
Abstract
Tau (Tubulin associated unit) protein is a major hallmark of Alzheimer’s disease (AD) and tauopathies. Tau is predominantly an axonal protein with a crucial role in the stabilization and dynamics of the microtubules. Since the discovery of Tau protein in 1975, research efforts were concentrated on the pathophysiological role of Tau protein in the context of the microtubules. Although, for more than three decades, different localizations of Tau protein have been discovered e.g., in the nuclear compartments. Discovery of the role of Tau protein in various cellular compartments especially in the nucleus opens up a new fold of complexity in tauopathies. Data from cellular models, animal models, and the human brain indicate that nuclear Tau is crucial for genome stability and to cope with cellular distress. Moreover, it’s nature of nuclear translocation, its interactions with the nuclear DNA/RNA and proteins suggest it could play multiple roles in the nucleus. To comprehend Tau pathophysiology and efficient Tau-based therapies, there is an urgent need to understand whole repertoire of Tau species (nuclear and cytoplasmic) and their functional relevance. To complete the map of Tau repertoire, understanding of various species of Tau in the nucleus and cytoplasm, identification if specific transcripts of Tau, isoforms and post-translational modifications could foretell Tau’s localizations and functions, and how they are modified in neurodegenerative diseases like AD, is urgently required. In this review, we explore the nuclear face of Tau protein, its nuclear localizations and functions and its linkage with Alzheimer’s disease.
Similar content being viewed by others

Background
Tau protein is a member of microtubule-associated proteins (MAPs) family that are crucial modulators of microtubule dynamics in the cell [1, 2]. In the brain, Tau is a major regulator of neuronal microtubules assembly and stabilization required for morphogenesis and axonal transport. It is also expressed in glial cells [3] and in non-neural cells, such as fibroblasts and lymphocytes [4, 5].
Tau protein is mainly localized in axons in the mature neurons, but other cellular localizations of Tau protein has also been reported e.g., in the nucleus (nucleolus) [6], mitochondria [7], at the plasma membrane [8], soma [9], dendrites [10], synapses [11] and extracellular vesicles [12]. This vast localization pattern of Tau protein suggests that in addition to its main function as a regulator of microtubule dynamics, it has diverse pathophysiological roles in the cell [6, 13].
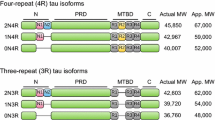
Tau, a natively unfolded protein, largely soluble and exhibits a very less tendency for aggregation [14].Tau protein is encoded by MAPT, comprising of 16 exons located on 17q21 chromosome location [15]. In the human brain, Tau has six main isoforms which are generated by alternative splicing of exons 2, 3, and 10 [1, 16]. Tau isoforms vary depending on the number of N-terminal 29-residue inserts and presence or absence of microtubule binding domain (R2) [16]. Tau isoforms vary in terms of expression of R domains (R1, R2, R3, and R4) consisting of 31–32 distinct and similar amino acid motifs. Isoforms that express the R2 domain are known as 4R while those lacking the R2 are designated as 3R. Primarily, Tau isoforms are denoted as 0N3R, 0N4R, 1N3R, 1N4R, 2N3R, 2N4R, however they are also known by the residue number or the clone name [1, 17].
Since the discovery of Tau protein, research efforts were concentrated on it’s role in the context of microtubules, even though, multiple localizations of Tau protein have been reported [18]. Tau is highly abundant in neuronal axons [19], but under various pathophysiological conditions, it can also be found in the soma [9], the dendrites [10], and the nucleus [10, 20].
Subcellular fractionation of murine brain tissue has been performed to identify distinct localization patterns for Tau isoforms [21]. The isoform 0 N is enriched in cell bodies and axons with a slight staining in nuclei and dendrites. The isoform 1 N is predominantly detected in the soluble nuclear fraction (neuronal nuclei). It is also detectable in cell bodies and dendrites, but not in axons. The isoform 2 N is highly expressed in cell bodies and axons, with a detectable expression in dendrites and a very low signal in nuclei [21]. This data indicate significant differences in the expression of Tau isoforms in the murine brain that likely reflect different neuronal functions. However, the localization pattern of nuclear Tau is different between human and murine cell-types [22, 23], which might be the one reason that transgenic mice models do not recapitulate full spectrum of AD pathology. Interestingly, these murine Tau isoforms do not contain any nuclear localization signal (NLS) for their transport from the cytoplasmic to the nuclear compartment [21]. Furthermore, Tau exists in different conformations in the nucleus, and specific epitopes are accessible to different antibodies in a compartment-dependent manner [23]. Understanding of complete spectrum of nuclear Tau species and their mode of nucleocytoplasmic transport may shed light on the nuclear face of Tau under pathophysiological conditions.
The multiple localizations of Tau protein suggest condition- and subcellular microenvironment-dependent interactions of Tau protein [24, 25] with different subcellular compartments e.g., nucleus. This suggests that Tau is a multifunctional protein and its role in pathophysiology of the neurons needs to be regularly reviewed in the light of emerging discoveries. Here, we discuss the nuclear face of Tau at different levels starting from nuclear lamina down to the nucleolus, and how its nuclear face contributes to neurodegeneration in Alzheimer’s disease.
Nuclear lamina and Tau
Tau is a multifunctional protein and precise function of which depends on its localization. The localization of Tau protein to the inner side of the nuclear lamina [26, 27] and regulation of nuclear pore complex emphasizes a crucial role of Tau [28, 29] protein in maintaining nucleus integrity. The oligomeric form of Tau directly binds to lamin B receptor and lamin proteins, which lose their solubility upon oligomerization and also their nuclear membrane localization, ultimately leading to disruption of the nucleocytoplasmic interface [30]. Tau regulates nuclear Lamin B1 expression [31, 32]. Tau-dependent reduction of Lamin B1 leads to disruption of nuclear lamina [33]. The expression of full-length Tau or its truncated variant (Asp421-truncated Tau) in SH-SY5Y cells leads to formation of nuclear envelope indentations [34].
