
Abstract
Redox homeostasis refers to the balance between the production of reactive oxygen species (ROS) as well as reactive nitrogen species (RNS), and their elimination by antioxidants. It is linked to all important cellular activities and oxidative stress is a result of imbalance between pro-oxidants and antioxidant species. Oxidative stress perturbs many cellular activities, including processes that maintain the integrity of DNA. Nucleic acids are highly reactive and therefore particularly susceptible to damage. The DNA damage response detects and repairs these DNA lesions. Efficient DNA repair processes are therefore essential for maintaining cellular viability, but they decline considerably during aging. DNA damage and deficiencies in DNA repair are increasingly described in age-related neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. Furthermore, oxidative stress has long been associated with these conditions. Moreover, both redox dysregulation and DNA damage increase significantly during aging, which is the biggest risk factor for neurodegenerative diseases. However, the links between redox dysfunction and DNA damage, and their joint contributions to pathophysiology in these conditions, are only just emerging. This review will discuss these associations and address the increasing evidence for redox dysregulation as an important and major source of DNA damage in neurodegenerative disorders. Understanding these connections may facilitate a better understanding of disease mechanisms, and ultimately lead to the design of better therapeutic strategies based on preventing both redox dysregulation and DNA damage.
Similar content being viewed by others


Background
All types of cells in the human body require oxygen for their physiological functions. However, the brain displays particularly high rates of metabolic activity, and it consumes up to 20% of available oxygen, much more than other organs [1]. Oxygen is highly reactive with other molecules and oxidation refers to the transfer of electrons from an atom to oxygen, with the formation of a negative ion. Reduction is the opposite process, referring to a gain of electrons. The delicate balance between cellular oxidation and reduction reactions, referred to as the cellular ‘redox state’, must always be maintained. However, imbalance in the redox state leads to the formation of free radicals, including reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive sulphur species (RSS) [2, 3]. Low levels of ROS, RNS and RSS are necessary for proper functioning of fundamental cellular processes such as proliferation, host defence, signal transduction, and gene expression (4, 5). However, excessive amounts of ROS, RNS and RSS can be severely toxic to cells. To neutralize the destructive effects of these species, the cell employs antioxidant systems to minimize oxidative damage. The cellular redox state therefore represents an essential defence system that regulates numerous signalling pathways, including DNA repair, calcium metabolism, axonal transport and protein homeostasis (proteostasis) mechanisms such as protein folding and degradation [6]. However, dysregulation of redox conditions disrupts these processes and can lead to aberrant post-translational modification of redox-sensitive proteins [6].
Dysregulation of cellular redox conditions is a major source of DNA damage because redox homeostasis activates or inhibits key proteins involved in DNA repair. Eukaryotic cells have developed complex signalling mechanisms, together referred to as the ‘DNA damage response’ (DDR), to detect, signal and repair DNA damage and thus maintain genome integrity [7]. However, if DNA lesions remain unrepaired, the accumulating DNA damage induces various cell death mechanisms to eradicate those cells with imperfect genomes. Whilst the DDR itself has now been characterised in some detail, the relationship between DNA damage and the cellular redox state is poorly understood in comparison and has emerged relatively recently.
Neurodegenerative diseases are devastating conditions that result from chronic degeneration and death of specific types of neurons. Whilst most cell types are continuously replaced and thus can withstand the loss of cells displaying irreparable DNA damage by apoptosis, neurons are post-mitotic and therefore susceptible to DNA lesions throughout their lifespan. Hence, they are particularly susceptible to damage. In addition, compared to other cell types, neurons are remarkably vulnerable to redox dysregulation due to their excessive oxygen consumption, large size, and high rates of metabolism, which produces significant quantities of ROS and RNS [8]. Age-associated increases in redox dysfunction contribute to protein misfolding and aggregation, and are widely implicated in neurodegeneration [9]. Furthermore, aging is the most significant risk for neurodegenerative diseases, and redox homeostasis and the efficiency of DNA repair become significantly impaired during aging. Not surprisingly, dysregulation of the cellular redox state has been widely described in neurodegenerative conditions, including Alzheimer’s disease (AD) [10], Parkinson’s disease (PD) [11], amyotrophic lateral sclerosis (ALS) [12,13,14] and related condition frontotemporal dementia (FTD) [15], and Huntington’s disease (HD) [16]. Furthermore, impaired repair of DNA damage is now strongly linked to age-associated neurodegenerative diseases [17,18,19]. Moreover, there have been major advances in this field over the last five years. Hence in this review, we provide a comprehensive and updated appraisal of current knowledge relating redox dysfunction to DNA damage, and discuss how this is impacted in neurodegenerative disorders.
DNA damage
Preservation of genetic material is essential for the perpetuation of life [7], but DNA is continuously subject to both exogenous and endogenous threats [7, 20]. In fact, it has been estimated that every day most human cells are exposed to tens of thousands of DNA lesions [21, 22]. Unrepaired DNA damage leads to mutations, compromises cellular viability, and prevents the correct transfer of genetic material to the next generation [22]. Many cellular functions, including DNA replication and transcription, are dysregulated following failure to repair DNA [7, 20]. Conversely, genome abnormalities, mutations and cell death can result from hindered DNA replication or transcription [7, 23]. To protect the genome, cells use the DDR to prevent or tolerate distinct types of DNA damage [20, 21, 24].
The mammalian DDR involves several components: (a) mechanisms to repair DNA to minimise the damage and thus restore the fidelity of genetic material; (b) activation of DNA damage checkpoints to arrest the cell cycle, thus providing more time for DNA repair to prevent the transfer of damaged DNA to daughter cells; (c) induction of a transcriptional response to allow expression of specific genes; and (d) apoptosis, to eliminate critically damaged cells, and therefore protect the organism [20]. Below, we discuss how redox-regulated mechanisms control the functions of the DDR. For a more detailed discussion of specific DDR mechanisms, please see several excellent recent review articles on this topic [7, 25,26,27].
The cellular redox system
The cellular redox system involves the production of free radicals—highly reactive molecules containing an uneven number of electrons [28]—and the antioxidant processes that neutralize them. An imbalance of these reactive species leads to oxidative or nitrosative stress. ROS include hydroxyl radicals (·OH), superoxide (O2·−), singlet oxygen (1O2), hydroperoxyl radicals (·HO2) [28, 29] and hydrogen peroxide (H2O2) [30]. In addition, peroxyl radicals (ROO·) are carbon-centred free radicals that are also classified as ROS [31]. O2·− is the origin of most intracellular ROS, but it is transformed either to H2O2 by the activity of catalase, or to peroxynitrite (PN) (ONOO−) by reaction with nitric oxide ·NO [30]. RNS include NO-derived compounds, including nitric oxide (·NO), PN (ONOO−), and nitrogen dioxide (·NO2) [30]. RSS are commonly produced by the oxidation of thiols and disulphide into higher oxidation states and they include persulphate, polysulphide, and thiosulphate (S2O32−) [32] (Fig. 1). Free radicals can attack different cellular components in neurons, including DNA, proteins and lipids, rendering them susceptible to oxidative stress. The highly reactive ·OH radical in particular damages both heterocyclic DNA bases and the sugar moiety [33].

Lewis structures of free radicals. Free radicals are highly reactive molecules with an uneven number of electrons that have the potential to harm cells. ROS, including ·OH, O2·−, 1O2, ·HO2 and H2O2, are types of free radicals containing oxygen. RNS are highly active molecules derived from nitric oxide-derived compounds including ·NO, ONOO−, and ·NO2. RSS are a family of sulphur-based chemical compounds that include H2S and S2O32− that can oxidize and inhibit thiol-proteins and enzymes
Cells have established complex antioxidant systems to defend against oxidative insults, involving both enzymes and cofactors that maintain redox balance, and mechanisms to limit respiration in mitochondria. Endogenous antioxidant enzymes include superoxide dismutases (SOD), catalase, glutathione peroxidase (GPx), glutathione reductase (GR), and peroxiredoxins (Prxs) [34, 35]. Smaller antioxidant molecules include glutathione, coenzyme Q, ferritin, bilirubin, ascorbic acid (vitamin C), and α-tocopherol (vitamin E) [36]. The overall cellular redox state is determined by two cellular disulphide mechanisms, the thioredoxin (Trx) and glutaredoxins (Grx) systems [37]. Free radicals affect many cellular components (proteins, nucleic acids, lipids, and carbohydrates). However, in this review we will focus only on those molecules relevant to DNA damage (Fig. 2).

Mechanisms involved in maintaining cellular redox homeostasis. The cellular redox state is a sensitive balance between oxidation and reduction reactions, involving the production of free radicals and the antioxidant systems that neutralize them. H2O2 is generated by SOD enzymes CuZnSOD in the cytoplasm, and it also enters the cell from the extracellular space, which together enhance intracellular H2O2 levels. H2O2 can be safely decomposed by catalase into water (H2O) and oxygen (O2). The mitochondrial enzyme MnSOD also has dismutase activity, which detoxifies the free radical O2·− generated by mitochondrial respiration. The cellular redox state is regulated by the thioredoxin (Trx/TrxR) and glutaredoxins (Grx) systems, which modifies specific redox-sensitive proteins, thereby triggering related signalling events. The nuclear factor erythroid 2-related factor 2 (Nrf2) system is then activated, leading to an antioxidant response. The NADPH oxidase complex is inactive under normal circumstances but is activated during respiratory burst. Glutaredoxin 4 (GPx4) reduces lipid hydroxide (LOOH) to alcohol (LOH). GR glutathione reductase, NF-κB nuclear factor kappa B, NOS nitric oxide synthase, NADPH nicotinamide adenine dinucleotide phosphate
Cellular redox mechanisms
Both Grx and Trx enzymes belong to the Trx superfamily, whose members are characterised by the presence of an active-site Cys-X-X-Cys motif in a Trx-like fold [38]. These antioxidant enzymes regulate the activity of substrate proteins through alterations of the redox state of thiol groups within their active-site cysteines. These thiols can be either reduced, or oxidized, where the two cysteines form an intramolecular disulphide bond with substrate proteins. Grxs and Trxs are present in multiple organelles, including the nucleus, and they often shuttle between the nucleus and cytoplasm [39].
Grxs are small enzymes that use glutathione (GSH) as a co-factor to maintain their reduced state [40]. GSH is a tripeptide consisting of γ-l-glutamyl-l-cysteinyl-glycine, which is present at high concentrations in most cells, including neurons [41]. GSH exists in both reduced and oxidized (GSSG) states and GSH regulates the thiol-disulphide redox states of proteins by maintaining their sulfhydryl groups in a reduced form. Hence the ratio of GSH to GSSG determines the cellular redox status. In normal cells the GSH/GSSG ratio is > 100, whereas in conditions of oxidative stress, this ratio decreases to > 10. GSH also participates in many antioxidant defence reactions including the synthesis of nucleic acids [42]. A unique feature of Grxs is their ability to catalyse the addition of GSH to a substrate protein (glutathionylation), and the reverse reaction (deglutathionylation), which together can also regulate redox conditions. The Trx system also consists of nicotinamide adenine dinucleotide phosphate (NADPH), GR, GPx, and Grx [37]. NADPH is the fundamental reductant that maintains the redox states of both the Trx and Grx systems. DNA repair is dependent on GSH, since elevation of DNA damage is related to defects in GSH metabolism in mice [43].
The GSH pool of the nucleus is an important protective factor against DNA damage induced by oxidation [44]. It also protects nuclear proteins in this reducing environment, facilitating gene transcription throughout the cell cycle in dividing cells [45]. Elevated GSH levels result in deglutathionylation of DNA-repair proteins, and hence more repair and protection against DNA damage [46]. GSH and Grx are protective against oxidative DNA damage through the regulation of DNA repair enzymes [47, 48]. GSH is synthesized in neurons and protects DNA from oxidative stress in the brain [49]. However, the molecular functions of GSH in the neuronal nucleus and how GSH is transported to the nucleus in neurons, remain topics of debate.
The main function of Trx is the reduction of cysteines and cleavage of disulphide bonds in substrate proteins. In addition to Trx and NADPH, the Trx system also comprises thioredoxin reductase-1 (Txnrd1), which maintains Trx proteins in their reduced state via NADPH [12]. Similar to the Grx system, Trx has been implicated in DNA repair [50, 51]. Thioredoxin-1 (Trx1) is protective against oxidative DNA damage through the regulation of DNA repair enzymes [47]. In addition, Trx1 plays a major role in the reduction of apurinic/apyrimidinic endonuclease 1 (APE1) [52]. Impaired DNA repair and cell cycle arrest have been reported following impaired activity of 2′-deoxyribonucleotides in Txnrd1-deficient T cells, implying that Txnrd also functions in the DDR [53]. Overexpression of thioredoxin-interacting protein (TXNIP), a negative regulator of Trx [51], elevates oxidative DNA damage and shortens lifespan in Drosphilia, while downregulation of TXNIP increases resistance to oxidative stress and extends lifespan [51]. Under physiological conditions, cytosolic Trx1 interacts with apoptosis inducing factor (AIF), although this is disrupted following the induction of oxidative stress [54]. Furthermore, the interaction between AIF and DNA is impaired following localization of Trx1 in the nucleus, thus attenuating AIF-mediated DNA damage [54].
Protein disulphide isomerase (PDIA1, also known as PDI) is the prototype member of a large family of Trx proteins that possess two different activities: disulphide interchange/oxidoreductase function, involving oxidation, reduction and/or isomerisation of protein disulphide bonds, and general chaperone activity [55]. Hence, PDI catalyses the correct folding of misfolded or unfolded proteins into their native structure. PDI and Erp57, the family member with closest homologue to PDI, facilitate disulphide bond formation in almost all cellular proteins [56]. PDI is upregulated during the unfolded protein response (UPR), where it alleviates endoplasmic reticulum (ER) stress by enhancing protein folding [57]. However, whilst PDI is conventionally regarded as being localised in the ER, it has been detected in other cellular locations, including the nucleus [58, 59].
In addition to these specific enzyme systems, mitochondria are the major organelles that regulate redox reactions. They are the main site of energy production in cells via oxidative phosphorylation (OXPHOS) [60]. In fact, being the powerhouse of the cell, mitochondria provide approximately 80% of energy requirements, although they consume 90% of cellular oxygen [1]. Five distinct multiprotein complexes (I–V) comprise the mitochondrial OXPHOS system [61], and O2·− is generated primarily by complexes I and III [62].
Oxidative and nitrosative stress
Several organelles and cellular processes, as well as environmental agents, contribute to the generation of ROS. Under physiological conditions, ROS are beneficial because they are essential for many biological functions that depend on redox signalling. However, when redox conditions are dysregulated, they can be harmful. Mitochondria are the major source of oxidative stress, which can be detrimental by damaging both mitochondrial DNA (mtDNA) and proteins [63]. The mitochondrial genome is highly vulnerable to damage because unlike nuclear DNA, it is not protected by histones, and it is physically located close to the electron transport chain (ETC) [64, 65]. Hence, equivalent levels of free radicals can induce more lesions in mtDNA compared to nuclear DNA [66]. In addition, damage to mtDNA hinders expression of proteins involved in the ETC, dysregulating their activity, producing free radicals and disrupting mitochondrial functions [67].
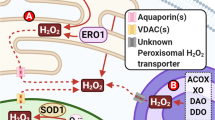
Other physiological processes and proteins can produce ROS, such as xanthine oxidoreductase (XOR). Mammalian XOR catalyzes the conversion of hypoxanthine to xanthine, and further to uric acid during purine metabolism, generating H2O2 [68]. XOR activity can therefore induce the generation of ROS, resulting in oxidative DNA damage and cell death [69]. Other mechanisms such as peroxisomal metabolism, anabolic processes and catabolic oxidation, can also produce ROS as by-products [70]. Neurons rely heavily on accurate DNA repair mechanisms and efficient DDR due to their high metabolic rate, but this can also generate ROS and hence oxidative DNA damage [71].
Similarly, RNS can be either destructive or favourable to cells depending on the conditions. Whilst they regulate important physiological processes, RNS also can be toxic by damaging metabolic enzymes and by reaction with superoxide, generating PN [72]. Furthermore, the interaction between NO– and O2·− creates the much more potent oxidant ONOO−, which influences whether NO induces physiological or pathological conditions [73]. PN binds to lipids, DNA, and proteins directly via oxidative reactions, or indirectly via radical-mediated mechanisms [73], and this can induce DNA damage [74].
ER stress can also induce redox dysfunction in cells [75, 76], which is increasingly linked to DNA damage [77, 78]. ER stress arises after accumulation of misfolded proteins in the ER, inducing the UPR [75]. Whilst these processes will not be discussed here, the reader is directed to several excellent recent review articles on this topic [75,76,77,78,79].
Types of DNA damage
DNA is a highly reactive molecule [20, 80] and therefore very susceptible to injury. DNA can be damaged in several different ways. This involves modification or loss of individual bases, breakage of one or both DNA strands, or DNA replication errors, including topoisomerase-mediated damage. Single-stranded breaks result in gaps in a single strand of the DNA double helix, and they arise frequently (tens of thousands per cell per day). They are generally accompanied by loss of a single nucleotide and by damaged 5′- and/or 3′-termini at the site of the break [81]. In contrast, damage to both strands of DNA results in a double-stranded break (DSB) which is considered to be the most toxic DNA injury, because it can lead to cell death if unrepaired, and to chromosomal translocations if mis-repaired [82].
Insults to DNA can be categorized as either endogenous or exogenous (environmental) depending on the source of damage, but a major source of both endogenous and exogenous DNA damage is oxidative stress [83]. The most common forms of DNA damage resulting from redox dysregulation include SSBs, oxidative modification of bases, and the formation of apurinic/apyrimidinic (AP) or abasic sites, which are regions of DNA lacking either a purine or a pyrimidine base. Oxidative DNA damage can also involve base mismatches, DSBs, and inter-strand crosslinks (ICLs) (Fig. 3). However, as ROS mainly induce SSBs, DSBs may be the result of conversion of SSBs and/or result of oxidized bases or abasic sites during the DNA repair process [84]. Elevated ROS and RNS can also induce DNA-DNA or DNA–protein cross-linking and sister chromatid exchange, and translocation in nuclear DNA [85, 86] (Fig. 3). Replication stress, oxygen radicals, ionizing radiation (IR), chemotherapeutics, ultraviolet (UV) light, and polycyclic aromatic hydrocarbons (PAHs), can also initiate oxidative DNA damage. Oxidized bases are resolved primarily by base excision repair (BER), whereas the DNA backbone is repaired by SSB repair or DSB repair pathways [82, 87]. Below we discuss the possible sources of redox-relevant DNA damage and the types of damage that can result, as well as the mechanisms that repair these forms of damage.

Types of oxidative DNA damage. Several types of stressors can lead to oxidative DNA damage. Replication stress is the major source of base mismatches in DNA, whereas free radicals primarily induce single-strand breaks (SSBs), and double-strand breaks (DSBs) to a lesser extent. Ionizing radiation and chemotherapeutics can induce both SSBs and DSBs, as well as interstrand crosslinks. DNA damage induced by UV radiation results in bulky DNA adducts
Exogenous DNA damage
Environmental conditions, including hypoxia, extreme temperatures (heat or cold) and oxidative stress, are important sources of exogenous DNA damage [88, 89]. Furthermore, many factors present in the environment can induce oxidative DNA damage. These include various types of radiation, chemical mutagens from food and other sources, industrial chemicals, and smoke.
UV radiation
UV radiation is one of the most powerful and carcinogenic environmental agents that interacts with DNA and can modify genomic integrity, either directly or indirectly [90]. UV radiation initiates the ‘preparation for oxidative stress’ antioxidant response, whereby antioxidants are upregulated [91] and the resulting minor redox imbalance leads to increased tolerance to additional oxidative insults. UV radiation also produces free radicals that attack the intracellular domains of ret tyrosine kinase, which is implicated in oncogenesis, leading to its dimerization and activation [92].
