
Abstract
Background
Greenhouse cultivation is mainly a monoculture system, which leads to severe soil degradation, with significant changes to the structure of the soil microbial community. The present study aimed to determine the effects of fertilizer and waterlogging (WL) on the microbial community of soil that was continuously monocropped with cucumber in a greenhouse for 3 years (2016–2019). Biolog EcoPlates™ and high-throughput sequencing were used to investigate the abundance, structure, and diversity of the soil microbial community.
Results
Compared with the initial total soluble salt content (8.65 g/kg), total soluble salt content after WL significantly decreased to 0.597 g/kg. Compared to control (CK) soil, the abundance of fungal species in the chemical fertilizer plus waterlogging (CFWL)-treated soil increased by 40.5%, while bacterial species abundance in the organic fertilizer plus waterlogging (OFWL)-treated soil increased by 10.7%. Furthermore, the relative abundance of Arthrobacter, Geobacter, and Bacillus was higher in OFWL-treated soil than CFWL-treated soil (p < 0.01).
Conclusions
The application of CFWL to greenhouse soil under continuous monoculture produces fungal-dominated soil, whereas OFWL application alone produces bacterial-dominated soil.
Graphical Abstract

Similar content being viewed by others
Introduction
Many studies examining land-use change have focused on the structure of soil microbial communities [1, 2]. Soil microorganisms can promote the decomposition of organic matter and the transformation of nutrients, thus playing functional roles in maintaining soil nutrient cycling, soil organic fertility, and soil ecosystem sustainability [3, 4]. Greenhouse cultivation is primarily a continuous monoculture system [5, 6], which leads to severe soil degradation, with significant declines in both the yield and quality of crops associated to changes to the structure of the soil microbial community [7,8,9]. Several studies have explored the use of agricultural inputs (such as quicklime [10], potassium [11], irrigation [12], and organic fertilizers [13]) to restore monoculture soil by regulating the structure of soil microbiota.
Fertilizers drive changes to the diversity of microbes in soil [14]. Many studies have demonstrated that fertilizers enhance the fertility and biodiversity of soil [15]. Chemical fertilizer application increases microbial biomass in cropping systems over the long term [16]. Chemical fertilizer plus waterlogging application increases the richness and diversity of the microbial community, whereas when each are applied individually fungal-dominated soil forms[4]. Organic manure plus fertilizer application with balanced N, P, and K content promotes the biomass, activity, and diversity of soil microbes, thus enhancing crop growth and production [4, 17]. Different fertilizer application strategies have distinct effects on the physicochemical and biological properties of soil, which, in turn, alter the soil microbial community [18, 19]. Previous reports showed that fertilizer application can enrich soil nutrients, but also cause secondary salinization [20], which is the main factor reducing the diversity of soil bacteria and fungi [21]. In contrast, irrigation is a major factor affecting salt leaching efficiency and water availability. In particular, water migration is the main driver for salt ion transport through the soil [22]. Irrigation influences the performance of soil microbial communities in terms of microbial growth, biomass, and composition [23,24,25]. Furthermore, Bastida et al. [12] reported that bacterial diversity and plant productivity are more sensitive to the quantity of water than its quality. Thus, waterlogging contributes to the increased availability of nutrients in the soil. For instance, a thin water layer promotes the abundance of fungi and bacteria in continuously cropped soil, thus improving the quality of degraded soil [26, 27]. However, it remains unclear whether the combined application of waterlogging with either synthetic or organic fertilization affects the structuring of bacterial and fungal communities in greenhouse soil subjected to continuous monoculture.
Greenhouses are suitable environments for crop growth and can protect crops from weather damage, which allows productivity goals to be met in terms of yield and crop quality [28,29,30]. Greenhouse cultivation occupies an important position in global agricultural production; however, obstacles associated with continuous cropping have seriously impacted its continued development [5]. Recent studies showed that the structure and function of soil microbial communities are sensitive indicators of soil quality [31]. Therefore, this study aimed to compare the effects of waterlogging alone and combined with different fertilizers on the abundance, structure, and diversity of bacterial and fungal communities in soil subject to continuous monoculture. The results are expected to provide useful information to support the remediation of greenhouse cultivation soil in monoculture systems.
Materials and methods
Treatments and soil sampling
The soil used in the present study was obtained from the Jiangsu Coastal Area Institute of Agricultural Sciences (33°35′N, 120°15′E). The soil was continuously cropped with cucumber from 2016 to 2019 (pH 8.19) in a greenhouse. In 2019, four treatments were applied to the soil in plots in the greenhouse after harvesting cucumber: control (CK, no fertilizer or waterlogging), waterlogging (WL, with a thin 5 cm water layer), chemical fertilizer (conventional rate of the total amount of chemical fertilizer was 750 kg·ha−1) plus waterlogging (CFWL), and organic fertilizer (conventional rate of the total amount of organic fertilizer was 1500 kg·ha−1) plus waterlogging (OFWL). The N, P2O5, and K2O levels in the chemical fertilizer were all 15% (Zhejiang Huidoli Fertilizer Technology Co., Ltd., Quzhou, China). In comparison, the N, P, and K levels in the organic fertilizer were all 5%, and the organic matter content was 45% (Shanxi Zhongnonghua Biotechnology Co., Ltd., Shanxi, China). At the end of the cucumber harvest, all plots were cleaned. The solid soluble fertilizers were dissolved in the fertilizer tank, and were then spread manually on the soil in the plots. The treatments were arranged in a completely randomized block design, which was repeated in triplicate. Fertilizers were applied to CFWL and OFWL treatments in this study in the amounts routinely used in the field. After two months of treatment, soil samples were taken from the surface layer (0–20 cm) at random sites, and were pooled together. A subsample was air-dried and passed through a 60-mesh filter to remove large debris, stones, and stubble, and was subsequently used to determine soluble salt content. A second subsample was placed in a sterile bag, packed in ice, and was returned to the laboratory within 12 h, where it was immediately frozen at –80 °C for the subsequent analysis of microbial parameters.