In Huntington’s disease and frontotemporal dementia, nuclear envelope indentations are filled with rod-like Tau deposits [35,36,37]. Likewise, Transgenic mice expressing mutant P301L-Tau shows disruption of NL [27]. One of the deleterious consequences of disruption of nuclear lamina is impairment of nucleocytoplasmic transport, which has been related to both overexpression of Tau and pathological Tau [27, 32, 38, 39]. The first indication of nuclear Tau in Alzheimer’s disease came from transmission electron microscopy (short paired helical filaments) in frontal lobe of AD cases [40]. Alterations in nuclear lamina are a characteristic of aging [41]. Indeed, mutations in the lamin A/C protein leads to the ‘’accelerated aging disorder Hutchinson-Gilford progeria syndrome’’ [42]. Alterations in nuclear lamina have been reported in AD brains [28, 32], where neurons exhibit a more complex structure of nuclear lamina.
Modifications in the nuclear lamina can initiate alterations in the DNA organization, through modification of nucleocytoplasmic transport. Iinteractions of Tau protein with the nuclear envelope contribute to defects in RNA and protein nucleocytoplasmic transport [27, 43]. Controlled nucleocytoplasmic exchange of cellular biomolecules—such as mRNA and rRNA, transcription regulators, nuclear and cytoplasmic proteins—is crucial for key functions of cell survival e.g., stress response, signal transduction and proteostasis [44,45,46]. How alterations in the nuclear lamina contribute to all these crucial processes (directly or indirectly) under health and disease require further investigations.
Nuclear speckles and Tau
Nuclear speckles (NSs) are membrane less organelles, which are sites of splicing factor storage and modifications, and are closely linked with RNA metabolism [47]. Nuclear Tau aggregates colocalize with nuclear speckles, and alter their composition and dynamics, in cellular models and in mouse brains [48]. In Alzheimer’s disease, pathological Tau drives ectopic accumulation of SRRM2, a core scaffold protein of nuclear speckles [49]. This depletion of SRRM2 may lead to altered splicing, transcription and translation, ultimately affecting neuronal physiology [50]. Two other nuclear speckles proteins MSUT2 and PABPN1 are also depleted from nucleus in cases of severe Alzheimer’s disease [51]. Furthermore, It has been reported that Tau aggregates particularly grow with endogenous mitotic interchromatin granules and cytoplasmic speckles [52] containing SRRM2 and PNN proteins. These evidence support the hijacking hypothesis for pathological-Tau in association with RNA to deplete critical workers from the nucleus (nuclear factors), affecting the biology of RNA in the nucleus. A similar sequestration of RNA binding proteins (e.g. TDP-43 and FUS) in cytosolic condensates, in amyotrophic lateral sclerosis has been associated with impaired nuclear RNA-processing [53,54,55]. Thus, the sequestration of RNA binding proteins and RNAs into pathological aggregates may signify a common pathophysiological feature in multiple neurodegenerative disorders affecting diverse cell types, with the depletion of crucial RNA processing factors from the nucleus, altering RNA processing, ultimately leading to altered gene expression. The identification of exact mechanism of speckle hijacking could highlight important aspects of Tau pathophysiology. Rescuing the SRRM2 splicing function by restoring its nuclear localization in the presence of Tau aggregates may mitigate neurodegeneration. The disruption of deleterious cytosolic Tau-RNA condensates using bait RNAs may restore nuclear localization of important nuclear factors. Clearly, urgent investigations are required to explore these hypotheses and their utility for therapeutic interventions.
Nucleolus and Tau
Nuclear studies have shown a crucial role of Tau protein in nucleolar structure conformation [4, 56]. Tau colocalizes with crucial nucleolar factors such as nucleolin, upstream binding transcription factor, and TIP5 in cellular models and human brain tissues [56, 57]. It has been found that nucleolar chaperons are reduced in different AD brain regions including nuclear Tau [58]. Tau enhances interactions of nuclear proteins like T cell intracellular antigen 1 (TIA1) with ribonucleoproteins, suggesting a role for it in rRNA gene metabolism [57]. The Tau protein localizes in the nucleolar organizer region (pericentromeric heterochromatin) and at the dense fibrillar regions as shown by immunofluorescence [22, 59, 60].
Tau can interact with both ribosomes and rRNA through its association with RNA binding proteins [61, 62]. In tauopathies, the interaction between Tau and ribosomes is pronounced [61], suggesting an impairment of its functioning. Translocation of phodphorylated-Tau into the nucleus results in nucleolar dispersion and p53-dependent apoptosis, both of which contributes to neurodegeneration [63].
Recently, non-phosphorylated Tau (at residues Ser 195, 198, 199 and 202) has been described as a bona fide nucleolar protein in neuronal cell lines and human brain tissue [57]. An association between Tau and TIP5 (a main player of heterochromatin stability and ribosomal DNA transcriptional repression) in SHSY5Y cells and human brain tissue [57] suggests a crucial role of Tau/TIP5 in the stabilization of repressive epigenetic marks on the ribosomal DNA. However, further investigations are required to study the precise role of Tau in nucleolar remodeling complex. Furthermore, nucleolar stress (induced by glutamate) redistributes nucleolar non-phosphorylated Tau in a similar way to other nucleolar factors e.g. fibrillarin, and induce a nuclear influx of phosphorylated Tau (Thr231) which shows a distinct localization from fibrillarin and nucleolar Tau [57]. These findings suggest that different species of Tau are present in the nucleus and play specific roles depending on type of cellular distress.