Usually UV radiation is divided into three categories based on the emission wavelength: UV-A (320–400 nm), UV-B (290–320 nm) and UV-C (190–290 nm) [90]. Both UVA and UVB radiation (to a lesser extent) induce oxidative DNA damage, unlike UV-C. UV radiation also induces DNA strand breaks and DNA–protein crosslinks [93], and UV-A radiation can target bases by photodynamic effects which involve the participation of singlet oxygen (1O2), and to a lesser extent, ·OH. To repair UV radiation-based damage, cells employ several defence mechanisms, including nucleotide excision repair (NER), homologous recombination, direct reversal of UV-damaged bases, and ICL repair [94, 95].
IR
IR is another source of exogenous DNA damage, which includes micro- and radio-waves, and alpha-, beta-, gamma-, and X-rays [96, 97]. IR is produced from the surroundings, including soil, rock, radon, medical devices and cosmic radiation [96]. Based on the quantity of energy transferred, IR radiation can be classified into either high linear energy transfer (LET), which refers to alpha radiation, or low LET, in the case of beta and gamma radiation [97].
Like UV radiation, DNA damage induced by IR can also be either direct or indirect (although it is mostly indirect) and associated with oxidative stress. Whilst IR directly induces DNA breaks, especially DSBs, it can also produce oxidative lesions in DNA by ROS, including the generation of abasic sites and SSBs [98], and by stimulating inducible nitric oxide synthase activity, thereby generating large amounts of ·NO. NO reacts with O2, producing ONOO−, which is highly invasive and induces DNA damage. Interestingly, SSBs formed by IR contain 3’ phosphate or 3’-phosphoglycolate ends instead of 3’ OH ends, and this differentiates them from other non-IR-induced SSBs [99]. DSBs can also be produced following adjacent sites damaged by IR that are present on both DNA strands [82]. IR-induced lesions are repaired by homologous recombination for DSBs [99], or by AP endonucleases, polynucleotide kinase phosphatase (PNKP) and tyrosyl DNA phosphodiesterase 1 (TDP1) for SSBs [100].
Chemical mutagens
In the sections below we discuss the mutagens known to induce oxidative DNA damage.
Alkylating agents
Alkylating agents are reagents that add alkyl groups to DNA bases, most commonly to guanine. Dietary ingredients, tobacco smoke, chemotherapeutic agents, burning biomass and industrial manufacturing are the foremost sources of exogenous alkylating agents [101], but this also includes sulphur and nitrogen mustards used in war [20]. Alkylation results in the formation of DNA adducts [20], including methyl methanesulfonate, ethyl methanesulfonate, methylnitrosourea and N-methyl-N’-nitro-N-nitrosoguanidine [102]. Nitrogenous base rings and the N3 of adenine and N7 of guanine are particularly susceptible to electrophilic alkylating agents, although all DNA bases are vulnerable [103]. The pathways involved in DNA repair induced by alkylated bases include BER, ICL and direct damage reversal pathways [102].
Aromatic amines
Aromatic amines are organic compounds containing an aromatic ring attached to an amine group that can induce oxidative DNA damage. They are present primarily in tobacco smoke, pesticides, motor fuels, and colourants. The most widely studied aromatic amines in vitro are 2-aminofluorene (AF) and N-acetyl-2-aminofluorene (AAF; an acetylated derivative of AF) [104]. Aromatic amines can be converted into esters and sulphates, modifying the C8 position of guanine, via alkylation by the activated P450 mono-oxygenase system, the primary cellular mechanism responsible for clearance of pharmacological compounds [105]. Oxidative DNA damage resulting from aromatic amines [106] is repaired by NER [107, 108].
PAHs
PAHs are non-polar hydrocarbons with two or more aromatic rings that are sources of DNA damage [20]. They include anthracene, naphthalene, pyrene, dibenzo [a,l] pyrene and benzo(a)pyrene [109]. The main sources of PAH in the environment are tobacco smoke, automobile exhaust fumes, incomplete combustion of organic materials, fossil fuels and overcooked food [110]. Similar to aromatic amines, exposure to PAHs promotes oxidative damage [111, 112], oxidative stress and lipid peroxidation [113]. The NER and BER pathways are involved in repair of PAH-induced DNA damage.
Endogenous DNA damage
DNA damage can also arise spontaneously from natural metabolic processes, and most endogenous DNA lesions are SSBs (75%). Oxidative lesions form a major component of this form of DNA damage [114, 115]. We discuss below the major types of endogenous DNA damage related to cellular redox processes.
Base modifications
ROS, particularly the OH· radical, directly attack both purine and pyrimidine bases and the deoxyribose sugar backbone of DNA [28]. The OH· radical removes hydrogen atoms and generates modified purine and pyrimidine base by-products and DNA–protein cross-links [28]. Approximately 20 different oxidized base adducts can be generated by oxidative DNA damage induced by ROS [116]. Pyrimidine bases modified by OH· can produce distinct adducts such as uracil glycol, 5-hydroxydeoxy uridine, thymine glycol 5-hydroxy deoxycytidine, 5-formyl uracil, cytosine glycol, 5,6-dihydrothyronine, 5-hydroxy-6-hydro-cytosine, 5-hydroxy-6-hydro uracil, uracil glycol, alloxan and hydantoin [116]. Other adducts—8-hydroxydeoxy guanosine, 8-hydroxy deoxy adenosine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine—can also be attacked by the purine adducts formed by OH·. Guanine is a prime target of oxidative DNA damage due to its lower reduction potential compared to other bases [83]. OH· radicals interact with the C4, C5 and C8 positions of the imidazole ring of guanine (G) to form several potentially mutagenic DNA lesions, including 8-oxoguanine (8-oxoG) [83]. This modified base pairs with adenine (A) instead of cytosine (C), leading to the frequent incorporation of mutations. PN can also interact with G to form 8-nitroguanine, which is used as a nitrosative DNA damage marker [83]. The deoxyribose sugar backbone of DNA can also form a number of free radical-induced adducts, including glycolic acid, 2-deoxytetrodialdose, erythrose, 2-deoxypentonic acid lactone, and 2-deoxypentose-4-ulose [116].
Base deamination
The deamination, or removal of an amino group from a base, is a major source of spontaneous DNA damage. In human cells, A, G, C, and 5-methyl cytosine (5mC) are capable of being deaminated, which convert to hypoxanthine, xanthine, uracil (U), and thymine (T), respectively [117]. Among these, 5mC becomes deaminated most frequently, followed by C [117], and single-stranded DNA (ssDNA) is the preferred target compared to double-stranded DNA (dsDNA) [118, 119]. Base deamination eventually induces mutations after successive DNA replication cycles. Oxidative stress is a major trigger of deamination and thus DNA damage [120,121,122]. However, deamination can also result from exposure to UV radiation, nitrate, sodium bisulphite and intercalating agents. DNA bases damaged by deamination are predominantly repaired by the BER pathway [123].
Abasic (apurinic/apyrimidinic) sites
The formation of an apurinic/apyrimidinic (AP), or abasic, site is one of the most frequent endogenous DNA lesions, particularly following oxidative stress [124]. Abasic sites result from the spontaneous hydrolysis or cleavage of N-glycosyl bonds, which link nitrogen bases to the sugar-phosphate backbone [20]. Human cells generate abasic sites at a higher frequency (approximately 1000 per day) compared to other organisms, which is further increased by high temperatures and extremes of pH, both acidic and basic [125]. Whilst oxidative stress promotes the formation of abasic sites, they are usually unstable and instantly convert into SSBs [126]. Abasic sites are principally repaired by BER, and sometimes by NER.
Topoisomerase (TOP)-mediated DNA damage
DNA TOP enzymes catalyze alterations of the topological state of DNA, and they are required for several important cellular functions, including DNA replication and transcription. The interweaved, supercoiled nature of the DNA double-helix can lead to topological problems and tension, but the introduction of transient breaks by TOPs allows the DNA strands to be rotated, thus relieving topological stress [127, 128]. However, this process can lead to endogenous DNA damage when these transient breaks are not repaired. Human TOPs are targets for some of the major chemotherapy drugs that function by inducing redox stress, producing ROS and lipid peroxidation products [129, 130]. TOP1 contains eight cysteines, two of which play a critical role in catalytic activity and are the target of thiol-reactive compounds [131].
There are two types of TOP enzymes: type I and type II, which act on SSBs and DSBs, respectively. In the case of topoisomerase type 1 (TOP1), temporary nicks wrap around TOP1-bound DNA, forming a complex that relaxes the DNA. TOP1 aligns the 5’-OH group of the DNA with the tyrosine-DNA phosphodiester bond to ligate the nicked ends and thus resolve the complex [132, 133]. Hence, stabilization of DNA breaks induces DNA damage, resulting in failure to properly align the strands [134]. Aberrant DNA morphology, including the presence of abasic sites and DNA adducts, can further stabilise the TOP1-DNA complex, creating DNA lesions [135]. Topoisomerase 2 (TOP2) enzymes resolve topological problems by a "two-gate" mechanism involving the hydrolysis of ATP [136]. They primarily induce DNA DSBs, but they can also induce SSBs [137] and oxidative DNA damage [137]. Three TOPs have been identified in mitochondria which are required by mtDNA. Furthermore, redox regulation of these TOPs may play a role in mitochondrial homeostasis [138].
DNA methylation
DNA methylation is an epigenetic process by which methyl groups are added to DNA, and it occurs most commonly to the C base, forming 5-methylcytosine, although it can also be added to G and A, resulting in the formation of N7-methylguanine, N3-methyladenine and O6-methylguanine [20]. DNA methylation regulates gene expression and therefore is a normal endogenous process, but it can also result in DNA damage. The endogenous production of choline, betaine and nitrosated bile salts, as well as exogenous factors such as smoking, diet, pollution and N-nitroso compounds, can induce DNA methylation [101]. The C base can be modified by oxidation, forming 5,6-dihydroxycytosine [139], which is necessary for DNA de-methylation [140]. This can result in base transition mutations, development of abasic sites and minor methyl lesions on DNA [20]. However, failure to remove these methyl groups induces DNA damage [20]. The BER pathway repairs these lesions by cleaving the glycosylic bonds of methylated bases.
Cross-linking DNA damage
Crosslinks of DNA are produced when two nucleotides form a covalent linkage, and it can be either intra-strand, within the same strand, or inter-strand, between opposite strands. Whereas intra-strand crosslinks are easily removed by NER, ICLs are extremely toxic lesions that prevent separation of the DNA strands, and as few as 20 unrepaired ICLs can kill mammalian cells [141]. ICL can be induced both by UVA and by chemical agents, including those used in chemotherapy, such as carboplatin and mitomycin C (MMC) [142]. Processes such as replication and transcription, where separation of the two DNA strands is essential, are inhibited by the presence of this irreversible covalent linkage, which can induce cell death [143]. The formation of ICLs requires two independent groups in an alkylating molecule that react with two bases present on opposite DNA strands. Oxidative stress and agents such as platinum compounds, MMC, psoralens, and nitrogen mustards induce ICLs [143]. ICLs are repaired by NER and other mechanisms [144].
DNA damage induced by lipid peroxidation
One of the consequences of excessive amounts of ROS and RNS is lipid peroxidation [145], whereby oxidants attack lipids containing one or more carbon–carbon double bonds [146]. This can induce DNA damage by the formation of reactive aldehydes, which produces mutagenic adducts in bases, particularly A and G [147, 148]. Among lipids, cholesterol esters, phospholipids, and triglycerides are particularly susceptible to oxidative modification because they contain polyunsaturated fatty acid (PUFA) side chains [30]. These PUFAs are extremely vulnerable to oxidation by free radical species, especially ·OH. Aldehyde products resulting from lipid peroxidation include 4-hydroperoxy-2-nonenal (4HNE) [149, 150], malondialdehyde (MDA) [151], and acrolein [152]. The oxidation of lipids is an important source of DNA damage [153, 154] and the reader is referred to several excellent reviews for more details [9, 155, 156].
DDR and DNA repair pathways
The DDR is an elaborate signalling network that detects, signals and repairs DNA lesions [20]. Specific DNA repair pathways are activated based on the type of damage induced [21, 157, 158], regulated by phosphatidylinositol 3-kinase (PI3K)-like kinase family members. These include ataxia telangiectasia mutated kinase (ATM) and ataxia telangiectasia and Rad3-related protein (ATR), that mediate DSB and SSB repair, respectively [159]. In dividing cells, the DDR arrests the cell cycle following DNA damage, both transiently by activating DNA damage checkpoints, and permanently by inducing cellular senescence [160]. Below we discuss the DNA repair pathways relevant to redox-relevant DNA damage.
BER pathway
BER is the most important mechanism that cells use to repair lesions formed following oxidative stress and redox dysregulation. It corrects damage to bases resulting from deamination, oxidation, or methylation, that have not significantly altered the arrangement of the DNA helix [161,162,163]. The nucleus is the main subcellular location where BER takes place, although it has also been detected in mitochondria [163].
DNA glycosylases play important roles in BER because they both detect and remove specific damaged or inappropriate bases, forming abasic sites whilst leaving the sugar phosphate backbone intact. At least 11 distinct types are involved in BER and 8-oxoG glycosylase (OGG1) initiates repair of the most common 8-oxoG lesions in both the nucleus and mitochondria [164]. The abasic site is then repaired by either ‘short-patch’ BER or ‘long-patch’ BER. In short-patch BER, which involves only a single nucleotide gap in the abasic site, DNA polymerase β (which is specific to BER) fills this gap, accompanied by the XRCC1/Ligase III complex [21]. In contrast, long-patch BER involves a repair tract of at least two (and up to 13) nucleotides, where the gap is sealed by Ligase I or proliferating cell nuclear antigen (PCNA) after resynthesis of DNA [144, 165]. BER is also implicated in the repair of SSBs through single-strand break repair (SSBR) [144]. Replication protein A (RPA) is required for each of the four major DNA repair pathways [166].
APE1 performs an important role in BER by acting as a nuclease, precisely cleaving the DNA backbone at the abasic site. During this process, APE1 is multifunctional because it displays endonuclease, 3′ phosphodiesterase, 3ʹ-to-5ʹ exonuclease, and RNA cleavage activities. Importantly, the exonuclease activity is required to remove DNA damage generated by ROS during oxidative stress, hence it is an essential component of BER. Moreover, APE1 forms a central link between redox regulation and DNA repair because it is the only DDR protein that can also regulate redox conditions. Hence it possesses two functions within the one protein (mediated by different domains), and thus it is also referred to as ‘redox effector factor 1,’ or ‘Ref-1’. APE1 also regulates multiple redox-regulated transcription factors, including nuclear factor kappa B (NF-κB) [167], STAT3, p53 [168], hypoxia inducible factor-1α [169], and cAMP-response element binding protein [169] (see also Sect. "AD" below).
NER pathway
NER is the central pathway responsible for the removal of large ssDNA adducts induced by UV irradiation, environmental mutagens, or chemotherapeutic agents [144, 170]. Moreover, NER also repairs lesions that result from oxidative stress [171]. There are two sub-pathways of NER: global genome NER (GG-NER) and transcription-coupled NER (TC-NER) [172] that differ in how they are initiated.
Unlike TC-NER, GG-NER is not induced during transcription. Specific proteins continuously scan the genome for distortions of the helix. Once detected, GG-NER is then initiated by either a complex of xeroderma pigmentosum complementation group C (XPC) and UV excision repair protein radiation sensitive 23 B (RAD23B) (XPC-RAD23B) alone, or in some cases, with UV-damaged DNA-binding protein [173]. In contrast, TC-NER is activated during transcription when RNA polymerase is stalled at a lesion with TC-NER specific proteins: Cockayne syndrome protein A, CSB, and XPA-binding protein 2. Once the lesion is identified, both TC-NER and GG-NER follow a similar mechanism, requiring transcription factor II H to excise and repair the lesion [170]. Bi-directional helicase reveals approximately 30 nucleotides in DNA during this process, stabilised by XPA and RPA. The lesion is removed by XPG and the DNA excision repair protein-1 (ERCC1)-XPF complex, which leaves a single-stranded gap. This is filled in by DNA replication proteins PCNA, RPA and DNA polymerases POL σ, κ, ε, and subsequently sealed by DNA ligases I or III [144]. The NER pathway is also involved in the early steps of ICL repair [144] (Fig. 4).

The major DNA repair pathways involved in correcting DNA damage induced by oxidative stress. BER is the predominant repair mechanism that removes oxidative DNA damage to bases, via two general pathways—short-patch and long-patch. Short-patch BER facilitates repair of a single nucleotide, whereas long-patch BER repairs two or more nucleotides. Although the nucleus is the main subcellular localisation where BER takes place, it has also been detected in mitochondria [163]. NER is the principal pathway responsible for the removal of large single-stranded DNA adducts induced by UV irradiation, environmental mutagens, or chemotherapeutic agents, but it is also implicated in repairing oxidative DNA damage. MMR is another pathway that repairs DNA damage induced by oxidative stress. MMR is responsible for the detection and repair of errors produced during DNA replication, involving the incorrect insertion, deletion or misincorporation of nucleotides. RPA Replication protein A; pol δ DNA polymerase δ; Exo1 exonuclease 1
Mismatch repair (MMR) pathway
The MMR pathway is responsible for the detection and repair of errors produced during DNA replication, involving the incorrect insertion, deletion or misincorporation of nucleotides. This prevents the permanent incorporation of mutations in dividing cells.
The MMR pathway comprises three linked, but different, protein subunits in humans: hMutSα, hMutSβ, and hMutLα. hMutSα is a heterodimer composed of hMSH2 and hMSH6, and it is constantly expressed, scanning the homoduplex DNA for errors [174]. Once mismatched bases are recognized, MMR is activated through upregulation of MSH2 and hMSH6. This complex then interacts with component proteins of the hMutLα heterodimer, MLH1 and PMS2, resulting in its binding to DNA lesions [165] to facilitate removal of the mismatched bases. RPA binds to nicked heteroduplex DNA to facilitate assembly of the MMR initiation complex [166]. Finally, the new DNA is synthesized (Fig. 4).
ICL repair pathway
ICL repair is highly conserved, and it maintains genomic stability during DNA replication. Two mechanisms are present in humans, recombination-dependent and recombination-independent pathways. Recombination-dependent ICL involves fanconi anemia (FA) proteins both in its detection and repair. FA is an autosomal recessive cancer syndrome characterized by progressive bone marrow failure and congenital anomalies, involving eight different complementation groups. Several FA proteins interact in a multi-subunit complex that repairs complex ICL lesions in DNA. The formation of an ICL prevents the DNA strand from unwinding and separating, which stalls the replication fork. A complex containing FA proteins FANCM and FAAP24 as well as the histone fold protein complex, binds to the stalled replication fork, which is then remodelled. This results in migration of the Holiday junction, unwinding of ssDNA [20], and activation of ATR-mediated Checkpoint kinase 1. This further activates FA proteins FANCI and FANCD2, which together form the ‘ID complex’. Phosphorylation of this complex by ATR results in its recruitment to the FA core where it becomes mono-ubiquitinated, and this subsequently recruits other DDR factors involved in ICL repair. Other DNA repair pathways, including homologous recombination, NER and translesion DNA synthesis, also contribute to the repair of ICL [22]. Failure to repair ICLs results in significant chromosomal abnormalities that promote tumorigenesis [175]. Moreover, following oxidative stress, ICLs can be generated. This has been reported for thymine radicals [176] and another major oxidative lesion (to A bases), oxoA, which can produce ICLs with the opposite base (G, A, C, T, I), to produce structurally diverse ICLs [177]. However, this mechanism has not been described for the most common oxidative lesion, 8-oxoG [177].