Total soluble salt analysis
Total soluble salt (TSS) content was determined according to Merkle and Dunkle with some modifications [32]. First, water-soluble salts were extracted from a 1:5 mixture of soil and distilled water (w/v). The sample mixture was oscillated for 10 min, after which it was filtered. The filtered water was treated with 15% H2O2 several times, and was allowed to digest. Then, the remaining material was placed in an oven at 105 °C until it reached a constant weight. The TSS content of the white crystalline powder was calculated using the following formula:
where m1 is the weight of the white crystalline powder (g), and m2 is the weight of the soil sample (kg).
Biolog EcoPlate™ analysis
Metabolic functional diversity induced by the different treatments was analyzed using Biolog EcoPlate™ [33]. Each 96-well EcoPlate™ contained 31 different sole carbon sources, with three groups in parallel and three negative controls. Fresh soil (10.0 g) was added to a triangular flask, after which 100 mL sterilized saline (0.85% NaCl, w/v) was added to the flask. The flask was then sealed with a sterile cotton stopper. The sample was shaken well (30 °C, 150 rpm, 20 min), and was then centrifuged at 500 g for 5 min. From the collected supernatant, an aliquot of 5 mL was mixed with 45 mL sterilized saline. Each well was filled with 150 μL of the diluted supernatant, and was incubated at room temperature (25 °C) for seven days in the dark. The absorbance of the sample was measured at 590 nm and 750 nm (Thermo Fisher Scientific, Waltham, MA, USA).
As different microbial species utilize different carbon sources [34], the carbon sources in the EcoPlate™ were classified into six biochemical categories: carbohydrates, amino acids, carboxylic acids, polymers, phenolic compounds, and amines [34, 35]. The depth of chroma directly reflected the ability of the microbial community to utilize the carbon source, and was measured using average well-color development (AWCD) according to the following formula:
where Ci is the optical density (OD) value at 590 nm minus that at 750 nm from triplicate wells, R is the OD value of the control well (no carbon source), and n is the total number of reactive wells.
Data were collected at 24, 48, 72, 96, 120, 144, and 168 h to calculate microbial diversity. The data collected at 96 h were also used to assess Shannon’s diversity (H), McIntosh’s equitability (U), and Simpson’s diversity (D) indices for species richness, dominance, and evenness in the microbial communities, respectively [17]. The three metabolic functional diversity indices were calculated using these three formulae:
where Pi is the average corrected absorbance of the ith well divided by the sum of the absorbance of all wells in a plate, and ni is the (Ci–R) value of the ith well.
Molecular analysis
DNA extraction of the soil samples was performed using the FastDNA SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s instructions. Genomic DNA degradation and contamination were monitored on 1% agarose gel and via spectrophotometry. The V3–V4 region of bacterial 16S rDNA genes was amplified using the primers 338F (5′-ACT CCT ACG GGA GGC AGC AG-3′) and 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′). The ITS region of fungal rDNA genes was amplified using the primers ITS1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) and ITS2R (5′-GCT GCG TTC TTC ATC GAT GC-3′). PCRs were performed in triplicate using an ABI GeneAmp® 9700 PCR thermocycler (Applied Biosystems, Foster City, CA, USA). Amplicons were sequenced by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) using the Illumina MiSeq platform (Illumina, San Diego, CA, USA). The raw 16S rRNA gene and ITS gene sequencing reads were demultiplexed and were then quality-filtered by fastp version 0.20.0 [36]. The reads were then merged with FLASH version 1.2.7 [37] using the following criteria: (i) 300 bp reads were truncated at any site receiving an average quality score of < 20 over a 50 bp sliding window, and any truncated reads shorter than 50 bp were discarded, reads containing ambiguous characters were also discarded. (ii) Only overlapping sequences longer than 10 bp were assembled according to their overlapping sequence; the maximum mismatch ratio of overlapping regions was 0.2; reads that could not be assembled were discarded. (iii) Samples were distinguished according to the barcode and primers, and sequence direction was adjusted; exact barcode matching was required, with only two nucleotide mismatches allowed in primer matching. Qualified sequences were clustered into operational taxonomic units (OTUs) at 97% similarity using the UPARSE software version 7.1 [38, 39]. Based on the OTU information, the richness and evenness of microbial species were evaluated using rank abundance curves. Venn diagrams were constructed to compare the shared and unique species among the treatments. Circos maps and heat maps were used to determine the structure of the bacterial and fungal communities at the phylum and genus levels, respectively.
