
Abstract
In this review, we discuss the synaptic aspects of Tau pathology occurring during Alzheimer’s disease (AD) and how this may relate to memory impairment, a major hallmark of AD. Whilst the clinical diagnosis of AD patients is a loss of working memory and long-term declarative memory, the histological diagnosis is the presence of neurofibrillary tangles of hyperphosphorylated Tau and Amyloid-beta plaques. Tau pathology spreads through synaptically connected neurons to impair synaptic function preceding the formation of neurofibrillary tangles, synaptic loss, axonal retraction and cell death. Alongside synaptic pathology, recent data suggest that Tau has physiological roles in the pre- or post- synaptic compartments. Thus, we have seen a shift in the research focus from Tau as a microtubule-stabilising protein in axons, to Tau as a synaptic protein with roles in accelerating spine formation, dendritic elongation, and in synaptic plasticity coordinating memory pathways. We collate here the myriad of emerging interactions and physiological roles of synaptic Tau, and discuss the current evidence that synaptic Tau contributes to pathology in AD.
Similar content being viewed by others

Introduction
This review is primarily focused on synaptic Tau in Alzheimer's disease. Studies that investigate the function of synaptic Tau have been obtained through various experimental models including overexpression of proteins, Tau with FTLD. We are aware of the current limitations of various animal models as they may not fully replicate human AD, however there is still much we can learn from these models especially when it comes to a molecular understanding of the disease, and we will therefore discuss results from animal models alongside results from tissue studies of AD patients.
An overview of Alzheimer’s disease, the most common form of dementia
Alzheimer’s disease (AD) is the most common form of dementia affecting 50 million people worldwide in 2018, a number predicted to triple by 2050 to affect over 152 million people. Alongside this, 25% of hospital beds in the UK are occupied by people aged over 65 and suffering from dementia (Alzheimer’s Research UK). Age is the greatest risk factor for developing the sporadic form of AD [82, 119, 176, 184], however there are also genetic and environmental risk factors [120, 217, 219] that contribute to familial or sporadic forms of AD, respectively. The pathophysiology of AD is complex and not fully understood as we will see through the course of this review article.
AD is diagnosed histologically in post mortem brains of patients by the presence of two types of aggregated proteins with little understanding of how these proteins interact with each other during the different stages of disease. Extracellular plaques of Amyloid-beta (Aβ) peptides and intracellular neurofibrillary tangles (NFTs) of microtubule-associated protein Tau (MAPT; Tau) are both hallmarks of AD. Due to the presence of these different protein pathologies in AD, the field has been divided for a long time between people believing in either Aβ or Tau being causal to AD pathology [322]. For example, scientists who suggested that Aβ was causative of AD thought that Tau and other pathology were secondary to the cascade triggered by Aβ [172].
In the past years though, some research has moved away from preventing Aβ pathology towards inhibiting Tau pathology as the distribution and density of Aβ positive plaques are variable between neuropathological stages of the disease and not informative of the cognitive status of the patient [144]. Cognitive decline is most closely associated to the load and progression of NFTs as compared to Aβ pathology [171, 326]. Therefore, Aβ may thus be considered as a catalyst of Tau pathology [35], with Tau being a more central player in AD progression. The latter is supported by the fact that there are currently more than 20 different Tauopathies [395]. Tau pathology spreads anterogradely and follows the disease progression, the so called ‘Braak stages’, which progress from I–IV based on brain regions burdened by NFTs. Although the locus coeruleus has previously been suggested to be the starting point [41, 43, 427], recent evidence suggests that Tau pathology begins in the transentorhinal/entorhinal regions [220]. Thus, symptoms of AD highly correlate with the progression of Tau pathology from the hippocampus to the cortex, beginning with memory dysfunction and later leading to other cognitive impairments including loss of executive functioning, language, and visuospatial skills [101, 144, 245, 362]. The hippocampus is an anatomical region of the brain responsible for spatial or contextually-based learning and memory and it is one of the earliest and most drastically affected areas, displaying atrophy, accumulation of Aβ plaques, and NFTs in AD [16,17,18]. The role of hippocampal neuron subtypes in learning and memory is defined by their characteristic calcium dynamics, a high degree of plasticity and the capacity to undergo synaptic remodeling into adulthood. It has also been believed that hippocampal neuron subtypes are a major source of human adult neurogenesis [4, 95, 233, 498] until recent controversy [420]. The properties of hippocampal neurons are thought to impart the selective vulnerability of these cells, as pathology drastically accelerates on reaching neurons in this region at early stages of AD [156].
Microtubule-associated protein tau (MAPT)
Full-length monomeric forms of Tau have long been seen as the ‘glue’ that binds and stabilises microtubules in axons, in concert with other microtubule-associated proteins, such as MAP2, which have homologous roles in neuronal dendrites [468]. Microtubule stability is important for cellular polarity and for antero- or retrograde cellular transport of vesicles and organelles to occur. However, as the full interactome of Tau is revealed, the ubiquity of Tau’s roles is being uncovered to show how Tau binds to a diverse range of molecules to elicit a multiplicity of functions. Before binding to microtubules, Tau is an intrinsically disordered protein which confers conformational and functional flexibility. Numerous Tau binding partners with diverse cellular functions have now been reported. Tau binds directly to DNA for DNA protection [58, 279, 432, 467], to calmodulin to regulate gene expression [24], at the cell membrane to support growth processes [257], to Fyn for synaptic activity [200, 338], to actin for crosslinking actin filaments [56] and to numerous other proteins with yet unknown functional consequences [276]. Missense mutations in MAPT, the gene coding for Tau, can result in familial forms of frontotemporal dementia but are not causative of AD [111, 197, 356]. The ability of Tau to bind and interact with such a diverse range of molecules, and thus taking up so many roles, stems from Tau being produced as six different splice variants [158], from its ability to be post-translationally modified, its diverse binding regions, and from it being prone to terminal truncations (for review see [3].
The complex structure of tau
Six isoforms of Tau are present in the adult human central nervous system, although Tau occurs as a larger isoform in the peripheral nervous system [153].
Figure 1a shows how the six different Tau isoforms arise from alternative splicing. N-terminus inserts, Exons 2 and 3, result in 0 N, 1 N or 2 N Tau, whereby exon 3 is never inserted independently of exon 2. Exclusion or inclusion of the microtubule binding repeat region (MTB), exon 10, results in 3 repeat (3R) or 4 repeat (4R)-Tau, respectively, altogether providing 0N3R-, 0N4R-, 1N3R-, 1N4R-, 2N3R-, 2N4R- Tau [10, 150, 151, 260]. The N-terminus projection region has been found capable of binding to synaptic vesicles, either through protein binding (Fig. 1b, [424, 503] or through direct membrane interactions [44, 265]. The proline-rich region and microtubule binding domain are capable of polymerising F-actin, a cytoskeletal protein that has various roles in neurons including remodelling dendritic spines upon synaptic stimulation [136, 177, 194]. The proline-rich region is also able to bind SH3 domains such as Fyn kinase, which is of interest for a post-synaptic role of Tau [371]. The microtubule binding repeat region, alongside binding and stabilising tubulin, can also bind to lipid membranes [142], and part of this region forms the core of aggregates [128, 129].

Tau is differentially expressed as six isoforms that contain multiple structural domains for diverse protein–protein interactions. a Alternative splicing of exons 2 and 3 (E2, E3) determines the N-terminus region, whereas exon 10 (E10) determines the number of repeat (R) regions and gives rise to 3R or 4R Tau. Overall six isoforms of Tau exist with their expression dependent upon age and anatomical location. b The N-terminus region of Tau is involved in membrane interactions and has been shown to bind to synaptic vesicles [44, 265, 301]. The microtubule binding repeat regions bind and stabilise microtubules [260]
Debates about the role of post-translational modifications and truncated forms of Tau are ongoing. In one study, it was shown that the major form of Tau in the pre-synaptic compartment is truncated at its C-terminal and therefore lacks the (aggregation-determining) microtubule binding domain region [416]. The release of this truncated form of Tau is increased upon synaptic activity [216]. However, a study investigating the level of truncation in AD patient brains has shown that there is a relative increase in N-terminal truncations of Tau in AD patients as compared to controls [505]. The longest isoform of Tau has a total of 85 possible phosphorylation sites that interact with multiple kinases and phosphatases [150, 151]. Phosphorylation can determine the conformation and protein–protein interactions of this intrinsically disordered protein and therefore the residues at which Tau is phosphorylated can differ between physiological versus pathological conditions (for a review see [209]. For example, phosphorylation can modulate binding dynamics of Tau to tubulin, biolipid, and Fyn kinase [200, 269, 317, 394] and thus forms part of Tau’s physiological role. For binding to microtubules, consecutive phosphorylation of Tau is required, one to allow efficient phosphorylation of the second ‘primed’ site [152]. However pathologically, hyperphosphorylation of Tau has been associated with the formation and growth of neurofibrillary tangles, as specific phosphorylation sites have been shown to readily enhance fibril formation [98].
Aggregation of tau
In the characteristic pathway leading to the formation of neurofibrillary tangles in AD, hyperphosphorylated monomeric Tau forms small soluble granular structures known as oligomers. These oligomers have been suggested to act as toxic species which form part in AD pathogenesis [55, 284, 345, 452]. Oligomers are seen as an intermediate structure, capable of inducing a conformational change in monomeric Tau which is then able to attach to the oligomeric structure. The latter then leads to the formation of stacked β-sheet-rich strands which grow to form insoluble paired helical filaments (PHFs) which consequently amalgamate into large NFTs [129, 164, 250]. It has been shown that PHF-like Tau can lead to loss of synaptic contacts which is known to occur in hippocampal neurons of hibernating animals but reversed upon awakening [15]. Hyperphosphorylation of Tau has also been reported to occur in mice suffering from hypothermia during anaesthesia [351]. Thus, the pathogenic versus protective role of larger, insoluble structures, that are less neurotoxic than soluble oligomers but confine intracellular space and prevent intracellular trafficking [213] as they grow in size, is still an ongoing debate in the field.
Pathological tau
How oligomeric Tau forms is still unclear. Potential pathways leading to Tau oligomerisation include Tau release from microtubules, poly-anionic induction factors, or uptake into low pH compartments [21, 231, 307]. As neuronal activity has been shown to increase the rate of Tau pathology [476], it is possible that activity-dependent pathways may also mediate its aggregation. This could be an age-dependent mechanism whereby neuronal activity over time causes the formation of pathological Tau species, and their propagation. Alternatively, high levels of network activity [355], lysosomal dysfunction [315], or cell death such as induced by traumatic brain injury [320, 376] may result in a higher concentration of Tau being released into the extracellular space. The latter then leads to endocytic uptake and aggregation of Tau at low pH [307]. Tau has been shown to cause membrane disruption allowing it to leak from endo/lysosomes [57]. It is also thought that the impaired endosomal sorting complex required for transport (ESCRT) III protein activity permits a leakage of Tau from endo/lysosomes into the cytosol [68]. Selectively, vulnerable cells may act as the primary site of aggregation. Tau released by these cells may consequently be propagated along synaptically-connected networks whereby they recruit endogenous Tau and result in AD symptoms only after several years in the brain of an AD patient [26, 134, 135, 167, 183, 226, 347]. Tau uptake through muscarinic receptors can alter calcium ion (Ca2+) homeostasis [157]. Many of the Tau uptake mechanisms are further increased upon phosphorylation [218, 304] or neuronal activity, which, along with how synaptic pathological Tau can perturb activity, will be discussed in the next sections [246, 303, 416, 462]. Tau is also able to form membrane pore-like amyloid structures (annular protofibrils) similarly to those seen by α-synuclein and Aβ, which have been suggested to allow uncontrolled release of aggregates, ions, or vesicles [50, 105, 252, 253, 343].
Oligomeric forms of Tau have been shown to impair synaptic function, the latter being an early marker preceding fibril formation, synaptic loss, axonal retraction and cell death [123, 241, 284, 297, 345, 353, 491]. Tau is also present at lower concentrations in the somatodendritic compartments, often considered as the loss of its physiological function as it requires the detachment of Tau from microtubules [200, 338]. Tau has previously been found in pre- and post- synaptic compartments of healthy human volunteers and AD patients, but in AD patients it is primarily found in its ubiquitinated and phosphorylated form [124, 438]. Pre- and post- synaptic forms of Tau pathology have been described without a clear mechanistic link between the two [200, 353, 503]. Since Tau is able to accelerate spine formation and dendritic elongation, and is involved in memory pathways [230, 391, 392, 496] it has recently been discussed whether Alzheimer’s disease may be described as a physiological to pathological shift of synaptic Tau function [200, 301].
Activity-dependence of tau pathology in the hippocampus
The release of soluble Tau from neurons, both in vivo and in vitro, can be regulated by neuronal activity, and is suggested to be a physiological process. It is not known whether Tau released by neurons is monomeric or oligomeric [355, 476, 484]. Wu et al. [476] investigated whether neural activity could increase the rate of the progression of Tau pathology by increasing the activity-dependent release of Tau to synaptically-connected neurons. To test this hypothesis, cells that expressed mutant P301L hTau aggregates were stimulated with picrotoxin and approximately 45% of the stimulated cells were shown to have internalised Tau as compared with 20% of unstimulated cells. Similar results were seen in vivo, where hippocampal cells that were optogenetically stimulated for 20 days showed greater accumulation of Tau in cell bodies, and increased hippocampal cell layer atrophy compared to unstimulated animals [476]. The study did, however not include experiments to link increased pathology with behavioural deficits related to AD to see whether neuronal stimulation and the Tau pathology it induced also caused an earlier or more pronounced behavioural phenotype. From the study above, it was also unclear whether neuronal stimulation was driving Tau seed formation or whether it only increased their propagation through synaptically connected cells.
The direct relationship between neuronal activity and Tau pathology still needs to be determined. From recent research it seems likely that there is a feedback mechanism whereby neuronal activity causes increased Tau pathology, which in turn alters neurotransmission, and feeds forward to further Tau aggregation and propagation. Interestingly, Amyloid β (Aβ) induced hyperexcitability has also been linked to catalysing Tau pathology [378]. Bright et al. [46] showed such a relationship that includes a link with Aβ production. Neuronal hyperactivity, which is able to regulate increased Tau translation and extracellular Tau secretion [235, 355], has been shown to increase Aβ production. Both Aβ and Tau have been related to neuronal hyperexcitability, and Tau has been linked to pro-convulsive effects [49, 53,54,55, 100, 175, 187, 200, 244, 311, 339, 374, 375]. The high frequency activity that occurs in the hippocampal formation for learning and memory, as for other activities, such as spatial exploration or sleep for example, may explain an increase in pathology reaching these networks and thus the increased vulnerability of hippocampal cells. However, studies showing network hypoactivity also exist [55, 290] and therefore more research is required to reconcile the role of Tau on neuronal activity and how this may affect memory impairment during the course of AD.
Relating tau pathology to models of memory impairment
Synapses were first hypothesised to be the primary site of memory simultaneously with their discovery by Ramón y Cajal (1894). The most well established model for activity-dependent synaptic strengthening was discovered when Lømo [278] found evoked responses to high frequency stimulation in the hippocampus that lasted for hours. Certain forms of neuronal activity, including the high frequency stimulation used by Lømo [278], result in the influx of Ca2+ ions into synapses. Ca2+ ions act as a 2nd messenger for phosphorylation-dependent signalling cascades, causing neurotransmitter release, structural plasticity of the cytoskeleton, and the incorporation or alteration of ion channels and their subunits. These alterations ultimately feedback to maintain an increased and sustained Ca2+ conductance and is known as long-term potentiation (LTP). Alongside LTP, its counterbalance that is induced by low frequency stimulation to decrease conductivity of synapses, long-term depression (LTD), was also discovered [423]. LTP and LTD have been heavily studied in the hippocampus where they may underlie declarative learning and memory [308, 312, 76]. Impairment to hippocampal-dependent memory function is seen as early symptom of AD, and correlates with Tau pathology in the hippocampus [16, 17, 42].
The next question that had to be addressed was which molecular mechanisms had occurred to maintain the enhanced synaptic response during LTP? Though a controversial field, three mechanisms have consistently shown to be important for the induction of LTP (Fig. 2). (1) The pre-synaptic mechanism increases the probability of neurotransmitter release by upregulating the number of release sites, or the concentration of cleft glutamate. (2) Post-synaptic mechanisms increases the single-AMPA receptor-conductance on binding glutamate, either by increasing their opening probability, or prolonging their mean open-time through phosphorylation or exchange of subunits. (3) An additional post-synaptic mechanism increases channel numbers by inserting receptor-containing vesicles into the plasma membrane, or by lateral diffusion of extrasynaptic regions [34]. The reversal of these mechanisms can instigate LTD. It needs to be noted here that both, LTP and LTD remain a means to model memory, and do not necessarily equate to human hippocampal memory. However, the above mechanisms involve cytoskeletal restructuring for controlling synaptic volume, for stabilising active zone synaptic densities, and for cycling and tethering of vesicles or proteins via cell membrane endo- and exocytosis or via recycling vesicles. Increased import of proteins into synapses, or local translation [84, 215], is also required. As the latter mechanisms are involved in memory formation, we thus think that LTP and LTD are a relevant model to study certain aspects of memory formation. Indeed, evidence that Tau can influence any of these mechanisms, either physiologically or in pathological conditions, would provide a direct molecular to behavioural link of how Tau may lead to memory impairment.

Key processes by which Tau could interfere with LTP induction to directly cause symptoms of memory impairment. Processes include channel activation or gating function for Ca2+ ion entry, pre-synaptic vesicle cycling, post-synaptic vesicle cycling for subunit exchange and channel insertion, recruitment of receptors tethered at extra synaptic sites, and cytoskeletal restructuring to coordinate these mechanisms and to control the synaptic volume
Tau may directly influence LTP and LTD. Tau knockout ameliorates Aβ induced deficits in LTP. Aβ oligomers show a fairly consistent impairment in LTP and enhancement of LTD [249, 402, 460]. Endogenous wild-type Tau expression, but not the N296H FTLD mutant form, is required for Aβ dependent impairment of LTP [453], and impairment of LTP by Tau or Aβ oligomers is dependent upon amyloid precursor protein expression [360, 464].
