
Abstract
Coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a life-threatening disease, especially in elderly individuals and those with comorbidities. The predominant clinical manifestation of COVID-19 is respiratory dysfunction, while neurological presentations are increasingly being recognized. SARS-CoV-2 invades host cells primarily via attachment of the spike protein to the angiotensin-converting enzyme 2 (ACE2) receptor expressed on cell membranes. Patients with Alzheimer’s disease (AD) are more susceptible to SARS-CoV-2 infection and prone to severe clinical outcomes. Recent studies have revealed some common risk factors for AD and COVID-19. An understanding of the association between COVID-19 and AD and the potential related mechanisms may lead to the development of novel approaches to treating both diseases. In the present review, we first summarize the mechanisms by which SARS-CoV-2 invades the central nervous system (CNS) and then discuss the associations and potential shared key factors between COVID-19 and AD, with a focus on the ACE2 receptor, apolipoprotein E (APOE) genotype, age, and neuroinflammation.
Similar content being viewed by others

Introduction
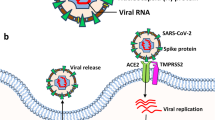
The outbreak of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has already resulted in more than 540 million infections and over 6 million deaths worldwide as of 27 June 2022 (https://covid19.who.int). The aged individuals and those with comorbidities are at a particularly high risk of poor outcomes [1]. SARS-CoV-2 is a positive-sense, single-stranded RNA virus with four major structural proteins: envelope, membrane, spike (S) and nucleocapsid phosphoprotein [2]. SARS-CoV-2 infects target cells primarily via interaction of the receptor-binding domain of the S protein with the cellular angiotensin-converting enzyme 2 (ACE2) receptor after activation of the S protein by transmembrane serine protease 2 (TMPRSS2) [3]. Thus, the expression and distribution of ACE2 is critical for SARS-CoV-2 infection. The S protein is composed of two functional subunits, S1 and S2; S1 is responsible for receptor binding, whereas S2 (the C-terminal domain) is specifically responsible for viral-cellular membrane fusion [4]. Although SARS-CoV-2 primarily targets the respiratory tract, causing fever, dry cough, sore throat, fatigue, and dyspnoea [5], the virus also results in dysfunction of multiple organ systems outside the lung, including the kidneys, liver, brain, heart, gastrointestinal tract and other organs [6,7,8,9], as ACE2 and other candidate receptors are also expressed in these tissues [10].
Emerging studies have revealed the neuroinvasive potential of SARS-CoV-2 [11, 12], with neurological manifestations ranging from lethargy, headache, loss of smell and taste, delirium, insomnia, brain inflammation, stroke, brain haemorrhage to cognitive impairment [13,14,15]. In fact, the central nervous system (CNS) complications have been observed in more than 30% of individuals with COVID-19 presenting with a higher infection severity [14]. Therefore, studies designed to provide insights into the invasion and effects of SARS-CoV-2 on the CNS are critical. Various studies using cultured cells, animal models, and brain tissues from patients who died of COVID-19, have independently revealed the capacity of SARS-CoV-2 to invade the CNS [16, 17]. Supporting evidence from a postmortem study indicated the presence of viral RNA and proteins in the brains of more than half (21 of 40) of German patients who died of COVID-19 [17], and this phenomenon was confirmed by other neuropathological case studies [18, 19]. In addition, the presence of SARS-CoV-2 in the cerebrospinal fluid (CSF) of infected individuals also confirms CNS infection [15, 20, 21]. Notably, by employing well-characterized human brain organoids, researchers have visualized widespread infectivity of SARS-CoV-2 and extensive death of virus-infected and nearby neuronal cells, and found that the viral infection can be abrogated by pretreatment with an ACE2 antibody or administration of CSF obtained from patients with COVID-19 [12]. Moreover, brain imaging studies have revealed the presence of multiple haemorrhagic lesions and changes in the brain structure of patients infected with SARS-CoV-2, even in milder cases [22,23,24]. Together, this evidence implies that SARS-CoV-2 has the ability to enter the CNS and cause neurological conditions.
Researchers have speculated that the CNS invasion may occur along nerves or through haematogenous spread [25]. CNS infection may lead to neuroinflammation, blood–brain barrier (BBB) disruption, and alterations in neurovascular and cognition functions, which are closely related to the risk and progression of neurodegenerative diseases [26]. Elderly individuals and those with comorbid conditions are more vulnerable to infection and may be subject to severe outcomes after SARS-CoV-2 infection. Individuals with dementia are three-fold more likely to contract severe COVID-19 condition (requiring hospitalization) than those without dementia [27, 28], and the mortality rate is 30% higher in patients who suffer dementia [29]. Alzheimer’s disease (AD) accounts for more than 60% of all dementia cases [30]. AD pathology is complex and determined by age, heredity, and environmental factors [31]. The main pathological manifestations of AD are the formation of extracellular amyloid-beta (Aβ) plaques and neurofibrillary tangles comprising abnormal tau, as well as neuroinflammation in the brain [32]. The morbidity and mortality rates for COVID-19 are increased in subjects with AD, which is possibly due to the AD-related pathological changes and factors, such as elevated proinflammatory molecules, an advanced age, BBB disruption, presence of APOE epsilon4 (APOE ε4) allele, diabetes mellitus, and lifestyle factors [33, 34]. Therefore, studies on the associations between AD and SARS-CoV-2 and the potential mechanisms are urgently needed. Here, we summarize the potential routes by which SARS-CoV-2 invades the CNS and then provide an overview of the associations and potential shared pathogenic mechanisms between SARS-CoV-2 and AD, with a focus on the ACE2 receptor, APOE genotype, age, and neuroinflammation, in order to advance the understanding of associations between COVID-19 and AD.
Potential routes of SARS-CoV-2 invasion into the CNS
The presence of viral RNA, proteins or particles in postmortem brain tissues and in the CSF of infected patients indicates the infiltration of SARS-CoV-2 in the CNS. Studies on the mechanisms by which SARS-CoV-2 invades the CNS, which are important for disease diagnosis, prognosis and interventional strategies, are ongoing. Researchers have proposed that SARS-CoV-2 enters the CNS mainly via the direct neuronal route or through haematogenous transport [35, 36].
The neuronal route relies on retrograde axonal transport of the virus from infected peripheral nerves. Evidence shows that SARS-CoV-2 potentially infects the CNS by travelling along olfactory axon bundles. The upper and rear part of the nasal cavity is the olfactory mucosa. The respiratory tract makes direct contact with the CNS through the olfactory sensory neurons. The cilia of the olfactory sensory neurons are present in the nasal cavity, and their axons extend into the olfactory bulb by passing through the ethmoid plate. Intact SARS-CoV-2 particles and viral RNA have been detected in the olfactory mucosa and in neuroanatomical regions that receive olfactory tract projections, indicating that the neuroinvasion occurs via the olfactory pathway [37]. SARS-CoV-2 has been observed to invade the CNS through this route in SARS-CoV-2-infected K18-hACE2 transgenic mice [38] and hamsters [39] (which express the human ACE2 receptor driven by the promoter of the human cytokeratin 18 gene), resulting in a life-threatening disease similar to severe SARS-CoV-2 infection [16, 40]. In those studies, viral RNA was detected in the brains of all animals five days after intranasal administration of SARS-CoV-2, and the severity of neuropathological deficits correlated well with the virus level in the brain. Consistently, in rhesus monkeys exposed to SARS-CoV-2 through intranasal inoculation, the viral RNA copies were sequentially detected in nasal mucosa, olfactory trigone, and entorhinal area, and the viral protein was detected in some functional brain areas, including the thalamus, entorhinal area, medulla oblongata, hippocampus and prepyriform cortex [41]. These findings suggest that SARS-CoV-2 may be transported to the CNS via the olfactory route. In fact, human coronavirus OC43, Middle East respiratory syndrome coronavirus, and SARS-CoV-1 have all been shown to infect the murine CNS via the reverse axonal transport route, invading olfactory sensory neurons in the nasal cavity and then being transported to other neural cells [42,43,44,45]. Given the similarities of the viral nucleic acid sequences of coronaviruses [46], SARS-CoV-2 may utilize a similar neural invasion mechanism. The persistence of anosmia and ageusia, typical symptoms of SARS-CoV-2 infection of the olfactory system, in a portion of patients even after recovery from infection, also supports this route of transport. Interestingly, olfactory dysfunction and structural alterations in the olfactory-related regions are early manifestations of several neurodegenerative diseases, such as AD and Parkinson's disease [47,48,49]. Some researchers have considered that the olfactory nerve is not likely the primary route through which SARS-CoV-2 accesses the brain [50], as human olfactory sensory neurons do not express or exhibit low expression of TMPRSS2 and ACE2, two key genes involved in SARS-CoV-2 entry [51]. Instead, proteins of the two genes are abundant in samples of the whole olfactory mucosa of humans [51] and in mouse olfactory epithelium [52]. Infection of the olfactory epithelium allows the virus to spread to horizontal basal cells as well as immature and mature olfactory neurons. In addition, the infected horizontal basal cells mature into olfactory sensory neurons [53], allowing the virus to reach the olfactory bulb through synaptic connections, thus providing the possibility of CNS infection (Fig. 1). Notably, in addition to ACE2, other receptors such as neuropilin-1 (NRP-1), a signalling protein, have also been discovered to serve as important SARS-CoV-2 receptors and enhance the viral infectivity [54, 55]. NRP-1 is expressed at high levels in the olfactory epithelium, olfactory tubules and paraolfactory gyri, further supporting its role in virus entry into olfactory epithelial cells [54, 56]. These studies indicate that the sustentacular cells are vulnerable to coronavirus entry and give rise to anosmia. Therefore, the olfactory pathway may constitute an important route of CNS invasion. In addition to the olfactory nerve, other peripheral nerves, including the vagus, trigeminus, and nasopharyngeal nerves, may be potential routes through which SARS-CoV-2 accesses the brain [57].