Statistical analysis
All assays were performed in triplicate. The results are presented as means and standard deviations (SD) of three independent experiments, which were calculated using Microsoft Excel and SPSS Statistics v. 17.0 software (IBM China Company Ltd., Beijing, China). One-way ANOVA and Duncan’s multiple range tests were performed with SPSS software version 22 (SPSS Inc., New York, USA) to evaluate differences in soil microbial diversity and gene copies among soil samples. In all tests, a p value < 0.05 was considered to be statistically significant. Correlations among carbon sources and between species were analyzed via principal component analysis (PCA) using SPSS. For Biolog data analysis, AWCD values were calculated from daily optical density readings for the three groups in parallel. Means (n = 3) of AWCD values were used to determine the ability of soil microbes to utilize different carbon sources among the four treatments, which reduced dimensionality by SPSS used for PCA.
Results
Soluble salt content
The initial soil TSS content was 8.65 g/kg (pH 8.19). In contrast, on day 60 of the soil treatments, TSS content significantly decreased (Table 1, p < 0.05). Compared to that in CK soil, the greatest reductions in TSS were detected in WL- and OFWL-treated soils, in which the mean TSS values were reduced to 0.597 g/kg and 0.599 g/kg, respectively (p < 0.05). Overall, OFWL and WL treatments induced a similar reduction in soil TSS content.
Dynamic features of carbon source metabolism
Biolog EcoPlate™ assays showed that the degree of change in AWCD was correlated with incubation time, with AWCD increasing in a similar manner in all soil groups. Over the initial 72 h, the AWCD values of all soil groups was relatively low, after which it increased and stabilized at 168 h (Fig. 1). CFWL-treated soil had the highest AWCD at 168 h (p < 0.05), indicating that the microbial community in this soil had the highest rate of substrate utilization.

Changes in average well color development values for soil microbial community. Differences between means marked by different letter (a–d) are significant within four soil groups (p < 0.05; Duncan's test)
PCA was conducted to determine the relative utilization of the six types of carbon sources in the soil groups (Fig. 2). PC1 accounted for 88.12% of overall variance, and the utilization of the six carbon source types was positively associated with it. The utilization of carbohydrates, amines, and phenolic compounds was positively associated with PC2. In contrast the utilization of carboxylic acids, amino acids, and polymers was negatively associated with PC2. The relative utilization was almost similar for all soil groups in the first three days (Fig. 2). After this period, the utilization of carboxylic acids, amino acids, and polymers was dominant the microbiota of OFWL- and WL-treated soils, whereas that of carbohydrates, amines, and phenolic compounds was dominant the microbiota of CK and CFWL-treated soils. Utilization rates of the six types of carbon sources differed significantly at different incubation times (p < 0.01), whereas those of carbohydrates, amines, and phenolic compounds differed significantly across treatments (p < 0.01; Table 2). However, the utilization of amino acids, carboxylic acids, and polymers did not differ significantly among the four soil groups (p > 0.05).

Principal component analysis of six groups of carbon sources by soil microbial community. For different treatments, number 1–7 represent the ability of the soil microbes to utilize different carbon sources from day 1 to 7
Diversity and evenness of microbial communities
Three diversity indices were calculated using the Biolog EcoPlate™ data to investigate the ecological characteristics of the soil microbial communities (Fig. 3). Shannon’s diversity (H), McIntosh’s equitability (U), and Simpson’s diversity (D) indices of the soil groups had the same trend at 96 h, with an overall order of CFWL > OFWL > WL > CK. H and U indices differed significantly among the soil groups (p < 0.05), whereas the D indices were not significantly different between CFWL- and OFWL-treated soils (p > 0.05). Thus, CFWL treatment had the highest microbial diversity.

Ecological diversity of soil microbial community in different treatments at 96 h. Differences between means marked by different letter (a–d) are significant within four soil groups (p < 0.05)
Species abundance of microbial communities
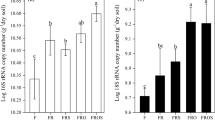
Rank abundance curves were used to elucidate the abundance and evenness of the bacterial and fungal community, which were reflected by the length of the abscissa (rank) and the curve trend, respectively (Fig. 4). Generally, bacterial OTUs had a similar curve trend in the four soil groups (Fig. 4A). However, the fungal rank abundance curve of CK soil exhibited a steep gradient (Fig. 4B), suggesting lower evenness of fungal species in CK soil compared to the soils of the other treatments. Moreover, the relative abundance of bacterial OTUs was the highest in the OFWL-treated soil (2172), followed by CFWL-treated (2161), WL-treated (2094), and CK soils (1951). In comparison, the relative abundance of fungal OTUs was the highest in the CFWL-treated soil (444), followed by OFWL-treated (402), WL-treated (353), and CK soils (316). These results indicated that fertilizer application with waterlogging increased the abundance of bacterial and fungal species in soil.

Bacterial (A) and fungal (B) Rank–Abundance curves in different treated soils
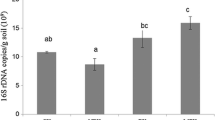
Amplicon sequencing generated 194,302 and 285,352 raw reads for bacteria and fungi in the soil, respectively. A total of 2705 bacterial OTUs were identified (Fig. 5A; Table 3), of which 1263 (46.7%) were shared among the four soil groups. A total of 720 fungal OTUs were identified, of which 146 fungal OTUs (20.3%) were shared among the four soil groups, indicating that fertilization changed the structure of the fungal community (Fig. 5B; Table 3). Furthermore, OFWL-treated soil (2172) had the highest species abundance of bacteria, whereas CFWL-treated soil (444) had the highest species abundance of fungi (p < 0.05). Compared to that in the CK soil, the bacterial species abundance in OFWL-treated soil increased by 10.7%, whereas fungal species abundance in CFWL-treated soil increased by 40.5%.