However, studies have shown varying effects dependent on age, disease model, and protocol used when understanding how Tau may alter electrophysiological properties of cells. Table 1. summarises how the effects of Tau on electrophysiological measurements have a large degree of variability depending upon the methods applied. The most consistent finding appears to be that exogenous oligomeric Tau impairs LTP, with monomeric Tau having no effect [123, 251, 352, 360, 434]. Fá et al. [123] showed that a 20 min treatment of CA3-CA1 hippocampal neurons with oligomeric 4R2N Tau before induction of LTP caused a marked reduction in LTP expression without affecting basal synaptic transmission. On the contrary, 4R1N monomeric Tau did not reduce LTP. Polydoro et al. [352] predicted that Tau impairs the induction rather than the expression of LTP as high frequency but not theta burst stimulation failed to induce LTP in a hTau mouse model. Another study in rTgP301L mice expressing mutant Tau showed an impairment to both basal transmission and LTP [190] consistent with two similar studies using P301S or hTau mice [352, 491]. One study has even shown improved cognitive performance and LTP in the dentate gyrus of young Tau-P301L mice, and suggested hyperphosphorylation of Tau to be the pathogenic cause of synaptic impairment [37]. Many other studies have also linked Tau pathology to poor cognitive performance at a behavioural level [14, 205, 365, 393, 435, 440], and the suppression of Tau expression with an amelioration of symptoms [391, 435].
In the studies listed in Table 1, no attempt was made to explain the molecular mechanism of how Tau impaired LTP. The relationship between Tau pathology and these activity-dependent mechanisms (Fig. 2) therefore requires further explanation. Very different results for how different forms of Tau can alter the electrophysiology of neurons can be seen in Table 1. One suggestion for the variation between models is the location and concentration of Tau expression, and the mutation site for the different forms of mutant Tau used. An example of mutations in different domains resulting in opposite electrophysiological functional effects is A152T [242] at the N-terminus projection domain, versus K280del in the second microtubule-binding repeat domain [316, 373]. A152T expressing mice show increased basal transmission with increased glutamate release, without changes to synaptic plasticity [92]. Mice overexpressing K280del show reduced basal transmission with reduced pre-synaptic vesicles, and impaired synaptic plasticity [91]. This, however, does not address how wildtype Tau in Alzheimer’s disease functions. An additional cause of variability may be when different Tau isoforms contribute differentially to pathology, though the relationship is unlikely to be so simple, for example 0 N and 1 N Tau result in similar electrophysiological phenotypes (Table 1, [190, 491, 503]. Different isoforms of Tau have different roles in dendrite and spine formation, and it has been argued that the pathological mis-sorting of Tau, from the axon, is dependent on the level of specific Tau isoforms, though this may also just be driven by overexpression [444, 496]. It is possible that these opposing phenotypes may arise from different binding affinities of various forms of Tau (such as mutant, phosphorylated, or other conformers or isoforms of Tau) to synaptic proteins, such as, for example, the vesicular protein synaptogyrin-3 [92, 93, 276, 301, 503].
The binding of tau to synaptic vesicles
In the pre-synaptic compartment, exocytosis of synaptic vesicles containing neurotransmitter is vital for the transmission of nerve impulses from the ‘pre-’ to ‘post-’ synaptic neuron via chemical synapses. To maintain a sustained release of neurotransmitters during periods of high synaptic activity, such as required for some forms of plasticity, a trafficking cycle occurs which can combine clathrin mediated endocytosis (CME) and the engagement of reserve pools of vesicles (for a review see [431]. The mediation of stages of this cycle are also highly Ca2+-dependent often due to Ca2+-dependent phosphorylation of synaptic proteins [94].
Tau is capable of mediating toxicity specifically via interactions with synaptic vesicle proteins and the prevention of vesicle release. Mutant (R406W, V337M or P301L) or phosphorylated Tau immobilises synaptic vesicles by preventing their release from F-actin. This reduced vesicle motility was hypothesised to occur through a mesh of immobilised vesicles formed by a crosslinking of the N-terminus of Tau with synaptogyrin-3 and its proline-rich and microtubule-binding domain binding to F-actin networks [136, 177, 194, 503]. The reduced vesicle mobility could be rescued by knock-down of synaptogyrin-3 or by depolymerisation of F-actin bundles [301, 503]. Deficits from this dysfunction, such as decreasing excitatory junction potential (EJP) amplitudes, are not seen from low frequency (0.2 Hz) stimulation that employ the recycling pool of vesicles for release, but only following high frequency (e.g. 10 Hz) stimulation requiring the reserve population of vesicles. Under high frequency stimulation, normal levels of release cannot be maintained and therefore result in impaired synaptic transmission. This work showed that this pathology only occurred with mutant FTLD or hyperphosphorylated Tau as opposed to wildtype Tau, which showed less synaptic colocalisation. However, it was also suggested that the formation of Tau multimers may also permit Tau to immobilise vesicles [503]. The above results are comparable to results on studies related to α-synuclein, which have shown that α-synuclein is equally capable to immobilise synaptic vesicles by aggregation [103, 461, 503]. As mentioned in Table 1, opposing effects of Tau have also been observed when measuring vesicle release probability. An increased release probability was shown to occur in 16 month-old mice expressing P30lL Tau in a subset of cells from the entorhinal cortex using a Tet-OFF system (rTgTauEC, [89, 353]. If mutations, phosphorylation or different conformations of Tau can alter its binding affinities with synaptic proteins, it could change the release probability of synaptic vesicles or influence the timing of other pathways required for the coordination of synaptic plasticity. Phosphorylation is known to alter binding properties and localisation of multiple other synaptic proteins including synapsin-1 [309], dynamin-1 [75], assembly of complexes to mediate Ca2+-dependent exocytosis [488], and post-synaptic AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) and NMDAR (N-methyl-D-aspartate receptor) subunits [180, 298]. An interesting question arises from Tau’s ability to bind synaptic vesicles as to whether it is acting as a static tether and scaffolding protein, or has an active role in a mechanism at the synaptic compartment.
Tau in the vesicle cycle of synaptic compartments
Bioinformatic analysis of Tau-interacting proteins based on co-immunoprecipitation studies by Liu et al. [276] show that many of these proteins are enriched in classes related to membrane trafficking and transportation, or metabolic activity (Fig. 3a). The functional annotation chart shows that these genes can be split into two functional groups with the highest enrichment scores (Fig. 3b). These clusters are related to metabolism and transport, and to synaptic processes. This suggests that Tau may have a role in membrane trafficking assisting in stabilising or transporting proteins. In the synapses, this could relate to processes such as CME and activity-dependent trafficking of membrane or proteins to support plasticity.

Bioinformatics analysis of Tau-interacting proteins suggests roles in scaffolding and transport with high enrichment at synapses and cell junctions. a Protein analysis through evolutionary relationships (PANTHER; [306] of proteins that bind Tau based on a co-immunoprecipitation study [276]. The proteins were classified according to their protein class. b Database for annotation, visualisation, and integrated discovery (DAVID GO annotation analysis [195, 196]. The two functional gene groups with the highest enrichment scores are shown for the 68 genes included in the annotation analysis
Clathrin mediated endocytosis is important for the internalisation of extracellular material and maintaining membrane homeostasis to balance exocytosis. CME requires the coordination of many endocytosis-related proteins for the formation of complexes at retrieved clathrin-coated pits on the membrane surface after calcium-dependent calcineurin is activated by neural activity [211, 478, 479]. At the pre-synaptic compartment, CME is the main mechanism through which the synaptic vesicle pool is replenished during physiological activity at the hippocampal synapse [162]. In neurons, it is estimated that ∼90% of all clathrin vesicles are involved in retrieval of synaptic vesicles [147]. At the post-synaptic compartment, CME regulates activity‐dependent endo- and exocytic trafficking of receptors [404]. CME is essential for activity-dependent AMPAR internalisation and LTD, and can therefore be upregulated by factors that induce synaptic depression such as NMDAR activation [12, 28, 114, 268, 288].
Postulating synaptic roles of Tau based on binding studies
To help us to better interpret the spatial distribution of these proteins, Fig. 4 shows proteins that have functional roles inside synaptic compartments, and have been shown to be capable of binding to monomeric Tau by co-immunoprecipitation studies [276]. Many of these proteins appear to be related to clathrin-mediated endocytosis, and vesicle cycling pathways in synapses. These proteins have been mapped onto pathways that occur in the synaptic compartments to come up with a potential role of endogenous Tau which subsequently may become impaired during the progression of AD pathogenesis (Fig. 4). It is important to note that while the binding partners of Tau have been described, the functional roles of these interactions have not been experimentally proven to be directly linked to Tau and must therefore be seen as discussion points. GluA2 and AMPARs are not known to be direct binding partners of Tau but have been added as a potential candidates, as Tau may modulate the latter by indirect interactions with PICK1 (Protein interacting with C kinase) [370]. The full list of synaptic proteins that Tau is capable of binding to, are listed in Supplementary Table 1.

A schematic of possible roles of endogenous Tau in synaptic plasticity. The figure shows proteins that have been found to interact with Tau (though not necessarily functionally). Many of these proteins appear to map to clathrin-mediated endocytosis pathways that relate to synaptic vesicle trafficking in the pre-synaptic compartment (a) or receptor trafficking in the post-syanptic compartment (b) and are vital for synaptic transmission. Image based on data from [276]. Tau is not known to directly bind PICK1, GluA2, though there is an NMDA-dependent interaction of this complex with phospho-Tau [370]. These proteins are therefore added to the figure in order to highlight specific pathways that have been previously mentioned in the literature [434]
Transferrin, AP2 and dynamin-1; possible role of tau in early stages of CME
Constitutive CME is required for plasma membrane protein and lipid turnover, endocytosis of activated growth-factor receptors, low-density lipoprotein and iron-saturated transferrin uptake [48, 79, 302]. Tau is capable of binding transferrin [276], which binds Fe3+ ions prior to clathrin-mediated uptake into cells via transferrin receptors. Transferrin receptor clustering is important for the initiation of clathrin coated pits for the earliest stages of CME to occur [275, 277]. Transferrin receptors are therefore also important for activity-dependent AMPAR internalisation that is dependent on CME and required for LTD as they recruit AP2 (adaptor protein 2). Tau is able to bind AP2, the protein responsible for clathrin pit formation [406, 448]. Loss of transferrin receptors also reduces LTP [264]. In the pre-synaptic compartment, following neuronal stimulation with KCl, Tau has been shown to relocate to the plasma membrane and to colocalise with, though not evidently bind, synaptic vesicle protein CSPɑ/DnaJC5 (Cysteine String Protein- ɑ) [503]. CSPɑ regulates endocytosis by binding dynamin-1, another protein capable of binding Tau that is involved in activity-dependent CME of synaptic vesicles through vesicle scission [126, 127, 386, 478, 479, 497].
PICK1 makes NMDAR-dependent interactions with endocytic proteins AP2 and dynamin. Following NMDAR stimulation, PICK1–AP2 interactions cluster AMPARs at endocytic zones, and PICK1 can polymerize dynamin-1 to undergo AMPAR endocytosis [130]. The preference of different Tau isoforms to bind to the proteins shown in Liu et al. [276] can be seen in Supplementary Table 1 [237].
Possible role of Tau in SNARE complex formation and exocytosis: syntaxin-1, synaptobrevin, NSF
CSPɑ is also essential for the high Ca2+-sensitivity of exocytosis as it mediates the release of anchored synaptic vesicles by formation of the Ca2+-sensitive SNARE complex (Fig. 4a; [65]. Like Tau, CSPɑ can bind synaptotagmin, and proteins involved in the SNARE complex including syntaxin-1 and synaptobrevin [121, 330, 400, 477]. SNARE complex assembly requires SNAP-25 (Soluble NSF Attachment Protein) and syntaxin-1 to bind to synaptobrevin to exert sufficient force for membrane fusion to occur and to release the vesicle contents into the synaptic cleft [417]. This assembly is disrupted in CSPɑ-deficient mice [403]. CSP-KO in itself can induce neurodegeneration, and in Drosophila prevents the release of neurotransmitters and causes early death [386, 451, 507].as CSPɑ is thought to induce the required structural conformation of SNAP-25 and prevent its degradation by the ubiquitin proteasome system (UPS), which degrades excess or damaged proteins [403] Tau is also able to bind the protein required for SNARE disassembly, NSF (N-ethylmaleimide sensitive fusion protein) (Fig. 4, [276, 417]. The role that CSP may play in Tau-mediated neurodegeneration is being questioned following the finding that CSP expression is downregulated in tauopathy models at timepoints that correspond to impaired synaptic function. In these models, CSPɑ was also found to be neuroprotective, whereby increased expression reduced neuronal loss [445].
CSPɑ/DnaJC5 bound to Hsc70 releases Tau from synapses in what is believed to be a physiological, activity-dependent mechanism [131]. It will be interesting to determine whether CSPɑ loss in tauopathies also reduces activity-dependent Tau release [355], and whether this leads to a clear phenotype. Other DnaJ proteins complex with Hsc70 for disaggregation [141, 331] or degradation [207] of aggregated proteins. Aggregated proteins can directly block CME through competition for Hsc70 [492]. It has been suggested that CSP may act as a chaperone to allow continuous and long-term use of proteins in the synaptic vesicle cycle [125].
ɑ-synuclein and 14–3-3ζ
The fatal phenotype caused by CSPɑ, that prevents vesicle release, is rescued by overexpression of ɑ-synuclein [66]. ɑ-synuclein is another pre-synaptic protein thought to have a role in the synaptic vesicle cycle including endocytosis [454], reclustering [327], and mobility [398, 461] but is found in Lewy body aggregates seen in Parkinson’s disease (for an overview on ɑ-synuclein induced synaptopathy see [45]. Tau can bind to both ɑ-synuclein and β-synuclein [204, 276]. Co-morbid ɑ-synuclein or Lewy-related pathology occur in more than 50% of AD brains, and ɑ-synuclein and Tau have synergistic effects on each other’s aggregation [145, 169, 272]. ɑ-synuclein and Tau are thought to form a membrane-bound complex with the actin cytoskeleton. Destabilisation of the cytoskeleton or the A30P ɑ-synuclein mutation linked to early-onset Parkinson’s disease reduces the formation of this complex [118, 243, 340]. ɑ-synuclein can also induce Tau phosphorylation at serine 262 to cause unbinding from actin and microtubules, and has been shown to be essential for Aβ42-induced Tau toxicity [56, 198, 204].
ɑ-synuclein shares functional homology with the highly conserved regulatory 14–3–3 proteins that are able to bind both ɑ-synuclein and Tau [276, 337]. Tau has also been found capable of binding to the zeta isoform of 14–3-3 proteins (14–3-3ζ), which are enriched in the hippocampus, especially in synapses, and thought to be involved in learning and memory pathways [27, 88, 276, 293, 412, 466]. Overexpression of 14–3-3ζ increases Tau phosphorylation at serine 262, actin unbinding, and depolymerisation of microtubules through the same pathway as ɑ-synuclein, and consequently leads to the degradation of synaptophysin by the UPS [204, 364]. 14–3-3 is also capable of phosphorylation-dependent binding to CSPɑ/DnaJC5 [359], and plays a role in priming exocytosis and enhancing vesicle release through structural rearrangements of the actin cytoskeleton [64, 382]. Alternatively, 14–3-3ζ can coordinate, together with other DnaJ-Hsc70 complexes, the resolubilization of heat-aggregated proteins [486].
V-ATPase
14–3–3ζ has an ATPase activity and helps to regulate vacuolar-type H+-ATPase (V-ATPase) activity [1, 5, 367]. Tau is able to bind to V-ATPase subunit A, required for the acidification of intracellular compartments for maintaining synaptic vesicle proton gradients, protein sorting, and receptor-mediated endocytosis. Loss of this protein impairs late stage exocytosis of synaptic vesicles. Mutations in V-ATPase subunits can cause epilepsy and parkinsonism [168, 240], cognitive impairment, and neurodegeneration [110].
Cytoskeletal plasticity: cofilin, troponomyosins and septin 7
Another interaction 14–3–3ζ can mediate, in concert with CaMKII (Ca2+/calmodulin-dependent protein kinase II) and in opposition with Ca2+/calmodulin-activated phosphatase calcineurin, is the dephosphorylation and activation of the actin organising protein cofilin [229, 500]. Tau can directly bind cofilin, CaMKII and calcineurin [273, 276, 490]. Cofilin can compete with Tau for tubulin binding which has been suggested to cause microtubule instability and promote tauopathies through increasing free Tau available for fibril formation [473]. The ability of both cofilin and Tau to bind to tubulin and actin suggests that they coordinate cytoskeletal plasticity pathways. Ca2+ entry through NMDARs can cause the indirect dephosphorylation and activation of cofilin through calcineurin. This causes cofilin to enter synaptic compartments and depolymerise F-actin to cause spine shrinkage. Overactivation of this pathway during stress can cause the transient cofilin-actin rod response that bundles actin and releases ATP. This response can occur in Alzheimer’s disease causing long-term F-actin bundles in axons and neurites [19, 287, 323]. In Tau-P301S mice, activated cofilin is also required for tauopathy, reduced synaptic integrity (as shown by depleted drebrin and synaptophysin, and LTP deficits; these deficits were rescued in mice having a 50% reduction in cofilin concentrations [473]. This reduction in cofilin also rescued loss of synaptic proteins and impairment to LTP in APP/PS1 mice [472]. Cofilin is important for spine dynamics during LTP and LTD, as well as for AMPAR trafficking, for example, following chemical induction of LTP, activated cofilin results in increased surface AMPARs [69, 165, 504].
Tropomyosin is another actin-associated protein that stabilises F-actin and that Tau is capable of binding to in vitro [276]. Tropomyosin recruits cofilins to F-actin and they help to determine the structure of pre-synaptic F-actin and the stiffness of the pre-synaptic membrane [31, 51, 430].
Septins are seen as the fourth filament protein in neurons alongside actin, tubulin, and neurofilaments. They help regulate synaptic vesicle trafficking and neurotransmitter release [296], and septin 7 interacts with the exocyst complex [193]. Septins can bind with actin during various stages of CME and endosomal sorting, which is required for the maintenance of mature synapses, and synaptic plasticity such that septin 7 expression is up-regulated during spatial memory formation [117, 457]., which is impaired in AD. In dendrites, septin 7 binds to the membrane of hippocampal neurons to regulate dendrite branching and spine morphology but it can also prevent the lateral diffusion of membrane proteins out of spines [122, 481]. Following phosphorylation, septin 7 stabilises post-synaptic density (PSD) protein PSD95 during spine maturation [483]. Several septins are also found in NFTs [232].