SSBR pathway
SSBs can arise directly, by breakdown of oxidized sugars or indirectly, through BER of oxidized bases, abasic sites or damaged/modified bases [161, 178, 179]. They pose severe risks to genetic stability and cell viability if not repaired promptly [81]. Therefore, cells have developed efficient mechanisms to repair SSBs, collectively known as SSBR [81], which is often considered to be a component of BER [180]. The overall SSBR process comprises four fundamental steps, involving SSB detection by ATR, DNA end processing, DNA gap filling and DNA ligation [81]. This also provides another mechanism to repair ROS-induced DNA damage, but its role is not as significant as BER, NER and MMR.
A growing body of evidence indicates that poly-ADP-ribose-polymerases (PARPs) perform a central function in SSBR [181,182,183,184]. The PARP enzyme family (consisting of 17 members) catalyzes the covalent transfer of polymers of ADP-ribose onto acidic residues in target proteins, using the redox substrate NAD + . This process, termed ‘PARylation’, is a post-translational modification that regulates multiple signalling mechanisms, including several DNA repair pathways. PARP1 and PARP2 are induced instantly after the formation of SSBs, which results in PARylation of DNA ligase III, DNA polymerase beta, XRCC1, and end-processing enzymes such as TDP1 and aprataxin, at sites of SSB damage [21]. Recruitment of chromatin remodelling factor ALC1 (amplified in liver cancer 1) and macroH2A1 at DNA damage sites is also PAR-dependent [22]. This leads to chromatin remodelling and the recruitment of complexes to facilitate DNA repair and chromatin modification [22]. Finally, DNA polymerase β, polynucleotide kinase (PNK) and nucleases APE1, aprataxin PNK-like factor (APLF) and aprataxin (APTX), seal the processed DNA [21]. The activation of PARP-1 following DNA damage and subsequent depletion of NAD + and then ATP, have been linked to energy failure, redox dysregulation, mitochondrial ROS production and apoptosis [185].
DSB repair pathway
There are two main pathways to repair DSBs in eukaryotes: non-homologous end joining (NHEJ) or homology directed repair (HDR) [26, 186, 187]. However, in terminally differentiated neurons, NHEJ is thought to be the most important mechanism [188] because HDR requires the presence of homologous sequences during the S and G2/M phases of the cell cycle, which is absent in neurons. Hence HDR functions only in the S-phase in undifferentiated or proliferating and neuronal stem/progenitor cells, although genome editing via HDR is possible in mature post-mitotic neurons [189]. Even low levels of ROS are known to produce complex DSBs and can produce mutations following error-prone NHEJ [189].
ATM-mediated DSB repair pathway
In both the homologous recombination and NHEJ pathways, DSBs are initially recognised by ATM. As for SSBR, activated PARP family members (PARP1 and PARP2) are rapidly recruited and bind to the MRN/ATM complex at DSBs [22]. This leads to recruitment of other DDR proteins [22] and activation of ATM. H2A histone family member X (H2AX) is then phosphorylated by ATM over a large region of DNA surrounding a DSB. Phosphorylated H2AX (γH2AX) forms visible foci in the nucleus that are widely used as an experimental marker of DSBs. Furthermore, γH2AX possesses important functions in the DDR by (i) assembling key substrates of ATM at sites containing damaged chromatin, including p53, p53-binding protein, and breast cancer gene 1 (BRCA1), and (ii) activating checkpoint proteins that arrest the cell cycle, such as Mdc1 (mediator of DNA damage checkpoint protein 1) and Chk2 [22, 190].
NHEJ pathway
There are two main mechanisms of NHEJ. Classical NHEJ results in ligation of DNA ends with no or little (1–3 bases) sequence homology, thus it can result in the production of deletion or insertion mutations [191]. In contrast, in alternative NHEJ, regions of DNA with micro-homologous sequences are ligated (4–20 bases) [191]. The molecular mechanisms involved in alternative NHEJ remain unclear, although it is thought to be a back-up system that is less efficient than classical NEHJ [192]. However alternative NHEJ has an even greater tendency to create mutations because the ligated products always contain deletions [192].
Classical NHEJ
Neurons remain arrested in the G0 phase of the cell cycle, but NHEJ repairs DNA throughout all phases of the cycle. NHEJ involves the detection of DNA ends, assembly and stabilization of the NHEJ complex, linking and then processing of the ends, and finally, attachment of the ends and disassembly of the NHEJ complex [193]. A specific kinase complex, DNA protein kinase (DNA-PK, another PIKK family member), performs a critical role in NHEJ. It consists of Ku, a heterodimer of Ku70 and Ku80 subunits, which directs the complex to DNA and activates the PI3K kinase activity of its catalytic subunit (DNA-PKcs). Upon DNA damage, Ku (Ku70/Ku80) creates a ring-like structure and rapidly binds to DNA strands at DSB sites [22]. Ku also facilitates the threading of PARP1 onto each broken DNA end and DNA end processing, and it also recruits nucleases to trim, and polymerases to fill in, the DNA ends [26]. DNA-PKcs substrates, such as X-ray repair cross-complementing protein 4 (XRCC4), become phosphorylated upon its activation, resulting in DNA end protection. Ultimately, DNA ligase IV complex and Cernunnos/XLF then ligate the released DNA ends [144]. However, nucleases ARTEMIS and APLF, along with PNK kinase/phosphatase, are responsible for processing the DNA ends that cannot be ligated [144]. In a small proportion of NHEJ (10%) repair events, ARTEMIS is phosphorylated by ATM, even though it is considered to be a DNA-PKcs substrate [144].
Alternative NHEJ
Whilst this end-joining mechanism is not well characterised, it does not involve the same machinery as classical NHEJ. Alternative NHEJ can result following primary DNA re-section by PARP and MRN when Ku or recombination factors are not available, and it requires the DNA end-processing factor, CtBP-interacting protein (CtIP). DSB breaks are sealed by microhomology-mediated base-pairing, followed by nucleolytic trimming of DNA flaps, DNA gap filling, and then ligation. In alternative NHEJ, XRCC1 and LIG III protect and ligate the DNA ends, respectively [22]. There is evidence that DNA polymerase θ is of critical importance for this mechanism [26].
Homologous recombination pathway
Homologous recombination is the most common form of HDR used to repair DSBs and ICLs. It uses a homologous sequence as a DNA template to repair the DSB, resulting in reformation of the original DNA sequence. Hence, it is less likely to introduce mutations compared to NHEJ. Following generation of the DSB, the MRN complex, consisting of MRE11, Rad50 and Nbs1, binds to DNA on either side of the DSB. Rad51 becomes recruited into DSB sites following activation of ATM by MRN [22]. DNA end resection follows, whereby the dsDNA is processed by the MRN complex, CtIP, PARP1, BRCA1 and other endonucleases, to remove nucleotides from the 5' end to produce short 3′ single-strands (30 bases) [22, 144]. This also prevents activation of classical NHEJ. MRE11 stabilizes the ends of DNA, and it facilitates the early steps of DNA end resection via its endonuclease and exonuclease activities [22, 194]. The two other members of the MRN complex, Rad50 and NBS1, promptly interact with MRE11 at the DNA ends of the DSBs and NBS1 binds to ATM by its C-terminal domain, which accelerates its recruitment to DSB sites [22]. RPA, which possesses a high affinity for ssDNA, binds to the 3’ ssDNA overhang and Rad51 then binds, forming a nucleoprotein filament. Rad51, aided by BRCA2, searches for a homologous sequence to the 3’ overhang on the sister chromatid. Once located, the nucleoprotein filament catalyses invasion of the strand. Base pairs on the homologous DNA strands are consecutively exchanged at specific regions, that move the branch point up/down the DNA sequence in a process called ‘branch migration’[195]. These regions are termed ‘Holliday junctions’, which contain four double-stranded DNA arms joined in a branched structure. The DNA strands with DSBs are thus replaced by a homologous sequence template on the sister chromatid [196].
Single-strand annealing (SSA)
SSA is another DSB repair pathway that is activated when a DSB occurs between two repeated sequences oriented in the same direction. It is considered to be midway between homologous recombination and NHEJ because it uses homologous repeats to bridge the DSB ends [196]. Next to the DSB, ssDNA regions are formed that extend to the repeat sequences so that complementary strands can anneal to each other. The annealed sequences are then processed by digestion of the ssDNA overhangs and filling in of the gaps. SSA plays a role in MRN exonuclease-mediated DNA end excision from both strands of the DSB site, until small homologous sequences on both strands are established [197].
Redox regulation, DNA repair and DNA damage
Increasing evidence indicates that the DDR is regulated by the cellular redox state, and vice versa. Furthermore, redox regulation also controls epigenetic mechanisms and other mechanisms related to DNA damage, such as the regulation of gene expression by transcription factors. As detailed below, there are several known links between redox regulation and maintenance of DNA repair mechanisms in normal cellular physiology, although some aspects are only just emerging. Below we provide an overview of these mechanisms in normal cells.
Proteins with possible dual functions in both DNA repair and redox regulation
APE1 is the classic example, given it possesses both redox activity and DNA repair functions in the nucleus [198], and it is protective following oxidative DNA damage to neurons. It functions as a redox effector factor for multiple transcription factors including AP1, p53 and HIF1-α. In addition to its well-established role in BER, the redox activity of APE1 is also implicated in NER [198, 199]. Similarly, whilst Ku is an essential component of NHEJ [200], it also possesses redox activity, which is essential for its binding to DNA [200]. The ability of Ku to bind to DNA damage sites decreases following induction of oxidative stress [200].
O-6-methylguanine-DNA methyltransferase (MGMT), which facilitates chemoresistance to alkylating agents, is one of the most specific DNA repair enzymes, and it is also redox-regulated [201]. MGMT replaces the highly mutagenic lesion O6-methylguanine with guanine, thus inhibiting mismatch errors and other mistakes that arise during transcription and DNA replication. During this process, MGMT transfers the methyl at O6 sites of damaged guanine nucleotides to cysteine residues [201]. S-nitrosylation at these active, essential cysteines can inhibit its enzymatic activity through a ‘suicide’ reaction [202]. Therefore, inhibition of MGMT can result following induction of oxidative stress [203].
In addition to direct effects on the DDR, redox regulation can modulate gene expression of DNA repair proteins [203]. Transcription factors with zinc-finger motifs or transition metal-binding regions are regulated by redox processes [204]. Both the Trx and Grx systems are upregulated by Nrf2, a transcription factor with a zinc-finger that controls many genes displaying antioxidant response elements and is a master regulator of the antioxidant response. Nrf2 is in turn regulated by thiol oxidation of kelch-like ECH-associated protein 1 [205].
Another zinc-binding transcription factor with multiple important roles in the DDR is p53 [206]. p53 is a multi-tasking protein activated by DNA damage that co-ordinates several DNA repair activities. Thus, it has been termed a “guardian of the genome” [207]. p53 participates in several DNA repair mechanisms and it controls DNA-damage checkpoints by halting the cell cycle, to allow time for repair. Cells lacking p53 are prone to mutations and genomic instability, hence it is also known as a tumor suppressor. Moreover, p53 is regulated by redox signalling, thus it has been referred to as a central ‘hub’ in redox homeostasis [208] because it also mediates antioxidant and pro-oxidant pathways [208]. In addition, it protects neurons from DNA-damaging agents [209]. Importantly, its function as a transcription factor and its ability to bind to DNA are dependent on cellular redox conditions. p53 contains 10 cysteine residues located in its DNA-binding region, three of which form the zinc fingers that are crucial for its correct folding. APE1 stimulates p53-dependent transcription [207, 210] and p53 activity is dependent on the Trx system, both directly and indirectly via APE1. The crosstalk between Trx and p53 involves TXNIP, which interacts with p53. TXNIP binds to and stabilises p53, and it detaches from Trx during oxidative stress. S-glutathionylation of cysteines in human p53 inhibits its DNA-binding property [211]. p53 also activates expression of several antioxidant genes, including sestrin (Sesn)1 and Sesn2 [212]. Interestingly, promoters of p53-regulated genes with antioxidant functions appear to be sensitive to low levels of p53, whereas pro-oxidant and pro-apoptotic p53 target genes are activated in response to higher p53 levels upon extensive stress [211].
There is increasing evidence that PDI proteins have a role in the DDR, particularly Erp57. Inhibition or knockdown of PDI family members (PDIA1 and PDIp) downregulates many DNA repair genes, including E2F transcription factor 1 and Rad51 [213, 214]. Similarly, Erp57 binds to DNA [215] and PDI/Erp57 immunoprecipitates with APE1 [41]. In addition, Erp57 can translocate into the nucleus where it binds to MSH6 and DNA polymerase δ, following oxidative stress [216]. ERp57, together with high-mobility group proteins 1 and 2 (HMGB1 and HMGB2), is part of a complex that recognises damaged DNA [217]. Erp57 also modulates phosphorylation of H2AX, and it relocates to the nucleus following DNA damage [218]. Downregulation of Erp57 significantly inhibits the phosphorylation of H2AX and induction of DDR following cytarabine treatment [218]. However, despite this evidence, a direct role for PDI proteins in DNA repair has not yet been described.
SOD1 is an important antioxidant protein expressed in the nucleus, as well as the cytoplasm and mitochondria [219,220,221], and recently a protective role for nuclear SOD1 against oxidative DNA damage was described [222]. Following oxidative stress, SOD1 is phosphorylated by Chk2, leading to its translocation to the nucleus and protection against DNA damage [222]. Nuclear SOD1 appears to regulate the expression of GSH during this process [223]. The nuclear localisation of SOD1 is enhanced in an ATM-dependent manner by its association with the Mec1/ATM effector, Dun1/Cds1 kinase, and phosphorylation of SOD1 at S60 and S99 [222]. Ribonucleotide reductase is a SOD1 transcriptional target [224] vital for the synthesis of deoxyribonucleosides and hence essential for DNA repair [225]. In addition, recent studies have shown that ATM also has a redox-sensor function in mitochondria, and can regulate oxidative stress by this mechanism.
Redox dysregulation and DNA repair in neurodegenerative diseases
The basic molecular mechanisms linking redox regulation to DNA damage remain unclear. However, improving our understanding of how these events are inter-related has important implications for diseases in which both processes are implicated in pathogenesis, such as neurodegenerative conditions [203].
There is now significant evidence linking defects in the DDR to neurodegenerative diseases [17] and the death of specific types of neurons is the underlying pathological feature of these conditions. However, the mechanisms by which neurons die in these conditions remain unclear, although it is well established that apoptosis plays a role [23]. Recently, many novel cell death pathways have been identified, and the Nomenclature Committee on Cell Death (NCDD) has developed guidelines to describe these mechanisms [226]. Importantly, several of these processes are induced by DNA damage, and there is increasing evidence that neurons die by at least some of these mechanisms. Parthanatos is a PARP-1-dependent cell death mechanism distinct from apoptosis or necrosis that can be induced by oxidative stress and DNA damage. It involves overactivation of PARP-1 leading to augmented production of long-chained and branched PAR polymers [23]. Importantly, multiple reports have implicated parthanatos in the death of neurons in several neurodegenerative diseases. Similarly, a role for p53 in neuronal death in neurodegenerative disorders has also been reported in several studies. For more details of these studies, and cell death mechanisms in relation to DNA damage in neurodegenerative disorders, please see a recent review [23].
It is also important to consider the role of glial cells in relation to oxidative DNA damage, neuroinflammation and neurodegeneration. When activated, microglia can produce several factors that are toxic to neurons, such as pro-inflammatory cytokines TNFα, PGE2, and INFγ and ROS (NO, H2O2, O2·−, NOO−), in response to diverse stimuli, including neuronal damage, misfolded proteins, and environmental toxins [227]. Microglial NOX2 is a major regulator of neurotoxicity by producing excessive ROS [228]. Through a complex antioxidant response, astrocytes also enhance the decomposition and clearance of free radicals produced by neurons and other cell types in the CNS [229]. Excessive free radicals can result in reactive astrogliosis, inducing neuroinflammation, which can lead to further oxidative stress [229]. Senescence is also strongly linked to DNA damage, and it is also implicated as a potential driver of neuroinflammation in neurodegenerative diseases [20].
Various environmental stressors are implicated as risk factors for neurodegenerative diseases, and interestingly they are also involved in aging, oxidative stress and/or DNA damage [230]. Of these, heavy metals (Pb, Cd, As, Hg, Cu, Zn and Fe) and pesticides (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP], paraquat, dieldrin, rotenone [231]) in particular are associated with neurotoxicity and induction of DNA damage via oxidative stress [232].
In the next part of the review, we will focus on neurodegenerative disorders and evidence linking redox dysregulation and DNA damage in each condition. Many of the relevant studies have employed animal models or murine cell lines. However, whilst these reports have provided interesting insights into the role of the DDR in these neurodegenerative diseases, it is important to also consider that there may be species differences in DNA repair mechanisms between mouse and human neurons [233, 234]. Human and mouse neurons possess different DNA repair kinetics and respond differently to oxidative stress. Furthermore, following DNA damage, differences in cell death, chromatin condensation, and activation of DDR sensor proteins are also evident [234].
AD
AD is the most common neurodegenerative disease [235]. It is characterised clinically by progressive memory loss with neuropsychiatric symptoms due to the degeneration of cortical neurons in the entorhinal cortex and hippocampus [236]. The pathological hallmarks of AD include the accumulation of cytoplasmic senile plaques composed of amyloid beta (Aβ) peptides (resulting from cleavage products of amyloid precursor protein) and the formation of neurofibrillary tangles (composed of hyper-phosphorylated tau) [237, 238]. Most AD cases are sporadic in nature. However, 5%–10% cases are familial with a predominately autosomal dominance inheritance pattern, consistent with polygenic origins and multifactorial pathogenic disease processes [239]. Several mechanisms have been implicated in the pathogenesis, including both oxidative stress and DNA damage [236, 240].
There is direct evidence linking redox dysregulation with DNA damage in AD. Increased levels (twofold) of DNA strand breaks were observed in the cerebral cortex of AD brains [241]. Higher levels of oxidative DNA damage, in the form of 8-OHG adducts and oxidized purine and pyrimidine bases, were detected in peripheral leukocytes of AD and mild cognitive impairment (MCI) patients compared to healthy controls [242,243,244]. Similar observations were made in AD lymphocytes [245]. Another study reported an age-dependent increase in the levels of 8-OHG in DNA in the cerebral cortex of AD patients. Consistent with this finding, elevated levels of 8-OHG, 8-hydroxyadenine (8-OHA) and 5-hydroxycytosine were detected in the total DNA of AD parietal lobe regions compared to matched controls [245]. The same study also observed higher levels of thymine glycol, 5-hydroxyuracil, 4,6-diamino-5-formamido-pyrimidine (FapyAde), and FaPyGua in several AD brain areas. In another study, significantly higher levels of 8-OHG, 8-OHA and 5-OHU were detected in the temporal and parietal lobes of AD compared to control patients [246]. Expression of adducts 8-OHG in RNA and 8-hydroxy-2’-deoxyguanosine (8-OHdG) in DNA, were also observed in late-stage AD compared to age-matched control patients [247].
Significantly more aldehydic by-products HNE and acrolein were detected in late-stage AD brains and CSF, including in the most vulnerable areas (hippocampus and superior and middle temporal gyri) of MCI and early-stage AD brains [248]. Moreover, higher levels of acrolein/guanosine adducts were also observed in the hippocampus of late-stage AD patients compared to controls [249]. Mutations in the gene encoding OGG1 have been identified in AD patients, resulting in reduced enzymatic activity [250]. Reduced levels of OGG are present in AD brains, implying that BER is defective in affected neurons [251]. Consistent with this notion, defective BER, diminished activity of DNA glycosylase and reduced DNA synthesis by DNA polymerase β have been reported in AD tissues [252].