Venn diagrams of bacterial (A) and fungal (B) OTU richness in different treated soils. Differences between means marked by different letter (a–d) are significant within four soil groups (p < 0.05)
Composition of microbial communities
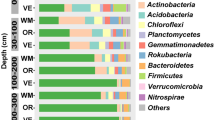
The relative abundances of bacterial and fungal phyla among the soil groups was compared (Fig. 6). The relative abundance was represented as the proportion of 16 s rRNA and ITS gene reads out of the total number of reads. The top six phyla in the soil bacterial communities were Proteobacteria (33.2–26.7%), Firmicutes (20.9–13.5%), Chloroflexi (19.3–11.9%), Actinobacteria (18.6–12.1%), Bacteroidetes (15.2–9.4%), and Acidobacteria (4.8–3.2%) (Fig. 6A). These results supported those in the bacterial community heatmap analysis at the phylum level (Fig. 7A), in which the soil microbial community in all treatments was dominated by Proteobacteria, Chloroflexi, Bacteroidetes, Firmicutes, Actinobacteria, and Acidobacteria. Proteobacteria was the dominant phylum in the soil bacterial communities, and had the highest relative abundance in CFWL-treated soil (33.2%) (p < 0.05). Firmicutes had the highest relative abundance in WL-treated soil (20.9%), whereas Chloroflexi had the highest relative abundance in OFWL-treated soil (19.3%) (p < 0.05). The highest relative abundance of Actinobacteria occurred in the CK-treated soil (18.6%) (Fig. 6A; p < 0.05). Ascomycota was the dominant phylum in all soil fungal communities (93.4–90.8%) (Figs. 6B, 7B). The relative abundances of certain fungal phyla increased in different soil groups, such as Ascomycota in the OFWL-treated soil, Basidiomycota in the WL-treated soil, and Chytridiomycota in the CFWL-treated soil (p < 0.05) (Fig. 6B).

Abundance of bacterial (A) and fungal (B) community at phyla level. Superscripts (a–d) differ significantly (p < 0.05) among different treated soils

Bacterial (A) and fungal (B) community composition analysed at the phyla level. The color of the square indicates the relative abundance of the phyla, ranging from high (red) to low (blue) (* indicates p ≤ 0.05, ** p ≤ 0.01)
A heat map was constructed to illustrate significant changes in genera among the soil groups (Fig. 8). For bacteria, the relative abundance of Lentimicrobium, Anaerolinea, Methylotenera, Geoalkalibacter, and Symbiobacterium was higher in the treated soils than CK soil (p < 0.01) (Fig. 8A). The relative abundance of Arthrobacter, Geobacter, and Bacillus was higher in the OFWL-treated soil compared to CFWL-treated soil (p < 0.01). Regarding fungi (Fig. 8B), the relative abundance of Cladosporium was the highest in CFWL-treated soil. The genus Cutaneotrichosporon appeared to be more abundant in treated soils than CK soil, while the opposite was observed for the genus Chromelosporium (p < 0.01). The relative abundance of Spizellomyces and Nectria was higher in CK and CFWL-treated soils than in WL- and OFWL-treated soils (p < 0.01).

Bacterial (A) and fungal (B) community composition analysed at the genus level. The color of the square indicates the relative abundance of the genus, ranging from high (red) to low (blue) (* indicates p ≤ 0.05, ** p ≤ 0.01)
Differences in the species composition of the soil microbial communities were analyzed by PCA (Fig. 9). CFWL and OFWL groups were clustered together at the species level for bacteria, indicating that the distribution of bacterial species was similar in these two treated soils. At the species level for fungi, WL and OFWL groups were located in close proximity, indicating a similar distribution of fungal species in WL- and OFWL-treated soils.

Principal component analysis of bacterial (A) and fungal (B) species level in each treated soil
Discussion
The present study showed that greenhouse cultivation soil subjected to continuous cucumber cropping had typical characteristics of saline soil (TSS 8.65 g/kg). Compared to the soil at the start, the TSS content in soil declined after applying WL or different fertilizers in combination with WL, especially in WL and OFWL treatments (p < 0.01). This result might be attributed to salt partly migrating to the deeper soil layers following persistent irrigation in continuously cropped soil. The highest TSS content was recorded in CK soil. Zhang et al. [40] reported that the diversity of microbes decreased with increasing soil salinity, with high salinity reducing microbial biomass [41]. Our result also verified that the relative abundance of microbial communities in CK soil was significantly lower than that in the soil under the other treatments (p < 0.05) (Figs. 1, 4). Examination of the pH in treated soils showed that, compared to CK soil, the pH range in the soils under the other treatments was narrow, spanning < 0.6 pH units (see Additional file 1: Table S1). Wang et al. [42] recently reported that, at the microscale level, pH (spanning < 1 pH unit) might not be the main factor determining the biogeographical distribution of soil microbial communities.
We used Biolog EcoPlate™ to assess the metabolic diversity of continuously cropped soil under different remediation treatments. AWCD values denoted the consumption of carbon sources by the microbial communities, and were used to indicate soil microbial activity [33].We found that CFWL-treated soil had the highest AWCD values (p < 0.05), indicating that the microbial community had the greatest ability to utilize carbon. The main carbon sources utilized by the microbial communities in CFWL-treated and CK soils were carbohydrates, amines, and phenolic compounds. In contrast, the main carbon sources utilized by the microbial communities in OFWL- and WL-treated soils were mainly carboxylic acids, amino acids, and polymers. The four soil groups displayed similar trends for the Shannon (p < 0.05), McIntosh (p < 0.05), and Simpson indices to those obtained for AWCD. Thus, the highest diversity of the microbial community was found in soil treated with CFWL, followed by OFWL, WL, and CK treatments.