Ca 2+ -dependent interactions: calcineurin, GAP-43/neuromodulin, neurogranin, neurochondrin, calmodulin, CaMKII, and CaMKv
Further to calcineurin activating cofilin, Tau is capable of binding calcineurin, GAP-43/neuromodulin, neurogranin, neurochondrin, calmodulin, CaMKII, and CaMKv (calmodulin kinase-like vesicle-associated), which have been shown to interact at synapses [36, 87, 266, 276, 349, 415, 490]. Calcineurin has long been known to regulate activity-dependent cytoskeletal remodelling; it is able to dephosphorylate Tau to polymerise and stabilise microtubules opposite to CaMKII [159]. Overexpression of Tau or Aβ oligomers have been shown to increase the activation of calcineurin [384, 487]. Calcineurin inhibition however, can rescue spine density and plasticity deficits in AD model mice [63, 366, 384]. Calcineurin can regulate the available concentration of calmodulin at the pre-synaptic compartment through dephosphorylation of GAP-43, which also causes actin capping [40, 178, 256]. GAP-43 can cause presynaptic membrane changes, and is thereby involved in neurotransmitter release, endocytosis [329], synaptic vesicle recycling, LTP, and spatial memory formation [96]. GAP-43 has a high affinity for calmodulin at low Ca2+ concentrations, sequestering calmodulin at the cell membrane until Ca2+ influx occurs [11]. Tau helps maintain this cytoplasmic concentration of calmodulin available through Ca2+-dependent binding to prevent it from entering the nucleus [24, 415]. Calmodulin is important for the activation of CaM-dependent kinases. CaMKII has been shown to phosphorylate Tau at sites including serine 262 that promotes microtubule unbinding [411] and is thought to be involved in the formation of fibrillar Tau [179, 490]. CaMKv is upregulated following AMPAR activity to cause actin rearrangement, and is said to be the ‘convergence point for the transduction of Ca2+ signals to the neuronal cytoskeleton’ [266]. The calmodulin/calcineurin pathway has also been show to directly modulate endocytosis through dephosphorylation of endocytic proteins including dynamin-1 [74, 75, 433].
MBP
Myelin basic protein (MBP) is the second most abundant protein in the central nervous system. Like Tau, MBP is an intrinsically disordered protein with multiple isoforms, and can be differentially phosphorylated suggesting that it has a role in neuronal signalling. Like Tau, MBP is also capable of binding tropomyosin, actin, microtubules, calmodulin, and clathrin [39, 106, 314, 358]. As MBP can act as a clathrin adaptor protein, it has been suggested that it may form a bridge between clathrin-coated vesicles and microtubules [358]. MBP can also polymerise, bundle and crosslink actin filaments and microtubules, and act as a tether for SH3-domain proteins to lipid membranes (such as for the SH3-domain of Fyn-kinase) [38, 39, 188]. Although Tau is able to bind MBP, Tau and MBP appear to have analogous roles in neurons versus oligodendrocytes, respectively. MBP is important for formation and stabilisation of the cytoskeleton in oligodendrocytes [113, 138]. MBP forms prion-like aggregates, in parallel to the accumulation of insoluble and phosphorylated Tau, and can occur due to reduced cholesterol levels and other lipids that cause MBP-membrane unbinding [133, 285, 470]. Like Tau, MBP can aggregate due to polyanionic factors such as lipids or lysosomal glycosaminoglycan (GAG) proteins [146].
Fyn
A small amount of Tau is found in dendrites and spines under physiological conditions and can be phosphorylated following NMDAR activation [317]. Phospho-Tau can facilitate the interaction of Fyn kinase, PSD95 and NMDARs to stabilise their position in the postsynaptic density [160, 201, 255, 313, 317]. The interaction of Tau and Fyn was previously predicted to cause the translocation of Tau to cholesterol-rich lipid rafts to act as a signalling protein [259]. The entry of Tau into synapses is also thought to regulate the activity-dependent transportation of synaptic proteins, including Fyn kinase, GluA1 and PSD95 [225, 317]. Transport of PSD proteins is required to allow synaptic plasticity [115, 425]. At the postsynaptic density, this complex has been implicated as the mechanism of Aβ -induced excitotoxicity caused during AD pathology through overactivation of NMDARs and phosphorylation of Tau at tyrosine 18 (Y18) [313, 389]. Y18 is also associated with the formation of insoluble Tau aggregates [47, 258]. Tau knockout has been shown to be neuroprotective by ameliorating Aβ-induced excitotoxicity, by causing the exclusion of Fyn from the post-synaptic compartment and by destabilisation of PSD-95 [200, 201]. Another recent paper has shown that post-synaptic FTLD-mutant Tau causes aberrant Fyn nanoclustering in hippocampal dendritic spines [338]. Fyn knockout causes impairments to LTP and spatial learning in mice, this is specific to Fyn as opposed to other nonreceptor tyrosine kinases [163]. Other than being linked with excitotoxicity, the phosphorylation of Tau at serine 396 has also been shown to be required for hippocampal LTD [370]. Although the exact mechanism was not described, it was shown that Tau is necessary for an activity-dependent molecular interaction between GluA2 and PICK1, both of which are required for the internalisation or stabilisation of intracellular pools of AMPARs [170, 280, 370, 443]. GluA2 subunits in AMPARs render them Ca2+ impermeable [52, 186, 418]. The GluA2 subunit can also bind NSF and AP2 for stabilisation versus internalisation [99, 262, 332, 336, 419, 480]. As NSF and AP2 binding sites on GluA2 overlap, they are thought to elicit different functions, which may explain the complexities of AMPAR trafficking [262]. As well as its involvement in the GluA2-PICK1 interaction, Tau has also been shown capable of binding to NSF and AP2 by co-immunoprecipitation studies [276]. The function of this binding may be related to NMDA-induced trafficking of AMPARs from synapses, whereby Tau deficiency results in reduced GluA2 subunits in the postsynaptic density during chemical LTD [434]. GluA2 also regulates metabotropic glutamate receptor-dependent LTD (mGluR-LTD) through a pathway involving cofilin-mediated actin reorganisation [506].
Tau can bind several proteins, interact with, or is directly involved in various stages of CME and synaptic trafficking, the proteins of which are also genetic risk factors for AD. These include the top three genetic risk factors, APOE [428], BIN1 [67, 217] and PICALM [174, 248]. Similarly, many genetic risk factors for AD have been linked to CME though these proteins may not be known to directly bind to Tau, and will therefore be discussed in more detail (Fig. 5).

The genetic risk factors for Alzheimer’s disease are involved in synaptic plasticity. Proteins that have genetic links to AD (italicised) are mapped into the pathways described for proteins that are capable of binding to Tau. Proteins used to demonstrate the pathway but that do not bind directly to Tau (purple) or are known genetic risk factors (italicised) are also shown
Possible roles of synaptic Tau based on interactions with proteins identified as genetic risk factors in Alzheimer’s disease
Another hint that CME may be closely associated with AD pathology comes from genetic mutations that can lead to late onset Alzheimer’s disease (LOAD) including PICALM [174, 248], EXOC3L2 (Exocyst complex component 3-like 2; [335, 401], BIN1 [217], MEF2 (myocyte enhancer factor-2; [61], KIBRA [83], RIN3 (Ras and Rab Interactor 3; [217], and Sorla [379]. Figure 5 illustrates how these genetic risk factor proteins may further interact with pathways involved in synaptic plasticity, or with the proteins previously discussed in Fig. 4.
PICALM
Picalm is an adaptor protein required for clathrin-mediated endocytosis by directly binding to clathrin, phosphatidylinositol, and AP2 to help form clathrin-coated pits on the cell membrane [109, 305, 441]. Overexpression or degradation of Picalm blocks endocytosis, and has also been related to the extent of Tau pathology [7, 8, 228, 385, 441]. Picalm may be involved in CME-mediated uptake of Tau species and is also found bound to fibrillar Tau [6, 475]. Increased concentrations of calpain-cleaved Picalm are found in the AD brain, which has been hypothesised to impair endocytic function in AD [6].
APP
Genetic mutations in the Amyloid precursor protein (APP), from which the fragment Amyloid-beta (Aβ1-42) arises, can be a direct cause of AD [149]. APP is only partially processed at the cell surface but requires CME for further processing and its synaptic activity-dependent release [73, 239, 247, 455]. APP has functional roles in regulating spine density whereby overexpression or knockdown of APP causes an increased or reduced number of spines, respectively [261]. Aβ can cause dysregulation of intracellular Ca2+ concentrations [13, 244], and synaptic depression, thought to occur by aberrant trafficking and excessive removal of AMPARs [166, 191]. This could induce LTD through NMDA receptor- and metabotropic glutamate receptor-mediated signaling [166, 281]. APP has been shown to bind to Tau [199] without any known role, a functional link between these proteins however occurs via Fyn kinase. As previously discussed, both Tau and Aβ have been related to neuronal excitability, however memory deficits, excitotoxic seizures, and seizure-induced premature mortality of mice overexpressing the Swedish mutation of APP (APPSwe) was reduced when nonreceptor tyrosine kinase, Fyn, was unable to enter post-synaptic compartments due to the expression of a truncated form of Tau [200]. For review on Aβ and trafficking see Perdigão et al. [346]. For reviews on synaptic mechanisms of Tau pathology see [107, 346].
BIN1, RIN3 and CD2AP
BIN1 is involved in endocytosis by binding to clathrin and AP2 during early endosome formation, and in the exocytosis of vesicles from recycling endosomes to the cell surface [57, 397]. Loss of BIN1 causes impaired presynaptic vesicle distribution and release, reduced synaptic density due to membrane trafficking, and an altered presynaptic protein clustering [381]. In the postsynaptic compartment, this loss also causes reduced surface expression of the GluA1 AMPAR subunit in the post-synaptic compartment and altered AMPAR-mediated synaptic transmission [397]. Overall, BIN1 knockout leads to deficits in synaptic transmission, and impaired spatial memory consolidation at the behavioural level [381]. Tau reduction has also been shown to reduce network hyperexcitability mediated by BIN1-interactions with L-type voltage-gated calcium channels (LVGCCs) [458]. BIN1 can rearrange the actin cytoskeleton and stabilise Tau-induced actin bundles [108]. BIN1 and Tau colocalise at the actin cytoskeleton [421] as has also be shown for BIN1 with the genetic risk factor protein RIN3 [217, 383] to mediate receptor-induced endocytosis and transport of vesicles from the plasma membranes to early endosomes [212]. BIN1 and RIN3 are both able to bind CD2AP (CD2 associated protein), a protein found in cases of sporadic AD [185, 324, 328, 383, 439]. This interaction has been related to regulating cholesterol, which has been linked to AD through genetic risk factors involved with cholesterol homeostasis [62, 295], and increased incidence of AD in hypercholesterolemia [295, 341, 407]. In Drosophila, loss of the CD2AP ortholog, cindr, causes a combination of endocytic and exocytic synaptic defects including impairments of synaptic vesicle recycling and release [334, 408] and enhanced Tau-induced neurodegeneration [408]. Loss of cindr causes defects in endocytosis as shown by depression of synaptic responses during high-frequency stimulation, as mutants are unable to sustain synaptic vesicle release [30, 173, 237]. It is thought that impairment to synaptic vesicle endocytosis may be through the ability of CD2AP to link the binding of clathrin and actin via cortactin [282, 334, 499]. CD2AP also binds actin, whereby its loss of function stabilises F-actin [210, 469]. Exocytosis of vesicles is also affected by cindr having a role in presynaptic Ca2+ homeostasis. This is thought to occur through binding 14–3-3ζ to regulate the UPS for activity-dependent proteostasis to control the degradation of proteins involved with plasticity [334]. It is possible that Tau influences this pathway as 14–3-3 can increase Tau aggregation and co-immunoprecipitation studies have shown that Tau can bind to 14–3-3ζ, though the functional relevance of this remains to be determined [182, 276]. The UPS has an important role in endocytosis, protein trafficking, the size of post-synaptic potentials and the formation of long-term memory [202, 422, 502]. Alpha-synuclein, synaptophysin, syntaxin1, SNAP-25, synapsin1, GluA2, PSD95 and plasma membrane calcium ATPase (PMCA) have been identified as pre-synaptic targets for the UPS [29, 71, 78, 283, 334, 344, 403, 471]. AMPAR subunits are targets for degradation by the UPS for LTD induction following uptake by CME [115]. This occurs through ubiquitination of PSD-95 which otherwise acts as tether for AMPARs and shields them from degradation [78]. Burbea et al. (2002) hypothesize that there is an intricate link between ubiquitination, clathrin-mediated endocytosis and UPS degradation, suggesting activity-dependent ubiquitin-conjugation of AMPARs to influence AMPARs at synapses [344]. Unregulated deubiquitination of synaptic proteins can also result in synaptic overgrowth and blocked release of synaptic vesicles [102]. Like Picalm, Bin1 and CD2AP depletion can cause impaired vesicle recycling or release and result in an accumulation of Aβ and other proteins inside of early endosomes [450]. This may also influence the aggregation of Tau, which is promoted at low pH inside of endolysosomal compartments [307].
MEF2C
MEF2C is a transcription factor that regulates hippocampal-dependent learning and memory through the control of dendritic spine density, miniature excitatory postsynaptic currents (mEPSCs) frequency, probability of vesicle release, and activity-dependent AMPAR trafficking through its presence in the pre- and post- synaptic compartments [20, 77, 221, 368]. MEF2C is therefore important for activity-dependent refinement of synaptic connectivity in homeostatic plasticity [20].
EXOC3L2
EXOC3L2 is a component of the exocyst, involved in the exocytosis of vesicles containing hormones, extracellular components, membrane lipids, and for the regulation of the readily releasable pool of synaptic vesicles via the binding of NSF and SNARE proteins including syntaxin1 [189, 354]. The activity-dependent addition of membrane to the synapse via the exocyst is required for synaptic plasticity [442]. The exocyst interacts with postsynaptic density proteins to regulate NMDAR and AMPAR trafficking and exocytosis at the postsynaptic membrane [143, 390]. Overall, the exocyst acts as an integrator between the secretory pathway and cytoskeleton, including septins, actin, and microtubules, to localise vesicles to release sites [192, 436, 456].
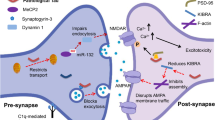
KIBRA
KIBRA is enriched in brain regions involved with memory such as the hippocampus and cortex, where it is found in the perinuclear and somatodendritic regions of neurons, particularly at postsynaptic densities. KIBRA acts as a postsynaptic scaffold protein that connects the cytoskeleton with signalling molecules [208]. KIBRA is capable of binding activity-dependent AMPAR regulators including NSF, PSD-95, PICK1, GluA1, GluA2 and GRIP1 (Glutamate receptor-interacting protein 1), and is involved in AMPAR recycling, through its ability to bind with the exocyst complex [286, 380]. By binding the exocyst, KIBRA can direct PKMζ (Protein kinase Mzeta), a brain-specific variant of PKCzeta that plays important roles in memory formation, to required locations which is why it has been hypothesised to be a ‘synaptic tagging’ protein [489]. PKMζ is necessary and sufficient for enhanced synaptic transmission during LTP maintenance and acts by increasing the number of postsynaptic AMPARs [270, 271]. PKMζ, but not other PKC isoforms, has been found in NFTs in brain regions specifically involved with memory loss in AD, whereas they are not found in NFTs of control brains without memory impairment [85]. As KIBRA is involved in AMPAR recycling, knockdown of KIBRA results in an increase of AMPAR recycling following NMDAR internalisation [286]. This mechanism is impaired by Tau in AD. Acetylated forms of Tau seen in AD brains (K274 and K281) promote memory loss by preventing the recruitment of KIBRA into post-synaptic compartments, causing impaired activity-dependent postsynaptic actin remodelling and AMPAR insertion [447].
PTK2B
PTK2B encodes Pyk2 (proline-rich tyrosine kinase 2), a susceptibility factor for AD [248]. Fyn kinase can activate Pyk2, which then binds and phosphorylates Tau [59, 263, 361]. Pyk2 interacts with NMDARs, dependent upon binding PSD95, to phosphorylate NR2 subunits and increase receptor conductance during the induction of LTP [238, 399, 463]. Pyk2 binding to PSD95 is activity-dependent as it requires activation of calmodulin by Ca2+ (Fig. 5, [25]. Pyk2 is fundamental to synaptic dysfunction triggered by Aβ as mice lacking Pyk2 were protected from synapse loss and memory impairment [388].
LDLR and ApolipoproteinE
The LDLR (low density lipoprotein receptor) has been linked to AD both through direct mutations and through interaction with ApoE (apolipoprotein E), the highest genetic risk factor for LOAD [81]. LDLR is involved in cholesterol uptake via CME. Cholesterol is essential for the maintenance of mature synapses to increase the number of synaptic vesicles and release sites, and overall release efficacy [148, 410]. Another low density lipoprotein receptor found at the postsynaptic density, LRP1, has been found to be the major receptor for monomeric or oligomeric Tau uptake, and can also cause age-dependent synaptic loss and neurodegeneration in a knockout mouse model [80, 275, 277, 300, 369]. Oligomeric hyperphosphorylated Tau can bind and be released from cells by HSPGs prior to binding LRP1 [80, 218, 304]. Apolipoprotein E deficient mice show heparan sulfate-enhanced low density lipoprotein (LDL) aggregates that are taken up by LRP1, causing cholesteryl ester accumulation in macrophages and production of atherosclerotic plaques [267, 289]. LRP1 is also responsible for the endocytosis and degradation of Aβ, or Aβ-ApoE complex, whereby amyloid pathology is enhanced by the APOE4 allele, dependent upon LRP1 uptake [90, 214, 437]. Alongside this, LRP1 is involved in the endocytosis of APP, which is required for its processing of Aβ peptides [234].