SARS-CoV-2 invades the CNS through the olfactory nerve. Once SARS-CoV-2 enters the nasal cavity, it contacts with olfactory epithelial cells, which express ACE2 and NRP-1 receptors at high levels, rendering the epithelium vulnerable to SARS-CoV-2 infection. The infected olfactory epithelial cells potentially transmit the virus to the surrounding cells, such as horizontal basal cells as well as immature and mature olfactory neurons. The infected horizontal basal cells also mature into olfactory sensory neurons. These neuronal cells extend to the apex, contact the air, and form small nerve bundles at the base before passing through the ethmoid plate and forming the olfactory nerve. Infected olfactory neurons are connected to neurons in the olfactory bulb through synapses, which allows the virus to spread from the olfactory nerve to the olfactory bulb via retrograde transport along axons. The olfactory bulb makes many connections throughout the brain, allowing the virus to spread quickly to other structures in the brain
In addition to the olfactory route, SARS-CoV-2 may also enter the CNS via the haematogenous route; however, in this route the virus must be able to cross the BBB to achieve entry into the CNS. The BBB, which is composed of endothelial cells, pericytes, and astrocytes, functions as an important supporting and protective interface that restricts the entry of circulating molecules, such as neurotoxic plasma components, leucocytes, and pathogens, into the brain [58]. SARS-CoV-2 infection of the lungs may lead to endothelial damage and increased capillary permeability [59, 60], which facilitates virus transfer from the lungs to the pulmonary microcirculation (Fig. 2a). SARS-CoV-2 in the blood will enter the cerebral circulation, where the slow blood flow may promote potential interactions of S protein with ACE2 [61] and NRP-1[62] on microvascular endothelial cells [35]. The presence of virus-like particles in the brain capillary endothelium in the frontal lobe of a patient who died from COVID-19 provides direct evidence for viral invasion into brain microvascular endothelial cells of the BBB [63]. Autopsy studies have revealed that SARS-CoV-2 disrupts the BBB. This has been confirmed in 2D static and 3D microfluidic engineered models of the human BBB [64]. In this model, the SARS-CoV-2 S protein activates the proinflammatory response in the brain endothelium and contributes to increased barrier permeability, which facilitates viral entry into the brain (Fig. 2b). In addition, viral RNA occasionally appears in the vascular wall, perivascular region, and brain microvascular endothelium of SARS-CoV-2-challenged K18-hACE2 transgenic mice [65]. Other mechanisms by which SARS-CoV-2 affects the BBB integrity include invasion of BBB epithelial cells and astrocytes, disruption of placodes or the actin cytoskeleton, and phosphorylation of tight junction proteins [66, 67]. SARS-CoV-2 also infects leukocytes and enters the BBB via these cells [68, 69]. Systemic virus dissemination in the CNS may also occur via exosomal cellular transport and lymphatic spread following infection, immune activation, and production of granulocyte macrophages [70]. Recently, researchers have observed that challenge with SARS-CoV-2 results in infection of the epithelium of the choroid plexus and leads to the breakdown of the blood-CSF barrier in an organoid model of the human choroid plexus, providing another entry route for SARS-CoV-2 into the brain [71]. Ultimately, the virus in the CSF must infect or penetrate the BBB to enter the brain. Notably, AD in particular has an early pathology associated with increased permeability and disruption of the BBB, which, together with reduced cerebral blood flow, can place individuals at a higher risk of contracting SARS-CoV-2 [72,73,74]. Moreover, even older individuals who are not yet symptomatic for AD (but who are perhaps very early in the pathological trajectory in developing AD) may experience some BBB degeneration and an early loss of pericytes, which promote increased vulnerability to the virus. After the virus enters brain cells, the viral life cycle begins, including genome replication, protein synthesis, assembly, maturation and virus release, ultimately leading to brain tissue infection and neurological symptoms (Fig. 2c). It has been widely demonstrated that vaccines are the most efficient tools for preventing COVID-19. Studies have reported that COVID-19 vaccines are safe and well-tolerated in patients with neurological disorders [75]. Nevertheless, a recent study reported that vaccinated patients with AD displayed a significantly increased overall risk for breakthrough infections (approximately 10.3%) than the matched older adults without dementia (approximately 5.6%) [76]. However, after further matching for comorbidities, such as heart diseases, cancers, type 2 diabetes, and hypertension, dementia patients no longer showed an obvious increase in risk of breakthrough infections. In addition, several rare neurological complications can occur in individuals who received COVID-19 vaccines (ChAdOx1nCoV-19 and BNT162b2), such as Guillain–Barré syndrome, Bell’s palsy and myasthenic disorders [77], whereas such complications are much more common among individuals with SARS-CoV-2 infections. Future studies are warranted to evaluate the possible use of vaccines in preventing the spread of viral infection throughout the CNS, as well as to evaluate the protective effects of vaccination on AD and the potential side effects.

SARS-CoV-2 infects the brain via the haematogenous route. a SARS-CoV-2 is a respiratory virus that spreads primarily through airborne droplets, causing infection of the lungs. Left panel: SARS-CoV-2 infection of the lungs may lead to endothelial damage and increased capillary permeability, which allow the transfer of SARS-CoV-2 from the lungs to the pulmonary microcirculation. Right panel: SARS-CoV-2 in the blood can enter the cerebral circulation, where the slow blood flow may allow the virus to damage the BBB. b Viruses in the cerebral circulation may infect and destroy microvascular endothelial cells, resulting in increased BBB permeability and facilitating viral entry into brain tissues. c SARS-CoV-2 infects brain cells via the interaction of S protein with ACE2, NRP1 and other potential receptors on microvascular endothelial cells after the S protein is primed by TMPRSS2. Once SARS-CoV-2 enters the cell, the viral life cycle begins, including genome replication, protein synthesis, virus assembly, maturation and release, ultimately leading to brain parenchyma infection and tissue damage
In conclusion, there are two major potential routes by which SARS-CoV-2 invades the brain: the olfactory pathway and haematogenous transport. Despite the evidence presented here, the primary invasion route, the mechanism by which SARS-CoV-2 spreads throughout the brain, and the mechanisms of SARS-CoV-2-induced damage to the CNS remain unclear and require further studies. Furthermore, how the coronavirus enters the bloodstream remains an open question.
Common risk factors and mechanisms of COVID-19 and AD
ACE2: a possible double-edged sword in AD
The transmembrane glycoprotein ACE2 is a vital component of the renin-angiotensin system (RAS) that is responsible for catalysing the conversion of angiotensin (Ang) II into Ang (1–7), thus protecting against the harmful effects of the ACE1/Ang-II axis [78]. Ang-II exerts vasoconstrictive, proinflammatory hypertrophy, and profibrotic effects, while Ang (1–7) exerts opposing effects, such as antiproliferative, anti-inflammatory, antiapoptotic, and mild vasodilatory effects, to protect against various cardiovascular diseases [79, 80]. In addition to acting as a negative regulator of RAS, ACE2 also plays a protective role in inflammatory lung diseases in other manners. Studies have identified that des-Arg9 bradykinin (DABK) is a biological substrate of ACE2 in airway epithelial cells, and the attenuation of ACE2 activity facilitates neutrophil infiltration, which is partially attributed to the modulation of DABK/bradykinin receptor B1 axis signalling [81]. Since the onset of the COVID-19 pandemic, ACE2 has been recognized as the main host cell receptor for SARS-CoV-2 infection. The amount and distribution of ACE2 may influence the risk of SARS-CoV-2 infection. As the respiratory tract is the dominant target of SARS-CoV-2, high levels of ACE2 have been detected in respiratory epithelial cells and pulmonary type II alveolar cells [82]. However, ACE2 expression is not restricted to the respiratory tract, as kidney proximal tubule cells, bladder urothelial cells, ileum and oesophageal epithelial cells, neurons and glial cells in the brain, and myocardial cells all exhibit high levels of ACE2 [61, 83,84,85,86,87]. As a result, the function and hyperactivity of the ACE2 receptor may increase the sensitivity of these target cells and organs to SARS-CoV-2 infection. ACE2 plays a key role in SARS-CoV-2 neuroinfection, but its expression in the context of AD remains controversial. A recent study reported that the ACE2 protein level is upregulated in the hippocampal tissues of patients with AD and that the change is not age- or sex-dependent, indicating a direct relationship between AD and ACE2 expression [88]. Another study detected ACE2 expression in multiple brain regions of a recently deceased patient with AD, and ACE2 expression was significantly increased in the temporal lobe and CA1 region of the hippocampus [89]. In addition, RNA-seq showed that the expression of ACE2 mRNA is elevated in cortical tissues from 5× FAD mice [90] but not significantly altered in the blood. However, previous studies have reported inconsistent findings for ACE2 expression and its effect on AD pathology. Immunohistochemical staining showed that ACE2 expression varies across different brain regions, decreased in the hippocampus, visual cortex, basal nucleus, amygdala, middle frontal gyrus and entorhinal cortex in response to AD pathology [91]. Specifically, ACE2 activity is obviously decreased in the postmortem brain tissues of patients with AD compared to those of age-matched controls, and is inversely related to Aβ levels and tau phosphorylation [92]. The decrease of ACE2 expression in the context of AD indicates the occurrence of RAS dysregulation and neurological damage in corresponding brain regions. Additional studies have also supported this hypothesis. For instance, elevated ACE2 activity in the brain induced by intraperitoneal administration of an established ACE2 activator (diminazene aceturate) reduces Aβ-related pathology and delays cognitive impairment in symptomatic (aged) Tg2576 mice and protects against the onset of cognitive decline in presymptomatic (younger) Tg2576 mice [93]. A previous study also found that a longer, neurotoxic species of Aβ (Aβ43) is converted into a shorter, less toxic or neuroprotective form of Aβ (Aβ40) by ACE and ACE2 [94] and that this conversion is reversed by the specific ACE2 inhibitor DX600. The researchers also found that ACE2 activity is decreased in the sera of patients with AD compared with the sera of age-matched controls, indicating a relationship between lower ACE2 activity and AD. Likewise, in a D-galactose-ovariectomized rat model of ageing and dementia, increased activation of ACE2 via administration of dimenazine ameliorates Aβ pathology in the brain and improves cognitive performance [95]. Therefore, during the initial stages of AD, maintaining ACE2 activity in the brain may be a protective factor, limiting the progression of AD pathology. Thus, researchers have speculated that the SARS-CoV-2 infection-induced downregulation of ACE2 expression in target cells [96] may inhibit the protective effect of ACE2 against AD. However, ACE2 levels in the CSF are similar between patients with AD and controls [97]. Furthermore, no significant difference in ACE2 activity was observed in the cerebral cortex or hippocampus of SAMP8 mice, a non-transgenic animal model of sporadic AD, during disease progression [98]. Possible explanations for the inconsistency in results among these studies might be the differences in animal models used, brain areas selected, and detection methods employed.