DNA and Tau
In the nucleus, Tau is involved in DNA protection and chromosome stability [4, 57, 64]. In vitro studies indicate that binding of Tau protein with DNA can increase the melting temperature of DNA [65] and protect it against heat shock induced double strand breaks, providing an evidence that nuclear Tau (non-phosphorylated) is crucial to cope with early stress responses in continuous changing microenvironment of neurons [66]. Further research could investigate the molecular mechanisms underlying this protective function and its implications for neuronal resilience in the face of environmental stressors. Moreover, identifying pathways through which Tau interacts with heat shock response machinery can provide us with potentially novel targets for therapeutic interventions.
It has been shown that Tau knockout cortical neurons are more susceptible to heat stress-induced and hypothermia-induced DNA breakage as compared to their wild-type counterparts [67]. In addition to its function in DNA protection, it has been shown to play a role in the modulation of gene expression. Exploring the regulatory mechanisms by which Tau influences gene transcription, can shed light on its role in neuronal homeostasis and its potential involvement in neurodegenerative diseases. Indirectly, Tau could affect the gene transcription by some compensatory changes in the gene expression [67]. Investigating how the loss of Tau affects gene transcription and whether compensatory mechanisms exist could offer insights into the broader consequences of nuclear Tau dysfunction.
To date, 14 genes have been identified, for which transcription has been reported to be significantly increased after Tau depletion in quantitative real-time PCR and microarray analyses [68, 69]. Tau plays an important role as an epigenetic regulator of gene expression, an organizer of heterochromatin, a contributor to chromosomal stability, and processor or silencer of ribosomal RNA [69]. All these evidence suggest that Tau can regulate genomic functions in the nucleus. Considering the significant regulatory role of the nucleus in the maintenance of cellular homeostasis and the presence of Tau in the nucleus of the AD brain, we focused on its nuclear role specifically in Alzheimer’s disease.
Considering the substantial impact of nuclear processes on cellular balance and the observation of Tau in the brains of Alzheimer’s disease patients, it is plausible to propose that Tau’s nuclear functions have a distinctive role in AD. These functions could potentially underlie the alterations in gene expression, instability in chromosomal structure, and disruptions in protein synthesis seen in AD. A focused investigation into the precise implications of nuclear Tau in the context of AD may reveal valuable insights into the disease’s development and provide leads for therapeutic interventions.
Nuclear Tau and stress granules
Emerging evidence indicate that Tau protein is a regulator of biology of RNA binding proteins [70]. Tau promotes stress granule development and regulate the interactome of TIA-1, a main component of stress granules [13]. However, which species of Tau (nuclear or other?) participate in stress granule formation, remains enigmatic. As, TIA-1 protein, a classical marker of stress granules, upon stress exit from the nucleus to initiate stress granule formation. Plausibly, nuclear Tau may also translocate to cytoplasm upon stress and contribute to stress granule formation, which has been reported as nidus of Tau misfolding [71, 72]. Investigation of precise species of Tau (nuclear or cytosolic) involved in stress granule formation may shed important light on dual role of nuclear Tau and its nidus of misfolding.
Nuclear Tau and aging
The levels of different isoforms of Tau protein could change in different types of neurons during the development, aging or diseases (tauopathies) in mammals. In some diseases, there is a toxic gain of function of altered Tau, due to the hyperphosphorylation or aggregation [73, 74]. These phenotypic changes are mainly found in aging organisms. Aging is a risk factor for several neurodegenerative diseases like Alzheimer’s disease or Parkinson’s disease. With aging, there is an increase of neuronal vulnerability to oxidative damage, that could modify Tau protein [75, 76] facilitating its aggregation [77, 78]. An age-dependent accumulation of Tau aggregation has been reported in a C. elegans model [79]. Also during aging, mitochondrial changes in the brain occur [80]. Abnormal binding of a mitochondrial protein, DRP1 (involved in mitochondrial fission) to Tau protein promote neurodegeneration through mitochondrial dysfunction [81].
Previous studies have shown a decrease in soluble Tau with increased aging [82] in resected human brain tissue as well as in post-mortem brains [83]. On the contrary, phosphorylated Tau (Thr 212) increases during aging, in the nucleolus and pericentromeric heterochromatin of pyramidal neurons in the CA1 region, with the maximum accumulation in senescent cells [26]. However, this phosphorylated form of Tau decreases in AD, at its later stages [26]. Since Thr212 is a direct target of kinase GSK3β [84], and the activity of GSK3β increase during aging, and AD pathology [84,85,86], the increased nuclear phosphorylated Tau with aging may be attributed to the increase of GSK3β [6].
Elevated intron retention, an alternative splicing process in which introns remain within mature mRNA transcripts, has also been associated with aging brains and the development of Alzheimer’s disease [87,88,89]. Specifically, the retention of intron 11 results in the translation of a novel truncated Tau11i isoform. These Tau11i proteins aggregate in granular-like formations within the temporal lobes of AD patients. It is of significant importance to uncover the mechanisms governing these distinct splicing events, understand how Tau11i proteins contribute to the formation of pre-tangles, and elucidate their role in the pathogenesis of AD [90].
Tau can be secreted into the extracellular space and can be transferred to the neighboring cells [91] e.g. neurons, astrocytes, and microglia, in a prion-like fashion. Particularly, an age-dependent spread of Tau protein has been observed in mouse brain. Aged animals show enhanced spreading of Tau in the hippocampus and neighboring cortical regions with accumulation of prominent misfolded Tau in entorhinal cortex [92]. All these evidence indicate that alterations in Tau protein levels (proportions), post-translational modifications, and spreading occur with aging. Future investigations addressing the heterogeneity of Tau species and their PTMs with age, and under different stressful conditions could highlight important aspects of Tau pathophysiology in normal aging and tauopathies.