More excision repair cross-complementing gene products have been also identified in AD brains compared to controls, suggesting that DNA repair pathways are activated to counteract increased oxidative damage [253]. In addition, higher levels of SSBs and small increases in DSBs were observed in AD brains [254]. In contrast, reduced DNA repair of SSBs [255] or DSBs by DNA-PK-mediated NHEJ [256] were reported in AD brains compared to controls. Similarly, significantly low levels of MRE11 DNA repair complex proteins were identified in the neocortex of AD brains [257]. This would hamper the recognition of DNA damage and its subsequent repair, contributing to neuronal death in AD [248].
Impaired SOD1 activity has also been detected in AD animal models and post-mortem AD brains [258]. The expression of SOD1 and SOD2 is elevated in age-matched AD brain tissues compared to controls [259]; however, the activity of both enzymes decreases significantly in the same tissues [259]. Enhanced formation of Aβ plaques, neuroinflammation, tau phosphorylation, and consequent memory decline have also been observed in SOD1-deficient Tg2576 mice [258, 260].
Oxidative imbalance and mitochondrial dysfunction are observed in AD [261,262,263], together with oxidative stress-induced mtDNA damage. Significantly higher levels of 8-OHdG and 8-OHG were reported in AD brains compared to age-matched control samples [264,265,266]. Another study analysing oxidized nucleosides revealed three-fold more oxidative damage in mtDNA in AD patients [266]. In addition, sporadic mutations were detected in mtDNA of AD brain tissues [267, 268]. Similarly, mutations in mtDNA in the blood of AD patients and in the lymphoblastoid lines derived from the blood of AD patients have been reported [269].
Elevated levels of both PAR polymers and PARP-1 were detected in neurons of human AD brain tissues [270, 271]. In addition, overexpression of PARP-1 is observed in AD brains, largely in the frontal and temporal lobes [240], and the accumulation of Aβ peptides is preceded by oxidative stress and upregulation of PARP-1 in the hippocampus of adult rats [272]. Similarly, a study in SHSY-5Y cells revealed that upregulation of PARP-1 induces pathological features of AD such as deposition of Aβ and the formation of tau tangles [273]. Moreover, co-immunoreactivity of PARP/PAR with Aβ, tau and microtubule-associated protein 2 has been observed in human AD brain tissues [274, 275]. p53 is increased in the temporal cortex of AD patients [276, 277]. Expression of Aβ peptides triggers p53-mediated microglial apoptosis and microglial neurotoxicity [278]. p53 is also prone to aggregate and is a component of misfolded aggregates in a tau mouse model and in human AD brains [279]. Interestingly, p53-mediated DDR has been found to be impaired in AD [279]. Taken together, these studies provide evidence that redox imbalance is associated with DNA damage and inefficient DNA repair, which together contribute to neurodegeneration in AD.
PD
PD is the second most common neurodegenerative disorder [11]. It is characterised by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) accompanied by the formation of intraneuronal inclusions called Lewy bodies [11, 280, 281]. The majority of PD cases (95%) are sporadic while only 5% of cases are linked to mutations in specific genes [11]. Multiple lines of evidence implicate both oxidative stress and DNA damage as key mechanisms in disease pathogenesis [282,283,284,285].
ROS-induced DNA damage in the form of oxidized bases and impaired repair of SSBs has been implicated in PD etiology [84]. Studies have reported elevated levels of 8-OHdG, resulting from DNA oxidation, in PD brains [11, 282] and increased levels of 8-OHG in the SNpc of PD patients [282]. Similarly, more DNA damage, indicated by elevated levels of markers γH2AX and p53-binding protein foci, is present in dopaminergic neurons of two synucleinopathy PD mouse models [286]. Further in vitro studies with dopaminergic SH-SY5Y cell lines suggested that excessive oxidation is at least partially responsible for DDR activation observed in vivo [286]. In comparison to age-matched controls, the SNpc of PD patients displays increased SOD levels, whereas the activities of CAT, GPx and GR are similar as controls [287]. Reduced levels of GSH and altered GSH/GSSG ratio, resulting in more of the oxidized form, have been detected in the SNpc of PD brains [288]. Similarly, depletion of GSH is observed in patients with a pre-symptomatic form of PD, known as incidental Lewy body disease, compared to control subjects [289]. Under elevated oxidative stress conditions, reduction in GSH results in dopaminergic neuronal loss [290]. In addition, depletion of GSH results in increased NO and MPTP/MPP + toxicity in dopaminergic neurons in animal models of PD [291, 292]. Glutamyl cysteine ethyl ester and GSH ethyl ester, two precursors of GSH, increase GSH levels in neuronal cells both in vitro and in vivo and are protective against oxidative and nitrosative stress [293, 294]. Similarly, intracellular GSH levels are also rescued by thiol antioxidants such as α-lipoic acid in both in vitro and in vivo PD models [295, 296]. Depletion of the antioxidant vitamin C has also been detected in PD [297] and vitamin C levels in lymphocytes may be a potential biomarker of disease progression in PD [298]. Furthermore, cells with lower levels of uric acid (UA) are more vulnerable to oxidative damage [299] and individuals with low cellular uric acid levels may be at a greater risk of developing PD [300]. UA prevents 6-hydroxydopamine (6-OHDA)-induced oxidative damage in neuron-like PC12 cells and increases GSH and SOD1 [301]. Similarly, GSH levels, SOD1 activity and dopaminergic neuronal damage are rescued in a 6-OHDA rat model of PD following UA treatment [302]. SOD1 may be a first-line protection against enhanced ROS production in PD patients [303]. RNS, such as NO and its metabolite PN, may also cause DNA damage in PD [11] by reacting with superoxide anion radicals. NO can then generate more oxidatively active PN, which in turn may induce DNA fragmentation [74].
Recent studies have identified mtDNA damage in PD [304] and abasic sites in mtDNA of dopaminergic neurons in PD post-mortem human and rat brains [305], which precede the onset of neurodegeneration [305]. Significant accumulation of abasic sites in dopaminergic neurons, but not in cortical neurons, has been detected [305,306,307]. Elevated levels of ROS render dopaminergic neurons in the SNpc more prone to DNA damage and contribute to neurodegeneration [305, 308]. Consistent with this notion, BER activity is increased in the SNpc of PD patients [309,310,311]. Knockout mouse models lacking DNA repair enzymes (human MutT homolog 1, an oxidized purine nucleoside triphosphate; and OGG1) are more susceptible to dopaminergic toxins and age-related degeneration in the nigrostriatal system [312, 313]. Moreover, transgenic mouse models expressing a mitochondrial-targeted restriction enzyme causing mtDNA damage in dopaminergic neurons recapitulate many of the key features of PD, including motor phenotype, progressive loss of dopaminergic neurons in the SN and depletion of dopamine in the striatum [314]. Taken together, these studies imply that redox dysregulation can induce mtDNA damage in PD and may contribute to neurodegeneration. α-Synuclein is co-localized with γH2AX and PAR in human HAP1 cells and in transgenic α-synuclein mouse models [315]. Reducing α-synuclein levels using bleomycin results in higher DSBs and impaired DNA repair in these cells [315]. Moreover, α-synuclein knockout mice show increased DSB levels [315], suggesting that it may play a role in DNA repair. Interestingly, increased DNA damage and dopaminergic neuronal death have been observed in two PD mouse models [286].
P53-mediated selective cell death is also evident in PD. NO-induced, p53-mediated dopaminergic neuronal death has been observed in a mouse SNpc-derived cell line (SN4741) as well as in vivo models of PD [316]. The neurotoxin 6-OHDA is widely used to induce selective degeneration of dopaminergic and noradrenergic neurons and therefore, can imitate PD symptoms [23]. DNA damage induced by 6-OHDA treatment is linked to p53-mediated cell death of primary dopaminergic neurons [317]. Together, these lines of evidence suggest that DNA damage resulting from redox dysregulation may contribute to neurodegeneration in PD. Several studies also reported parthanatos in PD. MPTP induces neurodegeneration of dopaminergic neurons in SNpc, leading to PD symptoms [281]. Several studies have linked neurotoxicity of MPTP to parthanatos of dopaminergic neurons. MPTP treatment induces DNA fragmentation both in vivo and in vitro [318, 319]. Similarly, PARP upregulation-mediated toxicity to dopaminergic neurons is observed following MPTP administration in a mouse model [320] and inhibition of PARP significantly attenuates these toxic effects [321, 322]. Activation of PARP-1 and progressive loss of dopaminergic neurons by parthanatos have also been reported in a transgenic mouse model overexpressing aminoacyl tRNA synthase complex-interacting multifunctional protein 2, a parkin (E3 ubiquitin ligase) substrate [323]. MPTP-induced parthanatos requires neuronal NO synthase [320], suggesting a link between MPTP-induced PARP activation and subsequent ADP-ribose polymerisation as well as NO-induced DNA damage. Increased NO levels are also observed in nigral cells in PD [324, 325].
ALS
ALS is a fatal, rapidly progressing neurodegenerative disorder that affects motor neurons in the brain, brainstem, and spinal cord [326]. It is clinically, genetically and pathologically linked to FTD, which manifests as frontotemporal lobar degeneration [327]. Variants in more than 40 genes cause ALS, most common of which are those encoding SOD1, chromosome 9 open reading frame 72 (C9orf72), TAR DNA-binding protein-43 (TDP-43) and fused in sarcoma (FUS), which are all linked to both sporadic and familial forms of disease [328].
DNA damage is now increasingly implicated as an important pathophysiological mechanism in ALS [329,330,331], particularly with the identification of both TDP-43 and FUS as proteins with normal cellular functions in DNA repair [332,333,334]. Elevated levels of oxidative DNA damage are consistently observed in both sporadic and familial ALS patients [335, 336]. Moreover, DNA damage is associated with redox dysregulation in ALS. Increased levels of 8-OHdG have been detected in the motor cortex of sporadic ALS patients, and in the spinal cords of both sporadic and familial ALS patients [337, 338]. Similarly, analysis of plasma, urine and CSF of ALS patients revealed increased levels of 8-OHdG [338]. High levels of 8-OHdG have also been reported in the SOD1G93A transgenic mouse model [339]. Decreased levels of BER enzymes DNA polymerases α and β have been detected in motor neurons of SOD1G93A mice [340]. Furthermore, decreased mitochondrial activity of OGG1 and increased 8-OHdG levels have been detected in spinal motor neurons of sporadic ALS patients, indicating that impairment of redox function, resulting in oxidative stress, disrupts DNA repair in the mitochondria [341]. In addition, a polymorphism in OGG1, resulting in the substitution of serine with cysteine (Ser326Cys), reduces DNA activity and is associated with increased risk of sporadic ALS [342]. The levels of a common mitochondrial DNA deletion mutation (mtDNA4977) encoding a subunit III of the redox enzyme cytochrome oxidase, involved in OXPHOS [343], are higher in Brodmann area 4 of primary motor cortices in sporadic ALS patients compared to controls [343]. Moreover, increased abasic sites are also detected in spinal motor neurons of ALS patients compared to controls [330]. Likewise, the levels of 8-OHdG are increased in cells expressing SOD1-G37R and SOD1-G85R compared to wild-type SOD1 [344]. A meta-analysis examining the levels of blood oxidative stress markers in ALS patients reported increased levels of 8-OHdG, MDA (the end product of lipid peroxidation) and AOPP (advanced oxidation protein product, a marker of protein oxidation), and reduced levels of GSH, compared to healthy controls, which all reflect both DNA damage and redox dysfunction in ALS [345]. Hence, these data imply that DNA damage is closely associated with ALS and is linked to redox dysregulation.
Similar to the other neurodegenerative diseases, PARP1 hyperactivation and toxicity are implicated in ALS pathogenesis [346, 347]. Elevated PAR levels are observed in motor neurons of patients carrying a polyglutamine expansion in the gene encoding ataxin-2 and cases displaying the G4C2-hexanucleotide repeat expansion in C9orf72 [348]. PARP-1 expression is also increased in astrocytes in sporadic ALS patients [349] and it is widespread in the cerebellum, motor cortex and parietal cortex, reflecting increased activation [348, 350]. PARP-1 levels are also elevated in astrocytes in the spinal cord in mutant SOD1G93A transgenic mice [351]. Pharmacological inhibition of PARP inhibits the accumulation of stress-induced TDP-43 granules in the cytoplasm and toxicity in rat primary spinal cord cultures [348]. Furthermore, another study demonstrated that PARP1 knockout or treatment with PARP inhibitor olaparib reduces PAR levels and rescues TDP43-induced death in NSC-34 cells [352]. Inhibition of PARP-1 inhibits the ROS-induced cell death and suppresses mitochondrial ROS production via ATF4 and MAP kinase phosphatase-1 in human cell lines [353].
A significant increase in p53 expression has been detected in spinal cord tissues of ALS patients [354]. Similarly, increased nuclear p53 immunoreactivity was detected in the motor cortex and spinal ventral horn of post-mortem tissues from ALS patients [355] and in spinal motor neurons in SOD1G86R mice [356]. Importantly, p53 knockout or knockdown extends the lifespan of a mouse model expressing poly(PR), and protects against neurodegeneration in Drosophila models [357]. Strikingly, p53 knockout also inhibits DNA damage in poly(PR)-transduced cells and C9orf72-ALS iPSC-derived motor neurons. Increased DNA damage was observed following the ectopic expression of poly(GR)80 or (GR)80 in iPSC-derived control neurons[13] and pharmacological or genetical reduction of oxidative stress partially retrieved DNA damage [13]. The adverse role of poly(GR) on DNA damage has also been confirmed in neuronal cells in Drosophila [358]. Together, these studies indicate that p53 mediates the C9orf72-DPR-induced toxicity upstream of DNA damage, rather than downstream, implying that redox homeostasis is crucial for regulation of p53 function, and its modulation may protect against DNA damage [357].
Expression of the C9orf72 repeat expansion induces DNA damage in familial ALS patient tissues and in cellular models [13, 347, 359]. Moreover, poly(GR)80 aggregates induce DNA damage and increase ROS levels in iPSC motor neurons derived from C9orf72-ALS/FTD patients, linking redox dysregulation to DNA damage [13]. Similarly, induction of oxidative stress and upregulation of DNA damage markers γH2AX, ATR, GADD45, and p53 were observed in an age-dependent manner in iPSC-derived C9orf72 motor neurons [13]. In the same study, cellular toxicity was rescued following administration of a water-soluble antioxidant and vitamin E analog, Trolox, in C9orf72 iPSCs. Furthermore, in another study, myogenic progenitors derived from C9orf72 ALS patients demonstrated high susceptibility to oxidative stress and dysregulation of mitochondrial and DNA repair genes, leading to cellular toxicity [360]. Similarly, modifiers of poly(GR)100 toxicity induce dysregulation of mitochondrial NADPH and DNA damage repair-related pathways in yeast cells [361]. Another study demonstrated increased mtDNA due to increased ROS specifically in C9orf72 ALS patient-derived fibroblasts, but not in TDP-43 A382T fibroblasts [362].
R-loops associated with oxidative stress are increased in post-mortem spinal cord tissues of C9orf72 patients and in poly(GA)-transfected MRC5 cells [359]. Both DNA damage and cell death in poly(GA)-expressing cells can be partly rescued by overexpressing senataxin, which resolves R-loops [359, 363]. Interestingly, mutations in the senataxin gene cause an autosomal dominant form of ALS, ALS4 [364]. Interestingly, there is evidence that sentataxin regulates redox homeostasis. An N-terminal truncation mutant of Sen1, the yeast homolog of human senataxin, is critical for cell survival through regulation of redox homeostasis. This mutant also displays severe loss of mtDNA [365]. Importantly, the N-terminal substrate interaction and C-terminal RNA/DNA helicase domains are conserved in Sen1, implying that the same domains may perform a similar function in human senataxin. Furthermore, sentataxin has 31 cysteine residues involved in disulphide bonding via redox-regulated PDI [366]. Moreover, residue C1554, which is expected to engage in disulphide linkage with C1509, is mutated in a sporadic case of ALS4 [367]. These findings together suggest that dysregulated redox signalling, leading to ROS production, is associated with C9orf72 in ALS.
While wild-type SOD1 is protective against DNA damage, ALS-mutant SOD1G93A displays less nuclear localization and antioxidant activity and is not protective in cellular models [368]. SOD1G93A expression in cells deficient in aprataxin, which facilitates SSB and NHEJ DNA repair, sensitises cells to oxidative stress, exacerbates DNA repair deficiencies and increases the levels of heterochromatin [369]. Another study demonstrated more DNA damage in peripheral blood mononuclear cells (PBMCs) of sporadic ALS patients, which display high levels of aggregated SOD1 compared to controls. However, no DNA damage was observed in PBMCs expressing soluble SOD1 only [370]. Increased levels of oxidative DNA damage, DNA strand breaks, p53 activity and apoptosis were detected in cells expressing mutant SOD1G93A compared to wild-type SH-SY5Y cells [371]. However, in a more recent study, similar levels of DNA damage, assessed by the presence of γH2AX-positive foci, were detected in SOD1G93A and SOD1A4V patient-derived iPSC lines compared to isogenic controls [330].
TDP-43 is redox-regulated because the cellular redox conditions control its solubility and nuclear function [372]. We and others have demonstrated that TDP-43 normally functions in the repair of DSBs by NHEJ and associates with XRCC4 and ATP-dependent DNA Ligase 4 [329, 373]. However, this function is impaired by the ALS-associated mutations [332]. Moreover, GSH depletion by buthionine sulfoximine significantly increases mutant TDP-43M337V mislocalisation and inclusion formation in Neuro-2a cells [12]. Redox dysfunction results in oxidation and phosphorylation of TDP-43 by GADD34, which is induced by DNA damage, leading to cytoplasmic mislocalisation in HEK293T cells [374]. Neuronal cells expressing mutant TDP-43 (Q331K and M337V) exhibit shortened neurites, increased oxidative stress and lower levels of heme oxygenase HO-1, which regulates redox signalling [375]. Hence, as TDP-43 is important in DNA repair, mutations in TDP-43 could lead to DNA damage and induce redox dysfunction.
FUS is recruited to oxidative DNA damage sites in response to DNA SSB formation, where it facilitates the recruitment of XRCC1 and nuclear ligase III to regulate its ligation activity for optimal BER activity [376]. Loss of nuclear FUS results in defects in DNA nick ligation in motor neurons due to reduced recruitment of XRCC1/ligase III to DNA strand breaks in cellular models [376]. Interestingly, PARP-dependent DNA damage and apoptosis have been detected in human iPSCs over-expressing mutant FUS-NLS, which induces FUS mislocalisation to the cytoplasm [377]. PARP is also involved in the formation of aberrant phase transition of FUS from the liquid compartments to solid-like aggregates, a process which is redox-regulated, at DNA damage sites [378,379,380]. In addition, FUSR521C transgenic mice display both oxidative damage and defects in DNA ligation [381], implying that defects in DNA repair mechanisms and redox dysregulation are associated with FUS in ALS.
APE1 is implicated as a possible ALS-associated gene that is upregulated in motor neurons of ALS patients [382,383,384]. Furthermore, the motor cortex of ALS patients contains epigenetic hypomethylation of the APE1 promoter, and this region is vulnerable to DNA lesions induced by free radicals and intermediates [330]. In pre-symptomatic transgenic SOD1G93A mice, expression of APE1 is reduced in spinal motor neurons, indicating that a deficiency in DNA repair precedes motor neuron degeneration [385]. However, whether the APE1 redox and DNA repair activity are dysregulated in ALS is unknown.