In conclusion, high levels of ACE2 enhance SARS-CoV-2 infection, whereas ACE2 expression decreases once viral infection occurs, and the protective function of ACE2 is inhibited. At present, the changes in ACE2 expression that occur in the context of AD are controversial. Future studies using more animal models and larger sample sizes, especially specimens from more patients with AD, will help to clarify the expression of ACE2 in individuals with AD and the relationship between ACE2 and AD pathology. Additionally, the role of ACE2 in virus transmission throughout the brain requires further study.
APOE4: increased susceptibility to COVID-19
The manifestations of SARS-CoV-2 infection vary widely among individuals, from no symptoms to life-threatening conditions, suggesting that the host genetic background may influence the risk and severity of virus infection [99, 100]. An important genetic factor is the APOE genotype. ApoE is a multifaceted secreted protein synthesized by astrocytes in the CNS and by the liver in the periphery [101]. Human APOE encodes a 299-amino-acid protein with three major isoforms that differ at amino acid residues 112 and 158, i.e., apoE2 (Cys112, Cys158), apoE3 (Cys112, Arg158), and apoE4 (Arg112, Arg158). The amino acid differences exert profound effects on the apoE protein structure and function [102, 103]. APOE4 is the strongest genetic susceptibility factor for sporadic AD [104], accounting for more than 95% of all AD cases [105]. ApoE2 is protective, and apoE3 is neutral [106]. Previous studies have reported that APOE4 is related to the occurrence and severity of infections with viruses and pathogenic microorganisms, such as HIV-1 [107], herpes simplex [108] and hepatitis C [109]. Since the onset of the COVID-19 pandemic, an increasing number of studies have been conducted on the possible associations of APOE polymorphisms with SARS-CoV-2 infection and disease severity. Recently, Kuo et al. found that APOE ε4/ε4 homozygotes exhibit a more than two-fold higher susceptibility to COVID-19 [110, 111] and a four-fold higher risk of mortality than APOE ε3/ε3 homozygotes, according to data from the UK Biobank cohort [110, 111]. The association was not diminished even after controlling for APOE4-related diseases, including coronary artery disease, hypertension, diabetes, and dementia, suggesting that APOE4 may exert an independent effect on COVID-19 severity. Other studies have reported similar findings in Spanish and Iraqi subjects: the ε4 allele of the APOE gene increases the incidence and clinical severity of SARS-CoV-2 infection [112, 113]. Another study showed no obvious difference in the frequency of the ε4 allele between viral RNA-positive subjects and the control population in a Czech cohort [114]. When the data were further analysed after stratification according to the disease status, the results revealed a significantly higher frequency of the ε4 allele in symptomatic subjects, indicating that the ε4 allele may be associated with an increased risk of symptomatic COVID-19. Notably, a recent study on a Finnish population indicated that the ε4 carriers are prone to severe COVID-19 and prolonged mental fatigue, partially due to cerebrovascular damage [115]. The effect of APOE4 on the susceptibility to and the severity of COVID-19 may provide an explanation for the variations of severity of COVID-19 among different ethnic groups. The frequency of the ε4 allele is approximately 30%–40% in black Africans and approximately 7%–20% in Caucasians [116]. Studies have indicated that the burden and the mortality rate of COVID-19 are disproportionately higher in black persons/African Americans than in white persons [117, 118], and black individuals with dementia are more prone to contracting SARS-CoV-2 than white individuals with dementia [119]. In contrast, Gunanidhi D et al. [120] conducted a global epidemiological study on the correlation of apoE isoforms with COVID-19 morbidity and mortality and observed a significant but negative relationship between the ε4 allele and COVID-19 susceptibility. When the data were further analysed after stratification according to ethnicity, the researchers observed that the ε4 isoform protected against COVID-19 in Asians but not in Europeans, Africans or Americans.
Multiple molecular mechanisms may underlie the possible relationship of APOE polymorphisms with SARS-CoV-2 infection and COVID-19 severity, and some of the mechanisms remain speculative. Recently, an in vitro study revealed that the APOE4 genotype increases the rate of SARS-CoV-2 infection of human induced pluripotent stem cell (iPSC)-derived neurons and astrocytes, and that APOE4-expressing astrocytes produce a detrimental response compared with APOE3 carriers [26] (Fig. 3a). These results suggest that APOE4 may exert a causal effect on the severity of COVID-19. Another potential mechanism is that APOE4 is associated with increased BBB permeability [121], which allows the virus to enter the CNS more easily through BBB leakage and makes patients more susceptible to infection [122] (Fig. 3b). Moreover, APOE4 has been reported to promote production of proinflammatory cytokines (such as IL-6 and TNF-α) by macrophages in the CNS and the periphery in response to proinflammatory stimuli [123,124,125] (Fig. 3c). As the cytokine storm has been identified as a critical hallmark of severe COVID-19 [126], APOE4 may be responsible for the poor outcomes of patients with COVID-19 due to its effect on the host immune response. Additionally, compared to AD patients with the APOE ε3/ε3 genotype, those with the ε4/ε4 genotype exhibit decreased expression of several antiviral defence genes, such as IFITM2, IFITM3, IFNAR1 and LY6E [127] (Fig. 3d). Compared to the two other isoforms, APOE4 is genetically associated with reduced apoE levels, which increases the risk of coronavirus infection and disease progression, and this reduction is consistently associated with severe COVID-19 [128]. Notably, APOE4 may contribute to the infectivity of SARS-CoV-2 by regulating intracellular cholesterol levels. ApoE is an amphipathic protein that is responsible for regulating organismal lipid and cholesterol homeostasis [129]. Individuals carrying the ε4 allele exhibit elevated intracellular and circulating cholesterol levels [130], and a high cellular cholesterol level may facilitate SARS-CoV-2 infectivity by increasing the binding of the S protein to the ACE2 receptor [131, 132] (Fig. 3e). Moreover, a recent molecular docking study revealed that the receptor-binding domain of SARS-CoV-2 binds to the apoE protein, potentially leading to the exposure of the active site of apoE; thus, the virus may enter host cells via apoE-related metabolic pathways [133]. However, this interaction must be confirmed by experimental studies, and whether this binding effect depends on the apoE isoform remains to be determined. Additionally, the interactions between apoE and the ACE2 receptor should be investigated to determine the direct relationship between APOE and COVID-19. Pathologically, apoE has been shown to be coexpressed with ACE2 in alveolar epithelial cells [134], which are the primary targets of SARS-CoV-2, and treatment with rhACE2 or genetic ablation of ACE2 has been shown to modulate apoE-related physiological functions [135]. However, the correlation between apoE and ACE2 levels should be examined in the context of COVID-19. Additionally, APOE4 is a risk factor for several cardiovascular and neurological diseases, such as AD [102], atherosclerosis [136], and diabetes [137]. Individuals with these pre-existing comorbidities are more vulnerable to developing severe COVID-19 and having higher mortality rates following SARS-CoV-2 infection [138, 139] (Fig. 3f). Thus, although apoE may exert independent effects on the risk of infection and disease severity, these comorbidities should be considered.

APOE4 contributes to increased susceptibility to COVID-19. a APOE4 was shown to increase the infectivity of SARS-CoV-2 in iPSC-derived neurons and astrocytes. b APOE4 has been reported to be associated with increased BBB permeability. c ApoE4 is related to increased production of proinflammatory cytokines by macrophages in the periphery and by microglial cells in the CNS. d Compared to AD patients presenting the APOE ε3/ε3 genotype, AD patients with the ε4/ε4 genotype display decreased expression of several antiviral defence genes, such as IFITM2, IFITM3, IFNAR1 and LY6E. e Individuals carrying the ε4 allele exhibit elevated intracellular and circulating cholesterol levels, and a high level of cellular cholesterol may facilitate SARS-CoV-2 infection. f APOE4 is a risk factor for several cardiovascular and neurological diseases, and individuals with these pre-existing comorbidities are more vulnerable to infection and poor clinical outcomes. Thus, individuals with the APOE4 genotype are more susceptible to SARS-CoV-2 infection, which may lead to neurodegeneration
In summary, the inconsistency in the findings related to the association between APOE gene polymorphism and COVID-19 risk and severity may be due to the differences in the genetic characteristics and age of the included subjects, the case definitions and the number of subjects employed. Further research in a larger sample size and employing subjects with different ethnic backgrounds will help identify those who are at high risks of infection and severe disease, in order to help prioritize health care and preventive measures. In addition, the direct effects of apoE isoforms on COVID-19 severity and the related pathways require further investigations in patients, animals, and cells to better delineate the major underlying mechanisms, thus facilitating the development of targeted interventions.