The ApoE type 4 allele, the highest genetic risk factor for AD [81], causes impaired vesicle cycling to the cell surface resulting in intracellular cholesterol accumulation [181, 428]. This impaired cycling also traps AMPARs and NMDARs clustered with ApoE receptors inside of endocytic vesicles, causing synaptic dysfunction [70]. Aβ is capable of regulating the surface expression versus endocytosis of NMDARs, potentially through disruption to their binding with PSD95 [377, 414]. Impaired cycling by ApoE4 is thought to exacerbate Aβ induced endocytosis of AMPA and NMDARs, and ApoE4 knock-in mice show increased sensitivity to the blockade of LTP by oligomeric Aβ and by failing to restore Reelin signalling [70, 414, 449]. In a pathway suggested by Durakoglugil et al. [112], Aβ competes against nonreceptor tyrosine kinase signalling, predominantly by Fyn kinase, over activation or antagonism of the Reelin pathway. In the absence of phosphorylation, the microtubule binding region of Tau is capable of binding ApoE3 but not ApoE4, [429]. ApoE4 also increases Tau-mediated neurodegeneration as compared with other alleles or knockout of ApoE, which is neuroprotective [405].
There are many genetic risk factors linked to AD that involve proteins required for CME, vesicle cycling or exocytosis. Alongside PICALM, BIN1, and Apolipoprotein E, which directly bind Tau, a number of proteins link genetic risk factor proteins with synaptic proteins capable of binding Tau and which are linked to vesicle cycling pathways [67, 276, 421]. Such proteins, recurring through this review, may include AMPARs, 14–3-3ζ, NSF, PSD95, Fyn kinase and clathrin itself. Many of the proteins which are able to bind Tau, or are related to LOAD and familial AD, also bind to the actin cytoskeleton and may therefore act as a linker between structural and signalling roles required for synaptic plasticity and memory mechanisms.
Tau as a linker between CME, vesicle trafficking, and the cytoskeleton
Tau is able to simultaneously bind actin and microtubules, and induce the polymerisation of actin along microtubule tracks [116]. As Tau has been linked with physiological and pathological actin structures, it is worth discussing how these cytoskeletal arrangements may link with CME and previously discussed synaptic trafficking mechanisms.
CME is intricately linked to actin dynamics though the exact stage, location and function of these associations in mammalian cells have been strongly debated. It appears that actin is involved with the invagination of membrane and late stages of CME [139]. Actin has been suggested to play structural role and mechanical roles in exerting force during scission and constriction steps required for vesicle endo- and exocytosis [203, 363, 413]. Actin is the main cytoskeletal component of synaptic compartments and spines, and is thought to facilitate the cytoarchitectural changes required for synaptic plasticity [60, 104, 299].
Actin has also been suggested to have an active role in the segregation of vesicle populations to determine their retention or release at the membrane surface [72]. In the pre-synaptic compartment, loss of F-actin integrity has shown impairment to synaptic vesicle release or recycling in multiple studies [409, 459, 501]. This greatly reduces the number of synaptic vesicles in the stimulated condition due to the inability to retrieve vesicles from the plasma membrane [409]. Stabilisation of F-actin by phalloidin also prevents neurotransmitter release [32, 350]. Pre-synaptically, the bundling and stabilisation of actin by phalloidin are far more dramatic following the induction of action potentials, which cause the assembly of filamentous actin fibres, tethered with vesicles, from the endocytic zone to the periphery of the vesicle pool [409]. This may be similar to the effects seen when Tau is shown to cause increased resistance to depolymerising drugs by directly stabilising actin [136]. Decreased actin dynamics through actin bundling has previously been associated with senescence, whereas knockdown of the actin bundling protein SM22/transgelin increases longevity [161].
F-actin also determines the mobility of receptors between the cell surface and the cytoplasm [2]. In the post-synaptic compartment, F-actin stabilises receptors in dendritic spines, whereby its disruption decreases the number of NMDAR and AMPAR clusters. In hippocampal neurons, post-synaptic actin depolymerisation causes AMPAR endocytosis, similar to that induced by glutamate [2]. By contrast, stabilisation of F-actin can inhibit AMPAR internalisation [2]. The specific linker proteins that allow F-actin to facilitate these functions are not fully known. Much is still poorly understood about how CME and its role in vesicle cycling and plasticity links with actin and its mechanical and structural roles within the synaptic compartments. A number of proteins are responsible for actin dynamics, including the previously discussed proteins tropomyosin, cofilin, and adducin. Alongside the better known pathology of Aβ plaques and neurofibrillary tangles seen in AD, actin-depolymerizing factor (ADF)/cofilin-actin rods can also occur [310] which may be precursors to Hirano bodies, actin-rich inclusions that contain tropomyosin, Tau, and cofilin, among other proteins [140, 146].
As Tau is capable of binding the filament proteins septin7, tubulin and actin, Tau may act like a Velcro that reversibly positions structures into place for signalling pathways and to restructure proteins depending on the levels of synaptic activity. This role may balance the level of proteins available for function versus their degradation through cleavage by calpain and the UPS. This has already been discussed for the protein synapsin1, however numerous other proteins including actin, cortactin, NMDAR and AMPAR subunits, PSD95, SNAP-25, GAP-43, and GRIP are either targeted to scaffolding proteins such as PSD95 for stabilisation, or else marked by cleavage or ubiquitination for degradation [9, 33, 78, 348, 357, 457, 474, 494]. Reduced post-synaptic glutamate receptor localisation was proposed to be due to a depletion of PSD95 in the post-synaptic compartment, resulting in smaller postsynaptic densities following a reduction or mutation of Tau [325, 465]. The UPS is only responsible for the local degradation of a subset of synaptic proteins, and its function is regulated by synaptic activity or neural growth factors (NGF) to adjust the concentration of proteins important for synaptic function. This activity-dependent or NGF-dependent UPS function can thereby feedback to regulate neurotransmitter release and synapse elimination [206, 254, 372, 422].
The role of Tau trafficking, its link to endocytosis, cholesterol and the cytoskeleton
A question that emerges from the above findings is whether the presence of Tau at different locations in the cells are due to internal Tau translocations or due to Tau being released into the extracellular space and its re-uptake by neighbouring neurons. There have been many reports on Tau trafficking and its uptake mechanism. It is thus interesting to note that several endocytosis-related pathways are involved in Tau trafficking and thus may explain why Tau pathology is linked to these different pathways (for a review see [50]. Furthermore, there are several studies emerging highlighting the role of the extracellular and intravesicular environment on protein misfolding involving high sodium, zinc, and calcium ion concentrations and solvents [319, 426, 426], low pH [307], presence of glucosaminoglycans [227, 321] to name a few. Recent studies [80, 369] also highlights another potentially important factor, namely cholesterol. Cholesterol has long been seen as a player in AD and many other neurodegenerative diseases, such as PD, Nieman Pick’s disease Type C (NPC) and ALS (for a review see [294]. The increased membrane-associated cholesterol concentration in the brains of patients with sporadic AD correlates with the disease severity [86, 291, 482]. In ageing neurons cholesterol is mainly taken up by endocytosis, as opposed to cell-autonomous cholesterol synthesis [137], and thus extracellular Tau and cholesterol may end up in the same endosomal compartment. Increased accumulation of Tau and cholesterol in endosomes may interfere with the WASH complex, similar to what has been observed in VPS35 (vacuolar protein sorting 35) related to PD [495], and thus affect the actin skeleton and endosome-lysosome networks [97, 154, 155]. Impaired cholesterol transport would not only reduce cholesterol being supplied to other organelles such as the mitochondria and the plasma membrane and lipid rafts, but also reduce the number of synaptic vesicles being formed.
Conclusion
In AD, Tau is commonly discussed with regards to pre-synaptic [301, 503] versus post-synaptic [200] pathology, though little emphasis is put on mechanisms that may target common plasticity pathways such as synaptic protein and lipid trafficking, and vesicle cycling. In this review paper, we have highlighted synaptic proteins that Tau is capable of binding to, or genetic risk factor proteins, and mapped these to pathways that relate to plasticity mechanisms that would directly link Tau with impaired memory, a primary symptom of AD [23]. It is also important to note that changes to memory mechanisms occur even as a result of healthy ageing. In general, it has been shown that in older animals LTP is less robust and requires stronger input whereas LTD is enhanced [22, 318, 333, 446]. AD pathology may further hijack these mechanisms leading to symptoms of dementia.
A small amount of Tau has been detected at synapses under physiological conditions. Due to the activity-dependence of Tau translocation to synapses, it has been hypothesised that Tau may have a supporting structural role during development and plasticity [190, 132, 317, 392, 434, 438]. Tau has been suggested to coordinate microtubule and actin dynamics to allow structural alterations during activity, as Tau binds F-actin with a physiological function [136, 177, 493]. Tau has been found to be capable of binding to a number of proteins with roles associated with clathrin-mediated endocytosis (Fig. 4). It is plausible that Tau acts as either a tethering protein between vesicles, similarly to synapsin, at least during pathological conditions [301]. This binding may occur with microtubules, septin, actin-mediated mechanisms, or HSPG extracellular matrix for either supplying, stabilising, or transporting components required for plasticity. Phosphorylation-dependent mechanisms that change protein interactions and synaptic scaffolding may become impaired in pathways leading to NFTs [317]. Synaptic vesicles and exocyst cycling, and receptor targeting may be impacted during Tau pathology. Postsynaptic roles in Tau pathophysiology have been related to AMPAR or NMDAR localisation, trafficking or functioning [92, 190, 200, 313, 434, 465]. Hoover et al. [190] showed, using rTgP301L mice, that Tau mutation or hyperphosphorylation impaired trafficking or anchoring of AMPARs and NMDARs. Multiple indirect mechanisms of NMDAR or AMPAR-dependent impairment have been shown through changes to import Fyn kinase, PSD95, and KIBRA proteins into post-synaptic compartments [200, 447, 465].
Based on recent evidence from the literature, we hypothesise that Tau serves as a scaffold to bind the cytoskeleton and to regulate its interactions with key synaptic targets, particularly in coordinating CME at both the pre- and post- synaptic compartments. A similar role for Aβ in CME and clathrin-dependent membrane and protein trafficking pathways, which is known to affect synaptic vesicle endocytosis and exocytosis, has already been posited in AD [223, 346, 485], in schizophrenia and bipolar disorder [396]. Aβ42 oligomers are known to directly interact with Syntaxin 1a [485] Synaptophysin [387], or indirectly interfere with dynamin through NMDAR activation [222, 224], and Synapsin1 [274, 292, 342]. Although we have listed many possible pathways by which Tau may mediate its role at the synapse based on its binding ability, not all of these may have functional relevance or be directly related to AD pathology. These pathways may however highlight the intricacies of the dysfunction that may occur, or at least show the complexity of the etiology and progression of AD.
Outstanding questions
Does Tau have physiological roles in the pre- and/or post-synaptic compartments for pathways related to vesicle cycling and protein trafficking for plasticity, or is its localisation in synaptic compartments purely pathological?
What is the phenomenon that causes the conversion of monomeric to multimeric Tau species? Does Tau aggregation impair any physiological roles of synaptic Tau and if so how and at what point(s) during the aggregation pathway? Is pathology to pathways involving physiological Tau directly responsible for memory impairment seen in AD?
At what stage does pathological phosphorylation of Tau occur and how does this deter from physiological phosphorylation pathways and normal function?
Is the presence of Tau at different locations in cells due to internal Tau translocations or due to Tau being released into the extracellular space and its re-uptake by neighbouring neurons? And therefore, how does the extracellular environment and Tau uptake into the endo/lysosomal pathway affect Tau location and pathology?
Of the proteins discussed in this review as being capable of binding Tau, which of these interactions have functional roles inside of neurons? Are these interactions affected by multimeric Tau and could they be therapeutically targeted?
Abbreviations
- Aβ:
-
Amyloid-beta
- AD:
-
Alzheimer’s disease
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- CME:
-
Clathrin mediated endocytosis
- LOAD:
-
Late onset Alzheimer’s disease
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- MAPT:
-
Microtubule-associated protein Tau
- MTB:
-
Microtubule binding repeat region
- NFT:
-
Neurofibrillary tangle
- NMDAR:
-
N-methyl-D-aspartate receptor
- UPS:
-
Ubiquitin proteasome system
References
Al-bataineh M et al (2004) AMPK regulates the vacuolar proton-ATPase via 14–3–3 proteins (1109.11). FASEB J. https://doi.org/10.1096/FASEBJ.28.1_SUPPLEMENT.1109.11
Allison DW et al (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18(7):2423–2436. https://doi.org/10.1523/jneurosci.18-07-02423.1998
Alquezar C, Arya S, Kao AW (2021) ‘Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol. https://doi.org/10.3389/fneur.2020.595532
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124(3):319–335. https://doi.org/10.1002/cne.901240303
Alzamora R et al (2013) AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am J Physiol Renal Physiol 305(7):F943. https://doi.org/10.1152/ajprenal.00303.2013
Ando K et al (2013) Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol 125(6):861–878. https://doi.org/10.1007/s00401-013-1111-z
Ando K et al (2016) ‘Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and pick disease. Neurobiol Dis 94:32–43. https://doi.org/10.1016/j.nbd.2016.05.017
Ando K et al (2020) Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol 139(4):773–789. https://doi.org/10.1007/s00401-020-02125-x
Ando K, Kudo Y, Takahashi M (2005) Negative regulation of neurotransmitter release by calpain: a possible involvement of specific SNAP-25 cleavage. J Neurochem 94(3):651–658. https://doi.org/10.1111/j.1471-4159.2005.03160.x
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human.tau. gene. Biochemistry 31(43):10626–10633. https://doi.org/10.1021/bi00158a027
Andreasen TJ et al (1983) Purification of a novel calmodulin binding protein from bovine cerebral cortex membranes. Biochemistry 22(20):4615–4618. https://doi.org/10.1021/bi00289a001
Anggono V, Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. https://doi.org/10.1016/j.conb.2011.12.006
Arbel-Ornath M et al (2017) Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener 12(1):27. https://doi.org/10.1186/s13024-017-0169-9
Arendash GW et al (2004) Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer’s Disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res 1012(1–2):29–41. https://doi.org/10.1016/j.brainres.2004.02.081
Arendt T et al (2003) Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci 23(18):6972–6981. https://doi.org/10.1523/jneurosci.23-18-06972.2003
Arriagada PV et al (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42(3 Pt 1):631–639. https://doi.org/10.1212/wnl.42.3.631
Arriagada PV, Marzloff K, Hyman BT (1992) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42(9):1681–1688. https://doi.org/10.1212/wnl.42.9.1681
Ball MJ et al (1985) A new definition of alzheimer’s disease: a hippocampal dementia. Lancet 325(8419):14–16. https://doi.org/10.1016/S0140-6736(85)90965-1
Bamburg JR et al (2010) ADF/Cofilin-actin rods in neurodegenerative diseases. Curr Alzheimer Res 7(3):241–250. https://doi.org/10.2174/156720510791050902
Barbosa AC et al (2008) MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Nat Acad Sci 105(27):9391–9396. https://doi.org/10.1073/pnas.0802679105
Barghorn S, Mandelkow E (2002) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry 41(50):14885–14896. https://doi.org/10.1021/bi026469j
Barnes CA, Rao G, Houston FP (2000) LTP induction threshold change in old rats at the perforant path-granule cell synapse. Neurobiol Aging 21(5):613–620. https://doi.org/10.1016/S0197-4580(00)00163-9
Barnett JH et al (2016) ‘The paired associates learning (PAL) test: 30 years of CANTAB translational neuroscience from laboratory to bedside in dementia research. Curr Top Behav Neurosci. https://doi.org/10.1007/7854_2015_5001
de Barreda EG, Avila J (2011) Tau regulates the subcellular localization of calmodulin. Biochem Biophys Res Commun 408(3):500–504. https://doi.org/10.1016/j.bbrc.2011.04.082
Bartos JA et al (2010) Postsynaptic clustering and activation of Pyk2 by PSD-95. J Neurosci 30(2):449–463. https://doi.org/10.1523/JNEUROSCI.4992-08.2010
Bateman RJ et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease . N Engl J Med 367(9):795–804. https://doi.org/10.1056/NEJMoa1202753
Baxter HC et al (2002) Immunolocalisation of 14–3-3 isoforms in normal and scrapie-infected murine brain. Neuroscience 109(1):5–14. https://doi.org/10.1016/S0306-4522(01)00492-4
Beattie EC et al (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3(12):1291–1300. https://doi.org/10.1038/81823
Bennett MC et al (1999) Degradation of α-synuclein by proteasome. J Biol Chem 274(48):33855–33858. https://doi.org/10.1074/jbc.274.48.33855
Bellen HJ, Tong C, Tsuda H (2010) 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci. https://doi.org/10.1038/nrn2839
Bernstein BW, Bamburg JR (1982) Tropomyosin binding to F-actin protects the F-actin from disassembly by brain actin-depolymerizing factor (ADF). Cell Motil 2(1):1–8. https://doi.org/10.1002/cm.970020102
Bernstein BW, Bamburg JR (1989) Cycling of actin assembly in synaptosomes and neurotransmitter release. Neuron 3(2):257–265. https://doi.org/10.1016/0896-6273(89)90039-1
Bi X et al (1997) Characterization of calpain-mediated proteolysis of GluR1 subunits of α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors in rat brain. J Neurochem 68(4):1484–1494. https://doi.org/10.1046/j.1471-4159.1997.68041484.x
Bliss TV, Collingridge GL (2013) Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain 6(1):5. https://doi.org/10.1186/1756-6606-6-5
Bloom GS (2014) Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71(4):505–508. https://doi.org/10.1001/jamaneurol.2013.5847
Boczek T et al (2015) Regulation of GAP43/calmodulin complex formation via calcineurin-dependent mechanism in differentiated PC12 cells with altered PMCA isoforms composition. Mol Cell Biochem 407(1–2):251–262. https://doi.org/10.1007/s11010-015-2473-4
Boekhoorn K et al (2006) Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci 26(13):3514–3523. https://doi.org/10.1523/JNEUROSCI.5425-05.2006
Boggs JM et al (2011) Myelin basic protein binds microtubules to a membrane surface and to actin filaments in vitro: Effect of phosphorylation and deimination. Biochim Biophys Acta Biomembranes 1808(3):761–773. https://doi.org/10.1016/j.bbamem.2010.12.016
Boggs JM, Rangaraj G (2000) Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry 39(26):7799–7806. https://doi.org/10.1021/bi0002129
Bolsover SR (2005) Calcium signalling in growth cone migration. Cell Calcium. https://doi.org/10.1016/j.ceca.2005.01.007
Braak H et al (2011) Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70(11):960–969. https://doi.org/10.1097/NEN.0b013e318232a379
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259. https://doi.org/10.1007/BF00308809
Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121(2):171–181. https://doi.org/10.1007/s00401-010-0789-4
Brandt R, Léger J, Lee G (1995) Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol 131(5):1327–1340. https://doi.org/10.1083/jcb.131.5.1327
Bridi JC, Hirth F (2018) Mechanisms of α-Synuclein induced synaptopathy in parkinson’s disease. Front Neurosci. https://doi.org/10.3389/fnins.2018.00080
Bright J et al (2015) Human secreted tau increases amyloid-beta production. Neurobiol Aging 36(2):693–709. https://doi.org/10.1016/j.neurobiolaging.2014.09.007
Briner A, Götz J, Polanco JC (2020) Fyn kinase controls tau aggregation in vivo. Cell Rep 32(7):108045. https://doi.org/10.1016/j.celrep.2020.108045
Brodsky FM et al (2001) Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. https://doi.org/10.1146/annurev.cellbio.17.1.517
Brown JT et al (2011) Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer’s disease. Neurobiol Aging 32(11):2109.e1-2109.e14. https://doi.org/10.1016/j.neurobiolaging.2011.05.025
Brunello CA et al (2020) Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci. https://doi.org/10.1007/s00018-019-03349-1
Bryce NS et al (2003) Specification of actin filament function and molecular composition by tropomyosin isoforms. Mol Biol Cell 14(3):1002–1016. https://doi.org/10.1091/mbc.E02-04-0244
Burnashev N et al (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. cell.com. https://www.cell.com/neuron/pdf/0896-6273(92)90120-3.pdf. Accessed 11 May 2020.
