain the immunosuppressive properties of CD8+ Treg cells. TGF-β and the transcription factor eomesodermin, which controls the follicular location of CD8+ Tregs, synergistically promote homeostasis in CD8+ Tregs [85]. In addition to influencing T cells, TGF-β regulates the activation, proliferation, and apoptosis of B cells. However, this effect of TGF-β on the B-cell-mediated antitumor immune response has not been well studied [86, 87].
Angiogenesis is a hallmark of cancer during its growth and distant metastasis. Besides, by suppressing the immune system, TGF-β also induces tumor angiogenesis. Increasing evidence shows that tumor angiogenesis is regulated by various cytokines, including TGF-β, IL-22, IL-1β [88,89,90,91]. Increased TGF-β expression in the TME is associated with tumor neovascularization in cancers [17, 92]. In endothelial cells, increasing TGF-β1/SMAD3-associated thrombospondin-4 mediated the effects of TGF-β1 on angiogenesis, resulting in tumor growth [93]. An in vivo study showed that increased TGF-β plasma concentrations are related to tumor vascularity [94]. During the tumor process, TGF-β stimulates angiogenesis by affecting TGF-β sequestration [95, 96]. In more detail, fibrillins play an essential role in matrix sequestering of TGF-β. The activation of TGF-β is closely related to integrin binding, which is upregulated upon TGF-β exposure. Moreover, the exposure of fibrillin-2 in the tumor endothelium directly induces tumor angiogenesis by affecting TGF-β sequestering by microfibrils, which results in the higher TGF-β concentration in the TME. Non-polymerized fibrillin-1, fibrillin-2, and fibrillin-containing microfibrils can indirectly bind and sequester TGF-β by interacting with LTBPs, which is the important component of TGF-β and promotes the binding to microfibrils in the ECM [97].
TGF-β and fibrosis
Overexpression of TGF-β correlated with the formation and development of fibrosis, which supports the fact that TGF-β is related to fibrotic diseases, such as pulmonary fibrosis, hepatic fibrosis, renal fibrosis, cardiac fibrosis, and systemic sclerosis [7, 98, 99] (Fig. 4). Macrophages are innate immune cells that have essential roles in tissue repair. TGF-β signaling is relevant to resident immune cells, including macrophages, which play critical roles and contribute to the development of fibrosis [100, 101]. TGF-β is crucial in regulating the recruitment and function of macrophages in fibrotic lesions. It functions as a chemoattractant for macrophages, leading to the recruitment of macrophages to fibrotic lesions [102, 103]. In turn, TGF-β induces the secretion of profibrotic cytokines by macrophages, thereby boosting TGF-β activities [104]. Besides. TGF-β also stimulates the expression of ECM proteins by macrophages [105, 106].

The functions of TGF-β in fibrosis. Tumor cells, endothelial cells, mesenchymal stem cells, cancer-associated fibroblasts, and macrophages can induce the production and secretion of TGF-β, which induces fibrosis, including pulmonary fibrosis, hepatic fibrosis, renal fibrosis, cardiac fibrosis and systematic sclerosis, through SMAD and non-SMAD pathways
ECM deposition is the main characteristic and initial process during fibrosis. TGF-β stimulates the activation and proliferation of fibroblasts, leading to ECM deposition and abnormal organ functions. During this physiological and pathological process, fibroblasts are the main cell types. TGF-β influences the biological behavior of fibroblasts, and low levels of TGF-β promote their proliferation [107]. In addition, TGF-β is chemoattractant for fibroblasts even at a relatively low concentration, which results in recruitment of fibroblasts to fibrotic sites after the activation of TGF-β [108]. Mammalian target of rapamycin (mTOR) signals in the noncanonical pathway are essential in enhancing protein synthesis and activating fibroblasts [109, 110]. Moreover, TGF-β induces epithelial-mesenchymal transition, which contributes to fibroblasts in fibrotic disease [111, 112]. Mechanistically, administration or expression of TGF-β induced fibrosis [113, 114], while inhibiting TGF-β receptor or SMAD signaling decreased the development of fibrosis [115, 116]. SMAD signals cooperated with other signals and transcription factors to promote fibrosis [117], as TGF-β/SMAD signals control the transcription of high-affinity DNA-binding factors, such as TCF/LEF and β-catenin. TCF/LEF and β-catenin are activated by WNT signals and adaptor protein 1 complexes. The adaptor protein 1 complexes are activated by the ERK, JNK, and MAPK pathways [4, 118, 119].
The increase in TGF-β activated fibroblasts, leading to the enhancement of protein synthesis and altering metabolic gene expression [120]. Genes that encode fibronectin and collagen Iα1 are potential transcriptional targets of TGF-β [121, 122]. Moreover, TGF-β promotes the expression of regulators and glycolytic enzymes of metabolism, which results in hyperglycolysis. Meanwhile, the expression of the transcription factor ATF4 increases protein synthesis to meet the crucial needs of collagen and ECM protein synthesis depending on SMAD and mTOR signals [123, 124].
The downstream target genes of TGF-β contributing to the formation of fibrosis is prominent. The crosstalk between the tyrosine kinase receptors and TGF-β signals induced a contractile protein expression signature. This signature leads to α-smooth muscle actin expression, which activates myofibroblast differentiation [125,126,127]. TGF-β signals are also associated with the expression of connective tissue growth factor (CTGF/CCN2), which plays an essential role in the expression of ECM proteins and the differentiation of myofibroblasts [128, 129]. TGF-β induces the expression of interleukin-11, which is a profibrotic cytokine secreted by fibroblasts and epithelial cells and contributes to myofibroblast differentiation, fibroblast activation, and ECM deposition [130, 131]. TGF-β signal also increases the expression of c-JUN, JUN-B, and JUN-D transcription factors, which heterodimerize with c-FOS and related proteins to form AP-1 transcription complexes, positioning them as drivers of fibrosis. AP-1 complexes are activated in response to ERK, JNK, and MAPK signals induced by TGF-β, thereby promoting fibrosis [132,133,134,135]. In addition, TGF-β/SMAD complexes cooperate with AP-1 complexes to increase target gene expression, including those encoding c-JUN, interleukin-11, fibronectin, and collagen Iα2, contributing to fibrosis [136, 137].
TGF-β differentiates cultured tubular epithelial cells into upregulated collagen cells and exhibits a distinct myofibroblast morphology [138,139,140]. Both canonical (SMAD3-dependent) and noncanonical signals mediate these differentiations [141,142,143,144]. TGF-β interacts with β-catenin, which regulates EMT via cAMP response element-binding protein [145]. In addition, bone morphogenic protein-7 (BMP-7) prevents TGF-β-induced EMT in epithelial cells by antagonizing TGF-β, inducing upregulation of α-SMA and downregulation of E-cadherin [139, 146]. TGF-β activates Jagged 1/Notch signals via SMAD and ERK pathways to initiate EMT [147]. TGF-β induces vascular endothelial cells to have mesenchymal characteristics [148,149,150]. Increased TGF-β signals promote endothelial-mesenchymal transdifferentiation, similar to EMT [151, 152]. Overexpression of TGF-β induces αvβ6 integrin-mediated activation of latent TGF-β in epithelial cells, which plays an essential role in the formation and development of fibrosis via mesenchymal traits [153,154,155]. Single-cell sequencing of pulmonary fibrotic lesions reveals that cells have suppressive epithelial features and potential mesenchymal characteristics, suggesting the contributions of EMT and endothelial-mesenchymal transdifferentiation to fibrosis [156, 157].
TGF-β and anemia
The TGF-β superfamily is associated with multiple ineffective erythropoiesis-induced anemias, including myelodysplastic syndrome, Fanconi anemia, β-thalassemia, cancer cachexia-related anemia, acquired aplastic anemia and sickle cell anemia [158,159,160,161,162,163]. In the hematopoietic system, the TGF-β pathway controls diversified biological processes, ranging from immune system homeostasis to hematopoietic stem cell proliferation, differentiation and self-renewal [164, 165]. The TGF-β/SMAD pathway plays an essential role in ineffective erythropoiesis, which is characterized by early-stage erythroid precursor expansion and late-stage precursor apoptosis [166, 167]. Various cells in the bone marrow niche produce TGF-β, including Schwann cells and megakaryocytes, to maintain the quiescence of hematopoietic stem cells [168, 169]. In addition, transcriptional intermediary factor 1γ (TIF1gamma) induces a differentiation response in hematopoietic stem cells, and SMAD4 mediates the antiproliferative response, whereas SMAD2/3 participates in both of these responses. Overall, SMAD2/3-SMAD4 and SMAD2/3-TIF1gamma are complementary effector arms in controlling hematopoietic cell fate through TGF-β signals [170, 171].
Myelodysplastic syndrome is a hematopoietic stem cell disease that manifests as bone marrow dysplasia and cytopenias because of impaired hematopoiesis [172, 173]. In myelodysplastic syndrome, TGF-β signaling controls the behavior of hematopoietic stem cells in the bone marrow niche. Moreover, the activation of TGF-β impairs the competitive advantage of normal hematopoietic stem cells, which actually contributes to the selection of early-stage myelodysplastic syndrome-genic clones [174, 175].
β-thalassaemia is a β-globin gene mutation that causes genetic disease, which is characterized by iron-loading anemia and ineffective erythropoiesis [176]. TGF-β is a negative regulatory factor in erythrocyte differentiation and maturation, similar to erythropoietin [177]. Hence, TGF-β is a possible target of β-thalassaemia and has been evaluated in clinical studies [178].
In addition to the aforementioned syndrome-inducing anemia, Fanconi anemia is a genetic DNA repair disorder that is characterized by progressive bone marrow failure and predisposition to malignancy [179]. TGF-β signal-mediated growth inhibition is one of the causes of bone marrow failure in Fanconi anemia by impairing the function of hematopoietic stem and progenitor cells [180, 181]. Hence, TGF-β is a potential target of Fanconi anemia.
TGF-β signaling and inflammatory diseases
TGF-β is supposed to act as a pro- or anti-inflammatory factor contributing to host defense which controls physiologic inflammation and immune response [182]. Overexpression and/or activation of TGF-β are observed in persistent inflammation. On the other hand, systemic routing of TGF-β can also prevent inflammatory pathogenesis through multiple mechanisms [183]. TGF-β maintains T cell tolerance to self and innocuous environmental antigens by influencing the differentiation and homeostasis of effector T cells and Tregs. The activity of TGF-β controls inflammatory response balance by targeting pathogens without evoking over immunopathology to healthy tissues [184].
TGF-β is essential in the development and progression of chronic respiratory diseases which is overexpressed in chronic inflammation, fibrosis and viral infection associated respiratory abnormities including asthma, chronic obstructive pulmonary disease and pulmonary fibrosis [185]. Moreover, TGF-β and SMAD4 mediated uncoupling protein-2 downregulation leads to Aspergillus protease associated inflammation in primary bronchial epithelial cells [186]. Besides, TGF-β is involved in the fluid homeostasis and fibrosis in the lung of COVID-19 patients, which may contribute to a potential immunotherapy strategy [187].
Dysregulated TGF-β signal is also observed in patients with inflammatory bowel disease, which is chronic intestinal inflammation, including ulcerative colitis and Crohn’s disease. The dysfunction of TGF-β signal transduction occurs in T-cells and dendritic cells, which leads to spontaneous colitis in vivo. Moreover, the immune homeostasis of host modulated by intestinal microbes depends on TGF-β production [188]. SMAD4 can restrain naive CD8+ T cells from becoming pathogenic for the gut to prevent inflammatory bowel disease in a TGF-β-independent manner [189]. However, the over expression of SMAD7 in inflammatory cells makes them unresponsive to TGF-β1 and negatively regulates gut inflammation [190]. Besides, TGF-β knockout mice present a phenotype with severe multiorgan inflammation [191].
TGF-β is also important in protecting keratinocytes from oxidative stress and involves in the wound healing process [192, 193]. The inhibition of TGF-β is demonstrated to accelerate wound closure and reduce scarring [194, 195]. Exogenous SMAD7 below an oncogenic level can mitigate wound healing and skin inflammation defects related to over activation of TGF-β and NF-κB [196].
TGF-β signaling and other diseases
In addition to the roles of TGF-β signals in cancers, fibrosis, anemia and inflammatory diseases, this signal is associated with the progression of other diseases. TGF-β family plays an essential role in the maintenance of normal blood vessel wall structure [197]. Mutations in TGF-β family components are associated with specific cardiovascular syndromes, such as primary pulmonary hypertension, and hereditary hemorrhagic telangiectasia [198, 199]. TGF-β family mutation associated specific hereditary vascular syndromes include Osler-Rendu-Weber disease, hereditary hemorrhagic telangiectasia, Loeys-Dietz syndrome, Shprintzen-Goldberg syndrome, and Marfan syndrome [200, 201].
Single-cell RNA sequencing reveals that TGF-β signal overexpression is the upstream driver of smooth muscle cells modulation which plays a pivotal role in promoting extracellular matrix substrate modulation and aortic aneurysm progression in Marfan syndrome [202, 203]. The SMAD signaling of TGF-β is essential in maintaining smooth muscle cell phenotype, while the noncanonical signaling pathway like ERK negatively regulates smooth muscle cell phenotype [204]. Moreover, dysregulated TGF-β signal transduction is related to nonhereditary disorders, including atherosclerosis and cardiac fibrosis, by influencing endothelial cells and smooth muscle cells proliferation, differentiation and migration [205].
The epigenetic alterations of TGF-β canonical and non-canonical pathways are related to thoracic ascending aorta dilatation and aortic aneurysm through remodeling of the vascular wall in Loeys-Dietz and Marfan’s syndromes [206, 207]. Aortic valve disease is characterized by elastic fiber fragmentation, fibrosis, and aberrant angiogenesis. Noncanonical TGF-β signals progressively increase over the progression of aortic valve disease, suggesting that TGF-β signals are possible targets in this disease [208]. In a cohort study, platelet expressed TGF-β1 plays a pivotal role in acute coronary syndromes and indicates a prognostic impact of TGF-β1 on clinical outcomes in patients with coronary artery disease [209].