Age: a primary common risk factor for COVID-19 and AD
Age is the greatest risk factor for AD [140], as the prevalence and mortality of AD increase substantially with age [141]. Approximately one in 10 people aged ≥ 65 years has AD, and the prevalence increases to 32% in those aged ≥ 85 years, among whom the per-year incidence rate is approximately 6.5% [142]. Age affects the pathology of AD in a number of ways, such as by causing dysregulation of innate immune responses, impairment of the BBB, and alterations in neuroinflammation [140]. Accumulating evidence has shown that elderly individuals, especially those with comorbidities such as chronic obstructive pulmonary disease, hypertension, cancers, and diabetes, are particularly vulnerable to SARS-CoV-2 infection and are more likely to develop severe illness [143,144,145]. Notably, a systematic study assessed the clinical, molecular, and immunological characteristics of 326 patients with COVID-19 from China and found that age, among other factors, is the most obviously associated with disease severity [146]. Early evidence from China indicated that the case-fatality ratio (CFR) of COVID-19 is less than 0.4% among patients aged 40 years and younger, 8% among patients aged 70–79 years and a startling 14.8% among those aged 80 years and older [147]. A consistent and more pronounced age-related trend in the CFR of COVID-19 has been reported in the Italian population: patients aged 40 years or younger have a CFR of less than 0.4%, whereas the CFR is markedly increased to 12.8% in patients aged 70–79 years and further increased to 20.2% in those aged ≥ 80 years [148]. In addition, the overall CFR in the Italian population (7.2%) was reported to be substantially higher than that observed in China (2.3%), possibly in part because that Italy has one of the highest percentages of elderly individuals worldwide, with an average life expectancy of 82.3 years and a proportion of 23% for individuals aged 65 years or older. Similar exponential increases in the CFR of COVID-19 with age have been reported in other countries. A meta-analysis of more than 600,000 patients with COVID-19 from the United Kingdom, China, Spain, Italy, and New York State reported that the mortality rate in patients aged 60–69 years was more than three times higher than that in those aged 50 to 59 years [149].
Several potential factors may account for the marked increase in the risk of SARS-CoV-2 infection and poor clinical outcomes in the elderly. First, age-related impairment and dysregulation of the host innate and adaptive immune systems may lead to decreased protection against virus infection and result in a state of chronic inflammation [150,151,152]. In addition, the levels of inflammatory markers, such as C-reactive protein (CRP), Toll-like receptor (TLR), TNF-α, IL-6, and IL-1β, generally increase with age [153, 154]. These changes result in the low-grade proinflammatory state observed in older populations [155], which may contribute to severe disease conditions. Specifically, increased serum CRP levels, which indicate an inflammatory state and are associated with advanced age, indicate a poor prognosis and increased risk of mortality in elderly patients with COVID-19 [156, 157]. Additionally, serum CRP levels have been used to monitor COVID-19 progression and patients’ response to COVID-19 treatments, such as tocilizumab [158]. Notably, loss of cerebrovascular integrity, a feature of ageing, may enable pathogens to enter the brains of older individuals [159]. Moreover, increases in the expression of ACE2, the major receptor for SARS-CoV-2, with age in several human tissues, such as the nasal epithelium [160], lung [145], and kidney [161], may increase the risk of S protein binding and the development of severe COVID-19 in older adults; however, this speculation remains controversial. Furthermore, intracellular cholesterol accumulates in type-2 pneumocytes and alveolar macrophages with ageing [162, 163]. As described above, a high cellular cholesterol level may facilitate SARS-CoV-2 infection by enhancing the S protein-ACE2 interaction [131, 132], consistent with the increase in poor COVID-19 outcomes in the aged population. Notably, age-related comorbidities, such as hypertension, cardiovascular disease, dementia, diabetes mellitus, kidney failure, cancer and metabolic syndrome, become more prevalent with ageing, and each of those pre-existing comorbidities increases the susceptibility to poor clinical outcomes after SARS-CoV-2 infection [164, 165]. In particular, memory impairment may affect the ability of elderly patients to comply with COVID-19 precautions, such as mask wearing, hand hygiene, and maintaining appropriate social distance. Together, these potential factors may individually or collectively render older individuals more vulnerable to coronavirus infection and lead to severe disease.
Age is clearly a major risk factor for COVID-19, possibly due to the age-related dysfunction of the immune system, inflammation and comorbidities. However, the molecular mechanisms of age-related increases in disease vulnerability and severity are currently poorly understood, which would be an important area of investigation.
Neuroinflammation: an important bridge between COVID-19 and AD
Neuroinflammation is recognized as another characteristic pathophysiology of AD [166]. Microglia and astrocytes are major sources of cytokines in individuals with AD [167]. Dysfunction of the immune system may promote the release of proinflammatory cytokines and result in synaptic damage, neuronal death, and inhibition of neurogenesis, which are related to the pathogenesis of AD [168]. Although accumulating evidence indicates that SARS-CoV-2 can enter the CNS, the presence of the virus in the brain may not critically correlate with the neurological conditions, as postmortem studies have noted occurrence of pronounced neuropathological changes even in patients with COVID-19 in whom the virus was not detected in the CNS [17, 19, 37, 169, 170]. Instead, extensive research on CSF and postmortem brain tissues has indicated that immune dysfunction and pronounced neuroinflammation within the CNS are the main driver of CNS damage and neurological symptoms in infected individuals. Neuroinflammation might result directly from coronavirus infection or from excessive peripheral inflammation. Both SARS-CoV-2 and proinflammatory mediators may promote BBB disruption, allowing the virus to cross the BBB, enter the CNS [171], and activate microglia and astrocytes, triggering a neuroinflammatory cascade that may contribute to the onset and progression of neurodegeneration [172,173,174] (Fig. 4).

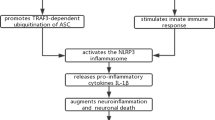
SARS-CoV-2 infection causes neurodegeneration by triggering neuroinflammation. SARS-CoV-2 infection causes activation of microglia and astrocytes, triggering a neuroinflammatory cascade. Activated microglia secrete inflammatory cytokines, which promote astrocyte activation and NLRP3 inflammasome formation. Binding of SARS-CoV-2 to ACE2 downregulates ACE2 expression, which may increase Ang II expression and subsequently activate AT1R in microglia, resulting in NF-κB expression and a proinflammatory response. On the other hand, downregulation of ACE2 activity is associated with decreased Ang (1–7) levels and subsequent MasR activity in astrocytes, which may lead to the production of ROS and proinflammatory factors in the CNS parenchyma and cerebral vessels. These processes result in Aβ deposition and tau phosphorylation, which ultimately lead to neurodegeneration and AD
Under physiological conditions, microglia act as resident phagocytes in the CNS parenchyma, which are critical for homeostasis, facilitate synaptogenesis, regulate synaptic pruning and myelination, and subsequently support neuronal survival [175, 176]. Under pathological conditions, microglia rapidly respond to CNS pathogens or excessive peripheral inflammation by expressing specific surface receptors, driving morphological changes, and producing proinflammatory molecules and reactive oxygen species (ROS), which exacerbate neuroinflammation [177]. Microglia are likely to be affected by SARS-CoV-2 entry into the brain, as NRP-1, an important receptor for the virus, has recently been shown to be expressed in microglia [54]. Several previous studies have suggested that the S protein of SARS-CoV-2 might function as a pathogen-associated molecular pattern (PAMP) to drive neuroinflammatory responses through activation of TLRs, which are widely expressed on macrophages and microglia and may function as pattern recognition receptors (PRRs) to recognize molecular sequences common to bacterial and viral pathogens [178,179,180]. Microglia have been shown to express a wide range of PRRs, including TLRs, NOD-like receptors, RAGE and scavenger receptors [181]. SARS-CoV-2 activates these receptors, possibly eliciting neuroinflammatory responses and contributing to disease progression and severity. Widespread microglial activation, which may lead to activation of lymphocytes and T cells [17], has been observed in brains of the majority of patients with COVID-19 [182]. SARS-CoV-2 infection-induced activation of microglia may increase the expression of TNF-α, IL-6, IL-1β, nitric oxide, and chemokine C–C motif ligand 2, which may further increase the AD-related tau pathology [183, 184]. In addition to being potential targets of coronaviruses, microglia are also highly susceptible to the effects of proinflammatory mediators. Studies have shown that microglial activation, microglial nodules and neuronophagia are present in postmortem brain tissues of the majority of patients with COVID-19 and that these changes are not directly due to SARS-CoV-2 infection but likely result from systemic inflammation [185], as the viral RNA or protein is not detected in the microglial nodules or neuronophagic cells.
Astrocytes are also important components of the intrinsic immune response in the CNS following viral infection. An increased plasma concentration of glial fibrillary acidic protein (GFAP), a hallmark of astrocyte activation, is commonly detected in patients with moderate and severe COVID-19 [186]. As components of the BBB, astrocytes are highly sensitive to peripheral inflammation. In addition to responding to signals from microglia, astrocytes also rapidly respond to proinflammatory cytokines secreted by endothelial cells. Astrocytes express inflammatory molecules that contribute to neuroinflammation and neurodegeneration upon exposure to activated proinflammatory microglia, including proinflammatory cytokines such as IL-6, IL-1β and TNF-α; PAMPs; and danger-associated molecular patterns [187]. These data clearly indicate that microglia and astrocytes participate in neuroinflammatory responses to viral infection of the CNS.