Furthermore, the present study applied high-throughput sequencing technology to derive meaningful information about the relationship between soil microbial diversity and soil function. We showed that soil bacterial biomass was more sensitive to the OFWL treatment than to the other treatments. The PCA results revealed similar bacterial community structures in OFWL- and CFWL-treated soils. Ai et al. [43] reported that Proteobacteria are effective rhizosphere and root colonizers, which was supported by our findings showing that the relative abundance of Proteobacteria was the highest in soils under all treatments, especially CFWL treated soil. Additionally, the relative abundance of Firmicutes was the highest in WL-treated soil, which was consistent with the findings of Sanchez et al. [44], who showed that Firmicutes was prevalent in the microbial communities of irrigated soil, regardless of the origin of water. Khonde et al. [45] also found that Proteobacteria and Firmicutes are able to endure environmental stresses in waterlogged soil. The present study showed that the relative abundance of phylum Chloroflexi was high in OFWL-treated soil. This phylum is large, and contains potential phototroph bacteria. It is widely distributed across various plant species, and is affected by temperature and moisture [46]. Species belonging to this phylum play an integral role in the biogeochemical chlorine cycle [47, 48]. The presence of Dehalococcoides-like Chloroflexi has been previously investigated in soils with different vegetative cover [48]. In the present study, the phylum Actinobacteria had high relative abundance in CK soil. Sall et al. [49] found that salinity reduced bacterial biomass, but increased the biomass of Actinobacteria. Bao et al. [50] found that the relative abundance of Actinobacteria increased with decreasing soil fertility. Actinobacteria are able to degrade plant residues, especially in less fertile soils [50, 51]. Furthermore, we found that the relative abundance of Lentimicrobium, Anaerolinea, Methylotenera, and Symbiobacterium was higher in the treated soils than in CK soil (p < 0.01). Wang et al. [52] reported that Lentimicrobium is required for the continuous removal of high concentrations of nitrate. Denitrifying microbial communities in the soil of agricultural soil ecosystems also respond to waterlogging [42]. Gschwend et al. [53] reported that Anaerolinea responded positively to waterlogging, with limited oxygen availability being identified as the main constraining factor of microbial communities in waterlogged soil. Methylotenera is reportedly the key bacterial genus involved in the degradation of cellular water pollutants by microbes [54]. A previous study showed that the abundance of Symbiobacterium, a genus of bacteria commensal with Bacillales, increased at the final stage of composting when organic matter was thoroughly degraded, irrespective of composting conditions [55]. We found that the abundance of Arthrobacter, Geobacter, and Bacillus was higher in OFWL-treated soil compared to CFWL-treated soil. Arthrobacter can oxidize manganese, which is difficult to remove from the environment [56]. Li and Zhou [57] reported that Geobacter plays a key role in regulating the emissions and biogeochemical cycling of soil-derived greenhouse gases. Finally, Bacillus is an important source of antibiotics, enzymes, and probiotics [58]. Our results showed that the application of fertilizer or waterlogging could select beneficial bacterial organisms in monoculture soil, especially under the OFWL treatment (Additional file 2).
The highest species abundance of fungi was detected in CFWL-treated soil (p < 0.05). PCA showed that the structure of the fungal community was similar in WL- and OFWL-treated soils. Soil fungal communities were mainly classified into five taxonomic phyla, with Ascomycota being the most dominant phylum in all soils. The relative abundance of Ascomycota, Basidiomycota, and Chytridiomycota was the highest in OFWL-, WL-, and CFWL-treated soil, respectively (p < 0.05). Ascomycota taxa might be better at colonizing a wide range of environments compared to other taxa. This group can utilize a higher number of resources than other taxa [59]. Basidiomycota is a group of fungi that has highly diverse ecological strategies, including ectomycorrhizas, plant pathogens, and saprotrophic fungi [60, 61]. Chytridiomycota species can survive variation in osmotic potential that may occur during the wetting and drying phases of soils [62]. Our results showed that the genus Cutaneotrichosporon had a higher abundance in treated soils than CK soil. The opposite trend was obtained for the genus Chromelosporium (p < 0.01). The genus Cutaneotrichosporon can convert lignocellulose into microbial lipids [63]. Lizarazo-Medina et al. [64] reported that the highest abundance of Chromelosporium was found in leaves and roots under greenhouse conditions. Furthermore, the highest relative abundance of Cladosporium was detected in CFWL-treated soil in the present study (p < 0.01). Iturrieta-González et al. [65] reported that Cladosporium includes fungi that are pathogenic to plants, animals, and humans. We also found that the abundance of the genera Nectria and Spizellomyces, which are reportedly plant pathogenic fungi [66, 67], was higher in CK and CFWL-treated soils compared to WL- and OFWL-treated soils (p < 0.01). These results concurred other recent studies demonstrating that the application of chemical fertilizer results in the excess growth of pathogenic fungi [68], whereas the application of organic fertilizer reduces their abundance [69]. However, the associations among soil fertility, crop productivity, and microbial diversity require further investigation (Additional file 3).