Besides, TGF-β family members play crucial roles in the development and homeostasis of connective tissue and skeletal system [210]. TGFBR1 or TGFBR2 mutations cause increased expression of TGF-β signaling, connective tissue growth factor and phosphorylation of SMAD2, which lead to a syndrome of altered cardiovascular, neurocognitive, craniofacial and skeletal development [211].
Therapies based on TGF-β signal transduction in disease
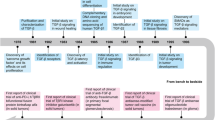
Novel strategies targeting TGF-β signaling transduction have been designed and evaluated clinically to treat cancers, sclerosis, and fibrosis. These strategies include neutralizing antibodies and ligand traps, small-molecule receptor kinase inhibitors targeting ligand–receptor signaling pathways, and antisense oligonucleotides to disrupt the production of TGF-β at the transcriptional level. In addition, some vaccines containing a TGF-β antisense transgene, downregulating TGF-β, also show promising therapeutic efficacy in cancer (Fig. 5).

Potential therapeutic strategies based on the TGF-β signaling pathway in disease. Antagonists targeting the TGF-β pathway, including neutralizing antibodies, ligand traps, small-molecule receptor kinase inhibitors, antisense oligonucleotides and vaccines, have recently been evaluated in clinical trials. Representative drugs are shown
Neutralizing antibodies
Fresolimumab
Fresolimumab (GC1008) is a human IgG4κ anti-TGF-β monoclonal antibody that neutralizes all TGF-β isoforms. This agent is safe and effective in a phase I study for advanced malignant melanoma and renal cell carcinoma. For efficacy, one melanoma patient achieves a partial response, and six patients are proven to have stable disease (NCT00356460) [13]. Fresolimumab potentially controls cutaneous lesions, such as cutaneous keratoacanthomas or squamous cell carcinomas [212]. In addition, administration of fresolimumab during radiotherapy is feasible for patients with metastatic breast cancer. Patients receiving 10 mg/kg fresolimumab have a longer median overall survival with a favorable systemic immune response than patients in the 1 mg/kg group (NCT01401062) [213].
In systemic sclerosis patients, administration of fresolimumab decreases disease-related biomarkers, including THBS1, COMP, SERPINE1, CTGF and other longitudinal pharmacodynamic biomarkers. Regarding efficacy, fresolimumab treatment improves clinical symptoms and decreases the infiltration of dermal myofibroblasts (NCT01284322) [214, 215]. In a clinical trial involving patients with primary focal segmental glomerulosclerosis, fresolimumab is well tolerated [216]. However, an additional phase II study is underpowered and does not achieve the primary or secondary endpoints. Thus, fresolimumab is appropriate for more evaluation in larger studies (NCT01665391) [217]. For osteogenesis imperfecta, a phase I study of fresolimumab is conducted in 8 patients. In this clinical trial, fresolimumab is associated with increases in lumbar spine areal bone mineral density in participants (NCT03064074) [218].
LY3022859
LY3022859 is a human anti-anti-TβRII IgG1 monoclonal antibody that inhibits the activation of receptor-mediated signals and has favorable antitumor efficacy for primary tumors and metastatic disease in tumor models [219]. A phase I study including patients with advanced solid tumors shows that the maximum tolerated dose of LY3022859 is not determined. During dose escalation, when the dose of LY3022859 is greater than 25 mg, patients have worsening symptoms, partially due to uncontrolled cytokine release (NCT01646203) [220].
SAR439459
SAR439459, a neutralizing antibody targeting all isoforms of TGF-β, is supposed to block TGF-β/SMAD signals. This agent also shows activity in reversing TGF-β-mediated NK-cell and T-cell suppression. An in vitro study shows that SAR439459 synergizes with an anti-PD1 antibody, resulting in enhancement of the T-cell response. Moreover, administration of SAR439459 prevents tumor growth by augmenting the proliferation of intertumoral CD8+ T cells, reducing their exhaustion, and evoking proinflammatory cytokines in syngeneic tumor models. This evidence supports the ongoing clinical exploration of SAR439459 in patients with solid tumors (NCT03192345) [221].
Other anti-TGF-β neutralizing antibodies, including ABBV151, NIS793, QLS31901, GT90001, and LY2382770, are undergoing clinical trials (Table 1).
TGF-β ligand traps
AVID200
TGF-β ligand traps are chimeric fusion proteins designed to restrain TGF-βs from binding to TGF-β receptors based on their ectodomain. AVID200 is a potent TGF-β1/TGF-β3 protein trap that enhances antitumor efficacy in a syngeneic 4 T1 triple-negative breast cancer model [86]. Currently, a phase I clinical trial of AVID200 has been conducted for advanced solid tumors (NCT03834662).
In fibrotic disease, administration of AVID200 decreases the proliferation of human mesenchymal stromal cells and reduced the phosphorylation of SMAD2 and the expression of collagen. Myelofibrosis mononuclear cells present increasing progenitor cells emerging after treatment with AVID200. In addition, AVID200 treatment reduces bone marrow fibrosis, increases bone marrow cellularity, and increases the numbers of murine progenitor and hematopoietic stem cells in a myelofibrosis mouse model [222]. AVID200 is supposed to promote the survival of murine/human fanconi anemia hematopoietic stem and progenitor cells in vitro by downregulating nonhomologous end-joining pathway-related genes and reducing DNA damage in vivo [223]. AVID200 also increases the hematopoietic colony formation of Shwachman-Diamond Syndrome patients’ bone marrow, leading to the improvement of bone marrow failure [224]. Currently, a clinical trial of AVID200 for systemic sclerosis has been launched (NCT03831438).
Bintrafusp alfa
Bintrafusp alfa (M7824) is a bifunctional fusion protein that contains the extracellular TGF-β trap fused to a human IgG monoclonal antibody against PD-L1. Bintrafusp alfa synergizes effectively with radiotherapy by modulating the TME to reverse cancer immune evasion. Combining bintrafusp alfa with radiotherapy increases tumor-infiltrating lymphocytes, attenuates radiotherapy-induced fibrosis, reconstitutes tumor immunity and regresses spontaneous lung metastases [12]. In addition, bintrafusp alfa shows safety and clinical activity in human papillomavirus (HPV)-associated cancers. The objective response rate is 30.5%, including five patients, with a disease control rate of 44.1% (NCT02517398, NCT03427411) [225]. Bintrafusp alfa is safe and enhances tumor antigen-specific immunity by reversing Treg immunosuppression and reducing myeloid cell tumor infiltration in patients with HPV-unrelated head and neck squamous cell carcinoma [226]. Several factors are associated with the clinical response during bintrafusp alfa therapy, including low levels of TGF-β1 expression and higher CD8+ T cell: MDSC ratios [227]. Bintrafusp alfa has promising antitumor efficacy in a phase I study involving patients with non-small cell lung cancer who are previously treated with platinum. The objective response rate in all patients is 21.3% (NCT02517398) [228]. In a phase I trial, bintrafusp alfa also has clinical activity for biliary tract cancer, with an objective response rate of 20%. In addition, the overall survival is 12.7 months [229]. In patients with advanced esophageal adenocarcinoma and esophageal squamous cell carcinoma, bintrafusp alfa shows clinical antitumor efficacy with a manageable safety profile. In patients with esophageal adenocarcinoma, the confirmed objective response rate is 20.0% (NCT02517398) [230]. Similarly, the confirmed objective response rate is 10.0% in patients with esophageal squamous cell carcinoma, with a median overall survival of 11.9 months (NCT02699515) [231]. Bintrafusp alfa also has antitumor efficacy in patients with pretreated advanced squamous cell carcinoma of the head and neck. The confirmed objective response rate is 13%, with 4 patients having stable disease (NCT02517398) [232]. In patients with advanced gastric and gastroesophageal junction cancer, the objective response rate to bintrafusp alfa is 16%, with a disease control rate of 26% [233]. In patients with advanced solid tumors who received bintrafusp alfa treatment, two of 23 patients have a partial response, for a disease control rate of 35.7% (NCT02699515) [234].
Luspatercept
Luspatercept (ACE-536, reblozyl) is an activin receptor type IIB fusion protein–ligand trap targeting TGF-β/SMAD signals. This agent has been used to treat anemia diseases, including beta-thalassemia, myelofibrosis, and myelodysplastic syndromes [235]. TGF-β/SMAD signals promote erythroid maturation by enhancing the differentiation of late-stage erythroblasts, thereby improving anemia [236]. Luspatercept impacts the bone marrow microenvironment, leading to a selective restoration of ineffective hematopoiesis [237]. In patients with transfusion-dependent lower-risk myelodysplastic syndrome, luspatercept shows clinical activity in a phase II (PACE-MDS) trial and a phase III (MEDALIST) trial, leading to US Food and Drug Administration approval in 2020 [238]. In a phase III (MEDALIST) trial, 38% of the patients treated with luspatercept have transfusion independence for 8 weeks and even longer (NCT02631070) [239,240,241]. In a phase II (PACE-MDS) trial, luspatercept is well tolerated and has clinical efficacy for patients with myelodysplastic syndromes inducing anemia (NCT01749514, NCT02268383) [242, 243]. In patients with myelodysplastic syndromes or myeloproliferative neoplasms who currently have no effective treatments, administration of luspatercept reduces the transfusion burden and improves the modified hematologic response-erythroid levels [244]. Luspatercept therapy has been demonstrated to strengthen the contribution of host immunity to disease biology in myelodysplastic syndromes with ring sideroblasts [245].
In patients with β-thalassemia after luspatercept therapy in a clinical trial, twenty-six of 64 patients achieve over a 20% reduction in red blood cell transfusion burden (NCT01749540 and NCT02268409) [246]. In a phase III (BELIEVE) trial for transfusion-dependent β-thalassemia, the transfusion burden is reduced after the administration of luspatercept (NCT02604433) [247]. Luspatercept is also supposed to be a potential strategy in patients with nontransfusion-dependent β-thalassemia [248]. In this phase II (BEYOND) trial in patients with nontransfusion-dependent β-thalassemia, 77% of patients after luspatercept therapy achieve an increase in hemoglobin concentration (NCT03342404) [249].
Sotatercept
Sotatercept (ACE-011), a TGF-β ligand trap, restrains late-stage negative regulators of erythropoiesis and improves ineffective erythropoiesis. For anemia caused by β-thalassemia, a phase II study demonstrated that sotatercept is clinically efficient and well tolerated. In nontransfusion-dependent patients, 18 of 30 (60%) achieve a hemoglobin increases of more than 1.0 g/dL, which is sustained for more than 3 months. In the transfusion-dependent β-thalassemia subgroup, four (100%) patients achieve a more than 20% transfusion-burden reduction (NCT01571635) [250].
For pulmonary arterial hypertension, a phase II (PULSAR, NCT03496207) study shows a reduction in pulmonary vascular resistance after sotatercept treatment [251]. The extension study revealed that 32 of 97 (30.8%) participants suffer serious treatment-related adverse events. Importantly, the placebo-crossed to sotatercept group is demonstrated to have improved both primary and secondary endpoints. The clinical effectiveness is well maintained in the patients with continued sotatercept [252].
For lower-risk myelodysplastic syndromes, especially in patients for whom previous erythropoiesis-stimulating agents failed, sotatercept is well tolerated and clinically effective. Thirty-six of 74 (49%) patients achieve hematological improvement-erythroid. Among them, 29 of 62 (47%) participants with a high transfusion burden achieve hematological improvement-erythroid, whereas seven of 12 (58%) patients with a low transfusion burden achieve hematological improvement-erythroid (NCT01736683) [253].
In patients with chemotherapy-induced anemia in advanced solid tumors, both clinical trials are terminated early because of the slow patient accrual. However, the existing results indicate that sotatercept is potentially effective with an acceptable safety profile when treated with chemotherapy-induced anemia (NCT00931606, NCT01284348) [254].
P144
P144 (Disetertide©) is a peptide inhibitor of TGF-β1. This inhibitor decreases the proliferation and invasiveness of glioblastoma cells. P144 increases apoptosis and anoikis by reducing SMAD2 phosphorylation, downregulating SK, and upregulating SMAD7 in vitro. Additionally, P144 impairs tumor growth and increases survival in a glioblastoma mouse model [255]. Besides, treatment with P144 results in a reduction in the mitotic-to-apoptotic ratio and angiogenesis, which are induced by TGF-β1. In addition, P144 abrogates EMT and the phenotypes of cancer stem cells, which decreases liver metastasis in patients with colorectal cancer [256]. P144 reduces tumor growth by reducing the infiltration of macrophages and increasing the intratumor levels of MCP-1 and VEGF [257]. The therapeutic applications of P144 are limited due to a lack of target selection, possible recognition by the immune system, and potential cytotoxicity on healthy cells. Encapsulation of P144 with nanoparticles facilitated its dissolution, improves its functionalization and improves its potential therapeutic applications in liver cancer [258].
P144 also has treatment efficacy in controlling fibrotic disease. Administration of P144 reduces radiation-induced fibrosis in soft tissue sarcoma by retaining the macro- and microscopic morphology of muscle, reducing extracellular matrix fibrosis and reducing SMAD2/3 phosphorylation [259]. P144 decreases renal fibrosis by blocking TGF-β1/SMAD3 signals and modulating the polarization of macrophages, suggesting its possible therapeutic potential in ischemia–reperfusion injury-induced renal fibrosis [260]. P144 decreases laser-induced choroidal neovascularization in a rat model [261]. P144 is also proposed to promote the maturation of scars, with the improvement of the morphology of hypertrophic scars in a mouse model [262]. P144 prevents the formation of an aortic aneurysm but not its progression in a Mafan syndrome mouse model. Hence, reducing the excess of active TGF-β signaling during the early stages of aortic disease progression is essential [263]. Furthermore, P144 inhibits TGF-β-dependent signals in cardiac fibroblasts, preventing myocardial fibrosis in spontaneously hypertensive rats [264]. P144 also inhibits NADPH oxidases and prevents kidney oxidative stress in spontaneously hypertensive rats [265].