Busche MA et al (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321(5896):1686–1689. https://doi.org/10.1126/science.1162844
Busche MA et al (2012) Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 109(22):8740–8745. https://doi.org/10.1073/pnas.1206171109
Busche MA et al (2019) Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci 22(1):57–64. https://doi.org/10.1038/s41593-018-0289-8
Cabrales Fontela Y et al (2017) Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat Commun 8(1):1–12. https://doi.org/10.1038/s41467-017-02230-8
Calafate S et al (2016) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17(4):931–940. https://doi.org/10.1016/j.celrep.2016.09.063
Camero S et al (2014) Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J Alzheimer’s Dis 39(3):649–660. https://doi.org/10.3233/JAD-131415
Canobbio I et al (2015) The focal adhesion kinase Pyk2 links Ca2+ signalling to Src family kinase activation and protein tyrosine phosphorylation in thrombin-stimulated platelets. Biochem J 469(2):199–210. https://doi.org/10.1042/BJ20150048
Carlisle HJ, Kennedy MB (2005) Spine architecture and synaptic plasticity. Trends Neurosci. https://doi.org/10.1016/j.tins.2005.01.008
Carmichael RE et al (2018) MEF2A regulates mGluR-dependent AMPA receptor trafficking independently of Arc/Arg3.1. Sci Rep. https://doi.org/10.1038/s41598-018-23440-0
Carter CJ (2007) Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. https://doi.org/10.1016/j.neuint.2006.07.007
Cavallucci V et al (2013) Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuromolecular Med 15(3):541–548. https://doi.org/10.1007/s12017-013-8241-2
Chamberlain LH et al (1995) Distinct effects of α-SNAP, 14–3-3 proteins, and calmodulin on priming and triggering of regulated exocytosis. J Cell Biol 130(5):1063–1070. https://doi.org/10.1083/jcb.130.5.1063
Chamberlain LH, Burgoyne RD (1998) Cysteine string protein functions directly in regulated exocytosis. Mol Biol Cell 9(8):2259–2267. https://doi.org/10.1091/mbc.9.8.2259
Chandra S et al (2005) α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123(3):383–396. https://doi.org/10.1016/j.cell.2005.09.028
Chapuis J et al (2013) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18(11):1225–1234. https://doi.org/10.1038/mp.2013.1
Chen JJ et al (2019) Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem 294(50):18952–18966. https://doi.org/10.1074/jbc.RA119.009432
Chen LY et al (2007) Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27(20):5363–5372. https://doi.org/10.1523/JNEUROSCI.0164-07.2007
Chen Y et al (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci USA 107(26):12011–12016. https://doi.org/10.1073/pnas.0914984107
Chin LS, Vavalle JP, Li A (2002) Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. J Biol Chem 277(38):35071–35079. https://doi.org/10.1074/jbc.M203300200
Cingolani LA, Goda Y (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. https://doi.org/10.1038/nrn2373
Cirrito JR et al (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 58(1):42–51. https://doi.org/10.1016/j.neuron.2008.02.003
Clayton EL et al (2009) The phospho-dependent dynamin-syndapin interaction triggers activity-dependent bulk endocytosis of synaptic vesicles. J Neurosci 29(24):7706–7717. https://doi.org/10.1523/JNEUROSCI.1976-09.2009
Clayton EL et al (2010) Dynamin i phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat Neurosci 13(7):845–851. https://doi.org/10.1038/nn.2571
Cohen NJ, Squire LR (1980) Preserved learning and retention of pattern-analyzing skill in amnesia: dissociation of knowing how and knowing that. Science (New York, NY) 210(4466):207–210. https://doi.org/10.1126/science.7414331
Cole CJ et al (2012) MEF2 negatively regulates learning-induced structural plasticity and memory formation. Nat Neurosci 15:1255. https://doi.org/10.1038/nn.3189
Colledge M et al (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40(3):595–607. https://doi.org/10.1016/S0896-6273(03)00687-1
Conner SD, Schmid SL (2003) Regulated portals of entry into the cell. Nature. https://doi.org/10.1038/nature01451
Cooper JM et al (2020) LRP1 mediates tau endocytosis in a process that is modulated by apolipoprotein E. Alzheimer’s Dementia 16(S3):e045959. https://doi.org/10.1002/alz.045959
Corder E et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923. https://doi.org/10.1126/science.8346443
Coria F et al (1993) Prevalence of age-associated memory impairment and dementia in a rural community. J Neurol Neurosurg Psychiatry 56(9):973–976. https://doi.org/10.1136/jnnp.56.9.973
Corneveaux JJ et al (2010) Evidence for an association between KIBRA and late-onset Alzheimer’s disease. Neurobiol Aging 31(6):901–909. https://doi.org/10.1016/j.neurobiolaging.2008.07.014
Cracco JB et al (2005) Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus 15(5):551–556. https://doi.org/10.1002/hipo.20078
Crary JF et al (2006) Atypical protein kinase C in neurodegenerative disease I: PKMζ aggregates with limbic neurofibrillary tangles and AMPA receptors in Alzheimer disease. J Neuropathol Exp Neurol 65(4):319–326. https://doi.org/10.1097/01.jnen.0000218442.07664.04
Cutler RG et al (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101(7):2070–2075. https://doi.org/10.1073/pnas.0305799101
Dateki M et al (2005) Nervous System Specific Neurochondrin Gene Disruption Neurochondrin negatively regulates CaMKII phosphorylation and nervous system specific gene disruption results in epileptic seizure. J Biol Chem 280(21):20503–20508. https://doi.org/10.1074/jbc.M414033200
Davis RL (1996) Physiology and biochemistry of Drosophila learning mutants. Physiol Rev. https://doi.org/10.1152/physrev.1996.76.2.299
de Calignon A et al (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73(4):685–697. https://doi.org/10.1016/j.neuron.2011.11.033
Deane R et al (2004) LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43(3):333–344. https://doi.org/10.1016/j.neuron.2004.07.017
Decker JM et al (2015) Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++) dysregulation. Acta Neuropathol Commun 3:23. https://doi.org/10.1186/s40478-015-0193-3
Decker JM et al (2016) The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR 2B receptor-mediated excitotoxicity. EMBO Rep 17(4):552–569. https://doi.org/10.15252/embr.201541439
Decker JM, Mandelkow EM (2019) Presynaptic pathophysiology encoded in different domains of tau—hyper-versus hypoexcitability? Adv Exp Med Biol. https://doi.org/10.1007/978-981-32-9358-8_8
DeLorenzo RJ, Freedman SD (1977) Calcium-dependent phosphorylation of synaptic vesicle proteins and its possible role in mediating neurotransmitter release and vesicle function. Biochem Biophys Res Commun 77(3):1036–1043. https://doi.org/10.1016/S0006-291X(77)80082-X
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11(5):339–350. https://doi.org/10.1038/nrn2822
Denny J (2006) Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr Neuropharmacol 4(4):293–304. https://doi.org/10.2174/157015906778520782
Derivery E et al (2009) The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev Cell 17(5):712–723. https://doi.org/10.1016/j.devcel.2009.09.010
Despres C et al (2017) Identification of the tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1708448114
Dev KK et al (1999) The protein kinase Cα binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology 38(5):635–644. https://doi.org/10.1016/S0028-3908(98)00230-5
DeVos SL et al (2013) Antisense reduction of tau in adult mice protects against Seizures. J Neurosci 33(31):12887–12897. https://doi.org/10.1523/JNEUROSCI.2107-13.2013
DeVos SL et al (2018) Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci. https://doi.org/10.3389/FNINS.2018.00267
DiAntonio A et al (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412(6845):449–452. https://doi.org/10.1038/35086595
Diao J et al (2013) ‘Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2:e00592. https://doi.org/10.7554/eLife.00592
Dillon C, Goda Y (2005) The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28(1):25–55. https://doi.org/10.1146/annurev.neuro.28.061604.135757
Ding TT et al (2002) Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41(32):10209–10217. https://doi.org/10.1021/bi020139h
Dobrowolski Z, Baryłko B, Drabikowski W (1986) Interaction of tropomyosin with myelin basic protein and its effect on the ATPase activity of actomyosin. Eur J Cell Biol 41(1):65–71
Dourlen P et al (2019) The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. https://doi.org/10.1007/s00401-019-02004-0
Dräger NM et al (2017) Bin1 directly remodels actin dynamics through its BAR domain. EMBO Rep 18(11):2051–2066. https://doi.org/10.15252/embr.201744137
Dreyling MH et al (1996) The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci USA 93(10):4804–4809. https://doi.org/10.1073/pnas.93.10.4804
Dubos A et al (2015) Conditional depletion of intellectual disability and Parkinsonism candidate gene ATP6AP2 in fly and mouse induces cognitive impairment and neurodegeneration. Hum Mol Genet 24(23):6736–6755. https://doi.org/10.1093/hmg/ddv380
Dumanchin C et al (1998) Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 7(11):1825–1829. https://doi.org/10.1093/hmg/7.11.1825
Durakoglugil MS et al (2009) Reelin signaling antagonizes β-amyloid at the synapse. Proc Natl Acad Sci USA 106(37):15938–15943. https://doi.org/10.1073/pnas.0908176106
Dyer CA et al (1995) Cytoskeleton in myelin-basic-protein deficient shiverer oligodendrocytes. Dev Neurosci 17(1):53–62. https://doi.org/10.1159/000111273
Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28(2):511–525. https://doi.org/10.1016/S0896-6273(00)00129-X
Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6(3):231–242. https://doi.org/10.1038/nn1013
Elie A et al (2015) Tau co-organizes dynamic microtubule and actin networks. Sci Rep 5(1):1–10. https://doi.org/10.1038/srep09964
Engmann O et al (2011) Cyclin-dependent kinase 5 activator p25 Is generated during memory formation and is reduced at an early stage in Alzheimer’s disease. Biol Psychiat 70(2):159–168. https://doi.org/10.1016/j.biopsych.2011.04.011
Esposito A et al (2007) α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis 26(3):521–531. https://doi.org/10.1016/j.nbd.2007.01.014
Evans DA et al (1989) Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA 262(18):2551–2556
Evans DA et al (1997) Education and other measures of socioeconomic status and risk of incident Alzheimer disease in a defined population of older persons. Arch Neurol 54(11):1399–1405. https://doi.org/10.1001/archneur.1997.00550230066019
Evans GJO, Morgan A (2002) Phosphorylation-dependent interaction of the synaptic vesicle proteins cysteine string protein and synaptotagmin I. Biochem J 364(2):343–347. https://doi.org/10.1042/BJ20020123
Ewers H et al (2014) A septin-dependent diffusion barrier at dendritic spine necks. PLoS ONE 9(12):e113916. https://doi.org/10.1371/journal.pone.0113916
Fá M et al (2016) Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep 6:19393. https://doi.org/10.1038/srep19393
Fein JA et al (2008) Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol 172(6):1683–1692. https://doi.org/10.2353/AJPATH.2008.070829
Fernández-Chacón R et al (2004) The synaptic vesicle protein CSPα prevents presynaptic degeneration. Neuron. 42(2): 237–251. https://doi.org/10.1016/S0896-6273(04)00190-4
Ferguson SM et al (2007) A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 316(5824):570–574. https://doi.org/10.1126/science.1140621
Ferguson SM, De Camilli P (2012) Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. https://doi.org/10.1038/nrm3266
Fichou Y et al (2019) The elusive tau molecular structures: Can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol Commun. https://doi.org/10.1186/s40478-019-0682-x
Fitzpatrick AWP et al (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547:185. https://doi.org/10.1038/nature23002
Fiuza M et al (2017) PICK1 regulates AMPA receptor endocytosis via direct interactions with AP2 α-appendage and dynamin. J Cell Biol. 216(10):3323–3338. https://doi.org/10.1083/jcb.201701034
Fontaine SN et al (2016) DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J 35(14):1537–1549. https://doi.org/10.15252/embj.201593489
Frandemiche ML et al (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci 34(17)
Frid K et al (2015) Aggregation of MBP in chronic demyelination. Ann Clin Transl Neurol 2(7):711–721. https://doi.org/10.1002/acn3.207
Friedhoff P et al (1998) A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA 95(26):15712–15717. https://doi.org/10.1073/pnas.95.26.15712
Frost B, Jacks RL, Diamond MI (2009) Propagation of Tau misfolding from the outside to the inside of a cell. J Biol Chem 284(19):12845–12852. https://doi.org/10.1074/jbc.M808759200
Fulga TA et al (2006) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9:139. https://doi.org/10.1038/ncb1528
Fünfschilling U et al (2007) Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. https://doi.org/10.1186/1471-2202-8-1
Galiano MR et al (2006) Myelin basic protein functions as a microtubule stabilizing protein in differentiated oligodendrocytes. J Neurosci Res 84(3):534–541. https://doi.org/10.1002/jnr.20960
Galletta BJ, Mooren OL, Cooper JA (2010) Actin dynamics and endocytosis in yeast and mammals. Curr Opin Biotechnol. https://doi.org/10.1016/j.copbio.2010.06.006
Galloway PG et al (1987) Hirano bodies contain tau protein. Brain Res 403(2):337–340. https://doi.org/10.1016/0006-8993(87)90071-0
Gao X et al (2015) Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol Cell 59(5):781–793. https://doi.org/10.1016/j.molcel.2015.07.012
Georgieva ER et al (2014) Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats. Biophys J 107(6):1441–1452. https://doi.org/10.1016/j.bpj.2014.07.046
Gerges NZ et al (2006) Dual role of the exocyst in AMPA receptor targeting and insertion into the postsynaptic membrane. EMBO J 25(8):1623–1634. https://doi.org/10.1038/sj.emboj.7601065
Giannakopoulos P, Herrmann FR, Bussière T et al (2003) ‘Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60(9):1495LP – 1500. https://doi.org/10.1212/01.WNL.0000063311.58879.01
Giasson BI et al (2003) Initiation and synergistic fibrillization of tau and alpha-synuctein. Science 300(5619):636–640. https://doi.org/10.1126/science.1082324
Gibson PH, Tomlinson BE (1977) Numbers of Hirano bodies in the hippocampus of normal and demented people with Alzheimer’s disease. J Neurol Sci 33(1–2):199–206. https://doi.org/10.1016/0022-510X(77)90193-9
Girard M et al (2005) Non-stoichiometric relationship between clathrin heavy and light chains revealed by quantitative comparative proteomics of clathrin-coated vesicles from brain and liver. Mol Cell Proteomics 4(8):1145–1154. https://doi.org/10.1074/mcp.M500043-MCP200
Go GW, Mani A (2012) Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med 19–28
Goate A et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349(6311):704–706. https://doi.org/10.1038/349704a0
Goedert M et al (1989) ‘Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8(2):393–399
Goedert M et al (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3(4):519–526. https://doi.org/10.1016/0896-6273(89)90210-9
Goedert M et al (1994) Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem J 301(3):871–877. https://doi.org/10.1042/bj3010871
Goedert M, Spillantini MG, Crowther RA (1992) ‘Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci USA 89(5):1983–1987. https://doi.org/10.1073/PNAS.89.5.1983
Gomez TS et al (2012) Trafficking defects in WASH-knockout fibroblasts originate from collapsed endosomal and lysosomal networks. Mol Biol Cell 23(16):3215–3228. https://doi.org/10.1091/mbc.e12-02-0101
Gomez TS, Billadeau DD (2009) A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell 17(5):699–711. https://doi.org/10.1016/j.devcel.2009.09.009
Gómez-Isla T et al (1996) ‘Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16(14):4491–4500
Gómez-Ramos A et al (2008) Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci 37(4):673–681. https://doi.org/10.1016/J.MCN.2007.12.010
Goode BL et al (2000) Structural and Functional Differences between 3-Repeat and 4-Repeat Tau Isoforms implications for normal tau function and the onset of neurodegenerative disease. J Biol Chem 275:38182–38189. https://doi.org/10.1074/jbc.M007489200
Goto S et al (1985) Dephosphorylation of microtubule-associated protein 2, τ factor, and tubulin by calcineurin. J Neurochem 45(1):276–283. https://doi.org/10.1111/j.1471-4159.1985.tb05504.x
Götz J et al (1995) Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 14(7):1304–1313
Gourlay CW et al (2004) A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol 164(6):803–809. https://doi.org/10.1083/jcb.200310148
Granseth B et al (2006) Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 51(6):773–786. https://doi.org/10.1016/j.neuron.2006.08.029
Grant SGN et al (1992) Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258(5090):1903–1910. https://doi.org/10.1126/science.1361685
Grundke-Iqbal I et al (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83(13):4913–4917. https://doi.org/10.1073/PNAS.83.13.4913
Gu J et al (2010) ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13(10):1208–1215. https://doi.org/10.1038/nn.2634
Guntupalli S, Widagdo J, Anggono V (2016) Amyloid-β-induced dysregulation of AMPA receptor trafficking. Neural Plast. https://doi.org/10.1155/2016/3204519
Guo JL, Lee VM-Y (2011) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286(17):15317–15331. https://doi.org/10.1074/jbc.M110.209296
Gupta HV et al (2015) A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2015.10.001
Hamilton RL (2006) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol 10(3):378–384. https://doi.org/10.1111/j.1750-3639.2000.tb00269.x
Hanley JG, Henley JM (2005) PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J 24(18):3266–3278. https://doi.org/10.1038/sj.emboj.7600801
Hanseeuw BJ et al (2019) Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol 76(8):915–924. https://doi.org/10.1001/jamaneurol.2019.1424
Hardy JA, Higgins GA (1992) ‘Alzheimer’s disease: the amyloid cascade hypothesis. Science. https://doi.org/10.1126/science.1566067
Harris KP, Littleton JT (2015) Transmission, development, and plasticity of synapses. Genetics 201(2):345–375. https://doi.org/10.1534/genetics.115.176529
Harold D et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088. https://doi.org/10.1038/ng.440
Hartley DM et al (1999) Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19(20):8876–8884. https://doi.org/10.1523/jneurosci.19-20-08876.1999
Hasegawa K, Homma A, Imai Y (1986) An epidemiological study of age-related dementia in the community. Int J Geriatr Psychiatry 1(1):45–55. https://doi.org/10.1002/gps.930010108
He HJ et al (2009) The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol 10(1):81. https://doi.org/10.1186/1471-2121-10-81
He Q, Dent EW, Meiri KF (1997) Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J Neurosci 17(10):3515–3524. https://doi.org/10.1523/jneurosci.17-10-03515.1997
Hector A et al (2020) Tau hyperphosphorylation induced by the anesthetic agent ketamine/xylazine involved the calmodulin-dependent protein kinase II. FASEB J 34(2):2968–2977. https://doi.org/10.1096/fj.201902135R
Hee JC et al (2004) Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci 24(45):10248–10259. https://doi.org/10.1523/JNEUROSCI.0546-04.2004
Heeren J et al (2004) Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem 279(53):55483–55492. https://doi.org/10.1074/jbc.M409324200
Hernández F, Cuadros R, Avila J (2004) Zeta 14–3-3 protein favours the formation of human tau fibrillar polymers. Neurosci Lett 357(2):143–146. https://doi.org/10.1016/j.neulet.2003.12.049
Hof PR, Giannakopoulos P, Bouras C (1996) The neuropathological changes associated with normal brain aging. Histol Histopathol 1075–1088
Hofman A et al (1991) The prevalence of dementia in europe: a collaborative study of 1980–1990 findings. Int J Epidemiol 20(3):736–748. https://doi.org/10.1093/ije/20.3.736
Hollingworth P et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43(5):429–436. https://doi.org/10.1038/ng.803
Hollmann M et al (1991) Ca2+ permeability of KA-AMPA--gated glutamate receptor channels depends on subunit composition. science.sciencemag.org. https://science.sciencemag.org/content/252/5007/851.short. Accessed 11 May 2020.