In addition, the systemic inflammatory response initiated by SARS-CoV-2 infection is partially mediated by hyperactivation of the NLRP3 inflammasome [188, 189]. Activation of the NLRP3 inflammasome decreases the phagocytosis of Aβ by microglia, thereby increasing the deposition of Aβ and subsequently facilitating the onset and development of AD pathology [190]. Hyperactivity of the intracellular NLRP3 inflammasome facilitates the polarization of microglia towards the M1 phenotype, resulting in Aβ deposition and increased cognitive deficits in transgenic AD mice [191]. Conversely, deficiency of the NLRP3 inflammasome results in the polarization of microglia towards the M2 phenotype, thus reducing the formation of Aβ plaques, inhibiting synaptic injury, and mitigating cognitive decline [192]. Activation of the NLRP3 inflammasome has also been reported to promote tau pathology, which promotes the occurrence and progression of AD [193]. Furthermore, hyperactivation of P2X7 receptors has been reported to be closely associated with the inflammatory process, as P2X7 receptors are stimulated by adenosine triphosphate (ATP) released from damaged cells, inducing inflammasome activation [194]. The increase of extracellular ATP level caused by SARS-CoV-2 infection may trigger hyperactivation of the P2X7 receptor, leading to stimulation of the NLRP3 inflammasome [195]. In this context, the P2X7 receptor may be an appealing target for prevention or treatment of neurological manifestations of COVID-19. In addition, comorbidities, such as obesity, heart disease, diabetes and hypertension, which are related to poor outcomes, are associated with pronounced inflammation in patients with COVID-19 [196].
Additionally, downregulation of ACE2 expression caused by binding of the SARS-CoV-2 S protein promotes cellular and tissue damage, possibly further aggravating neuroinflammation, oxidative stress and cerebrovascular endothelial injury. Oxidative stress and cyclooxygenase (COX)-1- or COX-2-mediated neuroinflammation are substantially inhibited in transgenic mice with neuron-specific overexpression of ACE2, resulting in improvement of the antioxidant status and nitric oxide homeostasis [197]. Therefore, pharmacological activation of ACE2 may exert beneficial effects on the neuropathological sequelae of SARS-CoV-2 infection [198,199,200]. Moreover, neuroinflammation might result from dysregulation of the RAS in the CNS. As described above, SARS-CoV-2 binding downregulates ACE2 expression, which may increase the expression of Ang II and subsequently activate AT1R in microglia, resulting in promotion of NF-κB expression and proinflammatory responses. On the other hand, decreased ACE2 activity is associated with decreases of Ang (1–7) level and MasR activity in astrocytes, which may lead to the production of ROS and proinflammatory factors in the parenchyma and cerebral vessels of the CNS. Neurodegeneration may subsequently occur. Furthermore, abnormalities in the levels of CSF biomarkers, including increased level of inflammatory mediators, have been observed in patients with COVID-19, suggesting that the increased cytokine and chemokine levels in the CSF following virus infection may contribute to neuroinflammation [201,202,203].
In addition to being a major contributor to cardiovascular diseases, hypertension has been demonstrated as a risk factor for AD [204]. Ang II receptor blockers (ARBs) are commonly used antihypertensive medications, and they have high affinity for AT1R, antagonizing the interaction between Ang II and its AT1R receptor. Regarding the effects of ARBs on AD, both in vitro and in vivo evidence has shown that treatment with ARBs can ameliorate most of the clinical risk factors of AD [205]. Although several studies failed to show restoration of spatial memory and learning deficiencies by the treatment [206, 207], an increasing number of studies reported that treatment with ARBs, such as losartan, candesartan, telmisartan, olmesartan or valsartan, is beneficial for memory and cognition in vivo [208,209,210]. In addition, administration of valsartan, losartan or telmisartan is capable of lessening the Aβ plaque burden [211, 212]. Evidence regarding the effect of ARBs on phosphorylated tau, another important hallmark of AD, is scarce. It seems that telmisartan is able to decrease the hippocampal content of hyperphosphorylated tau protein and neurofibrillary tangles [213, 214]. In addition to the aforementioned Aβ and tau, ARBs can also reduce neuroinflammation [206, 215]. Notably, a recent study investigated the effects of ARBs and ACE inhibitors (ACEIs) that are prescribed to treat COVID-19 in patients with AD or mild cognitive impairment (MCI). Although there was no significant relationship between ARB or ACEI use and the severity of COVID-19 among AD and MCI patients, the authors found that the ARBs were associated with a reduced risk for COVID-19 [216].
In conclusion, neuroinflammation is caused both directly by SARS-CoV-2 infection of the CNS and indirectly by peripheral inflammation via immune-to-brain signalling. However, further research assessing the driver of the major pathological processes, the duration of neuroinflammation after SARS-CoV-2 infection, and whether this neuroinflammation exerts long-term effects on the nervous system is warranted.
Other potential factors that mediate the association between COVID-19 and AD
In addition to the connections described above, other factors responsible for the relationship between AD and COVID-19 are being explored. According to a recent study, Aβ42 increases SARS-CoV-2 pseudovirus infection by strengthening the binding of the S1 subunit to the ACE2 receptor, suggesting that Aβ42 may play an important role in the development of severe COVID-19 [217]. Researchers also concluded that SARS-CoV-2 infection aggravates AD by exacerbating neurotoxicity and increasing the levels of Aβ, inflammation and oxidative stress [218, 219]. In addition, SARS-CoV-2-induced neuronal damage in 3D human brain organoids may be attributed to aberrant changes in the distribution and phosphorylation of tau [220]. Consistent with this study, SARS-CoV-2 infection activates biochemical pathways associated with tau pathology, which is one of the major factors that drive AD pathology [221]. Moreover, cognitive impairment and neuropsychiatric symptoms make it difficult for AD patients to understand and follow safety measures [14]. All these factors reinforce the potential link between COVID-19 and AD.
Conclusions and perspectives
The ongoing global COVID-19 pandemic caused by SARS-CoV-2 has extensively altered our daily lives. Infection is not only restricted to the respiratory system but also affects the CNS and leads to neurological manifestations. The morbidity and mortality rates of COVID-19 are increased in patients with AD. The mechanism by which the virus gains access to the CNS and why patients with AD are at higher risk of virus infection are not well understood. In this review, we summarize the current literature on the possible routes by which SARS-CoV-2 invade the CNS and further analyse the shared aetiological cofactors and potential mechanisms linking COVID-19 and AD, which may advance our understanding of the inherent relationship between the two diseases. Although people infected with SARS-CoV-2 frequently display neurological symptoms, multiple manifestations remain unrecognized. First, neurological symptomatology is not always present, and neurological symptoms are often ignored because the first goal is to combat respiratory symptoms. In addition, clarifying the mechanisms by which SARS-CoV-2 invades and spreads throughout the CNS is important for preventing and ameliorating neurological symptoms; however, the precise mechanism by which SARS-CoV-2 invades the CNS has not been completely elucidated. Moreover, researchers have not clearly determined whether the neuropathological alterations are directly due to the neuroinvasion by the virus or caused indirectly by the dysregulation of the immune system following infection. Emerging studies are focusing on the effect of the coronavirus on the onset and progression of AD. However, as it has not been long since the onset of the COVID-19 pandemic, whether SARS-CoV-2 infection contributes to long-term cognitive dysfunction and behavioural impairments in AD patients and triggers AD in infected individuals remain central questions. Therefore, long-term follow-up cohort studies designed to determine the long-term effects of COVID-19 on AD are urgently needed. Additionally, the relationships of AD-related pathological markers, risk factors and behavioural changes with COVID-19 must be further established. Moreover, as the COVID-19 pandemic continues, new SARS-CoV-2 mutants (e.g., delta and omicron strains) will continue to emerge, and the ability of these mutants to infect the CNS and their effect on AD must be elucidated. In summary, studies are imperatively needed to clarify the potential mechanisms underlying the elevated susceptibility and mortality rate of AD patients to COVID-19 and discover preventive strategies to minimize the risk of viral infection among patients with AD.
Availability of data and materials
Not applicable.
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- ACE2:
-
Angiotensin-converting enzyme 2
- AD:
-
Alzheimer’s disease
- CNS:
-
Central nervous system
- APOE:
-
Apolipoprotein E
- TMPRSS:
-
Transmembrane serine protease 2
- CSF:
-
Cerebrospinal fluid
- BBB:
-
Blood–brain barrier
- Aβ:
-
Amyloid-beta
- NRP-1:
-
Neuropilin-1
- RAS:
-
Renin-angiotensin system
- Ang:
-
Angiotensin
- CFR:
-
Case-fatality ratio
- CRP:
-
C-reactive protein
- TLR:
-
Toll-like receptor
- ROS:
-
Reactive oxygen species
- PAMPs:
-
Pathogen-associated molecular patterns
- PRRs:
-
Pattern recognition receptors
- GFAP:
-
Glial fibrillary acidic protein
- COX:
-
Cyclooxygenase
- ARBs:
-
Angiotensin II receptor blockers
- ACEIs:
-
ACE inhibitors (ACEIs)
- MCI:
-
Mild cognitive impairment
References
Liu Y, Mao B, Liang S, Yang JW, Lu HW, Chai YH, et al. Association between age and clinical characteristics and outcomes of COVID-19. Eur Respir J. 2020;55(5).
Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19(3):141–54.
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894-904 e899.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281-292 e286.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506.
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):819–24.
Bertolini A, van de Peppel IP, Bodewes F, Moshage H, Fantin A, Farinati F, et al. Abnormal liver function tests in patients with COVID-19: relevance and potential pathogenesis. Hepatology. 2020;72(5):1864–72.
Barros Camargo L, Quintero Marzola ID, Cardenas Gomez JC, Mendoza Daza LT, Quintana Pajaro L. Acute kidney injury associated with COVID-19: another extrapulmonary manifestation. Int Urol Nephrol. 2020;52(7):1403–4.
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol. 2020;19(9):767–83.
Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020;16(7): e9610.
Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dube M, et al. Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses. 2019;12(1).
Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. 2021;218(3).
Iadecola C, Anrather J, Kamel H. Effects of COVID-19 on the nervous system. Cell. 2020;183(1):16-27 e11.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan. China JAMA Neurol. 2020;77(6):683–90.
Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry. 2020;7(10):875–82.