Nuclear Tau in Alzheimer’s disease
Tau accumulation is a major pathological marker in several neurodegenerative diseases. Recently, it was found that hyper-acetylation of Tau at residue 174 (Tau-K174ac) increases its nuclear accumulation which is triggered by DNA damage signalling or SIRT6 shortage [69]. In Alzheimer’s disease, genomic instability has been reported, and SIRT6’s role in this context may have implications for disease progression. Targeting the acetylation of Tau at residue 174 or restoring SIRT6 levels could mitigate Tau accumulation and potentially offer therapeutic strategies for Alzheimer’s disease. Research may focus on how this acetylation event influences Tau pathology and developing interventions aimed at modulating these factors. Presence of higher nuclear Tau (Tau-K174ac), increased nucleolin and decreased SIRT6 levels have been reported in the AD cases [93]. The Tau-K174ac toxicity has been attributed to its nuclear accumulation and nucleolar dysfunction [93]. In a recent study, the role of nuclear Tau (AT8 epitope) has been reported in the onset of Alzheimer’s disease. There was decreased immunoreactivity in senile neurons, as compared to younger one’s [94]. Furthermore, the findings from this study suggest nuclear Tau’s involvement in the abnormal activation of cell cycle in differentiated cells [94].
Nulcear translocation of Tau occurs via importin-α/β pathway [29]. Hyperphosphorylated Tau in the nucleus interferes with important cellular processes such as nucleocytoplasmic transport and mislocalisation of nuclear factors, contributing to cell death [27, 29, 95]. Further investigation could explore the specific mechanisms through which Tau disrupts these processes and its implications in neurodegenerative diseases, such as Alzheimer’s disease.
It is also of great importance to consider that the role of nuclear Tau in neurodegenerative diseases could be through stress-dependent inhibition of nuclear Tau function because of aggregation and hyperphosphorylation [57, 96]. Research could examine how stress-related factors impact nuclear Tau’s functionality and whether this contributes to disease pathogenesis. Phosphorylation and aggregation of Tau protein are among the major post translational modifications responsible for the formation of paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) [97]. The binding of hyperphosphorylated Tau to the DNA alters its conformation and integrity leading to nucleosomal disorganization and altered gene expression [98]. Interestingly a recent study demonstrated a significantly increased Ca2+ nuclear concentration with the hyperphosphorylation of Tau. This may propagate a self-perpetuating loop to cause neurodegeneration [99]. Phosphorylation and aggregation of Tau, leading to the formation of paired helical filaments and neurofibrillary tangles, could be a key mechanism through which Tau disrupts nuclear functions. Investigating the impact of hyperphosphorylated Tau on DNA conformation, nucleosomal organization, and gene expression could provide insights into its role in neurodegeneration.
Moreover, the role of Tau protein as a chromatin modifier in Alzheimer’s disease and aging has been discovered. As compared to the younger neurons the aged neurons have increased levels of AT100 (p-The212-Ser214) immunoreativity in the dentate gyrus and hippocampal CA1 region [26, 100]. As the age progresses, positivity zone increases in intensity and frequency specifically near the nuclear membrane and nucleolus [101]. As the AD progresses, depletion of nuclear Tau is more evident with maximum depletion at late stages. At the late AD stage, AT100 immunopositivity can exclusively be found in the neurofibrillary tangles (NFTs) [58]. At initial stages, Tau exits the neuronal nucleus completely [58, 102] and causes global chromatin relaxation which consequently leads to abnormal transcription of various heterochromatin genes and dysregulation of euchromatin gene [31, 103]. Research may delve into the molecular pathways linking Tau’s nuclear exit with chromatin alterations and the subsequent transcriptional dysregulation. This information can eventually provide a great insight into factors causing abnormal transcription of heterochromatin genes which could further be intervened to stop these abnormalities in terms of therapeutic interventions.
The ultimate transcriptional silencing leads to decondensation of heterochromatin at perinuclear and pericentromeric regions [26, 27]. With the AD progression, disrupted nucleo-cytoplasm transport results in the mislocalization of many nuclear proteins in the cytoplasm [27, 95]. Investigating the specific genes affected by this silencing and their role in AD pathogenesis can offer insights into disease mechanisms.
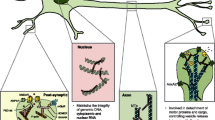
The crucial role of Tau protein on the regulation of genome has been validated by microarray hybridization (ChiP-on-chip) assays and genome-wide immunoprecipitation (ChiP) [104]. Nuclear depletion of Tau contributes to an impairment of perinuclear heterochromatin in AD [26]. Exploring the mechanisms by which this impairment relates to AD-related epigenetic changes and its consequences on gene expression can provide us with valuable information for better disease interventions for diagnostics and therapeutics. A strong interaction of Tau with intergenic and intronic parts of DNA, coding for long noncoding RNA (lncRNA), has been reported [104]. Investigating the specific lncRNAs involved, their functions, and their role in the regulation of chromatin and gene expression may uncover novel pathways in AD pathogenesis. Tau and lncRNAs both regulate transcription of chromatin and gene expression indirectly, these processes are remarkably deregulated in AD [105, 106]. Nucleolar depletion of Tau disturbs the tRNA synthesis and destabilizes rDNA loci [64]. Altogether nuclear Tau is essential for transcriptional regulation, NAD’s stability, and regulated functioning of the nucleolus [26].The role of Tau in nuclear pore complex dysfunction points out its probable contribution towards Tau-induced neurotoxicity in AD and tauopathies. Investigating the mechanisms through which Tau disrupts nuclear pore complex function and its implications for cellular homeostasis can provide insights into disease progression. The nuclear functions of Tau and their disruption in AD is summarized in Fig. 1.