Conclusions
In conclusion, this study demonstrated that the application of fertilizers or waterlogging treatment can increase the richness, diversity, and abundance of bacterial and fungal communities in greenhouse soil subjected to continuous monoculture. The CFWL treatment induced the highest abundance of fungal soil species, resulting in the excessive growth of pathogenic fungi. However, the OFWL treatment induced the highest abundance of bacterial soil species. Fungi have a competitive advantage over many bacteria because of their ability to adapt to changes in nutrient input; thus, the microbial community of CFWL-treated soil had the greatest ability to utilize carbon. Overall, coupling fertilizers (especially organic fertilizer) with short-term waterlogging could facilitate the development of healthy soil microbiota (bacterial-dominated) in greenhouse cultivation soil under monoculture systems. However, the mechanism underlying the different microbial patterns following waterlogging with either synthetic or organic fertilization treatment must be elucidated.
Availability of data and materials
The data sets used or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CK:
-
Control
- WL:
-
Waterlogging
- CFWL:
-
Chemical fertilizer plus waterlogging
- OFWL:
-
Organic fertilizer plus waterlogging
- AWCD:
-
Average well color development
- PCA:
-
Principal component analysis
References
Kaiser K, Wemheuer B, Korolkow V, Wemheuer F, Nacke H, Schöning I, Schrumpf M, Daniel R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci Rep. 2016;6:33696.
Millard P, Singh BK. Does grassland vegetation drive soil microbial diversity? Nutr Cycl Agroecosys. 2010;88(2):147–58.
Zornoza R, Acosta JA, Bastida F, Domínguez SG, Toledo DM, Faz A. Identification of sensitive indicators to assess the interrelationship between soil quality, management practices and human health. Soil. 2015;1(1):173–85.
Tao R, Hu B, Chu G. Impacts of organic fertilization with a drip irrigation system on bacterial and fungal communities in cotton field. Agric Syst. 2020;182: 102820.
Li DP, Wu ZJ, Liang CH, Chen LJ. Characteristics and regulation of greenhouse soil environment. Chin J Ecol. 2004;23:192–7.
Zhang XY, Tian YQ, Liu J, Gao LH. Changes of soil biological characters beneath greenhouse cucumber under different cultivation systems. Chinese J Appl Ecol. 2009;20(4):829.
Aparicio V, Costa JL. Soil quality indicators under continuous cropping systems in the Argentinean Pampas. Soil Tillage Res. 2007;96(1–2):155–65.
Zhang M, He Y, Zhou W, Ai L, Xie Y. Effects of continuous cropping of codonopsis tangshen on rhizospheric soil bacterial community as determined by pyrosequencing. Diversity. 2021;13(7):317.
Wang S, Cheng J, Li T, Liao YC. Response of soil fungal communities to continuous cropping of flue-cured tobacco. Sci Rep. 2020;10(1):19911.
You CH, Jiang LF, Xi FH, Wang WW, Li MJ, Xu ZB, Gu L, Wang FJ, Zhang ZY. Comparative evaluation of different types of soil conditioners with respect to their ability to remediate consecutive tobacco monoculture soil. Int J Agric Biol. 2015;17(5):969–75.
Yang YH, Chen DM, Ji Y, Wen HJ, Wang HB, Duan YQ, You CH, Guo XK, He HB, Lin WX. Effects of potassium application on functional diversities of microbes in rhizospheric soil of continuous cropped tobacco. Allelopathy J. 2011;27(2):185–92.
Bastida F, Torres IF, Romero-Trigueros C, Baldrian P, Větrovský T, Bayona JM, Alarcón JJ, Hernández T, García C, Nicolás E. Combined effects of reduced irrigation and water quality on the soil microbial community of a citrus orchard under semiarid conditions. Soil Biol Biochem. 2017;104:226–37.
Pang G, Cai F, Li RX, Zhao Z, Li R, Gu XL, Shen QR, Chen W. Trichoderma enriched organic fertilizer can mitigate microbiome degeneration of monocropped soil to maintain better plant growth. Plant Soil. 2017;416(1–2):181–92.
O’Donnell AG, Seasman M, Macrae A, Waite I, Davies J. Plants and fertilisers as drivers of change in microbial community structure and function in soils. Plant Soil. 2001;232:135–45.
Mäder P, Fliessbach A, Dubois D, Gunst L, Fried P, Niggli U. Soil fertility and biodiversity in organic farming. Science. 2002;296:1694–7.
Geisseler D, Scow KM. Long-term effects of mineral fertilizers on soil microorganisms – A review. Soil Biol Biochem. 2014;75:54–63.
Zhong WH, Gu T, Wei W, Zhang B, Lin XG, Huang QR, Shen WS. The effects of mineral fertilizer and organic manure on soil microbial community and diversity. Plant Soil. 2010;326(1–2):511–22.
Ai C, Liang G, Sun J, Wang X, Zhou W. Responses of extracellular enzyme activities and microbial community in both the rhizosphere and bulk soil to long-term fertilization practices in a fluvo-aquic soil. Geoderma. 2012;173:330–8.
Gomiero T, Pimentel D, Paoletti MG. Environmental impact of different agricultural management practices: conventional vs organic agriculture. Crit Rev Plant Sci. 2011;30(1–2):95–124.