HD
HD is a severe, rapidly progressing autosomal-dominant condition caused by expansion of CAG (encoding glutamine) repeats in the gene encoding huntingtin protein [386]. Translation of the polyglutamine repeat then produces an abnormally long protein. HD involves motor, psychiatric and cognitive symptoms and it results from degeneration of neurons in the striatum and other regions of the cerebral cortex. Unaffected individuals possess less than 35 polyglutamine repeats, whereas HD patients normally possess 36 to 120 repeats. Interestingly, the number of repeats correlates inversely with the age of disease onset, implying that disease is dependent on repeat length [387]. Similar to other neurodegenerative disorders, redox dysregulation and impaired DNA repair are implicated in the pathogenesis of HD.
DNA damage is strongly implicated in the etiology of HD [388]. Elevated DNA damage has been detected in human HD fibroblasts and HD mouse models [389], where it precedes the aggregation of mutant huntingtin [389]. More DNA damage has also been detected in HD patient PBMCs compared to controls [390] and another clinical study also reported increased DNA damage in prodromal HD in blood cells [391]. ATM was also identified as a modifier of HD-relevant phenotypes in a mouse model [392]. More recently, many genes involved in DNA repair were found to be important regulators of age of disease onset and severity in a large genome-wide association study, including FANCD2/FANCI-associated nuclease 1(FAN1) and ERCC3 (ERCC excision repair 3). Also, defects in DNA repair pathways, including inactivation of DNA mismatch repair genes such as MutS Homolog 3 (MSH3), were associated with modification of age of onset in multiple CAG repeat expansion diseases, suggesting that the CAG repeat itself is the cause of modification [393]. The most significant hit was FAN1, which is associated with ICL repair, and multiple MMR genes were also detected [388]. Moreover, several of the genes identified are also related to mitochondrial and redox signalling pathways [388].
There is also evidence for oxidative DNA damage in HD, as revealed by increased expression of 8-OHdG compared to controls, in both nuclear DNA and mtDNA [394], further linking DNA damage to redox dysregulation. HD patient fibroblasts also display deficient repair following oxidative DNA damage [389]. Furthermore, somatic expansion of the polyglutamine repeat has also been associated with BER, which is a naturally error-prone process. This involves the BER enzyme OGG1 and the removal of oxidized base lesions, resulting in somatic expansion of repeats by SSBs and strand slippage [386]. In addition, the extent of oxidative damage correlates positively with the expansion length [386, 387]. Furthermore, the somatic expansion process induces further oxidative damage and error-prone repair of this damage by the formation of longer repeats [386], forming a vicious, escalating oxidation–BER cycle. These data therefore imply that accumulation of oxidative DNA lesions due to dysfunction of mutant huntingtin in DNA repair in conditions of ROS may contribute to the onset of HD [389].
The normal huntingtin protein is also thought to function in DNA repair, where it is detected as part of the transcription-coupled repair (TCR) complex. TCR is a subtype of NER that rapidly removes specific types of DNA damage from transcribed strands of expressed genes, in contrast to non-transcribed strands. The TCR complex detects lesions and mediates repair during transcription. Mutant huntingtin impairs the function of components of the TCR complex, PNKP and ataxin-3, resulting in more DNA damage and ATM hyperactivation [395].
Huntingtin protein is also thought to repair damaged DNA following oxidative stress, revealing that redox dysregulation and DNA repair are intimately linked in HD [389]. Huntingtin localises and forms a scaffold at DNA damage sites via an ATM-dependent process in the presence of ROS. Huntingtin protein is a sensor of ROS and it relocates to the nucleus following oxidation of Met8 [396]. In the presence of ROS, liquid–liquid phase-separated droplets containing huntingtin colocalised with ATM are increased [396,397,398]. ATM is activated during oxidative stress [399] and inhibiting its activity delays disease progression in mouse HD models [392].
Several studies have reported oxidative damage, decreased levels of antioxidants, cysteine and vitamin C, and deposition of iron (Fe) in the cytoplasm and mitochondria, in cells and tissues from HD models and patients. Uptake of vitamin C is compromised in cellular and mouse models of HD, which precedes mitochondrial dysfunction [400]. The levels of GSH are dysregulated in the plasma and cortex of HD patients compared to controls [401, 402], although whether there is an increase or decrease in GSH is still under debate [403]. Nevertheless, both processes would perturb the cellular redox conditions in HD. There is also evidence for impaired cysteine metabolism in HD [16].
Whilst the HD repeat length is the main factor determining the age at disease onset, genetic modifiers also make a significant contribution to the variation in onset age [404]. The expanded CAG polyglutamine repeat is somatically unstable, and its length increases progressively over time in neurons, particularly in the striatum and cortex. Due to this somatic instability, larger increases in repeat length are associated with earlier disease onset [404]. Interestingly, somatic expansion of the HD CAG repeat is mediated by DNA damage, via a mechanism involving the introduction of mutations by MMR [388]. Similarly, in cellular and animal models, deficiency of MSH3, MSH2, MLH3, MLH1 or PMS2, or increased expression of FAN1, prevents the somatic expansion of CAG repeats [405, 406]. Similarly, transcriptome-wide association studies have revealed that lower expression of MSH3, and increased levels of FAN1, are associated with CAG repeat stability, later onset, and slower disease progression [407, 408]. However, it is unclear if this involves oxidative DNA damage. A more recent study showed that an interaction between FAN1 and MLH1, via a highly conserved SPYF motif at the N terminus of FAN1, is protective against somatic expansion by restricting the recruitment of MLHA by MSH3 [405]. This study suggested that FAN1 normally stabilizes CAG repeats, by both inhibiting formation of the MMR complex that promotes somatic repeat expansion and enhancing correct DNA repair via nuclease activity.
The impact of aging on redox regulation and DNA repair processes
The biggest risk factor for neurodegenerative diseases is aging [409, 410]. Importantly, age-related decline is also a critical socio-economic challenge world-wide due to the increasing aging population [411]. Thus, the incidence of neurodegenerative conditions is likely to increase significantly in the coming decades. Aging is a complex event characterised by damage to both proteins and DNA and during this process cells become senescent, whereby they lose the ability to grow and divide. Aging theories are associated with a decline in cellular function and the accumulation of damage, involving either programmed aging or failure accumulation. The latter theory states that aging is a consequence of the accumulation of damage to cellular components. Interestingly, both oxidative stress and DNA damage increase significantly during aging due to decreases in the efficiency of DNA repair and in the maintenance of redox homeostasis [409, 412]. Moreover, oxidative damage to DNA increases during aging, thus together these mechanisms are central contributors to the normal aging process. Similarly, during aging, the efficiency of proteostasis declines, leading to its ‘collapse’ [409, 413, 414]. While historically maintenance of the genome and the proteome were considered separate processes, emerging evidence reveals that they are much more inter-related than previously recognized [415, 416]. Hence, it is probable that redox dysregulation, DNA damage, and proteostasis decline together form a viscous cycle, driving or exacerbating the aging process [415, 416]. In addition, environmental factors such as UV and other forms of radiation, toxic chemicals, and heavy metals, are also strongly implicated in aging.
During normal aging, increased cellular GSSG/GSH ratio and more GSH oxidation are present, leading to increased levels of 8-OHdG [417]. The formation of 8-oxoG lesions increases with aging [248, 250, 418]. A recent study proposed a mechanism which links 8-OHdG elevation to aging [419]. This study demonstrated that histone deacetylase 1 activation results in attenuated accumulation of oxidative lesions in both aged mice and 5×FAD mice [419]. Mice with knockout of slc25a46 (solute carrier family 25 member 46), a nuclear gene encoding mitochondrial transmembrane protein, display premature aging phenotypes characterized by shortened life span, defective motor ability and redox imbalance in the brain, and neuropathy [420]. This finding implies that acceleration of redox dysregulation with aging may indirectly lead to increased neuronal loss and neurodegeneration. An interesting link between aging, neurodegeneration and NER has also been described, with a possible relationship to ALS. A mouse model with a hypomorphic mutation in Ercc1, Ercc1Δ/-, displays degeneration of motor neurons and shortened lifespan [421]. Together, these studies imply that aging, redox dysregulation, and DNA damage are closely associated and compound during neurodegeneration.
Genetic modifications to DNA repair proteins—what do they reveal about redox regulation, DNA damage and neurodegeneration?
This extensive evidence linking DDR defects to neurodegenerative diseases implies that neurons are highly susceptible to DNA damage, particularly oxidative DNA lesions. Furthermore, DSBs are now known to form and modify gene expression during physiological neuronal activity [422, 423], which may mediate neuronal plasticity [423]. However, it is also important to consider the relationship between genetic defects in DDR proteins and how this is related to neurodegeneration.
DNA repair syndromes are familial conditions characterized by mutations in DDR proteins. They include Cockayne syndrome, ataxia telangiectasia (AT), xeroderma, pigmentosum, trichothiodystrophy, and Nijmegen breakage syndrome [424]. Whilst heterogeneous, these disorders commonly display neurological features, particularly microcephaly and ataxia [425], as well as phenotypes associated with accelerated aging, the biggest risk factor for neurodegeneration. Furthermore, some of these syndromes also directly involve neuronal degeneration, such as AT, which displays progressive degeneration of both Purkinje and granule neurons in the cerebellum. Moreover, in some instances, the defect is almost exclusively neurological, such as ataxia with oculomotor apraxia type 1 (AOA1), AOA5 [426, 427], and spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) [425]. These findings thus raise the possibility that DNA damage arising in neurons can induce human diseases.
Further evidence for this hypothesis comes from the detection of single nucleotide polymorphisms (SNPs) in neurodegenerative disorders. SNPs involve substitution of a single nucleotide in DNA, and they arise frequently within the genome [428]. They have been detected in multiple DDR genes in neurodegenerative diseases, including those that repair oxidative DNA damage. In AD, SNPs in BER genes encoding APE1, OGG1, NEIL1, flap endonuclease 1, or DNA ligases I or III, have been associated with increased risk of disease [429, 430], although this was disputed in another study [431]. Similarly, in PD, APE1 and OGG1 polymorphisms have been observed [432, 433]. Furthermore, in ALS, a Ser326Cys polymorphism in OGG1 is associated with an increased risk of sporadic ALS [342], and several variants in APE1 have previously been described [383]. SNP modifiers in the HTT gene impair NF-κB binding and regulate DNA activity in HD patients [434]. However, investigations in larger populations and more extensive genetic studies are required to confirm the links between SNPs and neurodegenerative diseases. It also remains debatable whether these polymorphisms are independent disease risk factors.
While these observations strongly link DNA damage to human neurological diseases, it has been difficult to establish whether defects in the DDR directly cause neurodegeneration. Nevertheless, recent observations that proteins centrally involved in neurodegeneration also function in DNA repair—particularly TDP-43, FUS, and huntingtin—imply that genetic defects in the DDR directly induce neuronal degeneration and/or death. It is noteworthy that TDP-43, FUS, and huntingtin all function in the repair of oxidative DNA damage, implying that impairment in redox-dependent DNA repair mechanisms is capable of inducing human neurodegenerative diseases. In addition, a recent study provided further evidence for this hypothesis. Enhancing the repair of oxidative DNA damage by overexpressing APE1 and OGG1 was protective against neuronal apoptosis in mouse models [435]. Hence, this finding implies that the accumulation of oxidative DNA damage directly induces neuronal degeneration and death.
However, it should be noted that there are also significant differences between DNA repair syndromes and neurodegenerative disorders. DNA repair syndromes typically first appear in early childhood, whereas most neurodegenerative conditions present in mid-late adulthood. Indeed, microcephaly, which is present in a significant proportion of DNA repair syndromes, results from abnormal embryonic development. These differences, however, may reflect the nature of the specific mutation, the DDR pathway involved, and/or its degree of impact on DNA repair in neurons. Some genetic DDR defects may hasten the development of age-associated phenotypes to such an extent that DNA repair syndromes arise in childhood rather than in adulthood. In contrast, other defects may be less severe, and require the contribution of additional genetic or environmental factors before disease manifests, such as in neurodegenerative disorders. Consistent with this notion, recent studies have shown that neurodegeneration is a multistage process, whereby several sequential steps are required before disease manifests: six for ALS [436] and PD [437], and 14 for AD [438]. Thus, SNPs or genetic defects combined with environmental factors, oxidative stress and aging, may together be required for the development and progression of neurodegenerative diseases [439, 440]. Furthermore, it is possible that defects in DNA repair can both directly cause neurodegeneration and/or contribute to neuronal death in combination with other factors.
Interestingly, DNA repair syndromes displaying mutations in SSBR genes commonly result in neurodegeneration. AOA1, AO4, SCAN1, and AO5 display variations in genes encoding proteins that function in BER or SSB repair (APTX, PNKP, TDP1 and XRCC1, respectively)[435]. These findings may reflect the post-mitotic nature of neurons. SSBs arising in cells actively undergoing the cell cycle often convert to DSBs, which can be subsequently repaired by error-free HR, unlike in neurons. Hence, it is possible that neurons are particularly sensitive to mutations affecting SSBR. Furthermore, mutations to some DNA repair genes are embryonically lethal in mice, particularly those encoding BER proteins. This includes APE1, thymine DNA glycosylase, and DNA ligase IV [441, 442]. This implies that oxidative DNA damage cannot be tolerated in neurons, thus the most severe mutations do not manifest in humans because they are eliminated from the gene pool. This further highlights the important relationship between oxidative DNA damage and neuronal viability.
Nevertheless, it remains unclear why mutations within proteins involved in the same DNA repair pathways can lead to widely different pathologies. It is possible that the selective vulnerabilities of distinct neuronal subtypes to different repair deficits play a role. An interesting observation is that neurodegeneration in AT, AOA1, and SCAN1 is restricted primarily to neurons localised in the cerebellum, whereas in AD, PD, ALS and HD, a more diverse range of neuronal subtypes are targeted. However, impairment in motor control and posture are present in neurodegenerative diseases and the role of the cerebellum in the regulation of motor control is well established. Moreover, cerebellar dysfunction is now implicated in PD, HD, AD [443] and ALS [444], implying that neurodegenerative disorders are not as distinct from DNA repair syndromes as they may initially appear.
Conclusion
The pathways linking redox dysfunction and DNA damage are complex and we are only just beginning to unravel their full complexity. The most well characterised of these mechanisms involve DNA damage induced by oxidative stress. Furthermore, it has also been recognised for some time that several proteins, primarily APE1 and p53, possess both redox activity and DNA repair functions. However, an increasing number of DNA repair proteins are now known to also possess redox activity. Similarly, a growing list of redox regulatory proteins is now known to function in the DDR. It is possible that some of these proteins act indirectly, by modulating redox conditions in the nucleus, or by modulating expression of DNA repair proteins. Likewise, several transcription factors that function in the DDR are known to be regulated by redox processes. However, it is also possible that these proteins may function directly in DNA repair, although these mechanisms remain poorly understood. Nevertheless, it is becoming apparent that there are much more extensive links between the DDR and redox signalling than previously recognised.
Neurons are terminally differentiated and thus cannot dilute the effects of DNA lesions by cell division like other cell types. Defective DNA repair therefore has potentially catastrophic effects on neurons and is increasingly implicated in neurodegeneration. Neurons are very metabolically active and thus generate high levels of ROS, and similarly redox dysregulation is also implicated in the pathogenesis of these disorders. Thus, DNA repair imposes additional energetic stress onto neurons given their high rates of metabolism. As energy is depleted, ROS by-products cause further damage. Both DNA damage and redox dysregulation become impaired during the aging process, which may promote neurodegeneration. In addition, impaired levels of GSH have been reported in various neurodegenerative disorders including AD, PD, HD, and ALS. However, while environmental factors are known to induce DNA damage and oxidative stress, the evidence for their role in many neurodegenerative conditions is conflicting [445]. Mitochondrial function also becomes impaired during normal human aging, and damage to both nuclear DNA and mtDNA is implicated in neurodegeneration. Redox regulation controls epigenetic mechanisms, which may also contribute to neurodegeneration. Together, these factors are likely to combine and provide an ever-increasing threat to neurons, which struggle to maintain their integrity over time. Eventually, even minor impairments in DNA repair or redox dysregulation can have serious consequences and lead to neurodegeneration (Fig. 5).

Types of DNA damage induced by oxidative stress in neurodegenerative diseases. DNA damage can induce diverse cell death mechanisms such as apoptosis, autophagy-dependent cell death, necrosis, parthanatos, ferroptosis, pyroptosis and lysosome-dependent cell death. However, not all these mechanisms are associated with neurodegenerative diseases
Understanding the poorly characterised connections between oxidative stress and DNA damage is necessary for a better understanding of disease mechanisms in neurodegenerative diseases, given this is a primary source of DNA lesions in neurons. Moreover, there are no known mechanisms to repair oxidative (or other) DNA damage in neurodegenerative disorders. Hence, improving our knowledge of these relationships may ultimately lead to the design of better therapeutic strategies, based on preventing both redox dysregulation and DNA damage.
Availability of data and materials
Not applicable.
Abbreviations
- ROS:
-
Reactive oxygen species
- RNS:
-
Reactive nitrogen species
- DDR:
-
DNA damage response
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- HD:
-
Huntington’s disease
- RSS:
-
Reactive sulphur species
- FTD:
-
Frontotemporal dementia
- SOD:
-
Superoxide dismutases
- GPx:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- Prxs:
-
Peroxiredoxins
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- NOS:
-
Nitric oxide synthase
- GSH:
-
Glutathione
- TrxR:
-
Thioredoxin reductase
- APE1:
-
Apurinic/apyrimidinic endonuclease 1
- TXNIP:
-
Thioredoxin-interacting protein
- AIF:
-
Apoptosis inducing factor
- PDI:
-
Protein disulphide isomerase
- UPR:
-
Unfolded protein response
- OXPHOS:
-
Oxidative phosphorylation
- mtDNA:
-
Mitochondrial DNA
- XOR:
-
Xanthine oxidoreductase
- DSB:
-
Double-stranded break
- ICL:
-
Inter-strand crosslink
- PAH:
-
Polycyclic aromatic hydrocarbon
- BER:
-
Excision repair
- NER:
-
Nucleotide excision repair
- PNKP:
-
Polynucleotide kinase phosphatase
- TDP1:
-
Tyrosyl DNA phosphodiesterase 1
- 8-oxoG:
-
8-Oxoguanine
- TOP:
-
Topoisomerase
- MMC:
-
Mitomycin C
- PUFA:
-
Polyunsaturated fatty acid
- MDA:
-
Malondialdehyde
- PI3K:
-
Phosphatidylinositol 3-kinase
- ATM:
-
Ataxia telangiectasia mutated kinase
- ATR:
-
Rad3-related protein
- OGG1:
-
8-OxoG glycosylase
- PCNA:
-
Proliferating cell nuclear antigen
- GG-NER:
-
Global genome NER
- TC-NER:
-
Transcription-coupled NER
- RPA:
-
Replication protein A
- ERCC1:
-
Excision repair protein-1
- FA:
-
Fanconi anemia
- SSBR:
-
Single-strand break repair
- PARPs:
-
Poly-ADP-ribose-polymerases
- PNK:
-
Polynucleotide kinase
- HDR:
-
Homology directed repair
- NHEJ:
-
Non-homologous end joining
- H2AX:
-
H2A histone family member X
- CtIP:
-
CtBP-interacting protein
- MGMT:
-
O-6-methylguanine-DNA methyltransferase
- MCI:
-
Mild cognitive impairment
- 8-OHA:
-
8-Hydroxyadenine
- 8-OHdG:
-
8-Hydroxy-2’-deoxyguanosine
- SNpc:
-
Substantia nigra pars compacts
- UA:
-
Uric acid
- 6-OHDA:
-
6-Hydroxydopamine
- C9orf72:
-
Chromosome 9 open reading frame 72
- TDP-43:
-
TAR DNA-binding protein-43
- FUS:
-
Fused in sarcoma
- PBMC:
-
Peripheral blood mononuclear cell
- XRCC4:
-
X-ray repair cross-complementing protein 4
- FAN1:
-
FANCD2/FANCI-associated nuclease 1
- ERCC3:
-
ERCC excision repair 3
- MSH3:
-
MutS Homolog 3
- TCR:
-
Transcription-coupled repair
- AT:
-
Ataxia telangiectasia
- AOA1:
-
Ataxia oculomotor apraxia type 1
- SCAN1:
-
Spinocerebellar ataxia with axonal neuropathy 1
References
Watts ME, Pocock R, Claudianos C. brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mol Neurosci. 2018;11.