Other TGF-β ligand traps, including ACE-1334, TST005, and JS201, are under evaluation in clinical trials (Table 1).
Small-molecule receptor kinase inhibitors
Vactosertib
Currently, some small-molecule receptor kinase inhibitors of TGF-β signals are undergoing clinical trials to treat cancer and fibrosis. Vactosertib (TEW-7197, EW-7197) is a small-molecule kinase inhibitor of TGF-βRI that has promising antitumor and antifibrotic potential [11, 266, 267]. Vactosertib inhibits hepatic, renal, and pulmonary fibrosis by blocking both TGF-β1/SMAD2/3 and reactive oxygen species (ROS) signals [268]. The combination of vactosertib with radiation has a favorable antimetastatic efficacy in breast cancer [269]. Vactosertib prevents ulcerative colitis-associated inflammation and fibrosis, protecting against postsurgical adhesion formation by downregulating proinflammatory and profibrotic genes, inhibiting oxidative stress, decreasing inflammatory cell infiltration, and inhibiting excessive collagen deposition [270,271,272]. The combination of vactosertib with imatinib mesylate, a tyrosine kinase inhibitor, delays chronic myeloid leukemia relapse and prolongs survival by eliminating leukemia-initiating cells [273]. Vactosertib potently inhibits breast cancer lung metastasis by inhibiting SMAD/TGF-β signals and enhancing the activity of cytotoxic T cells [274]. Clinical trials based on vactosertib are undergoing in melanoma, lung cancer, urothelial carcinoma, gastric cancer, and colorectal cancer (Table 2).
Galunisertib
Galunisertib (LY2157299) is another small-molecule inhibitor that selectively binds to TGF-βRI, inhibiting kinase activity [275]. Galunisertib exerts antifibrotic effects on dermal fibroblasts by attenuating the expression of fibrotic genes and increasing the expression of antifibrotic genes such as decorin and MMP1 [276]. Galunisertib is a potential prophylactic drug for treating traumatic heterotopic ossification by intercepting TGF-β/SMAD2/3 signals [277]. What is more, galunisertib shows a prominent antifibrotic potential in liver fibrosis by inhibiting phosphorylation of SMAD2, blocking the production and maturation of collagens, and promoting the degradation of collagens [278, 279].
Galunisertib overcomes stemness-derived aggressiveness by reducing the expression of CD44 and THY1 in hepatocellular carcinoma [280]. A phase IB study of galunisertib plus ramucirumab for advanced hepatocellular carcinoma shows that the combination therapy displays favorable pharmacokinetics, with a disease control rate of 12.5% [281]. A pilot study of galunisertib combined with stereotactic body radiotherapy in patients with advanced hepatocellular carcinoma shows good tolerability and is associated with antitumor activity. Two out of 15 patients achieve a partial response [282]. In a phase II study, galunisertib plus sorafenib results in prolonged overall survival [283]. In patients with unresectable pancreatic cancer, galunisertib plus gemcitabine improves overall survival [284]. In another phase IB clinical trial of patients with pancreatic cancer, galunisertib is coadministered with durvalumab, showing tolerable adverse events but limited clinical activity, with progression-free survival of 1.87 months [285].
Galunisertib is supposed to suppress the activation of SMAD2 in neuroblastomas and activate NK cells, restore NK cytotoxic activity, and increase the efficacy of dinutuximab with activated NK cells against neuroblastoma tumors [286]. For recurrent glioblastoma, the combination of galunisertib and lomustine fails to demonstrate improved overall survival compared with the group receiving monotherapy [287]. In a phase II study of galunisertib for myelodysplastic syndromes, 10 out of 41 patients achieve hematologic improvement erythroid response, 18 patients have erythroid response and nine of 28 transfusion-dependent patients achieve hematologic improvement [288]. Other clinical trials targeting solid tumors, including hepatocellular carcinoma, breast cancer, and glioma, are ongoing (Table 2).
LY3200882
LY3200882 is an orally selective next-generation potent adenosine triphosphatase competitive TGF-βRI small-molecule inhibitor that has promising antitumor efficacy [289, 290]. Codelivery of LY3200882 and programmed cell death protein ligand 1 (PD-L1) siRNA boosts antitumor immunotherapy by downregulating the expression of ECM, promoting the infiltration of effector T cells, resulting in enhanced tumor antigen presentation and reversing the immunosuppressive microenvironment in triple-negative breast cancer [289]. LY3200882 effectively inhibits liver metastases by increasing the infiltration of CD8+ cytotoxic T cells and inhibiting the recruitment of immunosuppressive cells such as MDSCs in colorectal mouse models [290]. A phase I study showed that LY3200882 is well tolerated, with preliminary antitumor activity in advanced cancer. Four patients with grade 4 glioma have partial responses. In patients with advanced pancreatic cancer, 6 out of 12 patients have partial responses, and 3 patients are stable disease. In this trial, the overall disease-control rate of LY3200882 plus gemcitabine and nab-paclitaxel is 75% [291].
SHR-1701
SHR-1701 is a bifunctional fusion protein that is a PD-L1 monoclonal antibody fused with the extracellular TGF-βRII domain. This agent has promising antitumor efficacy in advanced cervical cancer [292, 293]. Among 32 patients with cervical cancer, the objective response rate is 15.6%, and the disease control rate is 50.0%. Notably, as assessed by imRECIST, the median PFS is 4.1 months, and the 12-month overall survival rate is 54.6% (NCT03774979) [293]. Moreover, patients with lung cancer suffering from persistent lymphopenia after chemotherapy are sensitive to SHR-1701 [294].
Cilengitide
αvβ integrin is a major local activator of latent TGF-β. Genetically and pharmacologically targeting αvβ integrin inhibits the TGF-β signals and suppresses tumor metastasis [295,296,297]. Cilengitide, a selective cyclic RGD pentapeptide antagonist of αvβ3 and αvβ5 integrin, has been demonstrated to modulate the attachment and viability of glioma cells and induce autophagy-mediated cell death [298, 299]. In a phase III study of cilengitide plus standard treatment for patients with glioblastoma, combinational therapy does not improve the outcomes [300]. In this trial, the authors recommend a different continuous infusion schedule that is more appropriate according to the pharmacokinetics [301]. In a phase II study with two cilengitide regimens plus standard treatment for patients with glioblastoma, the median overall survival is 16.3 months in the cilengitide arm and 14.5 months in the intensive cilengitide arm (NCT00813943) [302]. However, in another phase II trial, cilengitide plus metronomic temozolomide, procarbazine, and standard radiotherapy does not improve survival in patients with glioblastoma [303].
Cilengitide is supposed to enhance the inhibition of erlotinib on TGF-β1-induced EMT and phosphorylation of SMAD2/3 [304, 305]. In a phase I study, continuous infusion of cilengitide plus chemoradiotherapy for patients with stage III non-small cell lung cancer is potentially tolerable. Four out of 9 patients have a complete response, and 4 patients have a partial response [306]. In another phase II study that combined cilengitide with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small cell lung cancer patients, the progression-free survival is 6.8 months in the cilengitide group versus 5.6 months in the control group. The median overall survival is 13.6 versus 9.7 months compared with the control group (NCT00842712) [307].
Cilengitide has been demonstrated to downmodulate the invasiveness of melanoma cells by targeting αvβ5 integrin [308]. Cilengitide is well tolerated but has limited antitumor efficacy as a monotherapy for metastatic melanoma [309, 310]. Cilengitide enhances the effectiveness of anti-PD1 treatment and produces a more robust antitumor immune response by decreasing STAT3 phosphorylation and reducing tumor PD-L1 expression in a melanoma mouse model [311]. In addition, activation of POSTN releases TGF-β1 from the ECM and initiates the POSTN/TGF-β1 positive feedback loop. Cilengitide plus lenvatinib suppresses tumor cell growth in a hepatocellular carcinoma mouse model [312]. Generally, cilengitide, combined with paclitaxel, is well tolerated and has antitumor activity in patients with advanced solid tumors [313].
Cilengitide treatment decreases adhesion to vitronectin and fibronectin and reduces the expression of TGF-β-induced fibronectin genes, as well as the accumulation of mRNAs for fibronectin and collagen type I. However, cilengitide does not inhibit the development of pulmonary fibrosis in vivo [314]. Pharmacological inhibition of integrin utilizing cilengitide in vivo decreases angiogenesis but worsens biliary and septal fibrosis, despite its antifibrogenic effect on hepatic stellate cells [315, 316].
Other small-molecule inhibitors targeting TGF-β signaling pathways, such as GFH018 and PF06952229, are under clinical evaluation for patients with lung cancer, breast cancer, and prostate cancer (Table 2).
Antisense oligonucleotides (ASOs)
Trabedersen
Trabedersen (AP12009) is a synthetic phosphorothioate antisense oligodeoxynucleotide blocking the production of TGF-β2. Trabedersen has therapeutic potential in malignant brain tumors, skin tumors, pancreatic cancer, and colorectal cancer [317]. Trabedersen reduces the secretion of TGF-β2, inhibits cell proliferation and migration, and reverses TGF-β2-mediated immunosuppression in pancreatic cancer. In addition, trabedersen significantly inhibits tumor growth and lymph node metastasis in pancreatic cancer [318]. In a phase II clinical trial for recurrent high-grade glioma, superior efficacy is observed for trabedersen over chemotherapy. This positive risk-benefit assessment demonstrates its clinical use in high-grade glioma [319].
ISTH0036
ISTH0036 is an antisense oligonucleotide selectively targeting TGF-β2 signals [320]. In a phase I study involving patients with primary open angle glaucoma who receive trabeculectomy, single-dose ISTH0036 administration at the time of trabeculectomy results in intraocular pressure values persistently less than 10 mmHg during the three-month postoperative period [15]. Future phase II clinical trials are needed to assess repeat dosing for up to one year for constant antifibrotic effects. It is critical for clinical trials to assess the efficacy of ISTH0036 as an antifibrotic agent that inhibits glaucoma pathophysiological mechanisms by selectively suppressing TGF-β2 [321].
Other TGF-β antisense oligonucleotides, including TASO-001, STP705, and TRK250, are under evaluation in clinical trials (Table 2).
Vaccine-based therapy
Belagenpumatucel-L
Belagenpumatucel-L (Lucanix™) is a nonviral gene-modified allogeneic whole tumor cell vaccine expressing the antisense strand of the TGF-β2 gene. This approach is well tolerated in a phase II study of Belgel-L for non-small cell lung cancer [322]. During therapy, baseline circulating tumor cells are associated with overall survival [14]. In a phase III trial for non-small cell lung cancer, there is a difference in overall and progression-free survival between the belagenpumatucel-L group and the placebo group [323]. As the phase III trial of belagenpumatucel-L for stage III/IV non-small cell lung cancer does not meet the primary end point, further studies are needed to select patients who may benefit from this vaccine [324].
Vigil
Vigil (Gemogenovatucel-T, FANG™, IND14205) is an autologous compound consisting of a plasmid encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) and a bifunctional short hairpin RNAi (bishRNAi) targeting furin convertase, leading to downregulation of TGF-β1 and β2. There is a phase I study for advanced cancer, and this trial shows that Vigil is successful in 42 of 46 patients, of whom 27 receive over one vaccine. There are no serious adverse events after treatment [325]. The three-year follow-up of Vigil in 12 patients with metastatic advanced Ewing’s sarcoma reveals a one-year survival of 73% for Vigil-treated patients compared to 23% in the placebo group. The overall survival is 17.2 months between the Vigil (median overall survival of 731 days) and placebo groups (median overall survival of 207 days) [326].
In advanced ovarian cancer, an induction of the circulating activated T-cell population is observed in the Vigil group [327]. In the phase IIB (VITAL) trial for stage III/IV ovarian cancer, utilizing Vigil as maintenance immunotherapy is well tolerated. However, the primary endpoint is not met after the treatment (NCT02346747) [328]. However, Vigil is demonstrated to have clinical benefit for ovarian cancer with homologous recombination proficient. The recurrence-free and overall survival are improved in the Vigil group compared to the placebo group [329]. The three-year follow-up of Vigil for patients with homologous recombination-proficient ovarian cancer still shows durable activity in both recurrence-free and overall survival [330]. The gene expression profile suggests that Vigil’s overall survival benefit is correlated with elevated expression of MHC-II and positive IFN-γ ELISPOT in patients with recurrent ovarian cancer [331]. When combined with atezolizumab in relapsed ovarian cancer patients, the median overall survival is not reached. However, patients harboring BRCAwt suggest an improved overall survival benefit. Thus, a continued investigation of combination therapy with Vigil-1st and atezolizumab is needed for patients with BRCAwt [332, 333].
Conclusions and perspectives
The multifunctional cytokine TGF-β regulates inflammatory progression, differentiation, proliferation, and wound healing during homeostasis. Dysregulated TGF-β promotes EMT and immunosuppression during tumorigenesis and fibrosis. Therefore, there is increasing interest in targeting TGF-β signals. In addition, TGF-β-targeted therapies, including neutralizing antibodies and TGF-β ligand traps for ligand elimination, small-molecule receptor kinase inhibitors, ASOs and vaccine-based therapy, have achieved comparable results in preclinical trials to treat tumors, fibrosis, and other diseases. However, few of these anti-TGF-β compounds are in phase III clinical trials because of the different roles of TGF-β in different cancer stages and the poor stability and side effects of anti-TGF-β drugs [3].