Holth JK et al (2013) Tau loss attenuates neuronal network hyperexcitability in mouse and drosophila genetic models of epilepsy. J Neurosci 33(4):1651–1659
Homchaudhuri L et al (2009) Influence of membrane surface charge and post-translational modifications to myelin basic protein on its ability to tether the Fyn-SH3 domain to a membrane in vitro. Biochemistry 48(11):2385–2393. https://doi.org/10.1021/bi8022587
Hong WJ, Lev S (2014) Tethering the assembly of SNARE complexes. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2013.09.006
Hoover BR et al (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68(6):1067–1081. https://doi.org/10.1016/J.NEURON.2010.11.030
Hsieh H et al (2006) AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Elsevier. https://www.sciencedirect.com/science/article/pii/S0896627306008725. Accessed 21 Dec 2018.
Hsu SC et al (1996) The mammalian brain rsec6/8 complex. Neuron 7(6):1209–1219. https://doi.org/10.1016/S0896-6273(00)80251-2
Hsu SC et al (1998) Subunit composition, protein interactions, and structures of the mammalian brain sec6/8 complex and septin filaments. Neuron 20(6):1111–1122. https://doi.org/10.1016/S0896-6273(00)80493-6
Hsueh YP (2012) Neuron-specific regulation on F-actin cytoskeletons: the role of CTTNBP2 in dendritic spinogenesis and maintenance. Commun Integr Biol 5(4):334–336. https://doi.org/10.4161/cib.20364
Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13. https://doi.org/10.1093/nar/gkn923
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. https://doi.org/10.1038/nprot.2008.211
Hutton M et al (1998) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393(6686):702–704. https://doi.org/10.1038/31508
Iijima K, Gatt A, Iijima-Ando K (2010) Tau Ser262 phosphorylation is critical for Aβ42-induced tau toxicity in a transgenic Drosophila model of Alzheimer’s disease. Hum Mol Genet 19(15):2947–2957. https://doi.org/10.1093/hmg/ddq200
Islam K, Levy E (1997) Carboxyl-terminal fragments of β-amyloid precursor protein bind to microtubules and the associated protein tau. Am J Pathol 151(1):265–271
Ittner LM et al (2010) Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142(3):387–397. https://doi.org/10.1016/j.cell.2010.06.036
Ittner LM, Götz J (2010) Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12:67. https://doi.org/10.1038/nrn2967
Jarome TJ, Helmstetter FJ (2013) ‘The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol Learn Mem. https://doi.org/10.1016/j.nlm.2013.03.009
Jeng RL, Welch MD (2001) Cytoskeleton actin and endocytosis—no longer the weakest link. Curr Biol. https://doi.org/10.1016/S0960-9822(01)00410-9
Jensen PH et al (1999) α-synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem 274(36):25481–25489. https://doi.org/10.1074/jbc.274.36.25481
Van der Jeugd A et al (2012) Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol 123(6):787–805. https://doi.org/10.1007/s00401-012-0987-3
Jiang X et al (2010) A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J Neurosci 30(5):1798–1809. https://doi.org/10.1523/JNEUROSCI.4965-09.2010
Jinwal UK et al (2013) Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J 27(4):1450–1459. https://doi.org/10.1096/fj.12-220889
Johannsen S et al (2008) Temporal-spatial expression and novel biochemical properties of the memory-related protein KIBRA. Neuroscience 155(4):1165–1173. https://doi.org/10.1016/j.neuroscience.2008.06.054
Johnson GVW, Stoothoff WH (2004) Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. https://doi.org/10.1242/jcs.01558
Johnson RI, Seppa MJ, Cagan RL (2008) The Drosophila CD2AP/CIN85 orthologue Cindr regulates junctions and cytoskeleton dynamics during tissue patterning. J Cell Biol 180(6):1191–1204. https://doi.org/10.1083/jcb.200706108
Jung N, Haucke V (2007) Clathrin-mediated endocytosis at synapses. Traffic 8(9):1129–1136. https://doi.org/10.1111/j.1600-0854.2007.00595.x
Kajiho H et al (2003) RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J Cell Sci. https://doi.org/10.1242/jcs.00718
Kanaan NM et al (2011) ‘Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci 31(27):9858LP – 9868. https://doi.org/10.1523/JNEUROSCI.0560-11.2011
Kanekiyo T et al (2013) Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci 33(49):19276–19283. https://doi.org/10.1523/JNEUROSCI.3487-13.2013
Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273(5280):1402–1406. https://doi.org/10.1126/science.273.5280.1402
Kanmert D et al (2015) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci 35(30):10851–10865. https://doi.org/10.1523/JNEUROSCI.0387-15.2015
Karch CM, Goate AM (2015) Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiat. https://doi.org/10.1016/j.biopsych.2014.05.006
Katsinelos T et al (2018) Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 23(7):2039–2055. https://doi.org/10.1016/j.celrep.2018.04.056
Katzman R (1993) Education and the prevalence of dementia and Alzheimer’s disease. Neurology. https://doi.org/10.1212/wnl.43.1_part_1.13
Kaufman SK et al (2018) Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol 136(1):57–67. https://doi.org/10.1007/s00401-018-1855-6
Kavalali ET, Klingauf J, Tsien RW (1999) Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci USA 96(22):12893–12900. https://doi.org/10.1073/pnas.96.22.12893
Kelly BL, Ferreira A (2006) β-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem 281(38):28079–28089. https://doi.org/10.1074/jbc.M605081200
Kelly BL, Ferreira A (2007) Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience 147(1):60–70. https://doi.org/10.1016/j.neuroscience.2007.03.047
Kelly BL, Vassar R, Ferreira A (2005) β-amyloid-induced dynamin 1 depletion in hippocampal neurons: a potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem 280(36):31746–31753. https://doi.org/10.1074/jbc.M503259200
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61(3):340–350. https://doi.org/10.1016/J.NEURON.2009.01.015
Kfoury N et al (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287(23):19440–19451. https://doi.org/10.1074/jbc.M112.346072
Kisilevsky R et al (2007) Heparan sulfate as a therapeutic target in amyloidogenesis: prospects and possible complications. Amyloid Amyloid 14(1):21–32. https://doi.org/10.1080/13506120601116419
Kim JA, Kim HL (2001) Cleavage of purified neuronal clathrin assembly protein (CALM) by caspase 3 and calpain. Exp Mol Med 33(4):245–250. https://doi.org/10.1038/emm.2001.40
Kim JS, Huang TY, Bokoch GM (2009) Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell 20(11):2650–2660. https://doi.org/10.1091/mbc.E09-02-0131
Kimura T et al (2014) ‘Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond Ser B Biol Sci 369(1633):20130144. https://doi.org/10.1098/rstb.2013.0144
King ME et al (1999) Ligand-dependent tau filament formation: implications for Alzheimer’s disease progression. Biochemistry 38(45):14851–14859. https://doi.org/10.1021/bi9911839
Kinoshita A et al (1998) Identification of septins in neurofibrillary tangles in Alzheimer’s disease. Am J Pathol 153(5):1551–1560. https://doi.org/10.1016/S0002-9440(10)65743-4
Kitamura T et al (2009) Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 139(4):814–827. https://doi.org/10.1016/j.cell.2009.10.020
Knauer MF, Orlando RA, Glabe CG (1996) Cell surface APP751 forms complexes with protease nexin 2 ligands is internalized via the low density lipoprotein receptor-related protein (LRP). Brain Res 740(1–2):6–14. https://doi.org/10.1016/S0006-8993(96)00711-1
Kobayashi S et al (2019) Enhanced tau protein translation by hyper-excitation. Front Aging Neurosci 11:322. https://doi.org/10.3389/fnagi.2019.00322
Koch G et al (2016) Reversal of LTP-like cortical plasticity in Alzheimer’s disease patients with tau-related faster clinical progression. J Alzheimer’s Dis 50(2):605–616. https://doi.org/10.3233/JAD-150813
Koh TW et al (2007) Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol 178(2):309–322. https://doi.org/10.1083/jcb.200701030
Köhr G, Seeburg PH (1996) Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol 492(2):445–452. https://doi.org/10.1113/jphysiol.1996.sp021320
Koo EH et al (1996) Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 109(5).
Korvatska O et al (2013) Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum Mol Genet 22(16):3259–3268. https://doi.org/10.1093/hmg/ddt180
Koss DJ et al (2016) Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol 132(6):875–895. https://doi.org/10.1007/s00401-016-1632-3
Kovacs GG et al (2011) Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol 30(1):3–10. https://doi.org/10.5414/NPP30003
Krüger R et al (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genetics 18(2):106–108. https://doi.org/10.1038/ng0298-106
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY et al (2008) Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59(2):214–225. https://doi.org/10.1016/j.neuron.2008.06.008
Kumar A, Tsao JW (2018) Alzheimer disease: REVUE, StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK499922/. Accessed 10 May 2021.
Lachenal G et al (2011) Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci 46(2):409–418. https://doi.org/10.1016/j.mcn.2010.11.004
Lah JJ, Levey AI (2000) Endogenous Presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol Cell Neurosci 16(2):111–126. https://doi.org/10.1006/mcne.2000.0861
Lambert JC et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41(10):1094–1099. https://doi.org/10.1038/ng.439
Lambert MP et al (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95(11):6448–6453
Lasagna-Reeves CA et al (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49(47):10039–10041. https://doi.org/10.1021/bi1016233
Lasagna-Reeves CA et al (2012) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2:700. https://doi.org/10.1038/srep00700
Lasagna-Reeves CA et al (2014) The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta Neuropathol Commun 2(1):1–13. https://doi.org/10.1186/2051-5960-2-56
Lasagna-Reeves CA, Glabe CG, Kayed R (2011) Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem 286(25):22122–22130. https://doi.org/10.1074/jbc.M111.236257
Latina V et al (2018) NGF-dependent changes in ubiquitin homeostasis trigger early cholinergic degeneration in cellular and animal AD-model. Front Cell Neurosci 12:487. https://doi.org/10.3389/fncel.2018.00487
Lau DHW et al (2016) Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol Commun 4(1):49. https://doi.org/10.1186/s40478-016-0317-4
Lautermilch NJ, Spitzer NC (2000) Regulation of calcineurin by growth cone calcium waves controls neurite extension. J Neurosci 20(1):315–325. https://doi.org/10.1523/jneurosci.20-01-00315.2000
Lee G et al (1998) Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci 111(21)
Lee G et al (2004) Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci 24(9):2304–2312. https://doi.org/10.1523/JNEUROSCI.4162-03.2004
Lee G (2005) Tau and src family tyrosine kinases. Biochim Biophys Acta Mol Basis Disease. https://doi.org/10.1016/j.bbadis.2004.09.002
Lee G, Neve RL, Kosik KS (1989) The microtubule binding domain of tau protein. Neuron 2(6):1615–1624. https://doi.org/10.1016/0896-6273(89)90050-0
Lee KJ et al (2010) Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 169(1):344–356. https://doi.org/10.1016/j.neuroscience.2010.04.078
Lee SH et al (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36(4):661–674. https://doi.org/10.1016/S0896-6273(02)01024-3
Li C, Götz J (2018) Pyk2 is a novel tau tyrosine kinase that is regulated by the tyrosine kinase fyn. J Alzheimer’s Dis 64(1):205–221. https://doi.org/10.3233/JAD-180054
Li H et al (2016) Transferrin receptor controls AMPA receptor trafficking efficiency and synaptic plasticity. Sci Rep. https://doi.org/10.1038/srep21019
Li K, Arikan MC, Andreadis A (2003) Modulation of the membrane-binding domain of tau protein: splicing regulation of exon 2. Brain Res 116(1–2):94–105. https://doi.org/10.1016/s0169-328x(03)00259-6
Liang Z et al (2016) The pseudokinase CaMKv is required for the activity-dependent maintenance of dendritic spines. Nat Commun 7(1):1–13. https://doi.org/10.1038/ncomms13282
Lillis AP et al (2015) LDL receptor-related protein-1 (LRP1) regulates cholesterol accumulation in macrophages. PLoS ONE. https://doi.org/10.1371/journal.pone.0128903
Lin JW et al (2000) Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 3(12):1282–1290. https://doi.org/10.1038/81814
Lindwall G, Cole RD (1984) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259(8):5301–5305
Ling DSF et al (2002) Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat Neurosci 5(4):295–296. https://doi.org/10.1038/nn829
Ling DSF, Benardo LS, Sacktor TC (2006) Protein kinase Mζ enhances excitatory synaptic transmission by increasing the number of active postsynaptic AMPA receptors. Hippocampus 16(5):443–452. https://doi.org/10.1002/hipo.20171
Lippa CF et al (1998) Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153(5):1365–1370. https://doi.org/10.1016/S0002-9440(10)65722-7
Litersky JM et al (1996) Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J 316(2):655–660. https://doi.org/10.1042/bj3160655
Liu A et al (2017) Regulation of neurotransmitter release by amyloid precursor protein through synapsin phosphorylation. Neurochem Res 44(3):683–691. https://doi.org/10.1007/s11064-017-2418-2
Liu AP et al (2010) Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J Cell Biol 191(7):1381–1393. https://doi.org/10.1083/jcb.201008117
Liu C et al (2016) Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J Biol Chem 291(15):8173–8188. https://doi.org/10.1074/jbc.M115.641902
Liu Q et al (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci 30(50):17068–17078. https://doi.org/10.1523/JNEUROSCI.4067-10.2010
Lømo T (1966) Frequency potentiation of excitatory synaptic activity in the dentate area of the hippocampal formation. Acta Physiol Scand 68(128)
Loomis PA et al (1990) ‘Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci USA 87(21):8422–8426. https://doi.org/10.1073/PNAS.87.21.8422
Lu W, Ziff EB (2005) PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron 47(3):407–421. https://doi.org/10.1016/j.neuron.2005.07.006
Lüscher C et al (1999) Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24(3):649–658. https://doi.org/10.1016/S0896-6273(00)81119-8
Lynch DK et al (2003) A cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J Biol Chem 278(24):21805–21813. https://doi.org/10.1074/jbc.M211407200
Ma Z et al (2005) Evidence that insulin secretion influences SNAP-25 through proteasomal activation. Biochem Biophys Res Commun 329(3):1118–1126. https://doi.org/10.1016/j.bbrc.2005.02.086
Maeda S et al (2006) Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci Res 54(3):197–201. https://doi.org/10.1016/J.NEURES.2005.11.009
Maier O et al (2009) Lovastatin induces the formation of abnormal myelin-like membrane sheets in primary oligodendrocytes. Glia 57(4):402–413. https://doi.org/10.1002/glia.20769
Makuch L et al (2011) Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron 71(6):1022. https://doi.org/10.1016/J.NEURON.2011.08.017
Maloney MT et al (2005) β-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid β: a feedforward mechanism for alzheimer’s disease. J Neurosci 25(49):11313–11321. https://doi.org/10.1523/JNEUROSCI.3711-05.2005
Man HY et al (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25(3):649–662. https://doi.org/10.1016/S0896-6273(00)81067-3
Maor I et al (2000) Macrophage-released proteoglycans enhance LDL aggregation: studies in aorta from apolipoprotein E-deficient mice. Atherosclerosis 150(1):91–101. https://doi.org/10.1016/S0021-9150(99)00390-1
Marinković P et al (2019) In vivo imaging reveals reduced activity of neuronal circuits in a mouse tauopathy model. Brain 142(4):1051–1062. https://doi.org/10.1093/brain/awz035
Marquer C et al (2014) Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol Neurodegener 9(1):60. https://doi.org/10.1186/1750-1326-9-60
Marsh J et al (2017) Synapsin I phosphorylation is dysregulated by beta-amyloid oligomers and restored by valproic acid. Neurobiol Dis 106:63–75. https://doi.org/10.1016/j.nbd.2017.06.011
Martin H et al (2002) Subcellular localisation of 14–3-3 isoforms in rat brain using specific antibodies. J Neurochem 63(6):2259–2265. https://doi.org/10.1046/j.1471-4159.1994.63062259.x
Martín MG, Pfrieger F, Dotti CG (2014) Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep 15(10):1036–1052. https://doi.org/10.15252/embr.201439225
Martins IJ et al (2009) Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J Neurochem. https://doi.org/10.1111/j.1471-4159.2009.06408.x
Marttinen M et al (2015) Synaptic dysfunction and septin protein family members in neurodegenerative diseases. Mol Neurodegener. https://doi.org/10.1186/s13024-015-0013-z
Masliah E et al (2001) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56(1):127–129. https://doi.org/10.1212/WNL.56.1.127
Matsuda S, Mikawa S, Hirai H (1999) Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem 73(4):1765–1768. https://doi.org/10.1046/j.1471-4159.1999.731765.x
Matus A (2000) Actin-based plasticity in dendritic spines. Science. https://doi.org/10.1126/science.290.5492.754
May P et al (2004) ‘Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol 24(20):8872–8883. https://doi.org/10.1128/mcb.24.20.8872-8883.2004
McInnes J et al (2018) Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97(4):823-835.e8. https://doi.org/10.1016/j.neuron.2018.01.022
McPherson PS, Kay BK, Hussain NK (2001) Signaling on the endocytic pathway. Traffic. https://doi.org/10.1034/j.1600-0854.2001.002006375.x
Meldolesi J (2018) Exosomes and ectosomes in intercellular communication. Curr Biol. https://doi.org/10.1016/j.cub.2018.01.059
Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, Uronen R-L et al (2018) Secretion of Tau via an unconventional non-vesicular mechanism. Cell Rep 25(8):2027-2035.e4. https://doi.org/10.1016/j.celrep.2018.10.078
Meyerholz A et al (2005) Effect of Clathrin assembly lymphoid myeloid leukemia protein depletion on Clathrin coat formation. Traffic 6(12):1225–1234. https://doi.org/10.1111/j.1600-0854.2005.00355.x
Mi H et al (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47(D1):D419–D426. https://doi.org/10.1093/nar/gky1038