Metodiewa D, Kośka C. Reactive oxygen species and reactive nitrogen species: relevance to cyto(neuro)toxic events and neurologic disorders. An overview. Neurotox Res. 2000;1(3):197–233.
Corpas FJ, Barroso JB. Reactive sulfur species (RSS): possible new players in the oxidative metabolism of plant peroxisomes. Front Plant Sci. 2015;6.
Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95.
Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. 2016;2016:1245049.
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10(8):1343–74.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8.
Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ. A developmental redox dysregulation leads to spatio-temporal deficit of parvalbumin neuron circuitry in a schizophrenia mouse model. Schizophrenia Res. 2019;213:96–106.
Parakh S, Spencer DM, Halloran MA, Soo KY, Atkin JD. Redox regulation in amyotrophic lateral sclerosis. Oxid Med Cell Longev. 2013;2013: 408681.
Chen YY, Wang MC, Wang YN, Hu HH, Liu QQ, Liu HJ, et al. Redox signaling and Alzheimer’s disease: from pathomechanism insights to biomarker discovery and therapy strategy. Biomark Res. 2020;8(1):42.
Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. Biochim Biophys Acta. 2008;1780(11):1362–7.
Parakh S, Shadfar S, Perri ER, Ragagnin AMG, Piattoni CV, Fogolín MB, et al. The redox activity of protein disulfide isomerase inhibits ALS phenotypes in cellular and zebrafish models. iScience. 2020;23(5):101097.
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92(2):383–91.
Jagaraj CJ, Parakh S, Atkin JD. Emerging evidence highlighting the importance of redox dysregulation in the pathogenesis of amyotrophic lateral sclerosis (ALS). Front Cell Neurosci. 2021;14.
Wang H, Kodavati M, Britz GW, Hegde ML. DNA damage and repair deficiency in ALS/FTD-associated neurodegeneration: from molecular mechanisms to therapeutic implication. Front Mol Neurosci. 2021;14.
Paul BD, Snyder SH. Impaired redox signaling in Huntington’s disease: therapeutic implications. Front Mol Neurosci. 2019;12.
Welch G, Tsai LH. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022;23(6): e54217.
Pessina F, Gioia U, Brandi O, Farina S, Ceccon M, Francia S, et al. DNA damage triggers a new phase in neurodegeneration. Trends Genet. 2021;37(4):337–54.
Qin N, Geng A, Xue R. Activated or impaired: an overview of DNA repair in neurodegenerative diseases. Aging Dis. 2022;13(4):987–1004.
Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–63.
Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011;3(1): a000745.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204.
Shadfar S, Brocardo M, Atkin JD. The complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. Int J Mol Sci. 2022;23(5):2484.
Choi E-H, Yoon S, Koh YE, Seo Y-J, Kim KP. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp Mol Med. 2020;52(8):1220–9.
Gong F, Miller KM. Histone methylation and the DNA damage response. Mutat Res Rev Mutat Res. 2019;780:37–47.
Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506.
Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83(2):266–82.
Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30(1):11–26.
Nakai K, Tsuruta D. What are reactive oxygen species, free radicals, and oxidative stress in skin diseases? Int J Mol Sci. 2021;22(19).
Sbodio JI, Snyder SH, Paul BD. Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid Redox Signal. 2019;30(11):1450–99.
Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc Natl Acad Sci USA. 2018;115(23):5839–48.
Fukuto JM, Ignarro LJ, Nagy P, Wink DA, Kevil CG, Feelisch M, et al. Biological hydropersulfides and related polysulfides—a new concept and perspective in redox biology. FEBS Lett. 2018;592(12):2140–52.
Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic Res. 2012;46(4):382–419.
Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015;6:183–97.
Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247(1):1–11.
Marengo B, Nitti M, Furfaro AL, Colla R, Ciucis CD, Marinari UM, et al. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid Med Cell Longev. 2016;2016:6235641.
Ren X, Zou L, Zhang X, Branco V, Wang J, Carvalho C, et al. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid Redox Signal. 2017;27(13):989–1010.
Zhang H, Du Y, Zhang X, Lu J, Holmgren A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid Redox Signal. 2014;21(5):669–81.
Hanschmann E-M, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins–molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013;19(13):1539–605.
Brose J, La Fontaine S, Wedd AG, Xiao Z. Redox sulfur chemistry of the copper chaperone Atox1 is regulated by the enzyme glutaredoxin 1, the reduction potential of the glutathione couple GSSG/2GSH and the availability of Cu(I). Metallomics. 2014;6(4):793–808.
Grillo C, D’Ambrosio C, Scaloni A, Maceroni M, Merluzzi S, Turano C, et al. Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic Biol Med. 2006;41(7):1113–23.
Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27(9–10):916–21.
Rojas E, Valverde M, Kala SV, Kala G, Lieberman MW. Accumulation of DNA damage in the organs of mice deficient in gamma-glutamyltranspeptidase. Mutat Res. 2000;447(2):305–16.
Cotgreave IA. Analytical developments in the assay of intra- and extracellular GSH homeostasis: specific protein S-glutathionylation, cellular GSH and mixed disulphide compartmentalisation and interstitial GSH redox balance. BioFactors. 2003;17(1–4):269–77.
Chen J, Delannoy M, Odwin S, He P, Trush MA, Yager JD. Enhanced mitochondrial gene transcript, ATP, bcl-2 protein levels, and altered glutathione distribution in ethinyl estradiol-treated cultured female rat hepatocytes. Toxicol Sci. 2003;75(2):271–8.
Chatterjee A. Reduced glutathione: a radioprotector or a modulator of DNA-repair activity? Nutrients. 2013;5(2):525–42.
Yamawaki H, Berk BC. Thioredoxin: a multifunctional antioxidant enzyme in kidney, heart and vessels. Curr Opin Nephrol Hypertens. 2005;14(2):149–53.
Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2(4):811–20.
Miller VM, Lawrence DA, Mondal TK, Seegal RF. Reduced glutathione is highly expressed in white matter and neurons in the unperturbed mouse brain–implications for oxidative stress associated with neurodegeneration. Brain Res. 2009;1276:22–30.
Kim HL, Koedrith P, Lee SM, Kim YJ, Seo YR. Base excision DNA repair defect in thioredoxin-1 (Trx1)-deficient cells. Mutat Res. 2013;751–752:1–7.
Oberacker T, Bajorat J, Ziola S, Schroeder A, Röth D, Kastl L, et al. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett. 2018;592(13):2297–307.
Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA. 1997;94(8):3633–8.
Muri J, Heer S, Matsushita M, Pohlmeier L, Tortola L, Fuhrer T, et al. The thioredoxin-1 system is essential for fueling DNA synthesis during T-cell metabolic reprogramming and proliferation. Nat Commun. 2018;9(1):1851.
Shelar SB, Kaminska KK, Reddy SA, Kumar D, Tan CT, Yu VC, et al. Thioredoxin-dependent regulation of AIF-mediated DNA damage. Free Radic Biol Med. 2015;87:125–36.
Parakh S, Perri ER, Vidal M, Sultana J, Shadfar S, Mehta P, et al. Protein disulphide isomerase (PDI) is protective against amyotrophic lateral sclerosis (ALS)-related mutant Fused in Sarcoma (FUS) in in vitro models. Sci Rep. 2021;11(1):17557.
Ocklenburg T, Neumann F, Wolf A, Vogel J, Göpelt K, Baumann M, et al. In oxygen-deprived tumor cells ERp57 provides radioprotection and ensures proliferation via c-Myc, PLK1 and the AKT pathway. Sci Rep. 2021;11(1):7199.
Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6(1):28–32.
Ostermeier M, De Sutter K, Georgiou G. Eukaryotic protein disulfide isomerase complements Escherichia coli dsbA mutants and increases the yield of a heterologous secreted protein with disulfide bonds. J Biol Chem. 1996;271(18):10616–22.
Shadfar S, Vidal M, Parakh S, Laird AS, Atkin JD. Protein disulphide isomerase (PDI) is protective against several types of DNA damage, including that induced by amyotrophic lateral sclerosis-associated mutant TDP-43 in neuronal cells/ in vitro models. bioRxiv. 2021:2021.08.31.458441.
Huertas JR, Casuso RA, Agustín PH, Cogliati S. Stay fit, stay young: mitochondria in movement: the role of exercise in the new mitochondrial paradigm. Oxid Med Cell Longev. 2019;2019:7058350.
Sousa JS, D’Imprima E, Vonck J. Mitochondrial respiratory chain complexes. Subcell Biochem. 2018;87:167–227.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17(5):308–21.
Farge G, Falkenberg M. Organization of DNA in mammalian mitochondria. Int J Mol Sci. 2019;20(11):2770.
Nadalutti CA, Ayala-Peña S, Santos JH. Mitochondrial DNA damage as driver of cellular outcomes. Am J Physiol Cell Physiol. 2022;322(2):C136–50.
Rong Z, Tu P, Xu P, Sun Y, Yu F, Tu N, et al. The mitochondrial response to DNA damage. Front Cell Dev Biol. 2021;9.
Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst). 2006;5(2):145–52.
Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine oxidoreductase: one enzyme for multiple physiological tasks. Redox Biol. 2021;41: 101882.
Huang CC, Chen KL, Cheung CHA, Chang JY. Autophagy induced by cathepsin S inhibition induces early ROS production, oxidative DNA damage, and cell death via xanthine oxidase. Free Radic Biol Med. 2013;65:1473–86.
De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19(3):169–85.
Orii KE, Lee Y, Kondo N, McKinnon PJ. Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci USA. 2006;103(26):10017–22.
Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001;11(2):66–75.
Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424.
Carr AC, McCall MR, Frei B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: reaction pathways and antioxidant protection. Arterioscler Thromb Vasc Biol. 2000;20(7):1716–23.
Chong WC, Shastri MD, Eri R. Endoplasmic reticulum stress and oxidative stress: a vicious nexus implicated in bowel disease pathophysiology. Int J Mol Sci. 2017;18(4).
Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21(3):396–413.
González-Quiroz M, Blondel A, Sagredo A, Hetz C, Chevet E, Pedeux R. When endoplasmic reticulum proteostasis meets the DNA damage response. Trends Cell Biol. 2020;30(11):881–91.
Bolland H, Ma TS, Ramlee S, Ramadan K, Hammond EM. Links between the unfolded protein response and the DNA damage response in hypoxia: a systematic review. Biochem Soc Trans. 2021;49(3):1251–63.
Bhattarai KR, Riaz TA, Kim H-R, Chae H-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med. 2021;53(2):151–67.
Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA damage—How and why we age? eLife. 2021;10:e62852.
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619–31.
Cannan WJ, Pederson DS. Mechanisms and consequences of double-strand DNA break formation in chromatin. J Cell Physiol. 2016;231(1):3–14.
Smith JA, Park S, Krause JS, Banik NL. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem Int. 2013;62(5):764–75.
Hegde ML, Hegde PM, Rao KS, Mitra S. Oxidative genome damage and its repair in neurodegenerative diseases: function of transition metals as a double-edged sword. J Alzheimers Dis. 2011;24 Suppl 2(0 2):183–98.
Crawford DR, Suzuki T, Sesay J, Davies KJ. Analysis of gene expression following oxidative stress. Methods Mol Biol. 2002;196:155–62.
Davies KJ. Oxidative stress: the paradox of aerobic life. Biochem Soc Symp. 1995;61:1–31.
Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–48.
Kantidze OL, Velichko AK, Luzhin AV, Razin SV. Heat stress-induced DNA damage. Acta Naturae. 2016;8(2):75–8.
Neutelings T, Lambert CA, Nusgens BV, Colige AC. Effects of mild cold shock (25°C) followed by warming up at 37°C on the cellular stress response. PLoS ONE. 2013;8(7): e69687.
Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980.
Geihs MA, Moreira DC, López-Martínez G, Minari M, Ferreira-Cravo M, Carvajalino-Fernández JM, et al. Commentary: ultraviolet radiation triggers “preparation for oxidative stress” antioxidant response in animals: Similarities and interplay with other stressors. Comp Biochem Physiol A Mol Integr Physiol. 2020;239: 110585.
Kato M, Iwashita T, Takeda K, Akhand AA, Liu W, Yoshihara M, et al. Ultraviolet light induces redox reaction-mediated dimerization and superactivation of oncogenic Ret tyrosine kinases. Mol Biol Cell. 2000;11(1):93–101.
Pfeifer GP, Besaratinia A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci. 2012;11(1):90–7.
Kciuk M, Marciniak B, Mojzych M, Kontek R. Focus on UV-induced DNA damage and repair-disease relevance and protective strategies. Int J Mol Sci. 2020;21(19):7264.
Eppink B, Tafel AA, Hanada K, van Drunen E, Hickson ID, Essers J, et al. The response of mammalian cells to UV-light reveals Rad54-dependent and independent pathways of homologous recombination. DNA Repair (Amst). 2011;10(11):1095–105.
Donya M, Radford M, ElGuindy A, Firmin D, Yacoub MH. Radiation in medicine: origins, risks and aspirations. Glob Cardiol Sci Pract. 2014;2014(4):437–48.
Mavragani IV, Nikitaki Z, Kalospyros SA, Georgakilas AG. Ionizing radiation and complex DNA damage: from prediction to detection challenges and biological significance. Cancers (Basel). 2019;11(11):1789.
Borrego-Soto G, Ortiz-López R, Rojas-Martínez A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet Mol Biol. 2015;38(4):420–32.
Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–50.
Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JNM. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci. 2011;36(5):262–71.
Katerji M, Duerksen-Hughes PJ. DNA damage in cancer development: special implications in viral oncogenesis. Am J Cancer Res. 2021;11(8):3956–79.
Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19(12):1580–94.
Kondo N, Takahashi A, Ono K, Ohnishi T. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids. 2010;2010: 543531.
Kriek E. Fifty years of research on N-acetyl-2-aminofluorene, one of the most versatile compounds in experimental cancer research. J Cancer Res Clin Oncol. 1992;118(7):481–9.
Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18(4):851–4.
Murata M, Tamura A, Tada M, Kawanishi S. Mechanism of oxidative DNA damage induced by carcinogenic 4-aminobiphenyl. Free Radic Biol Med. 2001;30(7):765–73.
Mu H, Kropachev K, Wang L, Zhang L, Kolbanovskiy A, Kolbanovskiy M, et al. Nucleotide excision repair of 2-acetylaminofluorene- and 2-aminofluorene-(C8)-guanine adducts: molecular dynamics simulations elucidate how lesion structure and base sequence context impact repair efficiencies. Nucleic Acids Res. 2012;40(19):9675–90.
Skipper PL, Kim MY, Sun HLP, Wogan GN, Tannenbaum SR. Monocyclic aromatic amines as potential human carcinogens: old is new again. Carcinogenesis. 2010;31(1):50–8.
Zelinkova Z, Wenzl T. The occurrence of 16 EPA PAHs in food—a review. Polycycl Aromat Compd. 2015;35(2–4):248–84.
Vu AT, Taylor KM, Holman MR, Ding YS, Hearn B, Watson CH. Polycyclic aromatic hydrocarbons in the mainstream smoke of popular US cigarettes. Chem Res Toxicol. 2015;28(8):1616–26.
Hrdina AIH, Kohale IN, Kaushal S, Kelly J, Selin NE, Engelward BP, et al. The parallel transformations of polycyclic aromatic hydrocarbons in the body and in the atmosphere. Environ Health Perspect. 2022;130(2): 025004.
Yu H. Environmental carcinogenic polycyclic aromatic hydrocarbons: photochemistry and phototoxicity. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2002;20(2):149–83.
Jeng HA, Pan CH, Diawara N, Chang-Chien GP, Lin WY, Huang CT, et al. Polycyclic aromatic hydrocarbon-induced oxidative stress and lipid peroxidation in relation to immunological alteration. Occup Environ Med. 2011;68(9):653–8.
Yu Y, Cui Y, Niedernhofer LJ, Wang Y. Occurrence, biological consequences, and human health relevance of oxidative stress-induced DNA damage. Chem Res Toxicol. 2016;29(12):2008–39.
Dizdaroglu M, Coskun E, Jaruga P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic Res. 2015;49(5):525–48.
Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med. 2002;32(11):1102–15.
Kumar S, Chinnusamy V, Mohapatra T. Epigenetics of modified DNA bases: 5-methylcytosine and beyond. Front Genet. 2018;9:640.
Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–15.
Yonekura S, Nakamura N, Yonei S, Zhang-Akiyama QM. Generation, biological consequences and repair mechanisms of cytosine deamination in DNA. J Radiat Res. 2009;50(1):19–26.
Saini N, Giacobone CK, Klimczak LJ, Papas BN, Burkholder AB, Li JL, et al. UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin. PLoS Genet. 2021;17(1): e1009302.
McKelvey SM, Horgan KA, Murphy RA. Chemical form of selenium differentially influences DNA repair pathways following exposure to lead nitrate. J Trace Elem Med Biol. 2015;29:151–69.
Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, Gordenin DA. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 2012;8(12): e1003149.
Stratigopoulou M, van Dam TP, Guikema JEJ. Base excision repair in the immune system: small DNA lesions with big consequences. Front Immunol. 2020;11.
Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–214.
Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11(19):3610–8.
Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33.
Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413.
McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutat Res. 2007;623(1–2):83–97.
Flor AC, Wolfgeher D, Wu D, Kron SJ. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017;3:17075.
Flor AC, Doshi AP, Kron SJ. Modulation of therapy-induced senescence by reactive lipid aldehydes. Cell Death Discov. 2016;2:16045.
Montaudon D, Palle K, Rivory LP, Robert J, Douat-Casassus C, line, et al. Inhibition of topoisomerase I cleavage activity by thiol-reactive compounds: importance of vicinal cysteines 504 and 505. J Biol Chem. 2007;282(19):14403–12.
Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279(5356):1534–41.
Carey JF, Schultz SJ, Sisson L, Fazzio TG, Champoux JJ. DNA relaxation by human topoisomerase I occurs in the closed clamp conformation of the protein. Proc Natl Acad Sci USA. 2003;100(10):5640–5.
Pommier Y, Cherfils J. Interfacial inhibition of macromolecular interactions: nature’s paradigm for drug discovery. Trends Pharmacol Sci. 2005;26(3):138–45.
Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat Neurosci. 2014;17(6):813–21.
Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9(5):327–37.
Li TK, Chen AY, Yu C, Mao Y, Wang H, Liu LF. Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev. 1999;13(12):1553–60.
Sobek S, Boege F. DNA topoisomerases in mtDNA maintenance and ageing. Exp Gerontol. 2014;56:135–41.
Ménézo Y, Dale B, Cohen M. DNA damage and repair in human oocytes and embryos: a review. Zygote. 2010;18(4):357–65.
Menezo YJR, Silvestris E, Dale B, Elder K. Oxidative stress and alterations in DNA methylation: two sides of the same coin in reproduction. Reprod Biomed Online. 2016;33(6):668–83.