Role of nuclear Tau in Alzheimer’s disease. Nuclear Tau is involved in genome stability and maintenance of nucleo-cytoplasmic transport. Depletion of nuclear Tau leads to conformational changes in the heterochromatin and makes DNA vulnerable to damage, disturbs rRNA synthesis and ribonucleotide pool balance. The cytosolic Tau aggregates sequester nuclear factors leading to their depletion from the nucleus. Tau-mediated Impairment of nuclear pore complex disrupts nucleo-cytoplasmic transport, ultimately contributing to Tau-induced neurotoxicity (Created with https://www.BioRender.com)
Conclusions
Since the discovery of Tau protein in 1975, research efforts were concentrated on the role of Tau protein in pathophysiology in the context of the microtubules dynamics and stabilization, even though, for more than three decades, different localizations of Tau protein have been discovered e.g., in the nuclear compartments. Discovery of the role of Tau protein in various cellular compartments especially in the nucleus opens up a new fold of complexity in tauopathies. Perturbations in the Tau protein (as happens in AD and other tauopathies) could alter its multiple functions in the nucleus enhancing genome vulnerability and neurodegeneration. Its interaction with several nuclear components strongly suggests that it could play multiple functions in the nucleus, although more investigations are required to find out a precise role of Tau in these processes. Nuclear tau heckles nuclear speckles leading to RNA splicing defects, nucleo-cytoplasmic transport deficits, chromatin organization disturbances and nucleolar organization deficits. A through identification of different species of Tau (nuclear and cytosolic), their mode of nucleo-cytoplasmic transport, neuronal microenvironment-mediated post-translational modifications of Tau protein are crucial aspects to understand the whole repertoire of Tau and its nidus of aggregation. Furthermore, to find out effective Tau-based therapies, there is an urgent need to understand precise functional relevance of these diverse cellular localizations of Tau protein and how it is altered during neurodegeneration.
Data Availability
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- CSF:
-
Cerebrospinal fluid
- NFT:
-
neurofibrillary tangles
- NSs:
-
nuclear speckles
- MAPT:
-
microtubule associated protein Tau
- MAPs:
-
microtubule-associated proteins
- NL:
-
Nuclear Lamina
- PHF:
-
paired helical filaments
- p-Tau:
-
phosphorylated Tau
- Tau:
-
tubulin associated unit
- T-Tau:
-
total Tau
References
Barbier P, Zejneli O, Martinho M et al (2019) Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci 10(JUL). https://doi.org/10.3389/fnagi.2019.00204
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. Published online 1975. https://doi.org/10.1073/pnas.72.5.1858
Narasimhan S, Changolkar L, Riddle DM et al (2020) https://doi.org/10.1084/jem_20190783
Rossi G, Dalprà L, Crosti F et al (2008) A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle Published Online. https://doi.org/10.4161/cc.7.12.6012
Ibáñez-Salazar A, Bañuelos-Hernández B, Rodríguez-Leyva I et al (2017) Oxidative stress modifies the levels and phosphorylation state of tau protein in human fibroblasts. Front Neurosci Published Online. https://doi.org/10.3389/fnins.2017.00495
Antón-Fernández A, Vallés-Saiz L, Avila J, Hernández F (2023) Neuronal nuclear tau and neurodegeneration. Neurosci Published Online. https://doi.org/10.1016/j.neuroscience.2022.07.015
Cieri D, Vicario M, Vallese F et al (2018) Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular ca 2 + handling. Biochim Biophys Acta - Mol Basis Dis Published Online. https://doi.org/10.1016/j.bbadis.2018.07.011
Pooler AM, Hanger DP (2010) Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans Published Online. https://doi.org/10.1042/BST0381012
Li C, Götz J (2017) Somatodendritic accumulation of Tau in Alzheimer’s Disease is promoted by fyn-mediated local protein translation. EMBO J Published Online. https://doi.org/10.15252/embj.201797724
Ittner A, Ittner LM (2018) Dendritic Tau in Alzheimer’s Disease. Neuron Published Online. https://doi.org/10.1016/j.neuron.2018.06.003
Robbins M, Clayton E, Kaminski Schierle GS (2021) Synaptic tau: a pathological or physiological phenomenon? Acta Neuropathol Commun Published Online. https://doi.org/10.1186/s40478-021-01246-y
Saman S, Kim WH, Raya M et al (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer Disease. J Biol Chem Published Online. https://doi.org/10.1074/jbc.M111.277061
Vanderweyde T, Apicco DJ, Youmans-Kidder K et al (2016) Interaction of tau with the RNA-Binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep Published Online. https://doi.org/10.1016/j.celrep.2016.04.045
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci Published Online. https://doi.org/10.1038/nrn.2015.1
Andreadis A Misregulation of tau alternative splicing in neurodegeneration and Dementia. Altern Splicing Dis Published Online 2006:89–107
Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci Published Online. https://doi.org/10.1146/annurev-neuro-072116-031153
Mandelkow EM, Mandelkow E (2012) https://doi.org/10.1101/cshperspect.a006247
Maina MB, Al-Hilaly YK, Serpell LC (2016) Nuclear tau and its potential role in alzheimer’s Disease. Biomolecules Published Online. https://doi.org/10.3390/biom6010009
Maccioni RB, Cambiazo V (1995) Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev Published Online. https://doi.org/10.1152/physrev.1995.75.4.835
Zempel H, Mandelkow E (2014) Lost after translation: Missorting of tau protein and consequences for Alzheimer Disease. Trends Neurosci Published Online. https://doi.org/10.1016/j.tins.2014.08.004
Liu C, Götz J (2013) Profiling murine tau with 0 N, 1 N and 2 N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1 N isoform being enriched in the nucleus. PLoS One Published Online. https://doi.org/10.1371/journal.pone.0084849
Loomis PA, Howardt TH, Castleberryt RP, Binder L 1 Identification of nuclear tau isoforms in human neuroblastoma cells (nucleolus/microtubule-associated proteins/Alzheimer disease/Down syndrome). Proc Natl Acad Sci USA. Published online 1990.