The role of TGF-β in tumorigenesis and progression is different and complex. Multiple types of research indicate that TGF-β becomes a tumor suppressor at an early stage. In contrast, at a late stage, overexpressed TGF-β promotes the formation of EMT, TME, immunosuppression, and CAFs. It is difficult but essential to determine whether a patient’s TGF-β is a promotor or a suppressor. More research should be ongoing to identify which tumor types or fibrosis could benefit from targeting TGF-β therapies. In addition, combination strategies could also solve cardiovascular adverse effects [334], poor stability in vivo [335], and some other side effects promoting poor therapeutics. In conclusion, progress in detecting the universal mechanisms of TGF-β in specific tumor subtypes and diverse stages of cancer, as well as other diseases, and exploring appropriate combination dosing regimens to reduce side effects are essential and prospective.
Availability of data and materials
The materials supporting our conclusion of this review are included within the article.
Abbreviations
- TGF-β:
-
Transforming growth factor beta
- ECM:
-
Extracellular matrix
- LAP:
-
Latency-associated peptide
- GARP:
-
Glycoprotein-A repetitions predominant
- LRRC32:
-
Leucine-rich repeat containing 32
- EMT:
-
Epithelial-to-mesenchymal transition
- JNK:
-
c-Jun N-terminal kinase
- NF-κB:
-
Nuclear factor kappa-B
- TRAF4/6:
-
Tumor necrosis factor-associated factor 4/6
- ERK:
-
Extracellular signal regulated kinase
- RREB1:
-
RAS-responsive element-binding protein 1
- PDGF:
-
Platelet-derived growth factor
- TME:
-
Tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- MDSCs:
-
Myeloid-derived suppressor cells
- DCs:
-
Dendritic cells
- CAFs:
-
Cancer-associated fibroblasts
- Tregs:
-
Regulatory T cells
- NK cells:
-
Natural killer cells
- IFN-γ:
-
Interferon-γ
- IL-2:
-
Interleukin-2
- PD-1:
-
Programmed death 1
- mTOR:
-
Mammalian target of rapamycin
- CTGF/CCN2:
-
Connective tissue growth factor
- BMP-7:
-
Bone morphogenic protein-7
- TIF1gamma:
-
Transcriptional intermediary factor 1γ
- HPV:
-
Human papillomavirus
- PD-L1:
-
Programmed cell death protein ligand 1
- ASOs:
-
Antisense oligonucleotides
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
References
Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237–53. https://doi.org/10.1038/s41571-021-00588-9.
Leppkes M, Neurath MF. Cytokines in inflammatory bowel diseases - Update 2020. Pharmacol Res. 2020;158:104835. https://doi.org/10.1016/j.phrs.2020.104835.
Györfi AH, Matei AE, Distler JHW. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018;68-69:8–27. https://doi.org/10.1016/j.matbio.2017.12.016.
Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal. 2019;12(570). https://doi.org/10.1126/scisignal.aav5183.
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–46. https://doi.org/10.1016/s0140-6736(16)00587-0.
Hao Y, Baker D, Ten Dijke P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci. 2019;20(11). https://doi.org/10.3390/ijms20112767.
Ong CH, Tham CL, Harith HH, Firdaus N, Israf DA. TGF-β-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur J Pharmacol. 2021;911:174510. https://doi.org/10.1016/j.ejphar.2021.174510.
Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, et al. TGF-β signalling and liver disease. FEBS J. 2016;283(12):2219–32. https://doi.org/10.1111/febs.13665.
Batlle E, Massagué J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity. 2019;50(4):924–40. https://doi.org/10.1016/j.immuni.2019.03.024.
Tauriello DVF, Sancho E, Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25–44. https://doi.org/10.1038/s41568-021-00413-6.
Jung SY, Hwang S, Clarke JM, Bauer TM, Keedy VL, Lee H, et al. Pharmacokinetic characteristics of vactosertib, a new activin receptor-like kinase 5 inhibitor, in patients with advanced solid tumors in a first-in-human phase 1 study. Investig New Drugs. 2020;38(3):812–20. https://doi.org/10.1007/s10637-019-00835-y.
Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, et al. Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39(10):1388–403.e10. https://doi.org/10.1016/j.ccell.2021.08.008.
Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. https://doi.org/10.1371/journal.pone.0090353.
Nemunaitis J, Nemunaitis M, Senzer N, Snitz P, Bedell C, Kumar P, et al. Phase II trial of Belagenpumatucel-L, a TGF-beta2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009;16(8):620–4. https://doi.org/10.1038/cgt.2009.15.
Pfeiffer N, Voykov B, Renieri G, Bell K, Richter P, Weigel M, et al. First-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF-β2), in subjects with open-angle glaucoma undergoing glaucoma filtration surgery. PLoS One. 2017;12(11):e0188899. https://doi.org/10.1371/journal.pone.0188899.
Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol. 2016;8(5). https://doi.org/10.1101/cshperspect.a021873.
Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9–34. https://doi.org/10.1038/s41571-020-0403-1.
Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316(6030):701–5. https://doi.org/10.1038/316701a0.
Grainger DJ, Mosedale DE, Metcalfe JC. TGF-beta in blood: a complex problem. Cytokine Growth Factor Rev. 2000;11(1–2):133–45. https://doi.org/10.1016/s1359-6101(99)00037-4.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8. https://doi.org/10.1038/nature25501.
Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. 2020;12(536). https://doi.org/10.1126/scitranslmed.aay8456.
Hinck AP, Mueller TD, Springer TA. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb Perspect Biol. 2016;8(12). https://doi.org/10.1101/cshperspect.a022103.
Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell. 2018;174(1):156–71.e16. https://doi.org/10.1016/j.cell.2018.05.027.
Robertson IB, Rifkin DB. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb Perspect Biol. 2016;8(6). https://doi.org/10.1101/cshperspect.a021907.
Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-β pathway. J Hematol Oncol. 2021;14(1):55. https://doi.org/10.1186/s13045-021-01053-x.
Metelli A, Salem M, Wallace CH, Wu BX, Li A, Li X, et al. Immunoregulatory functions and the therapeutic implications of GARP-TGF-β in inflammation and cancer. J Hematol Oncol. 2018;11(1):24. https://doi.org/10.1186/s13045-018-0570-z.
Metelli A, Wu BX, Riesenberg B, Guglietta S, Huck JD, Mills C, et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-β. Sci Transl Med. 2020;12(525). https://doi.org/10.1126/scitranslmed.aay4860.
Ma W, Qin Y, Chapuy B, Lu C. LRRC33 is a novel binding and potential regulating protein of TGF-β1 function in human acute myeloid leukemia cells. PLoS One. 2019;14(10):e0213482. https://doi.org/10.1371/journal.pone.0213482.
Purcell JW, Tanlimco SG, Hickson J, Fox M, Sho M, Durkin L, et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018;78(14):4059–72. https://doi.org/10.1158/0008-5472.can-18-0327.
Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020;10(2):232–53. https://doi.org/10.1158/2159-8290.cd-19-0644.
Cheifetz S, Hernandez H, Laiho M, ten Dijke P, Iwata KK, Massagué J. Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J Biol Chem. 1990;265(33):20533–8.
Luo K, Lodish HF. Signaling by chimeric erythropoietin-TGF-beta receptors: homodimerization of the cytoplasmic domain of the type I TGF-beta receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996;15(17):4485–96.
López-Casillas F, Wrana JL, Massagué J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73(7):1435–44. https://doi.org/10.1016/0092-8674(93)90368-z.
Wieser R, Wrana JL, Massagué J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995;14(10):2199–208. https://doi.org/10.1002/j.1460-2075.1995.tb07214.x.
Abdollah S, Macías-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997;272(44):27678–85. https://doi.org/10.1074/jbc.272.44.27678.
Heldin CH, Moustakas A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb Perspect Biol. 2016;8(8). https://doi.org/10.1101/cshperspect.a022053.
Shi X, Chen F, Yu J, Xu Y, Zhang S, Chen YG, et al. Study of interaction between Smad7 and DNA by single-molecule force spectroscopy. Biochem Biophys Res Commun. 2008;377(4):1284–7. https://doi.org/10.1016/j.bbrc.2008.10.145.
de Ceuninck van Capelle C, Spit M, Ten Dijke P. Current perspectives on inhibitory SMAD7 in health and disease. Crit Rev Biochem Mol Biol. 2020;55(6):691–715. https://doi.org/10.1080/10409238.2020.1828260.
Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–5. https://doi.org/10.1038/39369.
Humeres C, Shinde AV, Hanna A, Alex L, Hernández SC, Li R, et al. Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J Clin Invest. 2022;132(3). https://doi.org/10.1172/jci146926.
Troncone E, Marafini I, Stolfi C, Monteleone G. Transforming Growth Factor-β1/Smad7 in Intestinal Immunity, Inflammation, and Cancer. Front Immunol. 2018;9:1407. https://doi.org/10.3389/fimmu.2018.01407.
Zhang J, Cao L, Wang X, Li Q, Zhang M, Cheng C, et al. The E3 ubiquitin ligase TRIM31 plays a critical role in hypertensive nephropathy by promoting proteasomal degradation of MAP 3K7 in the TGF-β1 signaling pathway. Cell Death Differ. 2022;29(3):556–67. https://doi.org/10.1038/s41418-021-00874-0.
Fu L, Cui CP, Zhang X, Zhang L. The functions and regulation of Smurfs in cancers. Semin Cancer Biol. 2020;67(Pt 2):102–16. https://doi.org/10.1016/j.semcancer.2019.12.023.
Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J. 2005;386(Pt 3):461–70. https://doi.org/10.1042/bj20040738.
Liu S, González-Prieto R, Zhang M, Geurink PP, Kooij R, Iyengar PV, et al. Deubiquitinase Activity Profiling Identifies UCHL1 as a Candidate Oncoprotein That Promotes TGFβ-Induced Breast Cancer Metastasis. Clin Cancer Res. 2020;26(6):1460–73. https://doi.org/10.1158/1078-0432.ccr-19-1373.
Zhou F, Xie F, Jin K, Zhang Z, Clerici M, Gao R, et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017;36(11):1623–39. https://doi.org/10.15252/embj.201695372.
Zhang YE. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb Perspect Biol. 2017;9(2). https://doi.org/10.1101/cshperspect.a022129.
Ungefroren H, Witte D, Lehnert H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev Dyn. 2018;247(3):451–61. https://doi.org/10.1002/dvdy.24505.
Vardouli L, Moustakas A, Stournaras C. LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem. 2005;280(12):11448–57. https://doi.org/10.1074/jbc.M402651200.
Gunaratne A, Thai BL, Di Guglielmo GM. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor β-induced epithelial-to-mesenchymal transition. Mol Cell Biol. 2013;33(5):874–86. https://doi.org/10.1128/mcb.00837-12.
Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307(5715):1603–9. https://doi.org/10.1126/science.1105718.
Hamidi A, Song J, Thakur N, Itoh S, Marcusson A, Bergh A, et al. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci Signal. 2017;10(486). https://doi.org/10.1126/scisignal.aal4186.
Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–207. https://doi.org/10.1038/ncb1780.
Geng XQ, Ma A, He JZ, Wang L, Jia YL, Shao GY, et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-β/Smad and MAPK signaling pathways. Acta Pharmacol Sin. 2020;41(5):670–7. https://doi.org/10.1038/s41401-019-0324-7.
Luo K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017;9(1). https://doi.org/10.1101/cshperspect.a022137.
Torrealba N, Vera R, Fraile B, Martínez-Onsurbe P, Paniagua R, Royuela M. TGF-β/PI3K/AKT/mTOR/NF-kB pathway. Clinicopathological features in prostate cancer. Aging Male. 2020;23(5):801–11. https://doi.org/10.1080/13685538.2019.1597840.
Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26(17):3957–67. https://doi.org/10.1038/sj.emboj.7601818.
Jin S, Gao J, Qi Y, Hao Y, Li X, Liu Q, et al. TGF-β1 fucosylation enhances the autophagy and mitophagy via PI3K/Akt and Ras-Raf-MEK-ERK in ovarian carcinoma. Biochem Biophys Res Commun. 2020;524(4):970–6. https://doi.org/10.1016/j.bbrc.2020.02.028.
Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–39. https://doi.org/10.1038/cr.2008.328.
Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. 2020;577(7791):566–71. https://doi.org/10.1038/s41586-019-1897-5.
Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11(2):147–60. https://doi.org/10.1016/j.ccr.2006.11.023.
Xiong B, Gong LL, Zhang F, Hu MB, Yuan HY. TGF beta1 expression and angiogenesis in colorectal cancer tissue. World J Gastroenterol. 2002;8(3):496–8. https://doi.org/10.3748/wjg.v8.i3.496.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. https://doi.org/10.1038/ni.2703.
Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79(18):4557–66. https://doi.org/10.1158/0008-5472.can-18-3962.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131. https://doi.org/10.1186/s12943-021-01428-1.
Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220–7. https://doi.org/10.1016/j.it.2010.04.002.
O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67. https://doi.org/10.1038/s41571-018-0142-8.
Ginefra P, Lorusso G, Vannini N. Innate Immune Cells and Their Contribution to T-Cell-Based Immunotherapy. Int J Mol Sci. 2020;21(12). https://doi.org/10.3390/ijms21124441.
Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61. https://doi.org/10.1186/s13045-022-01282-8.
Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, et al. TGF-β transactivates EGFR and facilitates breast cancer migration and invasion through canonical Smad3 and ERK/Sp1 signaling pathways. Mol Oncol. 2018;12(3):305–21. https://doi.org/10.1002/1878-0261.12162.
Mancarella S, Krol S, Crovace A, Leporatti S, Dituri F, Frusciante M, et al. Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression. Cancers (Basel). 2019;11(10). https://doi.org/10.3390/cancers11101510.
Cantelli G, Crosas-Molist E, Georgouli M, Sanz-Moreno V. TGFΒ-induced transcription in cancer. Semin Cancer Biol. 2017;42:60–9. https://doi.org/10.1016/j.semcancer.2016.08.009.