Lawley PD, Phillips DH. DNA adducts from chemotherapeutic agents. Mutat Res. 1996;355(1–2):13–40.
Cecchini S, Masson C, La Madeleine C, Huels MA, Sanche L, Wagner JR, et al. Interstrand cross-link induction by UV radiation in bromodeoxyuridine-substituted DNA: dependence on DNA conformation. Biochemistry. 2005;44(51):16957–66.
Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11(7):467–80.
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43.
Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014: 360438.
Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944–72.
Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21(3):361–70.
VanderVeen LA, Hashim MF, Nechev LV, Harris TM, Harris CM, Marnett LJ. Evaluation of the mutagenic potential of the principal DNA adduct of acrolein. J Biol Chem. 2001;276(12):9066–70.
Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193–9.
Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44(10):625–33.
Taso OV, Philippou A, Moustogiannis A, Zevolis E, Koutsilieris M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann Res Hosp. 2019;3.
Dang TN, Arseneault M, Murthy V, Ramassamy C. Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr Mol Pharmacol. 2010;3(2):66–78.
Huang X, Ahn DU. Lipid oxidation and its implications to meat quality and human health. Food Sci Biotechnol. 2019;28(5):1275–85.
Niki E. Chapter 14—Dual stressor effects of lipid oxidation and antioxidants. In: Sies H, editor. Oxidative Stress: Academic Press; 2020;249–62.
Shields HJ, Traa A, Van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front Cell Dev Biol. 2021;9: 628157.
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843.
Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radic Biol Med. 2010;49(1):9–21.
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85.
Huang TT, Lampert EJ, Coots C, Lee JM. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat Rev. 2020;86: 102021.
Davalli P, Marverti G, Lauriola A, D’Arca D. Targeting oxidatively induced dna damage response in cancer: opportunities for novel cancer therapies. Oxid Med Cell Longev. 2018;2018:2389523.
Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18(1):27–47.
Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst). 2007;6(6):695–711.
Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5(4): a012583.
de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, et al. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61(14):5378–81.
Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193(1–2):3–34.
Zou Y, Liu Y, Wu X, Shell SM. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J Cell Physiol. 2006;208(2):267–73.
Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36(13):4327–36.
Leak RK, Li P, Zhang F, Sulaiman HH, Weng Z, Wang G, et al. Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid Redox Signal. 2015;22(2):135–48.
Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: More than a passive phenomenon? Antioxid Redox Signal. 2005;7(3–4):367–84.
Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5(10): a012609.
Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30(1):2–10.
Gillet LC, Schärer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106(2):253–76.
Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst). 2002;1(8):601–16.
Wagner VP, Webber LP, Salvadori G, Meurer L, Fonseca FP, Castilho RM, et al. Overexpression of MutSα complex proteins predicts poor prognosis in oral squamous cell carcinoma. Medicine. 2016;95(22): e3725.
Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–17.
Hong IS, Greenberg MM. Efficient DNA interstrand cross-link formation from a nucleotide radical. J Am Chem Soc. 2005;127(11):3692–3.
Rozelle AL, Cheun Y, Vilas CK, Koag M-C, Lee S. DNA interstrand cross-links induced by the major oxidative adenine lesion 7,8-dihydro-8-oxoadenine. Nat Commun. 2021;12(1):1897.
Demple B, DeMott MS. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene. 2002;21(58):8926–34.
Pogozelski WK, Tullius TD. Oxidative strand scission of nucleic acids: routes initiated by hydrogen abstraction from the sugar moiety. Chem Rev. 1998;98(3):1089–108.
Abbotts R, Wilson DM 3rd. Coordination of DNA single strand break repair. Free Radic Biol Med. 2017;107:228–44.
Durkacz BW, Shall S, Irwin J. The effect of inhibition of (ADP-ribose)n biosynthesis on DNA repair assayed by the nucleoid technique. Eur J Biochem. 1981;121(1):65–9.
James MR, Lehmann AR. Role of poly(adenosine diphosphate ribose) in deoxyribonucleic acid repair in human fibroblasts. Biochemistry. 1982;21(17):4007–13.
Lehmann AR, Broughton BC. Poly(ADP-ribosylation) reduces the steady-state level of breaks in DNA following treatment of human cells with alkylating agents. Carcinogenesis. 1984;5(1):117–9.
Schraufstatter IU, Hyslop PA, Hinshaw DB, Spragg RG, Sklar LA, Cochrane CG. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 1986;83(13):4908–12.
Murata MM, Kong X, Moncada E, Chen Y, Imamura H, Wang P, et al. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol Biol Cell. 2019;30(20):2584–97.
Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Ann Rev Genet. 2010;44:113–39.
Symington LS. Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol. 2016;51(3):195–212.
Baleriola J, Álvarez-Lindo N, de la Villa P, Bernad A, Blanco L, Suárez T, et al. Increased neuronal death and disturbed axonal growth in the Polμ-deficient mouse embryonic retina. Sci Rep. 2016;6:25928.
Sharma V, Collins LB, Chen TH, Herr N, Takeda S, Sun W, et al. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget. 2016;7(18):25377–90.
Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30(3):290–4.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol. 2016;231(1):15–24.
Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2(3):130–43.
Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85(4):509–20.
Zheng XF, Prakash R, Saro D, Longerich S, Niu H, Sung P. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst). 2011;10(10):1034–43.
Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113.
Blasiak J. Single-strand annealing in cancer. Int J Mol Sci. 2021;22(4).
Caston RA, Gampala S, Armstrong L, Messmann RA, Fishel ML, Kelley MR. The multifunctional APE1 DNA repair–redox signaling protein as a drug target in human disease. Drug Discov Today. 2021;26(1):218–28.
Kim H-S, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, et al. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat Res. 2015;779:96–104.
Andrews BJ, Lehman JA, Turchi JJ. Kinetic analysis of the Ku-DNA binding activity reveals a redox-dependent alteration in protein structure that stimulates dissociation of the Ku-DNA complex. J Biol Chem. 2006;281(19):13596–603.
Yu W, Zhang L, Wei Q, Shao A. O(6)-Methylguanine-DNA methyltransferase (MGMT): challenges and new opportunities in glioma chemotherapy. Front Oncol. 2019;9:1547.
Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011;32(7):973–7.
Mikhed Y, Görlach A, Knaus UG, Daiber A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015;5:275–89.
Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med. 1996;21(3):335–48.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Choi SH, Koh DI, Cho SY, Kim MK, Kim KS, Hur MW. Temporal and differential regulation of KAISO-controlled transcription by phosphorylated and acetylated p53 highlights a crucial regulatory role of apoptosis. J Biol Chem. 2019;294(35):12957–74.
Feroz W, Sheikh AMA. Exploring the multiple roles of guardian of the genome: P53. Egypt J Med Hum Genet. 2020;21(1):49.
Eriksson SE, Ceder S, Bykov VJN, Wiman KG. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J Mol Cell Biol. 2019;11(4):330–41.
Gudkov AV, Komarova EA. Pathologies associated with the p53 response. Cold Spring Harbor Perspect Biol 2010;2(7):a001180.
Guikema JEJ, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, et al. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204(12):3017–26.
Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46(26):7765–80.
Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11(12):1306–13.
Xu S, Liu Y, Yang K, Wang H, Shergalis A, Kyani A, et al. Inhibition of protein disulfide isomerase in glioblastoma causes marked downregulation of DNA repair and DNA damage response genes. Theranostics. 2019;9(8):2282–98.
Liu Y, Ji W, Shergalis A, Xu J, Delaney AM, Calcaterra A, et al. Activation of the unfolded protein response via inhibition of protein disulfide isomerase decreases the capacity for DNA repair to sensitize glioblastoma to radiotherapy. Cancer Res. 2019;79(11):2923–32.
Chichiarelli S, Ferraro A, Altieri F, Eufemi M, Coppari S, Grillo C, et al. The stress protein ERp57/GRP58 binds specific DNA sequences in HeLa cells. J Cell Physiol. 2007;210(2):343–51.
Krynetski EY, Krynetskaia NF, Bianchi ME, Evans WE. A nuclear protein complex containing high mobility group proteins B1 and B2, heat shock cognate protein 70, ERp60, and glyceraldehyde-3-phosphate dehydrogenase is involved in the cytotoxic response to DNA modified by incorporation of anticancer nucleoside analogues. Cancer Res. 2003;63(1):100–6.
Song D, Liu H, Wu J, Gao X, Hao J, Fan D. Insights into the role of ERp57 in cancer. J Cancer. 2021;12(8):2456–64.
Krynetskaia NF, Phadke MS, Jadhav SH, Krynetskiy EY. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response to chemotherapy-induced DNA damage. Mol Cancer Ther. 2009;8(4):864–72.
Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu Zn-SOD in mitochondria. J Biol Chem. 2001;276(42):38388–93.
Tanwir K, Amna, Javed MT, Shahid M, Akram MS, Ali Q. Chapter 32—Antioxidant defense systems in bioremediation of organic pollutants. In: Hasanuzzaman M, Prasad MNV, editors. Handbook of Bioremediation: Academic Press; 2021. p. 505–21.
Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–83.
Bordoni M, Pansarasa O, Dell'Orco M, Crippa V, Gagliardi S, Sproviero D, et al. Nuclear phospho-SOD1 protects DNA from oxidative stress damage in amyotrophic lateral sclerosis. J Clin Med. 2019;8(5).
Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XFS. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun. 2014;5:3446.
Das AB, Sadowska-Bartosz I, Königstorfer A, Kettle AJ, Winterbourn CC. Superoxide dismutase protects ribonucleotide reductase from inactivation in yeast. Free Radic Biol Med. 2018;116:114–22.
Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34(16):2011–21.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69(14):2409–27.
Zeng X, Ren H, Zhu Y, Zhang R, Xue X, Tao T, et al. Gp91phox (NOX2) in activated microglia exacerbates neuronal damage induced by oxygen glucose deprivation and hyperglycemia in an in vitro model. Cell Physiol Biochem. 2018;50(2):783–97.
Chen Y, Qin C, Huang J, Tang X, Liu C, Huang K, et al. The role of astrocytes in oxidative stress of central nervous system: a mixed blessing. Cell Prolif. 2020;53(3): e12781.
Migliore L, Coppedè F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat Res. 2009;674(1–2):73–84.
Shadfar S, Khanal S, Bohara G, Kim G, Sadigh-Eteghad S, Ghavami S, et al. Methanolic extract of boswellia serrata gum protects the nigral dopaminergic neurons from rotenone-induced neurotoxicity. Mol Neurobiol. 2022;59(9):5874–90.
Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci. 2015;9:124.
Johnson MA, Deng Q, Taylor G, McEachin ZT, Chan AWS, Root J, et al. Divergent FUS phosphorylation in primate and mouse cells following double-strand DNA damage. Neurobiol Dis. 2020;146: 105085.
Martin LJ, Chang Q. DNA damage response and repair, DNA methylation, and cell death in human neurons and experimental animal neurons are different. J Neuropathol Exp Neurol. 2018;77(7):636–55.
Shadfar S, Hwang CJ, Lim M-S, Choi D-Y, Hong JT. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch Pharm Res. 2015;38(12):2106–19.
Butterfield DA, Swomley AM, Sultana R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal. 2013;19(8):823–35.
Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5(3):278–86.
Wang WT, Tailor BA, Cohen DS, Huang X. Alzheimer’s pathogenesis, metal-mediated redox stress, and potential nanotheranostics. EC Pharmacol Toxicol. 2019;7(7):547–58.
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–27.
Lin X, Kapoor A, Gu Y, Chow MJ, Peng J, Zhao K, et al. Contributions of DNA damage to Alzheimer's disease. Int J Mol Sci. 2020;21(5).
Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol Aging. 1990;11(3):169–73.
Mecocci P, Polidori MC, Cherubini A, Ingegni T, Mattioli P, Catani M, et al. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch Neurol. 2002;59(5):794–8.
Mecocci P, Polidori MC, Ingegni T, Cherubini A, Chionne F, Cecchetti R, et al. Oxidative damage to DNA in lymphocytes from AD patients. Neurology. 1998;51(4):1014–7.
Migliore L, Fontana I, Trippi F, Colognato R, Coppedè F, Tognoni G, et al. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging. 2005;26(5):567–73.
Kadioglu E, Sardas S, Aslan S, Isik E, Esat KA. Detection of oxidative DNA damage in lymphocytes of patients with Alzheimer’s disease. Biomarkers. 2004;9(2):203–9.
Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem. 1998;71(5):2034–40.
Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19(6):1959–64.
Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007;35(22):7497–504.
Liu X, Lovell MA, Lynn BC. Development of a method for quantification of acrolein-deoxyguanosine adducts in DNA using isotope dilution-capillary LC/MS/MS and its application to human brain tissue. Anal Chem. 2005;77(18):5982–9.
Mao G, Pan X, Zhu BB, Zhang Y, Yuan F, Huang J, et al. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res. 2007;35(8):2759–66.
Lovell MA, Xie C, Markesbery WR. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000;855(1):116–23.
Weissman L, Jo DG, Sørensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, et al. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35(16):5545–55.
Hermon M, Cairns N, Egly JM, Fery A, Labudova O, Lubec G. Expression of DNA excision-repair-cross-complementing proteins p80 and p89 in brain of patients with Down Syndrome and Alzheimer’s disease. Neurosci Lett. 1998;251(1):45–8.
Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, et al. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch Biochem Biophys. 2006;449(1–2):143–56.
Boerrigter ME, van Duijn CM, Mullaart E, Eikelenboom P, van der Togt CM, Knook DL, et al. Decreased DNA repair capacity in familial, but not in sporadic Alzheimer’s disease. Neurobiol Aging. 1991;12(4):367–70.
Shackelford DA. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol Aging. 2006;27(4):596–605.
Jacobsen E, Beach T, Shen Y, Li R, Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res Mol Brain Res. 2004;128(1):1–7.
McLimans KE, Clark BE, Plagman A, Pappas C, Klinedinst B, Anatharam V, et al. Is cerebrospinal fluid superoxide dismutase 1 a biomarker of Tau but not amyloid-induced neurodegeneration in Alzheimer’s disease? Antioxid Redox Signal. 2019;31(8):572–8.
Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA. Increased expression but reduced activity of antioxidant enzymes in Alzheimer’s disease. J Alzheimers Dis. 1999;1(3):139–45.
Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem. 2011;286(52):44557–68.
Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70(3):357–60.
Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm (Vienna). 1998;105(8–9):855–70.
Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109 Suppl 1(Suppl 1):153–9.
Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol. 1996;149(1):21–8.
Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J Neurochem. 1997;68(5):2061–9.
Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36(5):747–51.
Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, et al. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23(2):471–6.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–23.
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–7.
Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain. 1999;122(2):247–53.
Strosznajder JB, Czapski GA, Adamczyk A, Strosznajder RP. Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol Neurobiol. 2012;46(1):78–84.
Strosznajder JB, Jeśko H, Strosznajder RP. Effect of amyloid beta peptide on poly(ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim Pol. 2000;47(3):847–54.
Martire S, Mosca L, d’Erme M. PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev. 2015;146–148:53–64.
Narne P, Pandey V, Simhadri PK, Phanithi PB. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin Cell Dev Biol. 2017;63:154–66.
Nair VD, McNaught KS, González-Maeso J, Sealfon SC, Olanow CW. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem. 2006;281(51):39550–60.
Chung YH, Shin C, Kim MJ, Lee B, Park KH, Cha CI. Immunocytochemical study on the distribution of p53 in the hippocampus and cerebellum of the aged rat. Brain Res. 2000;885(1):137–41.
Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 1997;232(2):418–21.
Davenport CM, Sevastou IG, Hooper C, Pocock JM. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J Neurochem. 2010;112(2):552–63.
Farmer KM, Ghag G, Puangmalai N, Montalbano M, Bhatt N, Kayed R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol Commun. 2020;8(1):132.
Shadfar S, Kim Y-G, Katila N, Neupane S, Ojha U, Bhurtel S, et al. Neuroprotective effects of antidepressants via upregulation of neurotrophic factors in the MPTP model of Parkinson’s disease. Mol Neurobiol. 2018;55(1):554–66.
Jung YY, Katila N, Neupane S, Shadfar S, Ojha U, Bhurtel S, et al. Enhanced dopaminergic neurotoxicity mediated by MPTP in IL-32β transgenic mice. Neurochem Int. 2017;102:79–88.
Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69(3):1196–203.
Andersen JK. Oxidative stress in neurodegeneration: Cause or consequence? Nat Med. 2004;10(Suppl):S18-25.
Beal MF. Bioenergetic approaches for neuroprotection in Parkinson's disease. Ann Neurol. 2003;53 Suppl 3:S39–47; discussion S-8.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):461–91.
Milanese C, Cerri S, Ulusoy A, Gornati SV, Plat A, Gabriels S, et al. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018;9(8):818.
Marttila RJ, Lorentz H, Rinne UK. Oxygen toxicity protecting enzymes in Parkinson's disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J Neurol Sci. 1988;86(2–3):321–31.
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–55.
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol. 1994;35(1):38–44.
Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, et al. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27(51):13997–4006.
Ibi M, Sawada H, Kume T, Katsuki H, Kaneko S, Shimohama S, et al. Depletion of intracellular glutathione increases susceptibility to nitric oxide in mesencephalic dopaminergic neurons. J Neurochem. 1999;73(4):1696–703.
Wüllner U, Löschmann P-A, Schulz JB, Schmid A, Dringen R, Eblen F, et al. Glutathione depletion potentiates MPTP and MPP+ toxicity in nigral dopaminergic neurones. NeuroReport. 1996;7(4):921–3.
Chinta SJ, Rajagopalan S, Butterfield DA, Andersen JK. In vitro and in vivo neuroprotection by gamma-glutamylcysteine ethyl ester against MPTP: relevance to the role of glutathione in Parkinson’s disease. Neurosci Lett. 2006;402(1–2):137–41.
Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Exp Neurol. 2007;203(2):512–20.
Bharat S, Cochran BC, Hsu M, Liu J, Ames BN, Andersen JK. Pre-treatment with R-lipoic acid alleviates the effects of GSH depletion in PC12 cells: implications for Parkinson’s disease therapy. Neurotoxicology. 2002;23(4–5):479–86.
Karunakaran S, Diwakar L, Saeed U, Agarwal V, Ramakrishnan S, Iyengar S, et al. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson’s disease: protection by alpha-lipoic acid. FASEB J. 2007;21(9):2226–36.
Foy CJ, Passmore AP, Vahidassr MD, Young IS, Lawson JT. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. QJM. 1999;92(1):39–45.
Ide K, Yamada H, Umegaki K, Mizuno K, Kawakami N, Hagiwara Y, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406–8.
Cipriani S, Desjardins CA, Burdett TC, Xu Y, Xu K, Schwarzschild MA. Urate and its transgenic depletion modulate neuronal vulnerability in a cellular model of Parkinson’s disease. PLoS ONE. 2012;7(5): e37331.
de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Ann Neurol. 2005;58(5):797–800.
Zhu T-G, Wang X-X, Luo W-F, Zhang Q-L, Huang T-T, Xu X-S, et al. Protective effects of urate against 6-OHDA-induced cell injury in PC12 cells through antioxidant action. Neurosci Lett. 2012;506(2):175–9.
Gong L, Zhang QL, Zhang N, Hua WY, Huang YX, Di PW, et al. Neuroprotection by urate on 6-OHDA-lesioned rat model of Parkinson’s disease: linking to Akt/GSK3β signaling pathway. J Neurochem. 2012;123(5):876–85.
de Farias CC, Maes M, Bonifácio KL, Bortolasci CC, de Souza NA, Brinholi FF, et al. Highly specific changes in antioxidant levels and lipid peroxidation in Parkinson’s disease and its progression: Disease and staging biomarkers and new drug targets. Neurosci Lett. 2016;617:66–71.