Kumari P, Rothan HA, Natekar JP, Stone S, Pathak H, Strate PG, et al. Neuroinvasion and encephalitis following intranasal inoculation of SARS-CoV-2 in K18-hACE2 mice. Viruses. 2021;13(1):132.
Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 2020;19(11):919–29.
Solomon IH, Normandin E, Bhattacharyya S, Mukerji SS, Keller K, Ali AS, et al. Neuropathological features of Covid-19. N Engl J Med. 2020;383(10):989–92.
Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020;383(6):590–2.
Jarius S, Pache F, Kortvelyessy P, Jelcic I, Stettner M, Franciotta D, et al. Cerebrospinal fluid findings in COVID-19: a multicenter study of 150 lumbar punctures in 127 patients. J Neuroinflammation. 2022;19(1):19.
Bellon M, Schweblin C, Lambeng N, Cherpillod P, Vazquez J, Lalive PH, et al. Cerebrospinal fluid features in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin Infect Dis. 2021;73(9):e3102–5.
Coolen T, Lolli V, Sadeghi N, Rovai A, Trotta N, Taccone FS, et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology. 2020;95(14):e2016–27.
Chougar L, Shor N, Weiss N, Galanaud D, Leclercq D, Mathon B, et al. Retrospective observational study of brain MRI findings in patients with acute SARS-CoV-2 infection and neurologic manifestations. Radiology. 2020;297(3):E313–23.
Douaud G, Lee S, Alfaro-Almagro F, Arthofer C, Wang C, McCarthy P, et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature. 2022. https://doi.org/10.1038/s41586-022-04569-5.
Swain O, Romano SK, Miryala R, Tsai J, Parikh V, Umanah GKE. SARS-CoV-2 neuronal invasion and complications: potential mechanisms and therapeutic approaches. J Neurosci. 2021;41(25):5338–49.
Wang C, Zhang M, Garcia G Jr, Tian E, Cui Q, Chen X, et al. ApoE-isoform-dependent SARS-CoV-2 neurotropism and cellular response. Cell Stem Cell. 2021;28(2):331-342 e335.
Atkins JL, Masoli JAH, Delgado J, Pilling LC, Kuo CL, Kuchel GA, et al. Preexisting comorbidities predicting COVID-19 and mortality in the UK Biobank Community Cohort. J Gerontol A Biol Sci Med Sci. 2020;75(11):2224–30.
Numbers K, Brodaty H. The effects of the COVID-19 pandemic on people with dementia. Nat Rev Neurol. 2021;17(2):69–70.
Bianchetti A, Rozzini R, Guerini F, Boffelli S, Ranieri P, Minelli G, et al. Clinical presentation of COVID19 in dementia patients. J Nutr Health Aging. 2020;24(6):560–2.
Degenhardt EK, Witte MM, Case MG, Yu P, Henley DB, Hochstetler HM, et al. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics. 2016;57(2):208–16.
Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32.
Mok VCT, Pendlebury S, Wong A, Alladi S, Au L, Bath PM, et al. Tackling challenges in care of Alzheimer’s disease and other dementias amid the COVID-19 pandemic, now and in the future. Alzheimers Dement. 2020;16(11):1571–81.
Rahman MA, Islam K, Rahman S, Alamin M. Neurobiochemical cross-talk between COVID-19 and Alzheimer’s disease. Mol Neurobiol. 2021;58(3):1017–23.
Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995–8.
Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature. 2020;583(7818):834–8.
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2021;24(2):168–75.
Chow YH, O’Brodovich H, Plumb J, Wen Y, Sohn KJ, Lu Z, et al. Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways. Proc Natl Acad Sci U S A. 1997;94(26):14695–700.
Golden JW, Li R, Cline CR, Zeng X, Mucker EM, Fuentes-Lao AJ, et al. Hamsters expressing human angiotensin-converting enzyme 2 develop severe disease following exposure to SARS-CoV-2. mBio. 2022. https://doi.org/10.1128/mbio.02906-21
Golden JW, Cline CR, Zeng X, Garrison AR, Carey BD, Mucker EM, et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight. 2020;5(19).
Jiao L, Yang Y, Yu W, Zhao Y, Long H, Gao J, et al. The olfactory route is a potential way for SARS-CoV-2 to invade the central nervous system of rhesus monkeys. Signal Transduct Target Ther. 2021;6(1):169.
Butowt R, Bilinska K. SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem Neurosci. 2020;11(9):1200–3.
McCray PB Jr, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 2007;81(2):813–21.
Jacomy H, Talbot PJ. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology. 2003;315(1):20–33.
Li K, Wohlford-Lenane C, Perlman S, Zhao J, Jewell AK, Reznikov LR, et al. Middle East respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect Dis. 2016;213(5):712–22.
Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 2020;1866(10): 165878.
Silva MME, Mercer PBS, Witt MCZ, Pessoa RR. Olfactory dysfunction in Alzheimer’s disease systematic review and meta-analysis. Dement Neuropsychol. 2018;12(2):123–32.
Zapiec B, Dieriks BV, Tan S, Faull RLM, Mombaerts P, Curtis MA. A ventral glomerular deficit in Parkinson’s disease revealed by whole olfactory bulb reconstruction. Brain. 2017;140(10):2722–36.
Sanjari Moghaddam H, Dolatshahi M, Salardini E, Aarabi MH. Association of olfaction dysfunction with brain microstructure in prodromal Parkinson disease. Neurol Sci. 2019;40(2):283–91.
Butowt R, Meunier N, Bryche B, von Bartheld CS. The olfactory nerve is not a likely route to brain infection in COVID-19: a critical review of data from humans and animal models. Acta Neuropathol. 2021;141(6):809–22.
Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. 2020;6(31).
Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R. Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci. 2020;11(11):1555–62.
Barnett EM, Cassell MD, Perlman S. Two neurotropic viruses, herpes simplex virus type 1 and mouse hepatitis virus, spread along different neural pathways from the main olfactory bulb. Neuroscience. 1993;57(4):1007–25.
Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–60.
Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Anton-Plagaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–5.
Davies J, Randeva HS, Chatha K, Hall M, Spandidos DA, Karteris E, et al. Neuropilin1 as a new potential SARSCoV2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID19. Mol Med Rep. 2020;22(5):4221–6.
Wan D, Du T, Hong W, Chen L, Que H, Lu S, et al. Neurological complications and infection mechanism of SARS-COV-2. Signal Transduct Target Ther. 2021;6(1):406.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 2020;382(23):2268–70.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1–13.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7.
Wang Y, Cao Y, Mangalam AK, Guo Y, LaFrance-Corey RG, Gamez JD, et al. Neuropilin-1 modulates interferon-gamma-stimulated signaling in brain microvascular endothelial cells. J Cell Sci. 2016;129(20):3911–21.
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol. 2020;92(7):699–702.
Buzhdygan TP, DeOre BJ, Baldwin-Leclair A, Bullock TA, McGary HM, Khan JA, et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol Dis. 2020;146: 105131.
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv Q, et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther. 2021;6(1):337.
Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol. 2020;77(8):1018–27.
Spindler KR, Hsu TH. Viral disruption of the blood-brain barrier. Trends Microbiol. 2012;20(6):282–90.
Guadarrama-Ortiz P, Choreno-Parra JA, Sanchez-Martinez CM, Pacheco-Sanchez FJ, Rodriguez-Nava AI, Garcia-Quintero G. Neurological aspects of SARS-CoV-2 infection: mechanisms and manifestations. Front Neurol. 2020;11:1039.
Pennisi M, Lanza G, Falzone L, Fisicaro F, Ferri R, Bella R. SARS-CoV-2 and the nervous system: from clinical features to molecular mechanisms. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21155475.
Alenquer M, Amorim MJ. Exosome biogenesis, regulation, and function in viral infection. Viruses. 2015;7(9):5066–83.
Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell. 2020;27(6):951-961 e955.
Miners JS, Kehoe PG, Love S, Zetterberg H, Blennow K. CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther. 2019;11(1):81.
Asby D, Boche D, Allan S, Love S, Miners JS. Systemic infection exacerbates cerebrovascular dysfunction in Alzheimer’s disease. Brain. 2021;144(6):1869–83.
Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136(4):507–23.
Valletta M, Canevelli M, D’Antonio F, Trebbastoni A, Talarico G, Campanelli A, et al. Prevalence and safety of COVID-19 vaccination in community-dwelling people with dementia: findings from a tertiary memory clinic in Italy. J Alzheimers Dis. 2022;87(4):1467–74.
Wang L, Davis PB, Kaelber DC, Xu R. COVID-19 breakthrough infections and hospitalizations among vaccinated patients with dementia in the United States between December 2020 and August 2021. Alzheimers Dement. 2022. https://doi.org/10.1002/alz.12669.
Patone M, Handunnetthi L, Saatci D, Pan J, Katikireddi SV, Razvi S, et al. Neurological complications after first dose of COVID-19 vaccines and SARS-CoV-2 infection. Nat Med. 2021;27(12):2144–53.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–4.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/Angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018;98(1):505–53.
Gupta D, Kumar A, Mandloi A, Shenoy V. Renin angiotensin aldosterone system in pulmonary fibrosis: Pathogenesis to therapeutic possibilities. Pharmacol Res. 2021;174: 105924.
Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, Prindle T, Fulton WB, Wang S, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L17–31.
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016-1035 e1019.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271-280 e278.
Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;39(10): e105114.
Gallagher PE, Chappell MC, Ferrario CM, Tallant EA. Distinct roles for ANG II and ANG-(1–7) in the regulation of angiotensin-converting enzyme 2 in rat astrocytes. Am J Physiol Cell Physiol. 2006;290(2):C420-426.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R373-381.
Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107(6):1482–94.
Ding Q, Shults NV, Gychka SG, Harris BT, Suzuki YJ. Protein expression of angiotensin-converting enzyme 2 (ACE2) is upregulated in brains with Alzheimer’s disease. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22041687.