Witte D, Zeeh F, Gädeken T, Gieseler F, Rauch BH, Settmacher U, et al. Proteinase-Activated Receptor 2 Is a Novel Regulator of TGF-β Signaling in Pancreatic Cancer. J Clin Med. 2016;5(12). https://doi.org/10.3390/jcm5120111.
Akhurst RJ, Fee F, Balmain A. Localized production of TGF-beta mRNA in tumour promoter-stimulated mouse epidermis. Nature. 1988;331(6154):363–5. https://doi.org/10.1038/331363a0.
Isufi I, Seetharam M, Zhou L, Sohal D, Opalinska J, Pahanish P, et al. Transforming growth factor-beta signaling in normal and malignant hematopoiesis. J Interf Cytokine Res. 2007;27(7):543–52. https://doi.org/10.1089/jir.2007.0009.
Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day CP, et al. TGF-β signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 2013;3(8):936–51. https://doi.org/10.1158/2159-8290.cd-12-0527.
Lian GY, Wang QM, Mak TS, Huang XR, Yu XQ, Lan HY. Inhibition of tumor invasion and metastasis by targeting TGF-β-Smad-MMP2 pathway with Asiatic acid and Naringenin. Mol Ther Oncolytics. 2021;20:277–89. https://doi.org/10.1016/j.omto.2021.01.006.
Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell. 2019;179(5):1177–90.e13. https://doi.org/10.1016/j.cell.2019.10.029.
Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. TGF-β suppresses type 2 immunity to cancer. Nature. 2020;587(7832):115–20. https://doi.org/10.1038/s41586-020-2836-1.
Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19-20):1267–84. https://doi.org/10.1101/gad.314617.118.
Schuster P, Lindner G, Thomann S, Haferkamp S, Schmidt B. Prospect of Plasmacytoid Dendritic Cells in Enhancing Anti-Tumor Immunity of Oncolytic Herpes Viruses. Cancers (Basel). 2019;11(5). https://doi.org/10.3390/cancers11050651.
McKarns SC, Schwartz RH. Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3. J Immunol. 2005;174(4):2071–83. https://doi.org/10.4049/jimmunol.174.4.2071.
Das L, Levine AD. TGF-beta inhibits IL-2 production and promotes cell cycle arrest in TCR-activated effector/memory T cells in the presence of sustained TCR signal transduction. J Immunol. 2008;180(3):1490–8. https://doi.org/10.4049/jimmunol.180.3.1490.
Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016;6(12):1366–81. https://doi.org/10.1158/2159-8290.cd-15-1347.
Mishra S, Liao W, Liu Y, Yang M, Ma C, Wu H, et al. TGF-β and Eomes control the homeostasis of CD8+ regulatory T cells. J Exp Med. 2021;218(1). https://doi.org/10.1084/jem.20200030.
Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017;9(6). https://doi.org/10.1101/cshperspect.a022236.
Tamayo E, Alvarez P, Merino R. TGFβ Superfamily Members as Regulators of B Cell Development and Function-Implications for Autoimmunity. Int J Mol Sci. 2018;19(12). https://doi.org/10.3390/ijms19123928.
Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci (Landmark Ed). 2009;14(13):4848–61. https://doi.org/10.2741/3573.
Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20(9):556–67. https://doi.org/10.1016/j.tcb.2010.06.006.
Protopsaltis NJ, Liang W, Nudleman E, Ferrara N. Interleukin-22 promotes tumor angiogenesis. Angiogenesis. 2019;22(2):311–23. https://doi.org/10.1007/s10456-018-9658-x.
Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. 2017;214(9):2695–713. https://doi.org/10.1084/jem.20160392.
Yang EY, Moses HL. Transforming growth factor beta 1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J Cell Biol. 1990;111(2):731–41. https://doi.org/10.1083/jcb.111.2.731.
Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, Habib N, et al. Thrombospondin-4 mediates TGF-β-induced angiogenesis. Oncogene. 2017;36(36):5189–98. https://doi.org/10.1038/onc.2017.140.
Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50(4):1140–51. https://doi.org/10.1002/hep.23118.
Fujio K, Komai T, Inoue M, Morita K, Okamura T, Yamamoto K. Revisiting the regulatory roles of the TGF-β family of cytokines. Autoimmun Rev. 2016;15(9):917–22. https://doi.org/10.1016/j.autrev.2016.07.007.
Biancheri P, Giuffrida P, Docena GH, MacDonald TT, Corazza GR, Di Sabatino A. The role of transforming growth factor (TGF)-β in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev. 2014;25(1):45–55. https://doi.org/10.1016/j.cytogfr.2013.11.001.
van Loon K, Yemelyanenko-Lyalenko J, Margadant C, Griffioen AW, Huijbers EJM. Role of fibrillin-2 in the control of TGF-β activation in tumor angiogenesis and connective tissue disorders. Biochim Biophys Acta Rev Cancer. 2020;1873(2):188354. https://doi.org/10.1016/j.bbcan.2020.188354.
Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. 2017;133:124–30. https://doi.org/10.1016/j.diabres.2017.08.018.
Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem. 2016;64(3):157–67. https://doi.org/10.1369/0022155415627681.
Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. https://doi.org/10.1038/nri3073.
Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, et al. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int J Mol Sci. 2018;19(5). https://doi.org/10.3390/ijms19051294.
Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44(3):450–62. https://doi.org/10.1016/j.immuni.2016.02.015.
Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84(16):5788–92. https://doi.org/10.1073/pnas.84.16.5788.
Wahl SM, McCartney-Francis N, Allen JB, Dougherty EB, Dougherty SF. Macrophage production of TGF-beta and regulation by TGF-beta. Ann N Y Acad Sci. 1990;593:188–96. https://doi.org/10.1111/j.1749-6632.1990.tb16111.x.
Simões FC, Cahill TJ, Kenyon A, Gavriouchkina D, Vieira JM, Sun X, et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat Commun. 2020;11(1):600. https://doi.org/10.1038/s41467-019-14263-2.
Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, et al. Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J Immunol. 2008;180(7):5036–44. https://doi.org/10.4049/jimmunol.180.7.5036.
Zhang Y, Alexander PB, Wang XF. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect Biol. 2017;9(4). https://doi.org/10.1101/cshperspect.a022145.
Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J Exp Med. 1987;165(1):251–6. https://doi.org/10.1084/jem.165.1.251.
Woodcock HV, Eley JD, Guillotin D, Platé M, Nanthakumar CB, Martufi M, et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat Commun. 2019;10(1):6. https://doi.org/10.1038/s41467-018-07858-8.
Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35(2):83–92. https://doi.org/10.1016/j.jdermsci.2003.12.006.
Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front Cell Dev Biol. 2020;8:260. https://doi.org/10.3389/fcell.2020.00260.
Katsuno Y, Derynck R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev Cell. 2021;56(6):726–46. https://doi.org/10.1016/j.devcel.2021.02.028.
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103(35):13180–5. https://doi.org/10.1073/pnas.0605669103.
Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–76. https://doi.org/10.1172/jci119590.
Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, et al. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol. 2002;160(3):1057–68. https://doi.org/10.1016/s0002-9440(10)64926-7.
Meng XM, Huang XR, Xiao J, Chen HY, Zhong X, Chung AC, et al. Diverse roles of TGF-β receptor II in renal fibrosis and inflammation in vivo and in vitro. J Pathol. 2012;227(2):175–88. https://doi.org/10.1002/path.3976.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. https://doi.org/10.1038/nrm3434.
Piersma B, Bank RA, Boersema M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front Med (Lausanne). 2015;2:59. https://doi.org/10.3389/fmed.2015.00059.
Hill CS. Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol. 2016;8(10). https://doi.org/10.1101/cshperspect.a022079.
Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. https://doi.org/10.1038/ncomms12260.
Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18(5):1345–56. https://doi.org/10.1093/emboj/18.5.1345.
Salazar KD, Lankford SM, Brody AR. Mesenchymal stem cells produce Wnt isoforms and TGF-beta1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L1002–11. https://doi.org/10.1152/ajplung.90347.2008.
Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G, et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β (1)-induced collagen biosynthesis. Sci Signal. 2019;12(582). https://doi.org/10.1126/scisignal.aav3048.
Si M, Wang Q, Li Y, Lin H, Luo D, Zhao W, et al. Inhibition of hyperglycolysis in mesothelial cells prevents peritoneal fibrosis. Sci Transl Med. 2019;11(495). https://doi.org/10.1126/scitranslmed.aav5341.
Vallée A, Lecarpentier Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019;9:98. https://doi.org/10.1186/s13578-019-0362-3.
Carthy JM. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J Cell Physiol. 2018;233(1):98–106. https://doi.org/10.1002/jcp.25879.
Fang S, Xu C, Zhang Y, Xue C, Yang C, Bi H, et al. Umbilical Cord-Derived Mesenchymal Stem Cell-Derived Exosomal MicroRNAs Suppress Myofibroblast Differentiation by Inhibiting the Transforming Growth Factor-β/SMAD2 Pathway During Wound Healing. Stem Cells Transl Med. 2016;5(10):1425–39. https://doi.org/10.5966/sctm.2015-0367.
Yanagihara T, Tsubouchi K, Gholiof M, Chong SG, Lipson KE, Zhou Q, et al. Connective-Tissue Growth Factor Contributes to TGF-β1-induced Lung Fibrosis. Am J Respir Cell Mol Biol. 2022;66(3):260–70. https://doi.org/10.1165/rcmb.2020-0504OC.
Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4(8):599–604. https://doi.org/10.1038/ncb826.
Allanki S, Strilic B, Scheinberger L, Onderwater YL, Marks A, Günther S, et al. Interleukin-11 signaling promotes cellular reprogramming and limits fibrotic scarring during tissue regeneration. Sci Adv. 2021;7(37):eabg6497. https://doi.org/10.1126/sciadv.abg6497.
Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552(7683):110–5. https://doi.org/10.1038/nature24676.
Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P, Mauviel A. Induction of the AP-1 members c-Jun and JunB by TGF-beta/Smad suppresses early Smad-driven gene activation. Oncogene. 2001;20(18):2205–11. https://doi.org/10.1038/sj.onc.1204347.
Wygrecka M, Zakrzewicz D, Taborski B, Didiasova M, Kwapiszewska G, Preissner KT, et al. TGF-β1 induces tissue factor expression in human lung fibroblasts in a PI3K/JNK/Akt-dependent and AP-1-dependent manner. Am J Respir Cell Mol Biol. 2012;47(5):614–27. https://doi.org/10.1165/rcmb.2012-0097OC.
Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131–6. https://doi.org/10.1038/ncb0502-e131.
Cui L, Chen SY, Lerbs T, Lee JW, Domizi P, Gordon S, et al. Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat Commun. 2020;11(1):2795. https://doi.org/10.1038/s41467-020-16466-4.
Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394(6696):909–13. https://doi.org/10.1038/29814.
Wong C, Rougier-Chapman EM, Frederick JP, Datto MB, Liberati NT, Li JM, et al. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor beta. Mol Cell Biol. 1999;19(3):1821–30. https://doi.org/10.1128/mcb.19.3.1821.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96. https://doi.org/10.1038/nrm3758.
Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964–8. https://doi.org/10.1038/nm888.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38. https://doi.org/10.1038/nrneph.2016.48.
Mariasegaram M, Tesch GH, Verhardt S, Hurst L, Lan HY, Nikolic-Paterson DJ. Lefty antagonises TGF-beta1 induced epithelial-mesenchymal transition in tubular epithelial cells. Biochem Biophys Res Commun. 2010;393(4):855–9. https://doi.org/10.1016/j.bbrc.2010.02.098.
Wang D, Dai C, Li Y, Liu Y. Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int. 2011;80(11):1159–69. https://doi.org/10.1038/ki.2011.255.
Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, et al. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol. 2004;164(4):1389–97. https://doi.org/10.1016/s0002-9440(10)63225-7.
Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27–36. https://doi.org/10.1091/mbc.12.1.27.
Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H, et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem. 2012;287(10):7026–38. https://doi.org/10.1074/jbc.M111.276311.
Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal. 2012;24(5):1031–6. https://doi.org/10.1016/j.cellsig.2011.12.024.
Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23(5):1155–65. https://doi.org/10.1038/sj.emboj.7600069.
Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int J Mol Sci. 2017;18(10). https://doi.org/10.3390/ijms18102157.
Li Z, Wang F, Zha S, Cao Q, Sheng J, Chen S. SIRT1 inhibits TGF-β-induced endothelial-mesenchymal transition in human endothelial cells with Smad4 deacetylation. J Cell Physiol. 2018;233(11):9007–14. https://doi.org/10.1002/jcp.26846.
Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, et al. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol. 2015;26(4):817–29. https://doi.org/10.1681/asn.2013101137.
Shu DY, Butcher E, Saint-Geniez M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int J Mol Sci. 2020;21(12). https://doi.org/10.3390/ijms21124271.
Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. 2017;76(5):924–34. https://doi.org/10.1136/annrheumdis-2016-210229.
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–28. https://doi.org/10.1016/s0092-8674(00)80545-0.
Kariya Y, Oyama M, Suzuki T, Kariya Y. αvβ3 Integrin induces partial EMT independent of TGF-β signaling. Commun Biol. 2021;4(1):490. https://doi.org/10.1038/s42003-021-02003-6.
Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, et al. Cryo-EM Reveals Integrin-Mediated TGF-β Activation without Release from Latent TGF-β. Cell. 2020;180(3):490–501.e16. https://doi.org/10.1016/j.cell.2019.12.030.
Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1(20):e90558. https://doi.org/10.1172/jci.insight.90558.
Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6(28). https://doi.org/10.1126/sciadv.aba1972.
Wang B, Wang Y, Chen H, Yao S, Lai X, Qiu Y, et al. Inhibition of TGFβ improves hematopoietic stem cell niche and ameliorates cancer-related anemia. Stem Cell Res Ther. 2021;12(1):65. https://doi.org/10.1186/s13287-020-02120-9.