Fu HD, Zhang GX, Zhang F, Sun ZP, Geng GM, Li TL. Effects of continuous tomato monoculture on soil microbial properties and enzyme activities in a solar greenhouse. Sustainability. 2017;9(2):317.
Gao YH, Lu XH, Gao RJ, Miao ZQ, Li SD. Cultural practices impact soil microbial communities and eliminate negative effects of cucumber monoculture. Conference of Chinese Society of Plant Pathology. 2019. https://doi.org/10.26914/c.cnkihy.2019.020562.
Sun JN, Yang RY, Zhu JJ, Pan YH, Yang M, Zhang ZH. Can the increase of irrigation frequency improve the rate of water and salt migration in biochar-amended saline soil? J Soils Sediments. 2019;19(12):4021–30.
Fierer N, Schimel J, Holden P. Influence of drying-rewetting frequency on soil bacterial community structure. Microb Ecol. 2003;45:63–71.
Meisner A, Baath E, Rousk E. Microbial growth responses upon rewetting soil dried for four days or one year. Soil Biol Biochem. 2013;66:188–92.
Placella S, Brodie EL, Firestone MK. Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc Natl Acad SCI U S A. 2012;109(27):10931–6.
Matin NH, Jalali M. The effect of waterlogging on electrochemical properties and soluble nutrients in paddy soils. Paddy Water Environ. 2016;15(2):443–55.
Zhao BM, Gu YF, Zhao J, Xu YF. Effect of different water flooded methods on soil of old orchard and growth of replanted peach seedling. Southeast Hortic. 2015;44:1–5.
Syed AM, Hachem C. Review of construction; geometry; heating, ventilation, and air-conditioning; and indoor climate requirements of agricultural greenhouses. J Biosyst Eng. 2019;44(1):18–27.
Kristin B, Gustaf B, Ove J, Mikaela G, Klara L, Jenny K. Greenhouse production contributes to pesticide occurrences in Swedish streams. Sci Total Environ. 2020;809: 152215.
Yano A, Cossu M. Energy sustainable greenhouse crop cultivation using photovoltaic technologies. Renew Sust Energ Rev. 2019;109:116–37.
Khan S, Hesham EL, Qiao M, Rehman S, He JZ. Effects of Cd and Pb on soil microbial community structure and activities. Environ Sci Pollut Res. 2010;17(2):288–96.
Merkle FG, Dunkle EC. Soluble salt content of greenhouse soils as a diagnostic aid. Amer Soc Agronomy. 1944;36:10–9.
Garland JL, Mills AL. Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl Environ Microbiol. 1991;57:2351–9.
Choi KH, Dobbs FC. Comparison of two kinds of BIOLOG microplates (GN and ECO) in their ability to distinguish among aquatic microbial communities. J Microbiol Methods. 1999;36:203–13.
Yu B, Chen ZY, Lu XX, Huang YT, Zhou Y, Zhang Q, Wang D, Li JY. Effects on soil microbial community after exposure to neonicotinoid insecticides thiamethoxam and dinotefuran. Sci Total Environ. 2020;725: 138328.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8.
Stackebrandt E, Goebel BM. Taxonomic Note: A Place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol. 1994;44(4):846–9.
Zhang KP, Shi Y, Cui XQ, Yue P, Li KH, Liu XJ, Tripathi BM, Chu HY. Salinity is a key determinant for soil microbial communities in a desert ecosystem. Msystems. 2019;4(1):e00225-e318.
Barin M, Aliasgharzad N, Olsson PA, Rasouli-Sadaghiani MH. Salinity-induced differences in soil microbial communities around the hypersaline Lake Urmia. Soil Res. 2015;53(5):494.
Wang Y, Uchida Y, Shimomura Y, Akiyama H, Hayatsu M. Responses of denitrifying bacterial communities to short-term waterlogging of soils. Sci rep. 2017;7:803.
Ai C, Liang G, Sun J, Wang X, He P, Zhou W, He X. Reduced dependence of rhizosphere microbiome on plant-derived carbon in 32-year long-term inorganic and organic fertilized soils. Soil Biol Biochem. 2015;80:70–8.
Sanchez O, Ferrera I, Garrido L, Gomez-Ramos MD, Fernandez-Alba AR, Mas J. Prevalence of potentially thermophilic microorganisms in biofilms from greenhouse-enclosed drip irrigation systems. Arch Microbiol. 2014;196(3):219–26.
Khonde N, Maurya DM, Chamyal LS, Archana G. Microbial activity and culturable bacterial diversity in sediments of the great rann of kachchh, western India. Pedosphere. 2014;24(1):45–55.
Hanada S. The Phylum Chloroflexi, the family Chloroflexaceae, and the related phototrophic families Oscillochloridaceae and Roseiflexaceae. Berlin: Springer Berlin Heidelberg; 2014. p. 515–532.
Krzmarzick MJ, Crary BB, Harding JJ, Oyerinde OO, Leri AC, Myneni SCB, Novak PJ. Natural niche for organohalide-respiring chloroflexi. Appl Environ Microbiol. 2012;78(2):393.
Reina RG, Leri AC, Myneni SCB. Cl K-edge X-ray spectroscopic investigation of enzymatic formation of organochlorines in weathering plant material. Environ Sci Technol. 2004;38(3):783–9.
Sall SN, Ndour NYB, Diedhiou-Sall S, Dick R, Chottee JL. Microbial response to salinity stress in a tropical sandy soil amended with native shrub residues or inorganic fertilizer. J Environ Manage. 2015;161:30–7.