Lu J, Li T, He RQ, Bartlett PF, Götz J (2014) Visualizing the microtubule-associated protein tau in the nucleus. Sci China Life Sci Published Online. https://doi.org/10.1007/s11427-014-4635-0
Levine ZA, Larini L, LaPointe NE, Feinstein SC, Shea JE Regulation and aggregation of intrinsically disordered peptides. Proc Natl Acad Sci U S A. Published online 2015. https://doi.org/10.1073/pnas.1418155112
Sotiropoulos I, Galas MC, Silva JM et al (2017) Atypical, non-standard functions of the microtubule associated Tau Protein. Acta Neuropathol Commun Published Online. https://doi.org/10.1186/s40478-017-0489-6
Gil L, Federico C, Pinedo F et al (2017) Aging dependent effect of nuclear tau. Brain Res Published Online. https://doi.org/10.1016/j.brainres.2017.09.030
Eftekharzadeh B, Daigle JG, Kapinos LE et al (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s Disease. Neuron Published Online. https://doi.org/10.1016/j.neuron.2018.07.039
Gil L, Niño SA, Capdeville G, Jiménez-Capdeville ME Aging and Alzheimer’s disease connection: Nuclear Tau and lamin A. Neurosci Lett. Published online 2021. https://doi.org/10.1016/j.neulet.2021.135741
Candia RF, Cohen LS, Morozova V, Corbo C, Alonso AD (2022) Importin-mediated pathological Tau Nuclear translocation causes disruption of the Nuclear Lamina, TDP-43 mislocalization and cell death. Front Mol Neurosci Published Online. https://doi.org/10.3389/fnmol.2022.888420
Jiang L, Wolozin B (2021) Oligomeric tau disrupts nuclear envelope via binding to lamin proteins and lamin B receptor. Alzheimers Dement Published Online. https://doi.org/10.1002/alz.054521
Frost B, Hemberg M, Lewis J, Feany MB (2014) Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci Published Online. https://doi.org/10.1038/nn.3639
Frost B (2016) Alzheimer’s Disease: an acquired neurodegenerative laminopathy. Nucleus Published Online. https://doi.org/10.1080/19491034.2016.1183859
Montalbano M, McAllen S, Sengupta U et al (2019) Tau oligomers mediate aggregation of RNA-binding proteins Musashi1 and Musashi2 inducing Lamin alteration. Aging Cell Published Online. https://doi.org/10.1111/acel.13035
Monroy-Ramírez HC, Basurto-Islas G, Mena R et al (2013) Alterations in the nuclear architecture produced by the overexpression of tau protein in neuroblastoma cells. J Alzheimer’s Dis Published Online. https://doi.org/10.3233/JAD-122401
Fernández-Nogales M, Cabrera JR, Santos-Galindo M et al (2014) https://doi.org/10.1038/nm.3617
Paonessa F, Evans LD, Solanki R et al (2019) Microtubules deform the Nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated Frontotemporal Dementia. Cell Rep Published Online. https://doi.org/10.1016/j.celrep.2018.12.085
Fernández-Nogales M, Santos-Galindo M, Merchán-Rubira J et al (2017) Tau-positive nuclear indentations in P301S tauopathy mice. Brain Pathol Published Online. https://doi.org/10.1111/bpa.12407
Diez L, Wegmann S Nuclear Transport Deficits in Tau-Related Neurodegenerative Diseases. Front Neurol. Published online 2020. https://doi.org/10.3389/fneur.2020.01056
Prissette M, Fury W, Koss M et al (2022) Disruption of nuclear envelope integrity as a possible initiating event in tauopathies. Cell Rep Published Online. https://doi.org/10.1016/j.celrep.2022.111249
Metuzals J, Robitaille Y, Houghton S, Gauthier S, Leblanc R (1988) Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s Disease. J Neurocytol Published Online. https://doi.org/10.1007/BF01216709
Kristiani L, Kim M, Kim Y Role of the Nuclear Lamina in Age-Associated Nuclear Reorganization and Inflammation. Cells. Published online 2020. https://doi.org/10.3390/cells9030718
Gonzalo S, Coll-Bonfill N Genomic instability and innate immune responses to self-DNA in progeria. GeroScience. Published online 2019. https://doi.org/10.1007/s11357-019-00082-2
Cornelison GL, Levy SA, Jenson T, Frost B (2019) Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell Published Online. https://doi.org/10.1111/acel.12847
Gama-Carvalho M, Carmo-Fonseca M (2001) The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett Published Online. https://doi.org/10.1016/S0014-5793(01)02487-5
Zhang K, Daigle JG, Cunningham KM et al (2018) Stress Granule Assembly disrupts nucleocytoplasmic transport. Cell Published Online. https://doi.org/10.1016/j.cell.2018.03.025
Xu L, Massagué J (2004) Nucleocytoplasmic shuttling of signal transducers. Nat Rev Mol Cell Biol Published Online. https://doi.org/10.1038/nrm1331
Galganski L, Urbanek MO, Krzyzosiak WJ (2017) Nuclear speckles: molecular organization, biological function and role in Disease. Nucleic Acids Res Published Online. https://doi.org/10.1093/nar/gkx759
Lester E, Ooi FK, Bakkar N et al (2021) Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron Published Online. https://doi.org/10.1016/j.neuron.2021.03.026
McMillan PJ, Strovas TJ, Baum M et al (2021) Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s Disease. Acta Neuropathol Commun Published Online. https://doi.org/10.1186/s40478-021-01219-1
Gorenberg EL, Shorter J Tau heckles speckles: a pathogenic mechanism in tauopathy? Neuron. Published online 2021. https://doi.org/10.1016/j.neuron.2021.04.022
Wheeler JM, McMillan P, Strovas TJ et al (2019) https://doi.org/10.1126/scitranslmed.aao6545
Lester E, Van Alstyne M, McCann KL et al Cytosolic condensates rich in polyserine define subcellular sites of tau aggregation. Proc Natl Acad Sci U S A. Published online 2023. https://doi.org/10.1073/pnas.2217759120