Liang XH, Rong L, He G, He H, Lin S, Yang Y, et al. Polymorphisms of the TGF-β1 gene and the risk of acquired aplastic anemia in a Chinese population. Ann Hematol. 2017;96(3):339–44. https://doi.org/10.1007/s00277-016-2886-5.
Zhou L, McMahon C, Bhagat T, Alencar C, Yu Y, Fazzari M, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011;71(3):955–63. https://doi.org/10.1158/0008-5472.can-10-2933.
Balouchi S, Gharagozloo M, Esmaeil N, Mirmoghtadaei M, Moayedi B. Serum levels of TGFβ, IL-10, IL-17, and IL-23 cytokines in β-thalassemia major patients: the impact of silymarin therapy. Immunopharmacol Immunotoxicol. 2014;36(4):271–4. https://doi.org/10.3109/08923973.2014.926916.
Río P, Bueren JA. TGF-β: a master regulator of the bone marrow failure puzzle in Fanconi anemia. Stem Cell Investig. 2016;3:75. https://doi.org/10.21037/sci.2016.09.17.
Adewoye AH, Nolan VG, Ma Q, Baldwin C, Wyszynski DF, Farrell JJ, et al. Association of polymorphisms of IGF1R and genes in the transforming growth factor- beta /bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin Infect Dis. 2006;43(5):593–8. https://doi.org/10.1086/506356.
Blank U, Karlsson S. TGF-β signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542–50. https://doi.org/10.1182/blood-2014-12-618090.
Zermati Y, Fichelson S, Valensi F, Freyssinier JM, Rouyer-Fessard P, Cramer E, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol. 2000;28(8):885–94. https://doi.org/10.1016/s0301-472x(00)00488-4.
Cazzola M. Ineffective erythropoiesis and its treatment. Blood. 2021. https://doi.org/10.1182/blood.2021011045.
Pan D, Schomber T, Kalberer CP, Terracciano LM, Hafen K, Krenger W, et al. Normal erythropoiesis but severe polyposis and bleeding anemia in Smad4-deficient mice. Blood. 2007;110(8):3049–55. https://doi.org/10.1182/blood-2007-02-074393.
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147(5):1146–58. https://doi.org/10.1016/j.cell.2011.09.053.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321–6. https://doi.org/10.1038/nm.3706.
Bewersdorf JP, Zeidan AM. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia. 2019;33(6):1303–12. https://doi.org/10.1038/s41375-019-0448-2.
He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massagué J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell. 2006;125(5):929–41. https://doi.org/10.1016/j.cell.2006.03.045.
Bataller A, Montalban-Bravo G, Soltysiak KA, Garcia-Manero G. The role of TGFβ in hematopoiesis and myeloid disorders. Leukemia. 2019;33(5):1076–89. https://doi.org/10.1038/s41375-019-0420-1.
Gañán-Gómez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia. 2015;29(7):1458–69. https://doi.org/10.1038/leu.2015.69.
Muench DE, Ferchen K, Velu CS, Pradhan K, Chetal K, Chen X, et al. SKI controls MDS-associated chronic TGF-β signaling, aberrant splicing, and stem cell fitness. Blood. 2018;132(21):e24–34. https://doi.org/10.1182/blood-2018-06-860890.
Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112(8):3434–43. https://doi.org/10.1182/blood-2008-02-139824.
Longo F, Piolatto A, Ferrero GB, Piga A. Ineffective Erythropoiesis in β-Thalassaemia: Key Steps and Therapeutic Options by Drugs. Int J Mol Sci. 2021;22(13). https://doi.org/10.3390/ijms22137229.
Parisi S, Finelli C, Fazio A, De Stefano A, Mongiorgi S, Ratti S, et al. Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia. Int J Mol Sci. 2021;22(2). https://doi.org/10.3390/ijms22020827.
Makis A, Voskaridou E, Papassotiriou I, Hatzimichael E. Novel Therapeutic Advances in β-Thalassemia. Biology (Basel). 2021;10(6). https://doi.org/10.3390/biology10060546.
Bartlett AL, Romick-Rosendale L, Nelson A, Abdullah S, Luebbering N, Bartlett J, et al. Tryptophan metabolism is dysregulated in individuals with Fanconi anemia. Blood Adv. 2021;5(1):250–61. https://doi.org/10.1182/bloodadvances.2020002794.
Zhang H, Kozono DE, O'Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-β Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell. 2016;18(5):668–81. https://doi.org/10.1016/j.stem.2016.03.002.
Tummala H, Dokal I. TGF-β Pathway Inhibition Signals New Hope for Fanconi Anemia. Cell Stem Cell. 2016;18(5):567–8. https://doi.org/10.1016/j.stem.2016.04.008.
Wahl SM. Transforming growth factor beta (TGF-beta) in inflammation: a cause and a cure. J Clin Immunol. 1992;12(2):61–74. https://doi.org/10.1007/bf00918135.
Chen W, Wahl SM. Manipulation of TGF-beta to control autoimmune and chronic inflammatory diseases. Microbes Infect. 1999;1(15):1367–80. https://doi.org/10.1016/s1286-4579(99)00249-x.
Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008;28(4):468–76. https://doi.org/10.1016/j.immuni.2008.03.003.
Lachapelle P, Li M, Douglass J, Stewart A. Safer approaches to therapeutic modulation of TGF-β signaling for respiratory disease. Pharmacol Ther. 2018;187:98–113. https://doi.org/10.1016/j.pharmthera.2018.02.010.
Kim YH, Lee SH. TGF-β/SMAD4 mediated UCP2 downregulation contributes to Aspergillus protease-induced inflammation in primary bronchial epithelial cells. Redox Biol. 2018;18:104–13. https://doi.org/10.1016/j.redox.2018.06.011.
Chen W. A potential treatment of COVID-19 with TGF-β blockade. Int J Biol Sci. 2020;16(11):1954–5. https://doi.org/10.7150/ijbs.46891.
Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol. 2017;52(7):777–87. https://doi.org/10.1007/s00535-017-1350-1.
Igalouzene R, Hernandez-Vargas H, Benech N, Guyennon A, Bauché D, Barrachina C, et al. SMAD4 TGF-β-independent function preconditions naive CD8+ T cells to prevent severe chronic intestinal inflammation. J Clin Invest. 2022;132(8). https://doi.org/10.1172/jci151020.
Monteleone G, Pallone F, MacDonald TT. Smad7 in TGF-beta-mediated negative regulation of gut inflammation. Trends Immunol. 2004;25(10):513–7. https://doi.org/10.1016/j.it.2004.07.008.
Becker C, Fantini MC, Neurath MF. TGF-beta as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev. 2006;17(1–2):97–106. https://doi.org/10.1016/j.cytogfr.2005.09.004.
Hameedaldeen A, Liu J, Batres A, Graves GS, Graves DT. FOXO1, TGF-β regulation and wound healing. Int J Mol Sci. 2014;15(9):16257–69. https://doi.org/10.3390/ijms150916257.
O'Kane S, Ferguson MW. Transforming growth factor beta s and wound healing. Int J Biochem Cell Biol. 1997;29(1):63–78. https://doi.org/10.1016/s1357-2725(96)00120-3.
Xiaojie W, Banda J, Qi H, Chang AK, Bwalya C, Chao L, et al. Scarless wound healing: Current insights from the perspectives of TGF-β, KGF-1, and KGF-2. Cytokine Growth Factor Rev. 2022;66:26–37. https://doi.org/10.1016/j.cytogfr.2022.03.001.
Cordeiro MF. Beyond Mitomycin: TGF-beta and wound healing. Prog Retin Eye Res. 2002;21(1):75–89. https://doi.org/10.1016/s1350-9462(01)00021-0.
Li F, Bian L, Iriyama S, Jian Z, Fan B, Luo J, et al. Smad7 Ameliorates TGF-β-Mediated Skin Inflammation and Associated Wound Healing Defects but Not Susceptibility to Experimental Skin Carcinogenesis. J Invest Dermatol. 2019;139(4):940–50. https://doi.org/10.1016/j.jid.2018.10.031.
Grainger DJ. TGF-beta and atherosclerosis in man. Cardiovasc Res. 2007;74(2):213–22. https://doi.org/10.1016/j.cardiores.2007.02.022.
Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–27. https://doi.org/10.1038/cr.2008.326.
Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–98. https://doi.org/10.1056/NEJMoa055695.
Takeda N, Hara H, Fujiwara T, Kanaya T, Maemura S, Komuro I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int J Mol Sci. 2018;19(7). https://doi.org/10.3390/ijms19072125.
Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–36. https://doi.org/10.1016/j.jacc.2015.01.040.
Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2020;40(9):2195–211. https://doi.org/10.1161/atvbaha.120.314670.
Dawson A, Li Y, Li Y, Ren P, Vasquez HG, Zhang C, et al. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes (Basel). 2021;13(1). https://doi.org/10.3390/genes13010095.
Pedroza AJ, Koyano T, Trojan J, Rubin A, Palmon I, Jaatinen K, et al. Divergent effects of canonical and non-canonical TGF-β signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med. 2020;24(3):2369–83. https://doi.org/10.1111/jcmm.14921.
Goumans MJ, Ten Dijke P. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb Perspect Biol. 2018;10(2). https://doi.org/10.1101/cshperspect.a022210.
Forte A, Galderisi U, Cipollaro M, De Feo M, Della CA. Epigenetic regulation of TGF-β1 signalling in dilative aortopathy of the thoracic ascending aorta. Clin Sci (Lond). 2016;130(16):1389–405. https://doi.org/10.1042/cs20160222.
Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation. 2016;133(24):2516–28. https://doi.org/10.1161/circulationaha.116.009762.
Munjal C, Opoka AM, Osinska H, James JF, Bressan GM, Hinton RB. TGF-β mediates early angiogenesis and latent fibrosis in an Emilin1-deficient mouse model of aortic valve disease. Dis Model Mech. 2014;7(8):987–96. https://doi.org/10.1242/dmm.015255.
Rath D, Chatterjee M, Müller I, Müller K, Böckmann C, Droppa M, et al. Platelet expression of transforming growth factor beta 1 is enhanced and associated with cardiovascular prognosis in patients with acute coronary syndrome. Atherosclerosis. 2014;237(2):754–9. https://doi.org/10.1016/j.atherosclerosis.2014.10.021.
MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-β Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb Perspect Biol. 2017;9(11). https://doi.org/10.1101/cshperspect.a022269.
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–81. https://doi.org/10.1038/ng1511.
Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI, et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother. 2015;64(4):437–46. https://doi.org/10.1007/s00262-015-1653-0.
Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin Cancer Res. 2018;24(11):2493–504. https://doi.org/10.1158/1078-0432.ccr-17-3322.
Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015;125(7):2795–807. https://doi.org/10.1172/jci77958.
Moulin A, Mathieu M, Lawrence C, Bigelow R, Levine M, Hamel C, et al. Structures of a pan-specific antagonist antibody complexed to different isoforms of TGFβ reveal structural plasticity of antibody-antigen interactions. Protein Sci. 2014;23(12):1698–707. https://doi.org/10.1002/pro.2548.
Trachtman H, Fervenza FC, Gipson DS, Heering P, Jayne DR, Peters H, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79(11):1236–43. https://doi.org/10.1038/ki.2011.33.
Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P, et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int Rep. 2017;2(5):800–10. https://doi.org/10.1016/j.ekir.2017.03.011.
Song IW, Nagamani SC, Nguyen D, Grafe I, Sutton VR, Gannon FH, et al. Targeting TGF-β for treatment of osteogenesis imperfecta. J Clin Invest. 2022;132(7). https://doi.org/10.1172/jci152571.
Zhong Z, Carroll KD, Policarpio D, Osborn C, Gregory M, Bassi R, et al. Anti-transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res. 2010;16(4):1191–205. https://doi.org/10.1158/1078-0432.ccr-09-1634.
Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(4):673–80. https://doi.org/10.1007/s00280-017-3245-5.
Greco R, Qu H, Qu H, Theilhaber J, Shapiro G, Gregory R, et al. Pan-TGFβ inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. Oncoimmunology. 2020;9(1):1811605. https://doi.org/10.1080/2162402x.2020.1811605.
Varricchio L, Iancu-Rubin C, Upadhyaya B, Zingariello M, Martelli F, Verachi P, et al. TGF-β1 protein trap AVID200 beneficially affects hematopoiesis and bone marrow fibrosis in myelofibrosis. JCI. Insight. 2021;6(18). https://doi.org/10.1172/jci.insight.145651.
Rodríguez A, Yang C, Furutani E, García de Teresa B, Velázquez M, Filiatrault J, et al. Inhibition of TGFβ1 and TGFβ3 promotes hematopoiesis in Fanconi anemia. Exp Hematol. 2021;93:70–84.e4. https://doi.org/10.1016/j.exphem.2020.11.002.
Joyce CE, Saadatpour A, Ruiz-Gutierrez M, Bolukbasi OV, Jiang L, Thomas DD, et al. TGFβ signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond Syndrome. J Clin Invest. 2019;129(9):3821–6. https://doi.org/10.1172/jci125375.
Strauss J, Gatti-Mays ME, Cho BC, Hill A, Salas S, McClay E, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with human papillomavirus-associated malignancies. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-001395.
Redman JM, Friedman J, Robbins Y, Sievers C, Yang X, Lassoued W, et al. Enhanced neoepitope-specific immunity following neoadjuvant PD-L1 and TGF-b blockade in HPV-unrelated head and neck cancer. J Clin Invest. 2022. https://doi.org/10.1172/jci161400.
Tsai YT, Strauss J, Toney NJ, Jochems C, Venzon DJ, Gulley JL, et al. Immune correlates of clinical parameters in patients with HPV-associated malignancies treated with bintrafusp alfa. J Immunother Cancer. 2022;10(4). https://doi.org/10.1136/jitc-2022-004601.
Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J Thorac Oncol. 2020;15(7):1210–22. https://doi.org/10.1016/j.jtho.2020.03.003.