Bao YY, Dolfing J, Guo ZY, Chen MW, Wu M, Li ZP, Lin XG, Feng YZ. Important ecophysiological roles of non-dominant Actinobacteria in plant residue decomposition, especially in less fertile soils. Microbiome. 2021;9(1):84.
Liu YR, Delgado-Baquerizo M, Wang JT, Hu HW, Yang ZM, He JZ. New insights into the role of microbial community composition in driving soil respiration rates. Soil Biol Biochem. 2018;118:35–41.
Wang HS, Chen N, Feng CP, Deng Y, Gao Y. Research on efficient denitrification system based on banana peel waste in sequencing batch reactors: Performance, microbial behavior and dissolved organic matter evolution. Chemosphere. 2020;253: 126693.
Gschwend F, Aregger K, Gramlich A, Walter T, Widmer F. Periodic waterlogging consistently shapes agricultural soil microbiomes by promoting specific taxa. Appl soil ecol. 2020;155:103623.
Yang H, Yang XN, Zhang GZ, Wang BS, Zhang X, Li J. Key bacteria for the microbial degradation of pollutants in cellar water. Huan Jing Ke Xue. 2018;39(10):4766–77.
Nakasaki K, Hirai H, Mimoto H, Quyen TNM, Koyama M, Takeda K. Succession of microbial community during vigorous organic matter degradation in the primary fermentation stage of food waste composting. Sci Total Environ. 2019;671:1237–44.
Wan WJ, Xue ZJ, Zhang ZW, He DL. Manganese oxidation characteristics and oxidation mechanism of a manganese-oxidizing bacterium Arthrobacter sp. HW-16. Huan Jing Ke Xue. 2017;38(5):2036–43.
Li T, Zhou QX. The key role of Geobacter in regulating emissions and biogeochemical cycling of soil-derived greenhouse gases. Environ Pollut. 2020;266: 115135.
Nair GR, Raja SS. Climate and soil properties influence species diversity of soil bacillus community in India. Microbiol Insights. 2018;11:117863611881036.
Egidi E, Delgado-Baquerizo M, Plett JM, Wang JT, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat Commun. 2019;10:2369.
Kjoller R, Rosendahl S. Cultivated and fallow fields harbor distinct communities of Basidiomycota. Fungal Ecol. 2014;9:43–51.
Lim YW, Kim BK, Kim C, Jung HS, Kim BS, Lee JH, Chun J. Assessment of soil fungal communities using pyrosequencing. J Microbiol. 2010;48:284–9.
Gleason FH, Midgley DJ, Letcher PM, Mcgee PA. Can soil Chytridiomycota survive and grow in different osmotic potentials? Mycol Res. 2006;110:869–87.
Wang LY, Wang DM, Zhang ZL, Cheng S, Liu BB, Wang CY, Li RG, Guo SX. Comparative glucose and xylose coutilization efficiencies of Soil-Isolated yeast strains identify Cutaneotrichosporon dermatis as a potential producer of lipid. ACS Omega. 2020;5(37):23596–603.
Lizarazo-Medina PX, Mendoza-Salazar MM, Gutiérrez-Gallo AI. Diversity of endophytic mycobiota in Cattleya percivaliana and Cattleya trianaei growing under greenhouse conditions. Actu Biol. 2015;37(102):307–18.
Iturrieta-González I, Garcia D, Gene J. Novel species of Cladosporium from environmental sources in Spain. MycoKeys. 2021;77:1–25.
Etebu E, Osborn AM. Molecular assays reveal the presence and diversity of genes encoding pea footrot pathogenicity determinants in Nectria haematococca and in agricultural soils. J Microbiol. 2010;106(5):1629–39.
Chen SC, Yu HJ, Zhou XG, Wu FZ. Cucumber (Cucumis sativus L.) seedling rhizosphere Trichoderma and Fusarium spp. communities altered by vanillic acid. Front Microbiol. 2018;9:2195.
Nie SA, Lei XM, Zhao LX, Brookes PC, Wang F, Chen CR, Yang WH, Xing SH. Fungal communities and functions response to long-term fertilization in paddy soils. Appl Soil Ecol. 2018;130:251–8.
Walters DR, Bingham IJ. Influence of nutrition on disease development caused by fungal pathogens: implications for plant disease control. Ann Appl Biol. 2007;151(18):307–24.
Acknowledgements
Not applicable.
Funding
This work was supported by the Innovation and Promotion Project of Jiangsu Forestry Science and Technology (grant number LYKJ [2020] 07).
Author information
Authors and Affiliations
Contributions
Conceptualization, methodology, writing—review and editing, LND; methodology, data curation, formal analysis, LYF and KXZ; methodology, resources, GNF, YXS and HSL; writing editing, LZ and CQL; resources, funding acquisition, HSL. All the authors approved the version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors listed have read the complete manuscript and have approved submission of the paper.
Competing interests
The authors declare no conflict of interest related to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. The pH in soil in different treatments of the 60th day.
Additional file 2.
The data of bacterial OUT in different treated soils.
Additional file 3.
The data of fungal OUT in different treated soils.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deng, L., Fu, L., Zhang, K. et al. Effects of fertilizer and waterlogging on the diversity and functioning of the microbial community in greenhouse cultivation soil. Chem. Biol. Technol. Agric. 9, 31 (2022). https://doi.org/10.1186/s40538-022-00298-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-022-00298-z