Jiang LL, Guan WL, Wang JY, Zhang SX, Hu HY (2022) RNA-assisted sequestration of RNA-binding proteins by cytoplasmic inclusions of the C-terminal 35-kDa fragment of TDP-43. J Cell Sci Published Online. https://doi.org/10.1242/jcs.259380
Lagier-Tourenne C, Polymenidou M, Hutt KR et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci Published Online. https://doi.org/10.1038/nn.3230
Polymenidou M, Lagier-Tourenne C, Hutt KR et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci Published Online. https://doi.org/10.1038/nn.2779
Sjöberg MK, Shestakova E, Mansuroglu Z, Maccioni RB, Bonnefoy E (2006) Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci Published Online. https://doi.org/10.1242/jcs.02907
Maina MB, Bailey LJ, Wagih S et al (2018) The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun Published Online. https://doi.org/10.1186/s40478-018-0565-6
Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. Published online 2016. https://doi.org/10.1111/bpa.12335
Wang Y, Loomis PA, Zinkowski RP, Binder LI (1993) A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J Cell Biol Published Online. https://doi.org/10.1083/jcb.121.2.257
Thurston VC, Zinkowski RP, Binder LI (1996) Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma Published Online. https://doi.org/10.1007/BF02510035
Meier S, Bell M, Lyons DN et al Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci. Published online 2016. https://doi.org/10.1523/JNEUROSCI.3029-15.2016
Banerjee S, Ferdosh S, Ghosh AN, Barat C (2020) Tau protein- induced sequestration of the eukaryotic ribosome: implications in neurodegenerative Disease. Sci Rep Published Online. https://doi.org/10.1038/s41598-020-61777-7
Roqanian S, Ahmadian S, Nabavi SM et al (2022) Tau nuclear translocation is a leading step in tau pathology process through P53 stabilization and nucleolar dispersion. J Neurosci Res Published Online. https://doi.org/10.1002/jnr.25024
Bou Samra E, Buhagiar-Labarchède G, Machon C et al (2017) A role for tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat Commun Published Online. https://doi.org/10.1038/s41467-017-00633-1
Camero S, Benítez MJ, Barrantes A et al (2014) Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J Alzheimer’s Dis Published Online. https://doi.org/10.3233/JAD-131415
Sultan A, Nesslany F, Violet M et al (2011) Nuclear Tau, a key player in neuronal DNA protection. J Biol Chem Published Online. https://doi.org/10.1074/jbc.M110.199976
Violet M, Delattre L, Tardivel M et al (2014) A major role for tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci Published Online. https://doi.org/10.3389/fncel.2014.00084
Gómez de Barreda E, Dawson HN, Vitek MP, Avila J (2010) Tau deficiency leads to the upregulation of BAF-57, a protein involved in neuron-specific gene repression. FEBS Lett Published Online. https://doi.org/10.1016/j.febslet.2010.03.032
Oyama F, Kotliarova S, Harada A et al (2004) Gem GTPase and tau: morphological changes induced by gem GTpase in CHO cells are antagonized by tau. J Biol Chem Published Online. https://doi.org/10.1074/jbc.M401634200
Maziuk BF, Apicco DJ, Cruz AL et al (2018) RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun Published Online. https://doi.org/10.1186/s40478-018-0574-5
Rai SK, Savastano A, Singh P, Mukhopadhyay S, Zweckstetter M (2021) Liquid–liquid phase separation of tau: from molecular biophysics to physiology and Disease. Protein Sci Published Online. https://doi.org/10.1002/pro.4093
Ash PEA, Lei S, Shattuck J et al TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc Natl Acad Sci U S A. Published online 2021. https://doi.org/10.1073/pnas.2014188118
Liu M, Dexheimer T, Sui D et al (2020) Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription Drugs linked to Alzheimer’s Disease and cognitive functions. Sci Rep Published Online. https://doi.org/10.1038/s41598-020-73680-2
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimer’s Dis Published Online. https://doi.org/10.3233/JAD-2012-129031
Yan SD, Chen X, Schmidt AM et al Glycated tau protein in Alzheimer Disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. Published online 1994. https://doi.org/10.1073/pnas.91.16.7787
Ledesma MD, Bonay P, Colaço C, Avila J (1994) Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem Published Online. https://doi.org/10.1016/s0021-9258(17)31849-5
Pérez M, Cuadros R, Smith MA, Perry G, Avila J (2000) Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal. FEBS Lett Published Online. https://doi.org/10.1016/S0014-5793(00)02323-1
Reynolds MR, Lukas TJ, Berry RW, Binder LI (2006) Peroxynitrite-mediated τ modifications stabilize preformed filaments and destabilize microtubules through distinct mechanisms. Biochem Published Online. https://doi.org/10.1021/bi052142h
Aquino Nunez W, Combs B, Gamblin TC, Ackley BD (2022) Age-dependent accumulation of tau aggregation in Caenorhabditis elegans. Front Aging Published Online. https://doi.org/10.3389/fragi.2022.928574
Swerdlow RH (2011) Brain aging, Alzheimer’s Disease, and mitochondria. Biochim Biophys Acta - Mol Basis Dis Published Online. https://doi.org/10.1016/j.bbadis.2011.08.012
DuBoff B, Götz J, Feany MB Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron. Published online 2012. https://doi.org/10.1016/j.neuron.2012.06.026
Mukaetova-Ladinska EB, Harrington CR, Roth M, Wischik CM (1996) Alterations in tau protein metabolism during normal aging. Dement Geriatr Cogn Disord Published Online. https://doi.org/10.1159/000106861