Yoo C, Oh DY, Choi HJ, Kudo M, Ueno M, Kondo S, et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with pretreated biliary tract cancer. J Immunother Cancer. 2020;8(1). https://doi.org/10.1136/jitc-2020-000564.
Tan B, Khattak A, Felip E, Kelly K, Rich P, Wang D, et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Esophageal Adenocarcinoma: Results from a Phase 1 Cohort. Target Oncol. 2021;16(4):435–46. https://doi.org/10.1007/s11523-021-00809-2.
Lin CC, Doi T, Muro K, Hou MM, Esaki T, Hara H, et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Patients with Esophageal Squamous Cell Carcinoma: Results from a Phase 1 Cohort in Asia. Target Oncol. 2021;16(4):447–59. https://doi.org/10.1007/s11523-021-00810-9.
Cho BC, Daste A, Ravaud A, Salas S, Isambert N, McClay E, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-000664.
Kang YK, Bang YJ, Kondo S, Chung HC, Muro K, Dussault I, et al. Safety and Tolerability of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Asian Patients with Pretreated Recurrent or Refractory Gastric Cancer. Clin Cancer Res. 2020;26(13):3202–10. https://doi.org/10.1158/1078-0432.ccr-19-3806.
Doi T, Fujiwara Y, Koyama T, Ikeda M, Helwig C, Watanabe M, et al. Phase I Study of the Bifunctional Fusion Protein Bintrafusp Alfa in Asian Patients with Advanced Solid Tumors, Including a Hepatocellular Carcinoma Safety-Assessment Cohort. Oncologist. 2020;25(9):e1292–e302. https://doi.org/10.1634/theoncologist.2020-0249.
Markham A. Luspatercept: First Approval. Drugs. 2020;80(1):85–90. https://doi.org/10.1007/s40265-019-01251-5.
Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408–14. https://doi.org/10.1038/nm.3512.
Wobus M, Mies A, Asokan N, Oelschlägel U, Möbus K, Winter S, et al. Luspatercept restores SDF-1-mediated hematopoietic support by MDS-derived mesenchymal stromal cells. Leukemia. 2021;35(10):2936–47. https://doi.org/10.1038/s41375-021-01275-5.
Kubasch AS, Fenaux P, Platzbecker U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021;5(5):1565–75. https://doi.org/10.1182/bloodadvances.2020002177.
Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N Engl J Med. 2020;382(2):140–51. https://doi.org/10.1056/NEJMoa1908892.
Garcia-Manero G, Mufti GJ, Fenaux P, Buckstein R, Santini V, Díez-Campelo M, et al. Neutrophil and platelet increases with luspatercept in lower-risk MDS: secondary endpoints from the MEDALIST trial. Blood. 2022;139(4):624–9. https://doi.org/10.1182/blood.2021012589.
Zeidan AM, Platzbecker U, Garcia-Manero G, Sekeres MA, Fenaux P, DeZern AE, et al. Longer-term benefit of luspatercept in transfusion-dependent lower-risk myelodysplastic syndromes with ring sideroblasts. Blood. 2022. https://doi.org/10.1182/blood.2022016171.
Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338–47. https://doi.org/10.1016/s1470-2045(17)30615-0.
Platzbecker U, Götze KS, Kiewe P, Germing U, Mayer K, Radsak M, et al. Long-Term Efficacy and Safety of Luspatercept for Anemia Treatment in Patients With Lower-Risk Myelodysplastic Syndromes: The Phase II PACE-MDS Study. J Clin Oncol. 2022:Jco2102476. https://doi.org/10.1200/jco.21.02476.
Komrokji RS, Platzbecker U, Fenaux P, Zeidan AM, Garcia-Manero G, Mufti GJ, et al. Luspatercept for myelodysplastic syndromes/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Leukemia. 2022;36(5):1432–5. https://doi.org/10.1038/s41375-022-01521-4.
Farrukh F, Chetram D, Al-Kali A, Foran J, Patnaik M, Badar T, et al. Real-world experience with luspatercept and predictors of response in myelodysplastic syndromes with ring sideroblasts. Am J Hematol. 2022;97(6):E210–e14. https://doi.org/10.1002/ajh.26533.
Piga A, Perrotta S, Gamberini MR, Voskaridou E, Melpignano A, Filosa A, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood. 2019;133(12):1279–89. https://doi.org/10.1182/blood-2018-10-879247.
Cappellini MD, Viprakasit V, Taher AT, Georgiev P, Kuo KHM, Coates T, et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2020;382(13):1219–31. https://doi.org/10.1056/NEJMoa1910182.
Platzbecker U, Morison JK. Luspatercept in patients with non-transfusion dependent β-thalassaemia. Lancet Haematol. 2022. https://doi.org/10.1016/s2352-3026(22)00256-3.
Taher AT, Cappellini MD, Kattamis A, Voskaridou E, Perrotta S, Piga AG, et al. Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): a phase 2, randomised, double-blind, multicentre, placebo-controlled trial. Lancet Haematol. 2022. https://doi.org/10.1016/s2352-3026(22)00208-3.
Cappellini MD, Porter J, Origa R, Forni GL, Voskaridou E, Galactéros F, et al. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study. Haematologica. 2019;104(3):477–84. https://doi.org/10.3324/haematol.2018.198887.
Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2021;384(13):1204–15. https://doi.org/10.1056/NEJMoa2024277.
Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J. 2022. https://doi.org/10.1183/13993003.01347-2022.
Komrokji R, Garcia-Manero G, Ades L, Prebet T, Steensma DP, Jurcic JG, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol. 2018;5(2):e63–72. https://doi.org/10.1016/s2352-3026(18)30002-4.
Raftopoulos H, Laadem A, Hesketh PJ, Goldschmidt J, Gabrail N, Osborne C, et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: results from two phase 2 studies. Support Care Cancer. 2016;24(4):1517–25. https://doi.org/10.1007/s00520-015-2929-9.
Gallo-Oller G, Vollmann-Zwerenz A, Meléndez B, Rey JA, Hau P, Dotor J, et al. P144, a Transforming Growth Factor beta inhibitor peptide, generates antitumoral effects and modifies SMAD7 and SKI levels in human glioblastoma cell lines. Cancer Lett. 2016;381(1):67–75. https://doi.org/10.1016/j.canlet.2016.07.029.
Zubeldia IG, Bleau AM, Redrado M, Serrano D, Agliano A, Gil-Puig C, et al. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1-targeting peptides P17 and P144. Exp Cell Res. 2013;319(3):12–22. https://doi.org/10.1016/j.yexcr.2012.11.004.
Díaz-Valdés N, Basagoiti M, Dotor J, Aranda F, Monreal I, Riezu-Boj JI, et al. Induction of monocyte chemoattractant protein-1 and interleukin-10 by TGFbeta1 in melanoma enhances tumor infiltration and immunosuppression. Cancer Res. 2011;71(3):812–21. https://doi.org/10.1158/0008-5472.can-10-2698.
Hanafy NAN, Fabregat I, Leporatti S, Kemary ME. Encapsulating TGF-β1 Inhibitory Peptides P17 and P144 as a Promising Strategy to Facilitate Their Dissolution and to Improve Their Functionalization. Pharmaceutics. 2020;12(5). https://doi.org/10.3390/pharmaceutics12050421.
Cruz-Morande S, Dotor J, San-Julian M. P144 a Transforming Growth Factor Beta Inhibitor Peptide, Generates Antifibrogenic Effects in a Radiotherapy Induced Fibrosis Model. Curr Oncol. 2022;29(4):2650–61. https://doi.org/10.3390/curroncol29040217.
Li D, Zhang J, Yuan S, Wang C, Chang J, Tong Y, et al. TGF-β1 peptide-based inhibitor P144 ameliorates renal fibrosis after ischemia-reperfusion injury by modulating alternatively activated macrophages. Cell Prolif. 2022:e13299. https://doi.org/10.1111/cpr.13299.
Recalde S, Zarranz-Ventura J, Fernández-Robredo P, García-Gómez PJ, Salinas-Alamán A, Borrás-Cuesta F, et al. Transforming growth factor-β inhibition decreases diode laser-induced choroidal neovascularization development in rats: P17 and P144 peptides. Invest Ophthalmol Vis Sci. 2011;52(10):7090–7. https://doi.org/10.1167/iovs.11-7300.
Qiu SS, Dotor J, Hontanilla B. Effect of P144® (Anti-TGF-β) in an "In Vivo" Human Hypertrophic Scar Model in Nude Mice. PLoS One. 2015;10(12):e0144489. https://doi.org/10.1371/journal.pone.0144489.
Arce C, Rodríguez-Rovira I, De Rycke K, Durán K, Campuzano V, Fabregat I, et al. Anti-TGFβ (Transforming Growth Factor β) Therapy With Betaglycan-Derived P144 Peptide Gene Delivery Prevents the Formation of Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Arterioscler Thromb Vasc Biol. 2021;41(9):e440–e52. https://doi.org/10.1161/atvbaha.121.316496.
Hermida N, López B, González A, Dotor J, Lasarte JJ, Sarobe P, et al. A synthetic peptide from transforming growth factor-beta1 type III receptor prevents myocardial fibrosis in spontaneously hypertensive rats. Cardiovasc Res. 2009;81(3):601–9. https://doi.org/10.1093/cvr/cvn315.
Baltanás A, Miguel-Carrasco JL, San José G, Cebrián C, Moreno MU, Dotor J, et al. A synthetic peptide from transforming growth factor-β1 type III receptor inhibits NADPH oxidase and prevents oxidative stress in the kidney of spontaneously hypertensive rats. Antioxid Redox Signal. 2013;19(14):1607–18. https://doi.org/10.1089/ars.2012.4653.
Jung SY, Yug JS, Clarke JM, Bauer TM, Keedy VL, Hwang S, et al. Population pharmacokinetics of vactosertib, a new TGF-β receptor type Ι inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2020;85(1):173–83. https://doi.org/10.1007/s00280-019-03979-z.
Song KM, Chung DY, Choi MJ, Ghatak K, Minh NN, Limanjaya A, et al. Vactosertib, a Novel, Orally Bioavailable Activin Receptor-Like Kinase 5 Inhibitor, Promotes Regression of Fibrotic Plaques in a Rat Model of Peyronie's Disease. World J Mens Health. 2020;38(4):552–63. https://doi.org/10.5534/wjmh.190071.
Park SA, Kim MJ, Park SY, Kim JS, Lee SJ, Woo HA, et al. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol Life Sci. 2015;72(10):2023–39. https://doi.org/10.1007/s00018-014-1798-6.
Choi J, Park J, Cho I, Sheen Y. Co-treatment with vactosertib, a novel, orally bioavailable activin receptor-like kinase 5 inhibitor, suppresses radiotherapy-induced epithelial-to-mesenchymal transition, cancer cell stemness, and lung metastasis of breast cancer. Radiol Oncol. 2022;56(2):185–97. https://doi.org/10.2478/raon-2022-0012.
Binabaj MM, Asgharzadeh F, Avan A, Rahmani F, Soleimani A, Parizadeh MR, et al. EW-7197 prevents ulcerative colitis-associated fibrosis and inflammation. J Cell Physiol. 2019;234(7):11654–61. https://doi.org/10.1002/jcp.27823.
Soleimani A, Asgharzadeh F, Rahmani F, Avan A, Mehraban S, Fakhraei M, et al. Novel oral transforming growth factor-β signaling inhibitor potently inhibits postsurgical adhesion band formation. J Cell Physiol. 2020;235(2):1349–57. https://doi.org/10.1002/jcp.29053.
Tsauo J, Song HY, Choi EY, Kim DK, Kim KY, Park JH, et al. EW-7197, an oral transforming growth factor β type I receptor kinase inhibitor, for preventing peritoneal adhesion formation in a rat model. Surgery. 2018;164(5):1100–8. https://doi.org/10.1016/j.surg.2018.07.005.
Naka K, Ishihara K, Jomen Y, Jin CH, Kim DH, Gu YK, et al. Novel oral transforming growth factor-β signaling inhibitor EW-7197 eradicates CML-initiating cells. Cancer Sci. 2016;107(2):140–8. https://doi.org/10.1111/cas.12849.
Son JY, Park SY, Kim SJ, Lee SJ, Park SA, Kim MJ, et al. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol Cancer Ther. 2014;13(7):1704–16. https://doi.org/10.1158/1535-7163.mct-13-0903.
Bueno L, de Alwis DP, Pitou C, Yingling J, Lahn M, Glatt S, et al. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur J Cancer. 2008;44(1):142–50. https://doi.org/10.1016/j.ejca.2007.10.008.
Peterson JM, Jay JW, Wang Y, Joglar AA, Prasai A, Palackic A, et al. Galunisertib Exerts Antifibrotic Effects on TGF-β-Induced Fibroproliferative Dermal Fibroblasts. Int J Mol Sci. 2022;23(12). https://doi.org/10.3390/ijms23126689.
Mao D, Mi J, Pan X, Zhao G, Rui Y. Galunisertib attenuates progression of trauma-induced heterotopic ossification via blockage of Smad2/3 signaling in mice. Eur J Pharmacol. 2022;928:175109. https://doi.org/10.1016/j.ejphar.2022.175109.
Luangmonkong T, Suriguga S, Bigaeva E, Boersema M, Oosterhuis D, de Jong KP, et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br J Pharmacol. 2017;174(18):3107–17. https://doi.org/10.1111/bph.13945.
Hammad S, Cavalcanti E, Werle J, Caruso ML, Dropmann A, Ignazzi A, et al. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch Toxicol. 2018;92(7):2297–309. https://doi.org/10.1007/s00204-018-2231-y.
Rani B, Malfettone A, Dituri F, Soukupova J, Lupo L, Mancarella S, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018;9(3):373. https://doi.org/10.1038/s41419-018-0384-5.
Harding JJ, Do RK, Yaqubie A, Cleverly A, Zhao Y, Gueorguieva I, et al. Phase 1b study of galunisertib and ramucirumab in patients with advanced hepatocellular carcinoma. Cancer Med. 2021;10(9):3059–67. https://doi.org/10.1002/cam4.3880.