Michel CH et al (2014) Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem 289(2):956–967. https://doi.org/10.1074/jbc.M113.515445
Milner B, Corkin S, Teuber H-L (1968) Further analysis of the hippocampal amnesic syndrome: 14-year follow-up study of HM. Neuropsychologia 6(3):215–234. https://doi.org/10.1016/0028-3932(68)90021-3
Milovanovic D et al (2018) A liquid phase of synapsin and lipid vesicles. Science 361(6402):604–607. https://doi.org/10.1126/science.aat5671
Minamide LS et al (2000) Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol 2(9):628–636. https://doi.org/10.1038/35023579
Minkeviciene R et al (2009) Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29(11):3453–3462. https://doi.org/10.1523/JNEUROSCI.5215-08.2009
Mishkin M (1978) Memory in monkeys severely impaired by combined but not by separate removal of amygdala and hippocampus. Nature 273(5660):297–298. https://doi.org/10.1038/273297a0
Miyamoto T et al (2017) Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener 12(1):41. https://doi.org/10.1186/s13024-017-0176-x
Modesti NM, Barra HS (1986) The interaction of myelin basic protein with tubulin and the inhibition of tubulin carboxypeptidase activity. Biochem Biophys Res Commun 136(2):482–489. https://doi.org/10.1016/0006-291X(86)90466-3
Mohamed N-V et al (2015) Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep 4(1):5715. https://doi.org/10.1038/srep05715
Momeni P et al (2009) Clinical and pathological features of an Alzheimer’s disease patient with the MAPT ΔK280 mutation. Neurobiol Aging 30(3):388–393. https://doi.org/10.1016/j.neurobiolaging.2007.07.013
Mondragón-Rodríguez S et al (2012) Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation. J Biol Chem 287(38):32040–32053. https://doi.org/10.1074/jbc.M112.401240
Moore CI, Browning MD, Rose GM (1993) Hippocampal plasticity induced by primed burst, but not long-term potentiation, stimulation is impaired in area CA1 of aged fischer 344 rats. Hippocampus 3(1):57–66. https://doi.org/10.1002/hipo.450030106
Moreira GG et al (2019) ‘Zinc binding to tau influences aggregation kinetics and oligomer distribution. Int J Mol Sci. https://doi.org/10.3390/ijms20235979
Mortimer JA et al (1985) Head injury as a risk factor for Alzheimer’s disease. Neurology 35(2):264–267. https://doi.org/10.1212/wnl.35.2.264
Motamedi-Shad N et al (2009) Kinetic analysis of amyloid formation in the presence of heparan sulfate. Faster unfolding and change of pathway. J Biol Chem 284(43):29921–29934. https://doi.org/10.1074/jbc.M109.018747
Mudher A, Lovestone S (2002) ‘Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci 25(1):22–26
Munsie LN, Truant R (2012) The role of the cofilin-actin rod stress response in neurodegenerative diseases uncovers potential new drug targets. BioArchitecture 2(6):204–208. https://doi.org/10.4161/bioa.22549
Naj AC et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436. https://doi.org/10.1038/ng.801
Nakazawa T et al (2001) Characterization of fyn-mediated tyrosine phosphorylation sites on GluRε2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J Biol Chem 276(1):693–699. https://doi.org/10.1074/jbc.M008085200
Nelson PT et al (2012) Correlation of alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. https://doi.org/10.1097/NEN.0b013e31825018f7
Nemani VM et al (2010) Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65(1):66–79. https://doi.org/10.1016/j.neuron.2009.12.023
Neuvonen M et al (2011) ‘SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsp3 promotes viral RNA replication. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1002383
Neve RL et al (1998) The neuronal growth-associated protein GAP-43 interacts with rabaptin-5 and participates in endocytosis. J Neurosci 18(19):7757–7767. https://doi.org/10.1523/jneurosci.18-19-07757.1998
Nie Z et al (1999) Overexpression of cysteine-string proteins in Drosophila reveals interactions with syntaxin. J Neurosci 19(23):10270–10279. https://doi.org/10.1523/jneurosci.19-23-10270.1999
Nillegoda NB et al (2015) Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 524(7564):247–251. https://doi.org/10.1038/nature14884
Nishimune A et al (1998) NSF binding to GluR2 regulates synaptic transmission. Neuron 21(1):87–97. https://doi.org/10.1016/S0896-6273(00)80517-6
Norris CM, Korol DL, Foster TC (1996) Increased susceptibility to induction of long-term depression and long- term potentiation reversal during aging. J Neurosci 16(17):5382–5392. https://doi.org/10.1523/jneurosci.16-17-05382.1996
Ojelade SA et al (2019) cindr, the drosophila homolog of the CD2AP Alzheimer’s disease risk gene, is required for synaptic transmission and proteostasis. Cell Rep 28(7):1799-1813.e5. https://doi.org/10.1016/j.celrep.2019.07.041
Olgiati P et al (2011) Genetics of late-onset Alzheimer’s disease: update from the alzgene database and analysis of shared pathways. Int J Alzheimer’s Dis. https://doi.org/10.4061/2011/832379
Osten P et al (1998) The AMPA receptor GluR2 C terminus can mediate a reversible, ATP- dependent interaction with NSF and α- and β-SNAPs. Neuron 21(1):99–110. https://doi.org/10.1016/S0896-6273(00)80518-8
Ostrerova N et al (1999) α-Synuclein shares physical and functional homology with 14–3-3 proteins. J Neurosci 19(14):5782–5791. https://doi.org/10.1523/jneurosci.19-14-05782.1999
Padmanabhan P et al (2019) Frontotemporal dementia mutant Tau promotes aberrant Fyn nanoclustering in hippocampal dendritic spines. Elife. https://doi.org/10.7554/eLife.45040
Palop JJ et al (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55(5):697–711. https://doi.org/10.1016/j.neuron.2007.07.025
Pankratz N et al (2009) Alpha-synuclein and familial Parkinson’s disease. Mov Disord 24(8):1125–1131. https://doi.org/10.1002/mds.22524
Pappolla MA et al (2003) Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurol Lippincott 61(2):199–205. https://doi.org/10.1212/01.WNL.0000070182.02537.84
Park D et al (2017) Activation of CaMKIV by soluble amyloid-β1–42 impedes trafficking of axonal vesicles and impairs activity-dependent synaptogenesis. Sci Signal. https://doi.org/10.1126/scisignal.aam8661
Patel N et al (2015) Ion channel formation by tau protein: implications for Alzheimer’s disease and tauopathies. Biochemistry 54(50):7320–7325. https://doi.org/10.1021/acs.biochem.5b00988
Patrick GN et al (2003) Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol 13(23):2073–2081. https://doi.org/10.1016/j.cub.2003.10.028
Patterson KR et al (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286(26):23063–23076. https://doi.org/10.1074/jbc.M111.237974
Perdigão C et al (2020) Intracellular trafficking mechanisms of synaptic dysfunction in Alzheimer’s disease. Front Cell Neurosci. https://doi.org/10.3389/fncel.2020.00072
Perl DP (2010) Neuropathology of Alzheimer’s disease. Mount Sinai J Med. https://doi.org/10.1002/msj.20157
Perrin BJ, Amann KJ, Huttenlocher A (2006) Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell 17(1):239–250. https://doi.org/10.1091/mbc.E05-06-0488
Petersen A, Gerges NZ (2015) Neurogranin regulates CaM dynamics at dendritic spines. Sci Rep 5(1):1–10. https://doi.org/10.1038/srep11135
Photowala H, Freed R, Alford S (2005) Location and function of vesicle clusters, active zones and Ca 2+ channels in the lamprey presynaptic terminal. J Physiol 569(1):119–135. https://doi.org/10.1113/jphysiol.2005.091314
Planel E et al (2007) Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci 27(12):3090–3097. https://doi.org/10.1523/JNEUROSCI.4854-06.2007
Polydoro M et al (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29(34):10741–10749. https://doi.org/10.1523/JNEUROSCI.1065-09.2009
Polydoro M et al (2014) Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol 127(2):257–270. https://doi.org/10.1007/s00401-013-1215-5
Polzin A et al (2002) Ral-GTPase influences the regulation of the readily releasable pool of synaptic vesicles. Mol Cell Biol 22(6):1714–1722. https://doi.org/10.1128/mcb.22.6.1714-1722.2002
Pooler AM et al (2013) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14(4):389–394. https://doi.org/10.1038/embor.2013.15
Poorkaj P et al (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43(6):815–825. https://doi.org/10.1002/ana.410430617
Potter DA et al (1998) Calpain regulates actin remodeling during cell spreading. J Cell Biol 141(3):647–662. https://doi.org/10.1083/jcb.141.3.647
Prasad K et al (1995) Purification of a new clathrin assembly protein from bovine brain coated vesicles and its identification as myelin basic protein. J Biol Chem 270(51):30551–30556. https://doi.org/10.1074/jbc.270.51.30551
Prescott GR et al (2008) Phosphorylation of cysteine string protein on Serine 10 triggers 14–3-3 protein binding. Biochem Biophys Res Commun 377(3):809–814. https://doi.org/10.1016/j.bbrc.2008.10.069
Puzzo D et al (2017) LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. Elife. https://doi.org/10.7554/eLife.26991
Qian D et al (1997) Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J Exp Med 185(7):1253–1259. https://doi.org/10.1084/jem.185.7.1253
Qian J, Hyman BT, Betensky RA (2017) Neurofibrillary tangle stage and the rate of progression of Alzheimer symptoms: modeling using an autopsy cohort and application to clinical trial design. JAMA Neurol 74(5):540–548. https://doi.org/10.1001/jamaneurol.2016.5953
Qualmann B, Kessels MM, Kelly RB (2000) Molecular links between endocytosis and the actin cytoskeleton. J Cell Biol. https://doi.org/10.1083/jcb.150.5.F111
Qureshi HY et al (2013) Overexpression of 14–3-3z promotes tau phosphorylation at Ser 262and accelerates proteosomal degradation of synaptophysin in rat primary hippocampal neurons. PLoS ONE 8(12):e84615. https://doi.org/10.1371/journal.pone.0084615
Ramsden M et al (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25(46):10637–10647. https://doi.org/10.1523/JNEUROSCI.3279-05.2005
Ramser EM et al (2013) Amyloid-β oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell 24(16):2494–2505. https://doi.org/10.1091/mbc.E12-12-0858
Ramteke MP et al (2014) Identification of a novel ATPase activity in 14–3-3 proteins—Evidence from enzyme kinetics, structure guided modeling and mutagenesis studies. FEBS Lett 588(1):71–78. https://doi.org/10.1016/j.febslet.2013.11.008
Rashid AJ, Cole CJ, Josselyn SA (2014) Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav 13(1):118–125. https://doi.org/10.1111/gbb.12058
Rauch JN et al (2020) LRP1 is a master regulator of tau uptake and spread. Nature 580(7803):381–385. https://doi.org/10.1038/s41586-020-2156-5
Regan P et al (2015) Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci 35(12):4804–4812. https://doi.org/10.1523/JNEUROSCI.2842-14.2015
Reynolds CH et al (2008) Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cγ1, Grb2, and Src family kinases. J Biol Chem 283(26):18177–18186. https://doi.org/10.1074/jbc.M709715200
Rinetti GV, Schweizer FE (2010) Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J Neurosci 30(9):3157–3166. https://doi.org/10.1523/JNEUROSCI.3712-09.2010
Rizzu P et al (1999) High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 64(2):414–421. https://doi.org/10.1086/302256
Roberson ED et al (2007) Reducing endogenous tau ameliorates amyloid -induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754. https://doi.org/10.1126/science.1141736
Roberson ED et al (2011) Amyloid-/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31(2):700–711. https://doi.org/10.1523/JNEUROSCI.4152-10.2011
Roberts GW (1988) Immunocytochemistry of neurofibrillary tangles in dementia pugilistica and Alzheimer’s disease: evidence for common genesis. Lancet 2(8626–8627):1456–1458. https://doi.org/10.1016/s0140-6736(88)90934-8
Roche KW et al (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4(8):794–802. https://doi.org/10.1038/90498
Rodriguez GA et al (2020) Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Aβ and tau pathology in the hippocampus. PLoS Biol 18(8):e3000851. https://doi.org/10.1371/JOURNAL.PBIO.3000851
Rogaeva E et al (2007) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39(2):168–177. https://doi.org/10.1038/ng1943
Rosse C et al (2009) An aPKC-Exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol 7(11):e1000235. https://doi.org/10.1371/journal.pbio.1000235
De Rossi P et al (2020) Neuronal BIN1 regulates presynaptic neurotransmitter release and memory consolidation. Cell Rep 30(10):3520-3535.e7. https://doi.org/10.1016/j.celrep.2020.02.026
Roth D, Burgoyne RD (1995) Stimulation of catecholamine secretion from adrenal chromaffin cells by 14–3-3 proteins is due to reorganisation of the cortical actin network. FEBS Lett 374(1):77–81. https://doi.org/10.1016/0014-5793(95)01080-X
Rouka E et al (2015) Differential recognition preferences of the three Src Homology 3 (SH3) domains from the adaptor CD2-associated Protein (CD2AP) and Direct Association with Ras and Rab Interactor 3 (RIN3). J Biol Chem 290(42):25275–25292. https://doi.org/10.1074/jbc.M115.637207
Rozkalne A, Hyman BT, Spires-Jones TL (2011) Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis 41(3):650–654. https://doi.org/10.1016/j.nbd.2010.11.014
Rudinskiy N et al (2009) Calpain hydrolysis of α- and β2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J Biol Chem 284(18):12447–12458. https://doi.org/10.1074/jbc.M804740200
Ruiz R et al (2008) Cysteine string protein-α is essential for the high calcium sensitivity of exocytosis in a vertebrate synapse. Eur J Neurosci 27(12):3118–3131. https://doi.org/10.1111/j.1460-9568.2008.06301.x
Russell CL et al (2012) Amyloid-β acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS ONE 7(8):e43201. https://doi.org/10.1371/journal.pone.0043201
Salazar SV et al (2019) Alzheimer’s disease risk factor Pyk2 mediates amyloid-β-induced synaptic dysfunction and loss. J Neurosci 39(4):758–772. https://doi.org/10.1523/JNEUROSCI.1873-18.2018
Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317. https://doi.org/10.1038/nrn1368
Sans N et al (2003) ‘NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol. https://doi.org/10.1038/ncb990
SantaCruz K et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733):476–481. https://doi.org/10.1126/science.1113694
Sapir T et al (2012) Tau’s role in the developing brain: implications for intellectual disability. Hum Mol Genet 21(8):1681–1692. https://doi.org/10.1093/hmg/ddr603
Schindowski K et al (2006) Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169(2):599–616. https://doi.org/10.2353/AJPATH.2006.060002
Schneider A et al (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into alzheimer paired helical filaments. Am Chem Soc. https://doi.org/10.1021/BI981874P
Schraen-Maschke S et al (2008) Tau as a biomarker of neurodegenerative diseases. Biomark Med. https://doi.org/10.2217/17520363.2.4.363
Schubert KO et al (2012) Hypothesis review: are clathrin-mediated endocytosis and clathrin-dependent membrane and protein trafficking core pathophysiological processes in schizophrenia and bipolar disorder. Mol Psychiatry. https://doi.org/10.1038/mp.2011.123
Schürmann B et al (2019) A novel role for the late-onset Alzheimer’s disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0407-3
Scott D, Roy S (2012) α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32(30):10129–10135. https://doi.org/10.1523/JNEUROSCI.0535-12.2012
Seabold GK et al (2003) Interaction of the tyrosine kinase Pyk2 with the N-methyl-D-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J Biol Chem 278(17):15040–15048. https://doi.org/10.1074/jbc.M212825200
Seagar M et al (1999) Interactions between proteins implicated in exocytosis and voltage-gated calcium channels. Philos Trans R Soc B Biol Sci. https://doi.org/10.1098/rstb.1999.0380
Seshadri S et al (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303(18):1832–1840. https://doi.org/10.1001/jama.2010.574
Shankar GM et al (2008) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842. https://doi.org/10.1038/nm1782
Sharma M, Burré J, Südhof TC (2011) CSPαpromotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol 13(1):30–39. https://doi.org/10.1038/ncb2131
Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science. https://doi.org/10.1126/science.1075333
Shi Y et al (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549(7673):523–527. https://doi.org/10.1038/nature24016
Shih W, Gallusser A, Kirchhausen T (1995) A clathrin-binding site in the hinge of the β2 chain of mammalian AP-2 complexes. J Biol Chem 270(52):31083–31090. https://doi.org/10.1074/jbc.270.52.31083
Shobab LA, Hsiung GYR, Feldman HH (2005) Cholesterol in Alzheimer’s disease. Lancet Neurol. https://doi.org/10.1016/S1474-4422(05)70248-9
Shulman JM et al (2011) Functional screening of Alzheimer pathology genome-wide association signals in drosophila. Am J Hum Genet 88(2):232–238. https://doi.org/10.1016/j.ajhg.2011.01.006
Shupliakov O et al (2002) Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA 99(22):14476–14481. https://doi.org/10.1073/pnas.212381799
Simons K, Ikonen E (2000) How cells handle cholesterol. Science. https://doi.org/10.1126/science.290.5497.1721
Sironi JJ et al (1998) Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett 436(3):471–475. https://doi.org/10.1016/S0014-5793(98)01185-5
Skoulakis EMC, Davis RL (1996) Olfactory learning deficits in mutants for leonardo, a Drosophila gene encoding a 14–3-3 protein. Neuron 17(5):931–944. https://doi.org/10.1016/S0896-6273(00)80224-X
Slepnev VI, De Camilli P (2000) Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat Rev Neurosci 1(3):161–172. https://doi.org/10.1038/35044540
Snyder EM et al (2005) Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci 8(8):1051–1058. https://doi.org/10.1038/nn1503
Sobue K et al (1981) The calmodulin-binding protein in microtubules is tau factor. FEBS Lett 132(1):137–140. https://doi.org/10.1016/0014-5793(81)80447-4
Sokolow S et al (2015) Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J Neurochem 133(3):368–379. https://doi.org/10.1111/jnc.12991
Söllner T et al (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362(6418):318–324. https://doi.org/10.1038/362318a0
Sommer B et al (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. cell.com. https://www.cell.com/cell/pdf/0092-8674(91)90568-J.pdf. Accessed 11 May 2020.