Zhao Y, Li W, Lukiw W. Ubiquity of the SARS-CoV-2 receptor ACE2 and upregulation in limbic regions of Alzheimer’s disease brain. Folia Neuropathol. 2021;59(3):232–8.
Lim KH, Yang S, Kim SH, Joo JY. Elevation of ACE2 as a SARS-CoV-2 entry receptor gene expression in Alzheimer’s disease. J Infect. 2020;81(3):e33–4.
Cui H, Su S, Cao Y, Ma C, Qiu W. The altered anatomical distribution of ACE2 in the brain with Alzheimer’s disease pathology. Front Cell Dev Biol. 2021;9: 684874.
Kehoe PG, Wong S, Al Mulhim N, Palmer LE, Miners JS. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimers Res Ther. 2016;8(1):50.
Evans CE, Miners JS, Piva G, Willis CL, Heard DM, Kidd EJ, et al. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020;139(3):485–502.
Liu S, Liu J, Miura Y, Tanabe C, Maeda T, Terayama Y, et al. Conversion of Abeta43 to Abeta40 by the successive action of angiotensin-converting enzyme 2 and angiotensin-converting enzyme. J Neurosci Res. 2014;92(9):1178–86.
Kamel AS, Abdelkader NF, Abd El-Rahman SS, Emara M, Zaki HF, Khattab MM. Stimulation of ACE2/ANG(1–7)/Mas axis by diminazene ameliorates Alzheimer’s disease in the D-galactose-ovariectomized rat model: role of PI3K/Akt pathway. Mol Neurobiol. 2018;55(10):8188–202.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90.
Rocha NP, Toledo A, Corgosinho LTS, de Souza LC, Guimaraes HC, Resende EPF, et al. Cerebrospinal fluid levels of angiotensin-converting enzyme are associated with amyloid-beta42 burden in Alzheimer’s disease. J Alzheimers Dis. 2018;64(4):1085–90.
Jiang T, Zhang YD, Zhou JS, Zhu XC, Tian YY, Zhao HD, et al. Angiotensin-(1–7) is reduced and inversely correlates with Tau hyperphosphorylation in animal models of Alzheimer’s disease. Mol Neurobiol. 2016;53(4):2489–97.
Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–8.
Velavan TP, Pallerla SR, Ruter J, Augustin Y, Kremsner PG, Krishna S, et al. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine. 2021;72: 103629.
Jackson RJ, Meltzer JC, Nguyen H, Commins C, Bennett RE, Hudry E, et al. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain. 2021. https://doi.org/10.1093/brain/awab478.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501–18.
Chen Y, Strickland MR, Soranno A, Holtzman DM. Apolipoprotein E: structural insights and links to Alzheimer disease pathogenesis. Neuron. 2021;109(2):205–21.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3.
Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–52.
Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20(1):68–80.
Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A. 2008;105(25):8718–23.
Linard M, Letenneur L, Garrigue I, Doize A, Dartigues JF, Helmer C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimers Dement. 2020;16(1):200–8.
Mueller T, Fischer J, Gessner R, Rosendahl J, Bohm S, van Bommel F, et al. Apolipoprotein E allele frequencies in chronic and self-limited hepatitis C suggest a protective effect of APOE4 in the course of hepatitis C virus infection. Liver Int. 2016;36(9):1267–74.
Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, et al. APOE e4 genotype predicts severe COVID-19 in the UK biobank community cohort. J Gerontol A Biol Sci Med Sci. 2020;75(11):2231–2.
Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, et al. ApoE e4e4 genotype and mortality With COVID-19 in UK Biobank. J Gerontol A Biol Sci Med Sci. 2020;75(9):1801–3.
Del Ser T, Fernandez-Blazquez MA, Valenti M, Zea-Sevilla MA, Frades B, Alfayate E, et al. Residence, clinical features, and genetic risk factors associated with symptoms of COVID-19 in a cohort of older people in Madrid. Gerontology. 2021;67(3):281–9.
Al-Jaf SMA, Niranji SS, Ali HN, Mohammed OA. Association of apolipoprotein e polymorphism with SARS-CoV-2 infection. Infect Genet Evol. 2021;95: 105043.
Hubacek JA, Dlouha L, Dusek L, Majek O, Adamkova V. Apolipoprotein E4 allele in subjects with COVID-19. Gerontology. 2021;67(3):320–2.
Kurki SN, Kantonen J, Kaivola K, Hokkanen L, Mayranpaa MI, Puttonen H, et al. APOE epsilon4 associates with increased risk of severe COVID-19, cerebral microhaemorrhages and post-COVID mental fatigue: a finnish biobank, autopsy and clinical study. Acta Neuropathol Commun. 2021;9(1):199.
Abondio P, Sazzini M, Garagnani P, Boattini A, Monti D, Franceschi C, et al. The genetic variability of APOE in different human populations and its implications for longevity. Genes. 2019. https://doi.org/10.3390/genes10030222.
Holmes L Jr, Enwere M, Williams J, Ogundele B, Chavan P, Piccoli T, et al. Black-White risk differentials in COVID-19 (SARS-COV2) transmission, mortality and case fatality in the United States: Translational epidemiologic perspective and challenges. Int J Environ Res Public Health. 2020. https://doi.org/10.3390/ijerph17124322.
Yancy CW. COVID-19 and African Americans. JAMA. 2020;323(19):1891–2.
Wang Q, Davis PB, Gurney ME, Xu R. COVID-19 and dementia: Analyses of risk, disparity, and outcomes from electronic health records in the US. Alzheimers Dement. 2021;17(8):1297–306.
Dhangadamajhi G, Mishra S, Mukherjee P. Association of ApoE isoforms with COVID-19 outcomes: a world-wide epidemiological study. Hum Cell. 2021;34(6):1932–3.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Burks SM, Rosas-Hernandez H, Alejandro Ramirez-Lee M, Cuevas E, Talpos JC. Can SARS-CoV-2 infect the central nervous system via the olfactory bulb or the blood-brain barrier? Brain Behav Immun. 2021;95:7–14.
Zhang H, Wu LM, Wu J. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. 2011;2011: 949072.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Konttinen H, Cabral-da-Silva MEC, Ohtonen S, Wojciechowski S, Shakirzyanova A, Caligola S, et al. PSEN1DeltaE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-derived microglia. Stem Cell Reports. 2019;13(4):669–83.
Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473–4.
Zhou Y, Xu J, Hou Y, Leverenz JB, Kallianpur A, Mehra R, et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimers Res Ther. 2021;13(1):110.
Xiong N, Schiller MR, Li J, Chen X, Lin Z. Severe COVID-19 in Alzheimer’s disease: APOE4’s fault again? Alzheimers Res Ther. 2021;13(1):111.
Huebbe P, Rimbach G. Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Age Res Rev. 2017;37:146–61.
Dallongeville J, Lussier-Cacan S, Davignon J. Modulation of plasma triglyceride levels by apoE phenotype: a meta-analysis. J Lipid Res. 1992;33(4):447–54.
Wang H, Yuan Z, Pavel MA, Jablonski SM, Jablonski J, Hobson R, et al. The role of high cholesterol in age-related COVID19 lethality. bioRxiv. 2021.
Zhu H, Zheng F, Li L, Jin Y, Luo Y, Li Z, et al. A Chinese host genetic study discovered IFNs and causality of laboratory traits on COVID-19 severity. iScience. 2021;24(10):103186.
Yin YW, Sheng YJ, Wang M, Ma YQ, Ding HM. Interaction of serum proteins with SARS-CoV-2 RBD. Nanoscale. 2021;13(30):12865–73.
Gkouskou K, Vasilogiannakopoulou T, Andreakos E, Davanos N, Gazouli M, Sanoudou D, et al. COVID-19 enters the expanding network of apolipoprotein E4-related pathologies. Redox Biol. 2021;41: 101938.
Jin HY, Chen LJ, Zhang ZZ, Xu YL, Song B, Xu R, et al. Deletion of angiotensin-converting enzyme 2 exacerbates renal inflammation and injury in apolipoprotein E-deficient mice through modulation of the nephrin and TNF-alpha-TNFRSF1A signaling. J Transl Med. 2015;13:255.
Johnson LA, Arbones-Mainar JM, Fox RG, Pendse AA, Altenburg MK, Kim HS, et al. Apolipoprotein E4 exaggerates diabetic dyslipidemia and atherosclerosis in mice lacking the LDL receptor. Diabetes. 2011;60(9):2285–94.
El-Lebedy D, Raslan HM, Mohammed AM. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2016;15:12.
Finch CE, Kulminski AM. The ApoE locus and COVID-19: are we going where we have been? J Gerontol A Biol Sci Med Sci. 2021;76(2):e1–3.
Nandy K, Salunke A, Pathak SK, Pandey A, Doctor C, Puj K, et al. Coronavirus disease (COVID-19): a systematic review and meta-analysis to evaluate the impact of various comorbidities on serious events. Diabetes Metab Syndr. 2020;14(5):1017–25.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–81.
Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020.
Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: the Baltimore longitudinal study of aging. Neurology. 2000;54(11):2072–7.
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–5.
Liu K, Chen Y, Lin R, Han K. Clinical features of COVID-19 in elderly patients: A comparison with young and middle-aged patients. J Infect. 2020;80(6):e14–8.
Santesmasses D, Castro JP, Zenin AA, Shindyapina AV, Gerashchenko MV, Zhang B, et al. COVID-19 is an emergent disease of aging. Aging Cell. 2020;19(10): e13230.
Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583(7816):437–40.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323(13):1239–42.
Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. 2020;323(18):1775–6.
Bonanad C, Garcia-Blas S, Tarazona-Santabalbina F, Sanchis J, Bertomeu-Gonzalez V, Facila L, et al. The effect of age on mortality in patients with COVID-19: A meta-analysis with 611,583 subjects. J Am Med Dir Assoc. 2020;21(7):915–8.