Reiss KA, Wattenberg MM, Damjanov N, Prechtel Dunphy E, Jacobs-Small M, Lubas MJ, et al. A Pilot Study of Galunisertib plus Stereotactic Body Radiotherapy in Patients with Advanced Hepatocellular Carcinoma. Mol Cancer Ther. 2021;20(2):389–97. https://doi.org/10.1158/1535-7163.mct-20-0632.
Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. 2019;10(7):e00056. https://doi.org/10.14309/ctg.0000000000000056.
Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208–14. https://doi.org/10.1038/s41416-018-0246-z.
Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9(3). https://doi.org/10.1136/jitc-2020-002068.
Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin Cancer Res. 2017;23(3):804–13. https://doi.org/10.1158/1078-0432.ccr-16-1743.
Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology. 2016;18(8):1146–56. https://doi.org/10.1093/neuonc/now009.
Santini V, Valcárcel D, Platzbecker U, Komrokji RS, Cleverly AL, Lahn MM, et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin Cancer Res. 2019;25(23):6976–85. https://doi.org/10.1158/1078-0432.ccr-19-1338.
Zhang P, Qin C, Liu N, Zhou X, Chu X, Lv F, et al. The programmed site-specific delivery of LY3200882 and PD-L1 siRNA boosts immunotherapy for triple-negative breast cancer by remodeling tumor microenvironment. Biomaterials. 2022;284:121518. https://doi.org/10.1016/j.biomaterials.2022.121518.
Li Z, Xu W, Yang J, Wang J, Wang J, Zhu G, et al. A Tumor Microenvironments-Adapted Polypeptide Hydrogel/Nanogel Composite Boosts Antitumor Molecularly Targeted Inhibition and Immunoactivation. Adv Mater. 2022;34(21):e2200449. https://doi.org/10.1002/adma.202200449.
Yap TA, Vieito M, Baldini C, Sepúlveda-Sánchez JM, Kondo S, Simonelli M, et al. First-In-Human Phase I Study of a Next-Generation, Oral, TGFβ Receptor 1 Inhibitor, LY3200882, in Patients with Advanced Cancer. Clin Cancer Res. 2021;27(24):6666–76. https://doi.org/10.1158/1078-0432.ccr-21-1504.
Miller KM, Friedman CF. Bifunctional Blockade: A Novel Immunotherapy Approach for Cervical Cancer. Clin Cancer Res. 2022. https://doi.org/10.1158/1078-0432.ccr-22-1779.
Feng J, Tang D, Wang J, Zhou Q, Peng J, Lou H, et al. SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-β, for recurrent or metastatic cervical cancer: a clinical expansion cohort of phase 1 study. Clin Cancer Res. 2022. https://doi.org/10.1158/1078-0432.ccr-22-0346.
Cheng B, Ding K, Chen P, Ji J, Luo T, Guo X, et al. Anti-PD-L1/TGF-βR fusion protein (SHR-1701) overcomes disrupted lymphocyte recovery-induced resistance to PD-1/PD-L1 inhibitors in lung cancer. Cancer Commun (Lond). 2022;42(1):17–36. https://doi.org/10.1002/cac2.12244.
Nolte M, Margadant C. Controlling Immunity and Inflammation through Integrin-Dependent Regulation of TGF-β. Trends Cell Biol. 2020;30(1):49–59. https://doi.org/10.1016/j.tcb.2019.10.002.
Brown NF, Marshall JF. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers (Basel). 2019;11(9). https://doi.org/10.3390/cancers11091221.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. https://doi.org/10.1038/nrc2748.
Maurer GD, Tritschler I, Adams B, Tabatabai G, Wick W, Stupp R, et al. Cilengitide modulates attachment and viability of human glioma cells, but not sensitivity to irradiation or temozolomide in vitro. Neuro-Oncology. 2009;11(6):747–56. https://doi.org/10.1215/15228517-2009-012.
Lomonaco SL, Finniss S, Xiang C, Lee HK, Jiang W, Lemke N, et al. Cilengitide induces autophagy-mediated cell death in glioma cells. Neuro-Oncology. 2011;13(8):857–65. https://doi.org/10.1093/neuonc/nor073.
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1100–8. https://doi.org/10.1016/s1470-2045(14)70379-1.
Stupp R, Picard M, Weller M. Does cilengitide deserve another chance?-Authors' reply. Lancet Oncol. 2014;15(13):e585–e86. https://doi.org/10.1016/s1470-2045(14)71121-0.
Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncology. 2015;17(5):708–17. https://doi.org/10.1093/neuonc/nou356.
Khasraw M, Lee A, McCowatt S, Kerestes Z, Buyse ME, Back M, et al. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J Neuro-Oncol. 2016;128(1):163–71. https://doi.org/10.1007/s11060-016-2094-0.
Jeong J, Kim J. Cyclic RGD Pentapeptide Cilengitide Enhances Efficacy of Gefitinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition and Invasion in Human Non-Small Cell Lung Cancer Cells. Front Pharmacol. 2021;12:639095. https://doi.org/10.3389/fphar.2021.639095.
Jeong J, Kim J. Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. Int J Mol Sci. 2022;23(7). https://doi.org/10.3390/ijms23073423.
Massabeau C, Khalifa J, Filleron T, Modesto A, Bigay-Gamé L, Plat G, et al. Continuous Infusion of Cilengitide Plus Chemoradiotherapy for Patients With Stage III Non-Small-cell Lung Cancer: A Phase I Study. Clin Lung Cancer. 2018;19(3):e277–e85. https://doi.org/10.1016/j.cllc.2017.11.002.
Vansteenkiste J, Barlesi F, Waller CF, Bennouna J, Gridelli C, Goekkurt E, et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: results of an open-label, randomized, controlled phase II study (CERTO). Ann Oncol. 2015;26(8):1734–40. https://doi.org/10.1093/annonc/mdv219.
Ruffini F, Graziani G, Levati L, Tentori L, D'Atri S, Lacal PM. Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of αvβ5 integrin. Int J Cancer. 2015;136(6):E545–58. https://doi.org/10.1002/ijc.29252.
Kim KB, Prieto V, Joseph RW, Diwan AH, Gallick GE, Papadopoulos NE, et al. A randomized phase II study of cilengitide (EMD 121974) in patients with metastatic melanoma. Melanoma Res. 2012;22(4):294–301. https://doi.org/10.1097/CMR.0b013e32835312e4.
Stojanović N, Dekanić A, Paradžik M, Majhen D, Ferenčak K, Ruščić J, et al. Differential Effects of Integrin αv Knockdown and Cilengitide on Sensitization of Triple-Negative Breast Cancer and Melanoma Cells to Microtubule Poisons. Mol Pharmacol. 2018;94(6):1334–51. https://doi.org/10.1124/mol.118.113027.
Pan X, Yi M, Liu C, Jin Y, Liu B, Hu G, et al. Cilengitide, an αvβ3-integrin inhibitor, enhances the efficacy of anti-programmed cell death-1 therapy in a murine melanoma model. Bioengineered. 2022;13(2):4557–72. https://doi.org/10.1080/21655979.2022.2029236.
Chen G, Wang Y, Zhao X, Xie XZ, Zhao JG, Deng T, et al. A positive feedback loop between Periostin and TGFβ1 induces and maintains the stemness of hepatocellular carcinoma cells via AP-2α activation. J Exp Clin Cancer Res. 2021;40(1):218. https://doi.org/10.1186/s13046-021-02011-8.
Haddad T, Qin R, Lupu R, Satele D, Eadens M, Goetz MP, et al. A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(6):1221–7. https://doi.org/10.1007/s00280-017-3322-9.
Ritzenthaler JD, Zhang M, Torres-Gonzalez E, Roman J. The Integrin Inhibitor Cilengitide and Bleomycin-Induced Pulmonary Fibrosis : Cilengitide and Bleomycin-Induced Pulmonary Fibrosis. Lung. 2020;198(6):947–55. https://doi.org/10.1007/s00408-020-00400-y.
Patsenker E, Popov Y, Stickel F, Schneider V, Ledermann M, Sägesser H, et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50(5):1501–11. https://doi.org/10.1002/hep.23144.
Patsenker E, Popov Y, Wiesner M, Goodman SL, Schuppan D. Pharmacological inhibition of the vitronectin receptor abrogates PDGF-BB-induced hepatic stellate cell migration and activation in vitro. J Hepatol. 2007;46(5):878–87. https://doi.org/10.1016/j.jhep.2006.11.011.
Vallières L. Trabedersen, a TGFbeta2-specific antisense oligonucleotide for the treatment of malignant gliomas and other tumors overexpressing TGFbeta2. IDrugs. 2009;12(7):445–53.
Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011;102(6):1193–200. https://doi.org/10.1111/j.1349-7006.2011.01917.x.
Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro-Oncology. 2011;13(1):132–42. https://doi.org/10.1093/neuonc/noq142.
Wosikowski K, Hasenbach K, Allais L, Fant P, Truchot N, Krampert M, et al. Preclinical profile of ISTH0036, a potent and selective antisense oligonucleotide targeting transforming growth factor beta 2 (TGF-β2) for the treatment of ophthalmic diseases. 2015.
Gupta A, Kafetzis KN, Tagalakis AD, Yu-Wai-Man C. RNA therapeutics in ophthalmology - translation to clinical trials. Exp Eye Res. 2021;205:108482. https://doi.org/10.1016/j.exer.2021.108482.
Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. 2006;24(29):4721–30. https://doi.org/10.1200/jco.2005.05.5335.
Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51(16):2321–9. https://doi.org/10.1016/j.ejca.2015.07.035.
Rijavec E, Biello F, Genova C, Barletta G, Maggioni C, Dal Bello MG, et al. Belagenpumatucel-L for the treatment of non-small cell lung cancer. Expert Opin Biol Ther. 2015;15(9):1371–9. https://doi.org/10.1517/14712598.2015.1073709.
Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, Lazar M, et al. Phase I trial of "bi-shRNAi (furin)/GMCSF DNA/autologous tumor cell" vaccine (FANG) in advanced cancer. Mol Ther. 2012;20(3):679–86. https://doi.org/10.1038/mt.2011.269.
Ghisoli M, Barve M, Mennel R, Lenarsky C, Horvath S, Wallraven G, et al. Three-year Follow up of GMCSF/bi-shRNA (furin) DNA-transfected Autologous Tumor Immunotherapy (Vigil) in Metastatic Advanced Ewing's Sarcoma. Mol Ther. 2016;24(8):1478–83. https://doi.org/10.1038/mt.2016.86.
Oh J, Barve M, Matthews CM, Koon EC, Heffernan TP, Fine B, et al. Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol. 2016;143(3):504–10. https://doi.org/10.1016/j.ygyno.2016.09.018.
Rocconi RP, Grosen EA, Ghamande SA, Chan JK, Barve MA, Oh J, et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020;21(12):1661–72. https://doi.org/10.1016/s1470-2045(20)30533-7.
Rocconi RP, Monk BJ, Walter A, Herzog TJ, Galanis E, Manning L, et al. Gemogenovatucel-T (Vigil) immunotherapy demonstrates clinical benefit in homologous recombination proficient (HRP) ovarian cancer. Gynecol Oncol. 2021;161(3):676–80. https://doi.org/10.1016/j.ygyno.2021.03.009.
Walter A, Rocconi RP, Monk BJ, Herzog TJ, Manning L, Bognar E, et al. Gemogenovatucel-T (Vigil) maintenance immunotherapy: 3-year survival benefit in homologous recombination proficient (HRP) ovarian cancer. Gynecol Oncol. 2021;163(3):459–64. https://doi.org/10.1016/j.ygyno.2021.10.004.
Rocconi RP, Stanbery L, Madeira da Silva L, Barrington RA, Aaron P, Manning L, et al. Long-Term Follow-Up of Gemogenovatucel-T (Vigil) Survival and Molecular Signals of Immune Response in Recurrent Ovarian Cancer. Vaccines (Basel). 2021;9(8). https://doi.org/10.3390/vaccines9080894.
Rocconi RP, Stevens EE, Bottsford-Miller JN, Ghamande SA, Elder J, DeMars LL, et al. Proof of principle study of sequential combination atezolizumab and Vigil in relapsed ovarian cancer. Cancer Gene Ther. 2022;29(3–4):369–82. https://doi.org/10.1038/s41417-021-00317-5.
Sliheet E, Robinson M, Morand S, Choucair K, Willoughby D, Stanbery L, et al. Network based analysis identifies TP53m-BRCA1/2wt-homologous recombination proficient (HRP) population with enhanced susceptibility to Vigil immunotherapy. Cancer Gene Ther. 2022;29(7):993–1000. https://doi.org/10.1038/s41417-021-00400-x.
Tauriello DVF, Sancho E, Batlle E. Overcoming TGFβ-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25–44. https://doi.org/10.1038/s41568-021-00413-6.
Carter NJ. Pirfenidone: in idiopathic pulmonary fibrosis. Drugs. 2011;71(13):1721–32. https://doi.org/10.2165/11207710-000000000-00000.
Acknowledgments
We appreciate BioRender.com for providing us with materials for making figures.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (No. 2022SCU12057), the China Postdoctoral Science Foundation (No. 2022 M712259), and the Fundamental Research Funds for the Central Universities (No. 2021SCU12017).
Author information
Authors and Affiliations
Contributions
Ye Zhang and Huashan Shi offered the main direction of this manuscript. Yan Tie, Fan Tang and Dandan Peng drafted the manuscript, illustrated the figures and made tables for the manuscript. Yan Tie, Fan Tang and Dandan Peng contributed equally to this work. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tie, Y., Tang, F., Peng, D. et al. TGF-beta signal transduction: biology, function and therapy for diseases. Mol Biomed 3, 45 (2022). https://doi.org/10.1186/s43556-022-00109-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-022-00109-9