Song I et al (1998) Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 21(2):393–400. https://doi.org/10.1016/S0896-6273(00)80548-6
Sorrells SF et al (2018) Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555(7696):377–381. https://doi.org/10.1038/nature25975
Sottejeau Y et al (2015) Tau phosphorylation regulates the interaction between BIN1’s SH3 domain and Tau’s proline-rich domain. Acta Neuropathol Commun 3:58. https://doi.org/10.1186/s40478-015-0237-8
Speese SD et al (2003) The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy ning with the ubiquitin-activating enzyme (E1), which transfers ubiquitin to a ubiquitin-conjugating enzyme (E2). The conjugating enzyme then cooperates with a. Curr Biol 13:899–910. https://doi.org/10.1016/s0960-9822(03)00338-5
Staubli U, Lynch G (1990) Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res 513(1):113–118. https://doi.org/10.1016/0006-8993(90)91096-y
Stefanoska K et al (2018) An N-terminal motif unique to primate tau enables differential protein-protein interactions. J Biol Chem 293(10):3710–3719. https://doi.org/10.1074/jbc.RA118.001784
Steiner P et al (2008) Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 60(5):788–802. https://doi.org/10.1016/J.NEURON.2008.10.014
Stephens AD, Zacharopoulou M et al (2020) Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun 11(1):1–15. https://doi.org/10.1038/s41467-020-16564-3
Stratmann K et al (2016) Precortical phase of Alzheimer’s disease (AD)-related tau cytoskeletal pathology. Brain Pathol 26(3):371–386. https://doi.org/10.1111/bpa.12289
Strittmatter WJ et al (1993) Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90(5):1977–1981. https://doi.org/10.1073/pnas.90.5.1977
Strittmatter WJ et al (1994) Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA 91(23):11183–11186. https://doi.org/10.1073/pnas.91.23.11183
Suchowerska AK et al (2017) Developmental profiling of tropomyosin expression in mouse brain reveals Tpm4.2 as the major post-synaptic tropomyosin in the mature brain. Front Cell Neurosci. https://doi.org/10.3389/fncel.2017.00421
Südhof TC (2004) The synaptic vesicle cyle. Annu Rev Neurosci 27:509–556. https://doi.org/10.1146/annurev.neuro.26.041002.131412
Sultan A et al (2011) Nuclear tau, a key player in neuronal DNA protection. J Biol Chem 286(6):4566–4575. https://doi.org/10.1074/jbc.M110.199976
Sun T et al (2010) The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J Neurosci 30(35):11838–11847. https://doi.org/10.1523/JNEUROSCI.1481-10.2010
Suzuki M, Kimura T (2017) Microtubule-associated tau contributes to intra-dendritic trafficking of AMPA receptors in multiple ways. Neurosci Lett 653:276–282. https://doi.org/10.1016/J.NEULET.2017.05.056
Sydow A et al (2011) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic tau mutant. J Neurosci 31(7):2511–2525. https://doi.org/10.1523/JNEUROSCI.5245-10.2011
Synek L, Sekereš J, Žárský V (2014) The exocyst at the interface between cytoskeleton and membranes in eukaryotic cells. Front Plant Sci. https://doi.org/10.3389/fpls.2013.00543
Tachibana M et al (2019) APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J Clin Invest 129(3):1272–1277. https://doi.org/10.1172/JCI124853
Tai H-C et al (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181(4):1426–1435. https://doi.org/10.1016/j.ajpath.2012.06.033
Tao QQ et al (2017) Decreased gene expression of CD2AP in Chinese patients with sporadic Alzheimer’s disease. Neurobiol Aging 56:212.e5-212.e10. https://doi.org/10.1016/j.neurobiolaging.2017.03.013
Tatebayashi Y et al (2002) Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci 99(21):13896–13901. https://doi.org/10.1073/pnas.202205599
Tebar F, Bohlander SK, Sorkin A (1999) Clathrin assembly lymphoid myeloid leukemia (CALM) protein: Localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell 10(8):2687–2702. https://doi.org/10.1091/mbc.10.8.2687
Teodoro RO et al (2013) Ral mediates activity-dependent growth of postsynaptic membranes via recruitment of the exocyst. EMBO J 32(14):2039–2055. https://doi.org/10.1038/emboj.2013.147
Terashima A et al (2008) An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57(6):872–882. https://doi.org/10.1016/j.neuron.2008.01.028
Thies E, Mandelkow E-M (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 27(11)
Tiwari SS et al (2015) Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer’s disease. Mol Brain 8(1):6. https://doi.org/10.1186/s13041-015-0096-z
Tombaugh GC et al (2002) Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats. J Neurosci 22(22):9932–9940. https://doi.org/10.1523/jneurosci.22-22-09932.2002
Tracy TE et al (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90(2):245–260. https://doi.org/10.1016/j.neuron.2016.03.005
Traub LM et al (1999) Crystal structure of the α appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc Natl Acad Sci USA 96(16):8907–8912. https://doi.org/10.1073/pnas.96.16.8907
Trommer BL et al (2005) ApoE isoform-specific effects on LTP: Blockade by oligomeric amyloid-β1-42. Neurobiol Dis 18(1):75–82. https://doi.org/10.1016/j.nbd.2004.08.011
Ubelmann F et al (2017) Bin1 and CD 2 AP polarise the endocytic generation of beta-amyloid. EMBO Rep 18(1):102–122. https://doi.org/10.15252/embr.201642738
Umbach JA et al (1994) Presynaptic dysfunction in drosophila csp mutants. Neuron 13(4):899–907. https://doi.org/10.1016/0896-6273(94)90255-0
Usenovic M et al (2015) Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci 35(42):14234–14250. https://doi.org/10.1523/JNEUROSCI.1523-15.2015
Vargas-Caballero M et al (2017) Wild-type, but not mutant N296H, human tau restores Aβ-mediated inhibition of LTP in Tau-/- mice. Front Neurosci 11:201. https://doi.org/10.3389/fnins.2017.00201
Vargas KJ et al (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34(28):9364–9376. https://doi.org/10.1523/JNEUROSCI.4787-13.2014
Vassar R et al (1999) β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741. https://doi.org/10.1126/science.286.5440.735
Vega IE, Hsu SC (2001) The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J Neurosci 21(11):3839–3848. https://doi.org/10.1523/jneurosci.21-11-03839.2001
Vinade L et al (2001) Activation of calpain may alter the postsynaptic density structure and modulate anchoring of NMDA receptors. Synapse 40(4):302–309. https://doi.org/10.1002/syn.1053
Voskobiynyk Y et al (2020) Alzheimer’s disease risk gene BIN1 induces Tau-dependent network hyperexcitability. Elife 9:1–25. https://doi.org/10.7554/eLife.57354
Wang D et al (2010) Drosophila twinfilin is required for cell migration and synaptic endocytosis. J Cell Sci 123(9):1546–1556. https://doi.org/10.1242/jcs.060251
Wang HW et al (2002) Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Research Brain Res 924(2):133–140. https://doi.org/10.1016/S0006-8993(01)03058-X
Wang L et al (2014) α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24(19):2319–2326. https://doi.org/10.1016/j.cub.2014.08.027
Wang Y et al (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. https://doi.org/10.1186/s13024-016-0143-y
Wang YT, Salter MW (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369(6477):233–235. https://doi.org/10.1038/369233a0
Wang Z et al (2017) Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J Neurosci 37(49):11947–11966. https://doi.org/10.1523/JNEUROSCI.2009-17.2017
Warmus BA et al (2014) Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J Neurosci 34(49):16482–16495. https://doi.org/10.1523/JNEUROSCI.3418-14.2014
Watanabe M et al (1994) ‘Molecular cloning of rat cDNAs for the ζ and θ subtypes of 14–3-3 protein and differential distributions of their mRNAs in the brain. Mol Brain Res 25(1–2):113–121. https://doi.org/10.1016/0169-328X(94)90285-2
Wei Y et al (2008) Binding to the minor groove of the double-strand, Tau protein prevents DNA damage by peroxidation. PLoS ONE 3(7):e2600. https://doi.org/10.1371/journal.pone.0002600
Weingarten MD et al (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72(5):1858–1862
Welsch T et al (2005) Association of CD2AP with dynamic actin on vesicles in podocytes. Am J Physiol Renal Physiol 289(558–5):F1134–F1143. https://doi.org/10.1152/ajprenal.00178.2005
Wheeler D et al (2008) A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 131(11):3092–3102. https://doi.org/10.1093/brain/awn190
Wheeler TC et al (2002) Regulation of synaptophysin degradation by mammalian homologues of Seven in Absentia. J Biol Chem 277(12):10273–10282. https://doi.org/10.1074/jbc.M107857200
Woo JA et al (2015) Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1-integrin conformers. Cell Death Differ 22(6):921–934. https://doi.org/10.1038/cdd.2015.5
Woo JAA et al (2019) Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun Biol 2(1):1–15. https://doi.org/10.1038/s42003-019-0359-9
Wu HY et al (2005) Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J Biol Chem 280(22):21588–21593. https://doi.org/10.1074/jbc.M501603200
Wu JW et al (2013) Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288(3):1856–1870. https://doi.org/10.1074/jbc.M112.394528
Wu JW et al (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19(8):1085–1092. https://doi.org/10.1038/nn.4328
Wu MN et al (1999) Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron 23(3):593–605. https://doi.org/10.1016/S0896-6273(00)80811-9
Wu XS et al (2014) Calcineurin is universally involved in vesicle endocytosis at neuronal and nonneuronal secretory cells. Cell Rep 7(4):982–988. https://doi.org/10.1016/j.celrep.2014.04.020
Wu Y et al (2014) A dynamin 1-, dynamin 3- and clathrinindependent pathway of synaptic vesicle recycling mediated by bulk endocytosis. Elife. https://doi.org/10.7554/eLife.01621.001
Xia J et al (1999) Clustering of AMPA receptors by the synaptic PD domain-containing protein PICK1. Neuron 22(1):179–187. https://doi.org/10.1016/S0896-6273(00)80689-3
Xie Y et al (2007) The GTP-binding protein septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr Biol 17(20):1746–1751. https://doi.org/10.1016/j.cub.2007.08.042
Xiong H et al (2008) Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol Dis 29(3):422–437. https://doi.org/10.1016/j.nbd.2007.10.005
Yadav S et al (2017) TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through septin7 phosphorylation. Neuron 93(2):379–393. https://doi.org/10.1016/j.neuron.2016.12.006
Yamada K et al (2014) ‘Neuronal activity regulates extracellular tau in vivo. J Exp Med 211(3):387–393. https://doi.org/10.1084/jem.20131685
Yang Y et al (2015) Amyloid-β oligomers may impair SNARE-mediated exocytosis by direct binding to syntaxin 1a. Cell Rep 12(8):1244–1251. https://doi.org/10.1016/j.celrep.2015.07.044
Yano M et al (2006) A novel function of 14-3-3 protein: 14-3-3ζ is a heat-shock-related molecular chaperone that dissolves thermal-aggregated proteins. Mol Biol Cell. Ame Soc Cell Biol 17(11):4769–4779. https://doi.org/10.1091/mbc.E06-03-0229
Yin Y et al (2016) Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc Natl Acad Sci USA 113(26):E3773–E3781. https://doi.org/10.1073/pnas.1604519113
Yokoyama CT, Sheng ZH, Catterall WA (1997) Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci 17(18):6929–6938. https://doi.org/10.1523/jneurosci.17-18-06929.1997
Yoshihama Y et al (2009) KIBRA co-localizes with protein kinase Mζ (PKMζ) in the mouse hippocampus. Biosci Biotechnol Biochem 73(1):147–151. https://doi.org/10.1271/bbb.80564
Yoshimura Y, Ichinose T, Yamauchi T (2003) Phosphorylation of tau protein to sites found in Alzheimer’s disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci Lett 353(3):185–188. https://doi.org/10.1016/j.neulet.2003.09.037
Yoshiyama Y et al (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53(3):337–351. https://doi.org/10.1016/J.NEURON.2007.01.010
Yu A et al (2014) Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc Natl Acad Sci USA 111(15):E1481–E1490. https://doi.org/10.1073/pnas.1321811111
Yu J-Z, Rasenick MM (2006) Tau associates with actin in differentiating PC12 cells. FASEB J 20(9):1452–1461. https://doi.org/10.1096/fj.05-5206com
Zakharov VV, Bogdanova MN, Mosevitsky MI (2005) Specific proteolysis of neuronal protein GAP-43 by calpain: characterization, regulation, and physiological role. Biochem Mosc 70(8):897–907. https://doi.org/10.1007/s10541-005-0200-6
Zavodszky E et al (2014) Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. https://doi.org/10.1038/ncomms4828
Zempel H et al (2017) Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292(29):12192–12207. https://doi.org/10.1074/jbc.M117.784702
Zhang Y-Q et al (2012) Identification of CSPα clients reveals a role in dynamin 1 regulation. Neuron 74(1):136–150. https://doi.org/10.1016/j.neuron.2012.01.029
Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132(4):645–660. https://doi.org/10.1016/J.CELL.2008.01.033
Zhao J et al (2013) CD2AP links cortactin and capping protein at the cell periphery to facilitate formation of lamellipodia. Mol Cell Biol 33(1):38–47. https://doi.org/10.1128/mcb.00734-12
Zhao JW et al (2012) Regulation of cofilin activity by CaMKII and calcineurin. . Am J Med Sci 344(6):462–472. https://doi.org/10.1097/MAJ.0b013e318244745b
Zhao L et al (2013) Drosophila cyfip regulates synaptic development and endocytosis by suppressing filamentous actin assembly. PLoS Genet. https://doi.org/10.1371/journal.pgen.1003450
Zhao Y, Hegde AN, Martin KC (2003) The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr Biol 13(11):887–898. https://doi.org/10.1016/S0960-9822(03)00332-4
Zhou L et al (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8:15295. https://doi.org/10.1038/ncomms15295
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44(5):749–757. https://doi.org/10.1016/j.neuron.2004.11.011
Zhou Y et al (2018) Relevance of phosphorylation and truncation of tau to the etiopathogenesis of Alzheimer’s disease. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2018.00027
Zhou Z et al (2011) GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J Neurosci 31(3):819–833. https://doi.org/10.1523/JNEUROSCI.3869-10.2011
Zinsmaier KE et al (1994) Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263(5149):977–980. https://doi.org/10.1126/science.8310297
Acknowledgements
Not applicable.
Funding
G.S.K.S. acknowledges funding from the Wellcome Trust (065807/Z/01/Z) (203249/Z/16/Z), the UK Medical Research Council (MRC) (MR/K02292X/1), Alzheimer Research UK (ARUK) (ARUK-PG013-14), Michael J Fox Foundation (16238) and Infinitus China Ltd. M.A.R acknowledges funding from the Engineering and Physical Sciences Research Council (EP/L015889/1).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Declarations of interest: none.
Availability of data and materials
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Table 1. The proteins discussed in this article that are capable of binding to Tau and have involvement in synaptic pathways.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Robbins, M., Clayton, E. & Kaminski Schierle, G.S. Synaptic tau: A pathological or physiological phenomenon?. acta neuropathol commun 9, 149 (2021). https://doi.org/10.1186/s40478-021-01246-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01246-y