Buneeva O, Fedchenko V, Kopylov A, Medvedev A. Mitochondrial dysfunction in Parkinson's disease: focus on mitochondrial DNA. Biomedicines. 2020;8(12).
Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, et al. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol Dis. 2014;70:214–23.
Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst). 2004;3(1):1–12.
Lauritzen KH, Dalhus B, Storm JF, Bjørås M, Klungland A. Modeling the impact of mitochondrial DNA damage in forebrain neurons and beyond. Mech Ageing Dev. 2011;132(8–9):424–8.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468(7324):696–700.
Arai T, Fukae J, Hatano T, Kubo S, Ohtsubo T, Nakabeppu Y, et al. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson’s disease. Acta Neuropathol. 2006;112(2):139–45.
Fukae J, Takanashi M, Kubo S, Nishioka K, Nakabeppu Y, Mori H, et al. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005;109(3):256–62.
Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann Neurol. 1999;46(6):920–4.
Cardozo-Pelaez F, Sanchez-Contreras M, Nevin AB. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem Int. 2012;61(5):721–30.
Yamaguchi H, Kajitani K, Dan Y, Furuichi M, Ohno M, Sakumi K, et al. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006;13(4):551–63.
Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci. 2011;31(48):17649–58.
Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019;9(1):10919.
Lee SJ, Kim DC, Choi BH, Ha H, Kim KT. Regulation of p53 by activated protein kinase C-delta during nitric oxide-induced dopaminergic cell death. J Biol Chem. 2006;281(4):2215–24.
Bernstein AI, Garrison SP, Zambetti GP, O’Malley KL. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegen. 2011;6(1):2.
Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77(4):1037–48.
Zhang J, Pieper A, Snyder SH. Poly(ADP-Ribose) synthetase activation: an early indicator of neurotoxic DNA damage. J Neurochem. 1995;65(3):1411–4.
Mandir AS, Przedborski S, Jackson-Lewis V, Wang Z-Q, Simbulan-Rosenthal CM, Smulson ME, et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA. 1999;96(10):5774–9.
Cosi C, Colpaert F, Koek W, Degryse A, Marien M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996;729(2):264–9.
Outeiro TF, Grammatopoulos TN, Altmann S, Amore A, Standaert DG, Hyman BT, et al. Pharmacological inhibition of PARP-1 reduces α-synuclein- and MPP+-induced cytotoxicity in Parkinson’s disease in vitro models. Biochem Biophys Res Commun. 2007;357(3):596–602.
Lee Y, Karuppagounder SS, Shin J-H, Lee Y-I, Ko HS, Swing D, et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013;16(10):1392–400.
Ayton S, Lei P, Hare DJ, Duce JA, George JL, Adlard PA, et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J Neurosci. 2015;35(8):3591–7.
Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, et al. Nitric oxide synthase and neuronal vulnerability in parkinson’s disease. Neuroscience. 1996;72(2):355–63.
Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, Atkin JD. Motor neuron susceptibility in ALS/FTD. Front Neurosci. 2019;13.
Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2020;14:42.
Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: Where are we now? Front Neurosci. 2019;13:1310.
Konopka A, Atkin JD. DNA damage, defective DNA repair, and neurodegeneration in amyotrophic lateral sclerosis. Front Aging Neurosci. 2022;14.
Kim BW, Jeong YE, Wong M, Martin LJ. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol Commun. 2020;8(1):7.
Bradley WG, Krasin F. A new hypothesis of the etiology of amyotrophic lateral sclerosis: the DNA hypothesis. Arch Neurol. 1982;39(11):677–80.
Konopka A, Whelan DR, Jamali MS, Perri E, Shahheydari H, Toth RP, et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol Neurodegen. 2020;15(1):51.
Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, et al. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16(10):1383–91.
Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DAQ, Gómez-Herreros F, et al. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2013;42(1):307–14.
Sun Y, Curle AJ, Haider AM, Balmus G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020;64(5):847–61.
Kok JR, Palminha NM, Dos Santos SC, El-Khamisy SF, Ferraiuolo L. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell Mol Life Sci. 2021;78(15):5707–29.
Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69(5):2064–74.
Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, O’Donnell H, et al. Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med. 2000;29(7):652–8.
Warita H, Hayashi T, Murakami T, Manabe Y, Abe K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res. 2001;89(1–2):147–52.
Murakami T, Nagai M, Miyazaki K, Morimoto N, Ohta Y, Kurata T, et al. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res. 2007;1150:182–9.
Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol. 2002;103(4):408–14.
Coppedè F, Mancuso M, Lo Gerfo A, Carlesi C, Piazza S, Rocchi A, et al. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neurosci Lett. 2007;420(2):163–8.
Dhaliwal GK, Grewal RP. Mitochondrial DNA deletion mutation levels are elevated in ALS brains. NeuroReport. 2000;11(11):2507–9.
Lee M, Hyun D, Jenner P, Halliwell B. Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J Neurochem. 2001;76(4):957–65.
Wang Z, Bai Z, Qin X, Cheng Y. Aberrations in oxidative stress markers in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Oxid Med Cell Longev. 2019;2019:1712323.
McGurk L, Rifai OM, Bonini NM. Poly(ADP-Ribosylation) in age-related neurological disease. Trends Genet. 2019;35(8):601–13.
Farg MA, Konopka A, Soo KY, Ito D, Atkin JD. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26(15):2882–96.
McGurk L, Mojsilovic-Petrovic J, Van Deerlin VM, Shorter J, Kalb RG, Lee VM, et al. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2018;6(1):84.
Kim SH, Henkel JS, Beers DR, Sengun IS, Simpson EP, Goodman JC, et al. PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. J Neuropathol Exp Neurol. 2003;62(1):88–103.
Kim SH, Engelhardt JI, Henkel JS, Siklós L, Soós J, Goodman C, et al. Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology. 2004;62(2):319–22.
Chung YH, Joo KM, Lee YJ, Shin DH, Cha CI. Reactive astrocytes express PARP in the central nervous system of SOD(G93A) transgenic mice. Brain Res. 2004;1003(1–2):199–204.
Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019;29(3):233–47.
Hocsak E, Szabo V, Kalman N, Antus C, Cseh A, Sumegi K, et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic Biol Med. 2017;108:770–84.
Ranganathan S, Bowser R. p53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol J. 2010;4:11–22.
Martin LJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 2000;7(6 Pt B):613–22.
González de Aguilar JL, Gordon JW, René F, de Tapia M, Lutz-Bucher B, Gaiddon C, et al. Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: evidence for the implication of the p53 signaling pathway. Neurobiol Dis. 2000;7(4):406–15.
Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell. 2021;184(3):689-708.e20.
Lopez-Gonzalez R, Yang D, Pribadi M, Kim TS, Krishnan G, Choi SY, et al. Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc Natl Acad Sci USA. 2019;116(19):9628–33.
Walker C, Herranz-Martin S, Karyka E, Liao C, Lewis K, Elsayed W, et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. 2017;20(9):1225–35.
Lynch E, Semrad T, Belsito VS, FitzGibbons C, Reilly M, Hayakawa K, et al. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis Model Mech. 2019;12(8):dmm039552.
Chai N, Gitler AD. Yeast screen for modifiers of C9orf72 poly(glycine-arginine) dipeptide repeat toxicity. FEMS Yeast Res. 2018;18(4).
Onesto E, Colombrita C, Gumina V, Borghi MO, Dusi S, Doretti A, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. 2016;4(1):47.
Cohen S, Puget N, Lin Y-L, Clouaire T, Aguirrebengoa M, Rocher V, et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat Commun. 2018;9(1):533.
Grunseich C, Patankar A, Amaya J, Watts JA, Li D, Ramirez P, et al. Clinical and molecular aspects of senataxin mutations in amyotrophic lateral sclerosis 4. Ann Neurol. 2020;87(4):547–55.
Sariki SK, Sahu PK, Golla U, Singh V, Azad GK, Tomar RS. Sen1, the homolog of human Senataxin, is critical for cell survival through regulation of redox homeostasis, mitochondrial function, and the TOR pathway in Saccharomyces cerevisiae. FEBS J. 2016;283(22):4056–83.
Dutta A, Hromas R, Sung P. Senataxin: A putative RNA: DNA helicase mutated in ALS4—emerging mechanisms of genome stability in motor neurons. Amyotrophic Lateral Sclerosis-Recent Advances and Therapeutic Challenges: IntechOpen; 2020.
Hirano M, Quinzii CM, Mitsumoto H, Hays AP, Roberts JK, Richard P, et al. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12(3):223–7.
Zhong Y, Wang J, Henderson MJ, Yang P, Hagen BM, Siddique T, et al. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife. 2017;6: e23759.
Carroll J, Page TKW, Chiang S-C, Kalmar B, Bode D, Greensmith L, et al. Expression of a pathogenic mutation of SOD1 sensitizes aprataxin-deficient cells and mice to oxidative stress and triggers hallmarks of premature ageing. Hum Mol Genet. 2015;24(3):828–40.
Cereda C, Leoni E, Milani P, Pansarasa O, Mazzini G, Guareschi S, et al. Altered intracellular localization of SOD1 in leukocytes from patients with sporadic amyotrophic lateral sclerosis. PLoS ONE. 2013;8(10):e75916-e.
Barbosa LF, Cerqueira FM, Macedo AFA, Garcia CCM, Angeli JPF, Schumacher RI, et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim Biophys Acta. 2010;1802(5):462–71.
Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VMY. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012;31(5):1241–52.
Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci USA. 2019;116(10):4696–705.
Goh CW, Lee IC, Sundaram JR, George SE, Yusoff P, Brush MH, et al. Chronic oxidative stress promotes GADD34-mediated phosphorylation of the TAR DNA-binding protein TDP-43, a modification linked to neurodegeneration. J Biol Chem. 2018;293(1):163–76.
Duan W, Li X, Shi J, Guo Y, Li Z, Li C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience. 2010;169(4):1621–9.
Wang H, Guo W, Mitra J, Hegde PM, Vandoorne T, Eckelmann BJ, et al. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun. 2018;9(1):3683.
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun. 2018;9(1):335.
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162(5):1066–77.
Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88(4):678–90.
Saito Y, Kimura W. Roles of phase separation for cellular redox maintenance. Front Genet. 2021;12.
Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Investig. 2014;124(3):981–99.
Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. NeuroReport. 1997;8(6):1337–40.
Olkowski ZL. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. NeuroReport. 1998;9(2):239–42.
Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5(1):36–53.
Li J, Song M, Moh S, Kim H, Kim D-H. Cytoplasmic restriction of mutated SOD1 impairs the DNA repair process in spinal cord neurons. Cells. 2019;8(12):1502.
Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447(7143):447–52.
Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. 1997;15(2):197–200.
Maiuri T, Suart CE, Hung CLK, Graham KJ, Barba Bazan CA, Truant R. DNA damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics. 2019;16(4):948–56.
Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WM, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017;26(2):395–406.
Askeland G, Dosoudilova Z, Rodinova M, Klempir J, Liskova I, Kuśnierczyk A, et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep. 2018;8(1):9817.
Castaldo I, De Rosa M, Romano A, Zuchegna C, Squitieri F, Mechelli R, et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann Neurol. 2019;85(2):296–301.
Lu XH, Mattis VB, Wang N, Al-Ramahi I, van den Berg N, Fratantoni SA, et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington's disease. Sci Transl Med. 2014;6(268):268ra178.
Lee J-M, Wheeler Vanessa C, Chao Michael J, Vonsattel Jean Paul G, Pinto Ricardo M, Lucente D, et al. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. 2015;162(3):516–26.
Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41(5):646–53.
Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. eLife. 2019;8:e42988.
DiGiovanni LF, Mocle AJ, Xia J, Truant R. Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum Mol Genet. 2016;25(18):3937–45.
Bowie LE, Maiuri T, Alpaugh M, Gabriel M, Arbez N, Galleguillos D, et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc Natl Acad Sci USA. 2018;115(30):E7081–90.
Hung CL, Maiuri T, Bowie LE, Gotesman R, Son S, Falcone M, et al. A patient-derived cellular model for Huntington’s disease reveals phenotypes at clinically relevant CAG lengths. Mol Biol Cell. 2018;29(23):2809–20.
Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210.
Acuña AI, Esparza M, Kramm C, Beltrán FA, Parra AV, Cepeda C, et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat Commun. 2013;4(1):2917.
Peña-Sánchez M, Riverón-Forment G, Zaldívar-Vaillant T, Soto-Lavastida A, Borrero-Sánchez J, Lara-Fernández G, et al. Association of status redox with demographic, clinical and imaging parameters in patients with Huntington’s disease. Clin Biochem. 2015;48(18):1258–63.
Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, et al. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J Neurol Sci. 1992;108(1):80–7.
Ribeiro M, Silva AC, Rodrigues J, Naia L, Rego AC. Oxidizing effects of exogenous stressors in Huntington’s disease knock-in striatal cells—protective effect of cystamine and creatine. Toxicol Sci. 2013;136(2):487–99.
Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18(16):3039–47.
Goold R, Hamilton J, Menneteau T, Flower M, Bunting EL, Aldous SG, et al. FAN1 controls mismatch repair complex assembly via MLH1 retention to stabilize CAG repeat expansion in Huntington’s disease. Cell Rep. 2021;36(9): 109649.
Miller CJ, Kim G-Y, Zhao X, Usdin K. All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. 2020;16(6):e1008902.
Goold R, Flower M, Moss DH, Medway C, Wood-Kaczmar A, Andre R, et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. 2019;28(4):650–61.
Flower M, Lomeikaite V, Ciosi M, Cumming S, Morales F, Lo K, et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. 2019;142(7):1876–86.
Barth E, Sieber P, Stark H, Schuster S. Robustness during aging-molecular biological and physiological aspects. Cells. 2020;9(8).
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–81.
Jin K, Simpkins JW, Ji X, Leis M, Stambler I. The critical need to promote research of aging and aging-related diseases to improve health and longevity of the elderly population. Aging Dis. 2015;6(1):1.
Poetsch AR. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput Struct Biotechnol J. 2020;18:207–19.
Santra M, Dill KA, de Graff AMR. Proteostasis collapse is a driver of cell aging and death. Proc Natl Acad Sci USA. 2019;116(44):22173–8.
Sabath N, Levy-Adam F, Younis A, Rozales K, Meller A, Hadar S, et al. Cellular proteostasis decline in human senescence. Proc Natl Acad Sci USA. 2020;117(50):31902–13.
Paull TT. DNA damage and regulation of protein homeostasis. DNA Repair (Amst). 2021;105: 103155.
Huiting W, Bergink S. Locked in a vicious cycle: the connection between genomic instability and a loss of protein homeostasis. Genome Instab Dis. 2021;2(1):1–23.
Asensi M, Sastre J, Pallardo FV, Lloret A, Lehner M, Garcia-de-la Asuncion J, et al. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999;299:267–76.
Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999;27(8):1935–42.
Pao P-C, Patnaik D, Watson LA, Gao F, Pan L, Wang J, et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat Commun. 2020;11(1):2484.
Gao L, Wang M, Liao L, Gou N, Xu P, Ren Z, et al. A Slc25a46 mouse model simulating age-associated motor deficit, redox imbalance, and mitochondria dysfunction. J Gerontol A Biol Sci Med Sci. 2021;76(3):440–7.
Dollé MET, Kuiper RV, Roodbergen M, Robinson J, de Vlugt S, Wijnhoven SWP, et al. Broad segmental progeroid changes in short-lived Ercc1(-/Δ7) mice. Pathobiol Aging Age Relat Dis. 2011;1.
Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161(7):1592–605.
Stott RT, Kritsky O, Tsai LH. Profiling DNA break sites and transcriptional changes in response to contextual fear learning. PLoS ONE. 2021;16(7): e0249691.
Cleaver JE. Defective repair replication of DNA in Xeroderma Pigmentosum. Nature. 1968;218(5142):652–6.
Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130(6):991–1004.
Hoch NC, Hanzlikova H, Rulten SL, Tétreault M, Komulainen E, Ju L, et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. 2017;541(7635):87–91.
O’Connor E, Vandrovcova J, Bugiardini E, Chelban V, Manole A, Davagnanam I, et al. Mutations in XRCC1 cause cerebellar ataxia and peripheral neuropathy. J Neurol Neurosurg Psychiatry. 2018;89(11):1230–2.
Chen HH, Petty LE, Bush W, Naj AC, Below JE. GWAS and beyond: using omics approaches to interpret SNP Associations. Curr Genet Med Rep. 2019;7(1):30–40.
Kwiatkowski D, Czarny P, Toma M, Korycinska A, Sowinska K, Galecki P, et al. Association between single-nucleotide polymorphisms of the hOGG1, NEIL1, APEX1, FEN1, LIG1, and LIG3 genes and Alzheimer’s disease risk. Neuropsychobiology. 2016;73(2):98–107.
Dinçer Y, Akkaya Ç, Mutlu T, Yavuzer S, Erkol G, Bozluolcay M, et al. DNA repair gene OGG1 polymorphism and its relation with oxidative DNA damage in patients with Alzheimer’s disease. Neurosci Lett. 2019;709: 134362.
Parildar-Karpuzoğlu H, Doğru-Abbasoğlu S, Hanagasi HA, Karadağ B, Gürvit H, Emre M, et al. Single nucleotide polymorphisms in base-excision repair genes hOGG1, APE1 and XRCC1 do not alter risk of Alzheimer’s disease. Neurosci Lett. 2008;442(3):287–91.
Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–6.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Bečanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci. 2015;18(6):807–16.
Martin LJ, Wong M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech Ageing Dev. 2017;161:149–62.
Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13(11):1108–13.
Le Heron C, MacAskill M, Mason D, Dalrymple-Alford J, Anderson T, Pitcher T, et al. A multi-step model of Parkinson’s disease pathogenesis. Mov Disord. 2021;36(11):2530–8.
Licher S, van der Willik KD, Vinke EJ, Yilmaz P, Fani L, Schagen SB, et al. Alzheimer’s disease as a multistage process: an analysis from a population-based cohort study. Aging (Albany NY). 2019;11(4):1163–76.
Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359(6375):555–9.
Lodato MA, Walsh CA. Genome aging: somatic mutation in the brain links age-related decline with disease and nominates pathogenic mechanisms. Hum Mol Genet. 2019;28(R2):R197-r206.
Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93(17):8919–23.
Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146(1):67–79.
Samson M, Claassen DO. Neurodegeneration and the cerebellum. Neurodegener Dis. 2017;17(4–5):155–65.
Pradat PF. The cerebellum in ALS: Friend or foe? J Neurol Neurosurg Psychiatry. 2021;92(11):1137.
Chatterjee N, Lin Y, Santillan BA, Yotnda P, Wilson JH. Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc Natl Acad Sci U S A. 2015;112(12):3764–9.
Acknowledgements
We would like to take this opportunity to thank Dr. Nima Sayyadi (Macquarie University) for providing valuable feedback on drawing chemical structures of the free radicals.
Funding
This work was supported by an Australian National Health and Medical Research Council (NHMRC) Dementia Teams Research Grant (1095215), Macquarie University MQCRF fellowship, Fight MND Foundation, and Motor Neuron Disease Research Australia.
Author information
Authors and Affiliations
Contributions
S.S and J.D.A. conceptualized the structure of the manuscript. S.S, S.P., M.S.J and J.D.A. wrote sections of the manuscript. S.S. designed and created the figures with BioRender. J.D.A. and S.P also conceived the article and edited the manuscript throughout for content and style consistency. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shadfar, S., Parakh, S., Jamali, M.S. et al. Redox dysregulation as a driver for DNA damage and its relationship to neurodegenerative diseases. Transl Neurodegener 12, 18 (2023). https://doi.org/10.1186/s40035-023-00350-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00350-4