Chen Y, Klein SL, Garibaldi BT, Li H, Wu C, Osevala NM, et al. Aging in COVID-19: vulnerability, immunity and intervention. Ageing Res Rev. 2021;65: 101205.
Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13(12):875–87.
Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183(4):996-1012 e1019.
Weatherhead JE, Clark E, Vogel TP, Atmar RL, Kulkarni PA. Inflammatory syndromes associated with SARS-CoV-2 infection: dysregulation of the immune response across the age spectrum. J Clin Invest. 2020;130(12):6194–7.
Cartier A, Cote M, Lemieux I, Perusse L, Tremblay A, Bouchard C, et al. Age-related differences in inflammatory markers in men: contribution of visceral adiposity. Metabolism. 2009;58(10):1452–8.
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
Dugue PA, Hodge AM, Ulvik A, Ueland PM, Midttun O, Rinaldi S, et al. Association of markers of inflammation, the kynurenine pathway and B vitamins with age and mortality, and a signature of inflammaging. J Gerontol A Biol Sci Med Sci. 2022;77(4):826–36.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13.
Conrozier T, Lohse A, Balblanc JC, Dussert P, Royer PY, Bossert M, et al. Biomarker variation in patients successfully treated with tocilizumab for severe coronavirus disease 2019 (COVID-19): results of a multidisciplinary collaboration. Clin Exp Rheumatol. 2020;38(4):742–7.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302.
Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA. 2020;323(23):2427–9.
Salah HM, Arthur JM, Mehta JL. Implications of renal ACE2 expression in the age of COVID-19. Eur Heart J. 2020;41(48):4589–91.
Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. 2019;10(1):963.
Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17(4):549–61.
Zhou Y, Yang Q, Chi J, Dong B, Lv W, Shen L, et al. Comorbidities and the risk of severe or fatal outcomes associated with coronavirus disease 2019: a systematic review and meta-analysis. Int J Infect Dis. 2020;99:47–56.
Holder K, Reddy PH. The COVID-19 effect on the immune system and mitochondrial dynamics in diabetes, obesity, and dementia. Neuroscientist. 2021;27(4):331–9.
Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18(12):759–72.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–72.
Solomon T. Neurological infection with SARS-CoV-2-the story so far. Nat Rev Neurol. 2021;17(2):65–6.
Schaller T, Hirschbuhl K, Burkhardt K, Braun G, Trepel M, Markl B, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518–20.
Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP, et al. Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front Cell Neurosci. 2017;11:216.
Abate G, Memo M, Uberti D. Impact of COVID-19 on Alzheimer’s disease risk: viewpoint for research action. Healthcare. 2020. https://doi.org/10.3390/healthcare8030286.
Wang H, Tang X, Fan H, Luo Y, Song Y, Xu Y, et al. Potential mechanisms of hemorrhagic stroke in elderly COVID-19 patients. Aging. 2020;12(11):10022–34.
Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–43.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–8.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–51.
Chen Z, Zhong D, Li G. The role of microglia in viral encephalitis: a review. J Neuroinflammation. 2019;16(1):76.
Frank MG, Nguyen KH, Ball JB, Hopkins S, Kelley T, Baratta MV, et al. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav Immun. 2022;100:267–77.
Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021;31(7):818–20.
Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829–38.
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3(10):136.
Poloni TE, Medici V, Moretti M, Visona SD, Cirrincione A, Carlos AF, et al. COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol. 2021;31(5): e12997.
Olajide OA, Iwuanyanwu VU, Adegbola OD, Al-Hindawi AA. SARS-CoV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol Neurobiol. 2022;59(1):445–58.
Joly-Amado A, Hunter J, Quadri Z, Zamudio F, Rocha-Rangel PV, Chan D, et al. CCL2 overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model of tauopathy. Front Immunol. 2020;11:997.
Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK, et al. COVID-19 neuropathology at Columbia university irving medical center/New York Presbyterian hospital. Brain. 2021;144(9):2696–708.
Kanberg N, Ashton NJ, Andersson LM, Yilmaz A, Lindh M, Nilsson S, et al. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology. 2020;95(12):e1754–9.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al (2021) Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 218(3).
Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. 2022;606(7914):585–93.
Luciunaite A, McManus RM, Jankunec M, Racz I, Dansokho C, Dalgediene I, et al. Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem. 2020;155(6):650–61.
Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, et al. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019;38(17): e101064.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–73.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in infection and inflammation. Immunity. 2017;47(1):15–31.
Ribeiro DE, Oliveira-Giacomelli A, Glaser T, Arnaud-Sampaio VF, Andrejew R, Dieckmann L, et al. Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol Psychiatry. 2021;26(4):1044–59.
Aschman T, Schneider J, Greuel S, Meinhardt J, Streit S, Goebel HH, et al. Association between SARS-CoV-2 infection and immune-mediated myopathy in patients who have died. JAMA Neurol. 2021;78(8):948–60.
Sriramula S, Xia H, Xu P, Lazartigues E. Brain-targeted angiotensin-converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase-mediated inflammation. Hypertension. 2015;65(3):577–86.
Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RA, et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51(5):1312–7.
Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187(6):648–57.
Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension. 2014;63(4):774–82.
Farhadian S, Glick LR, Vogels CBF, Thomas J, Chiarella J, Casanovas-Massana A, et al. Acute encephalopathy with elevated CSF inflammatory markers as the initial presentation of COVID-19. Res Sq. 2020.
Eden A, Kanberg N, Gostner J, Fuchs D, Hagberg L, Andersson LM, et al. CSF biomarkers in patients with COVID-19 and neurologic symptoms: a case series. Neurology. 2021;96(2):e294–300.
Rustenhoven J, Drieu A, Mamuladze T, de Lima KA, Dykstra T, Wall M, et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell. 2021;184(4):1000-1016 e1027.
Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347(9009):1141–5.
Gouveia F, Camins A, Ettcheto M, Bicker J, Falcao A, Cruz MT, et al. Targeting brain Renin-Angiotensin System for the prevention and treatment of Alzheimer’s disease: Past, present and future. Ageing Res Rev. 2022;77: 101612.
Trigiani LJ, Royea J, Lacalle-Aurioles M, Tong XK, Hamel E. Pleiotropic benefits of the angiotensin receptor blocker candesartan in a mouse model of Alzheimer disease. Hypertension. 2018;72(5):1217–26.
Papadopoulos P, Tong XK, Imboden H, Hamel E. Losartan improves cerebrovascular function in a mouse model of Alzheimer’s disease with combined overproduction of amyloid-beta and transforming growth factor-beta1. J Cereb Blood Flow Metab. 2017;37(6):1959–70.
Ababei DC, Bild V, Ciobica A, Lefter RM, Rusu RN, Bild W. A Comparative study on the memory-enhancing actions of oral renin-angiotensin system altering drugs in scopolamine-treated mice. Am J Alzheimers Dis Other Demen. 2019;34(5):329–36.
Braszko JJ, Wincewicz D, Jakubow P. Candesartan prevents impairment of recall caused by repeated stress in rats. Psychopharmacology. 2013;225(2):421–8.
Nakagawa T, Hasegawa Y, Uekawa K, Senju S, Nakagata N, Matsui K, et al. Transient mild cerebral ischemia significantly deteriorated cognitive impairment in a mouse model of Alzheimer’s disease via angiotensin AT1 receptor. Am J Hypertens. 2017;30(2):141–50.
Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, et al. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117(11):3393–402.
Torika N, Asraf K, Cohen H, Fleisher-Berkovich S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav Immun. 2017;64:80–90.
Khalifa M, Safar MM, Abdelsalam RM, Zaki HF. Telmisartan protects against aluminum-induced Alzheimer-like pathological changes in rats. Neurotox Res. 2020;37(2):275–85.
Kurata T, Lukic V, Kozuki M, Wada D, Miyazaki K, Morimoto N, et al. Telmisartan reduces progressive accumulation of cellular amyloid beta and phosphorylated tau with inflammatory responses in aged spontaneously hypertensive stroke resistant rat. J Stroke Cerebrovasc Dis. 2014;23(10):2580–90.
Torika N, Asraf K, Roasso E, Danon A, Fleisher-Berkovich S. Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J Neuroimmune Pharmacol. 2016;11(4):774–85.
Wang Y, Li M, Kazis LE, Xia W. Clinical outcomes of COVID-19 infection among patients with Alzheimer’s disease or mild cognitive impairment. Alzheimers Dement. 2022;18(5):911–23.
Hsu JT, Tien CF, Yu GY, Shen S, Lee YH, Hsu PC, et al. The effects of Abeta1–42 binding to the SARS-CoV-2 spike protein S1 subunit and angiotensin-converting enzyme 2. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22158226.
Chiricosta L, Gugliandolo A, Mazzon E. SARS-CoV-2 Exacerbates beta-amyloid neurotoxicity, inflammation and oxidative stress in Alzheimer’s disease patients. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms222413603.
Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148–60.
Ramani A, Muller L, Ostermann PN, Gabriel E, Abida-Islam P, Muller-Schiffmann A, et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020;39(20): e106230.
Reiken S, Sittenfeld L, Dridi H, Liu Y, Liu X, Marks AR. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers Dement. 2022. https://doi.org/10.1002/alz.12558.
Acknowledgements
Not applicable.
Funding
This review was supported by the National Natural Science Foundation of China (81671181) and the Project funded by China Postdoctoral Science Foundation (2022M710848).
Author information
Authors and Affiliations
Contributions
F.C. and YT.C. drafted the manuscript. YX.W. designed the figures. QW.K. reviewed the manuscript and critically revised it for important intellectual content. LL.C. designed and reviewed the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, F., Chen, Y., Wang, Y. et al. The COVID-19 pandemic and Alzheimer’s disease: mutual risks and mechanisms. Transl Neurodegener 11, 40 (2022). https://doi.org/10.1186/s40035-022-00316-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-022-00316-y