Chatterjee S, Sealey M, Ruiz E et al (2023) Age-related changes in tau and autophagy in human brain in the absence of neurodegeneration. PLoS One Published Online. https://doi.org/10.1371/journal.pone.0262792
Timm T, Balusamy K, Li X, Biernat J, Mandelkow E, Mandelkow EM (2008) Glycogen synthase kinase (GSK) 3β directly phosphorylates serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J Biol Chem Published Online. https://doi.org/10.1074/jbc.M706596200
Souder DC, Anderson RM (2019) An expanding GSK3 network: implications for aging research. GeroScience Published Online. https://doi.org/10.1007/s11357-019-00085-z
Lauretti E, Dincer O, Praticò D (2020) Glycogen synthase kinase-3 signaling in Alzheimer’s Disease. Biochim Biophys Acta - Mol Cell Res Published Online. https://doi.org/10.1016/j.bbamcr.2020.118664
Adusumalli S, Ngian ZK, Lin WQ, Benoukraf T, Ong CT (2019) Increased intron retention is a post-transcriptional signature associated with Progressive aging and Alzheimer’s Disease. Aging Cell Published Online. https://doi.org/10.1111/acel.12928
García-Escudero V, Ruiz-Gabarre D, Gargini R et al A new non-aggregative splicing isoform of human tau is decreased in Alzheimer’s Disease. Acta Neuropathol. Published online 2021. https://doi.org/10.1007/s00401-021-02317-z
Ngian ZK, Tan YY, Choo CT et al Truncated tau caused by intron retention is enriched in Alzheimer’s Disease cortex and exhibits altered biochemical properties. Proc Natl Acad Sci U S A. Published online 2022. https://doi.org/10.1073/pnas.2204179119
Ong CT (2023) Enrichment of novel tau isoform with altered biochemical properties in Alzheimer’s Disease. Neural Regen Res Published Online. https://doi.org/10.4103/1673-5374.371359
Zhang H, Cao Y, Ma L, Wei Y, Li H (2021) Possible mechanisms of tau spread and toxicity in Alzheimer’s Disease. Front Cell Dev Biol Published Online. https://doi.org/10.3389/fcell.2021.707268
Wegmann S, Bennett RE, Delorme L et al (2019) Experimental evidence for the age dependence of tau protein spread in the brain. Sci Adv Published Online. https://doi.org/10.1126/sciadv.aaw6404
Portillo M, Eremenko E, Kaluski S et al (2021) SIRT6-CBP-dependent nuclear tau accumulation and its role in protein synthesis. Cell Rep Published Online. https://doi.org/10.1016/j.celrep.2021.109035
Bruno F, Sturiale V, Brancato D et al (2023) Nuclear tau as an early molecular marker of Alzheimer’s Disease. https://doi.org/10.3390/ecb2023-14131
Lu T, Aron L, Zullo J et al (2014) REST and stress resistance in ageing and Alzheimer’s Disease. Nat Published Online. https://doi.org/10.1038/nature13163
Cruz A, Verma M, Wolozin B (2019) The Pathophysiology of Tau and Stress Granules in Disease. In: Advances in Experimental Medicine and Biology.; https://doi.org/10.1007/978-981-32-9358-8_26
Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: implications for Alzheimer’s Disease. Neurochem Int Published Online. https://doi.org/10.1016/j.neuint.2010.12.023
Padmaraju V, Indi SS, Rao KSJ (2010) New evidences on Tau-DNA interactions and relevance to neurodegeneration. Neurochem Int Published Online. https://doi.org/10.1016/j.neuint.2010.04.013
Wei YP, Ye JW, Wang X et al (2018) Tau-Induced Ca2+/Calmodulin-Dependent protein Kinase-IV activation aggravates Nuclear Tau Hyperphosphorylation. Neurosci Bull Published Online. https://doi.org/10.1007/s12264-017-0148-8
Rutten BPF, Schmitz C, Gerlach OHH et al The aging brain: Accumulation of DNA damage or neuron loss? Neurobiol Aging. Published online 2007. https://doi.org/10.1016/j.neurobiolaging.2005.10.019
Mansuroglu Z, Benhelli-Mokrani H, Marcato V et al (2016) Loss of tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep Published Online. https://doi.org/10.1038/srep33047
Gil L, Niño SA, Chi-Ahumada E et al (2020) Perinuclear lamin a and nucleoplasmic lamin B2 characterize two types of hippocampal neurons through Alzheimer’s Disease progression. Int J Mol Sci Published Online. https://doi.org/10.3390/ijms21051841
Gil L, Niño SA, Guerrero C, Jiménez-Capdeville ME (2021) Phospho-tau and chromatin landscapes in early and late alzheimer’s Disease. Int J Mol Sci Published Online. https://doi.org/10.3390/ijms221910283
Benhelli-Mokrani H, Mansuroglu Z, Chauderlier A et al (2018) https://doi.org/10.1093/nar/gky929
Luo Q, Chen Y (2016) Long noncoding RNAs and Alzheimer’s Disease. Clin Interv Aging Published Online. https://doi.org/10.2147/CIA.S107037
Riva P, Ratti A, Venturin M (2016) The long non-coding RNAs in neurodegenerative Diseases: Novel mechanisms of Pathogenesis. Curr Alzheimer Res Published Online. https://doi.org/10.2174/1567205013666160622112234
Acknowledgements
Not applicable.
Funding
We acknowledge support by the Open Access Publication Funds/transformative agreements of the Göttingen University.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
NY designed the review outline, wrote the manuscript and did the literature search. IZ designed the review outline, did the literature search, and revised the manuscript. TS and AY contributed to the literature search and write-up. All authors made substantial contributions to discussion of the content and reviewed and/or edited the article before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Younas, N., Saleem, T., Younas, A. et al. Nuclear face of Tau: an inside player in neurodegeneration. acta neuropathol commun 11, 196 (2023). https://doi.org/10.1186/s40478-023-01702-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01702-x